

Abstract
Background
Here, the study aims to explore the effect of PM2.5 exposure on atherosclerosis in rats.
Materials and Methods
32 Wistar rats were selected in our study. An atherosclerosis model was established. All rats were evenly divided into four groups, including normal control group (NC), model control group (MC), model PM2.5 group (PM2.5) and model Atorvastatin group (Atorvastatin). The rats in NC and model control group were treated with saline 1 ml/kg body weight by tail intravenous injection, while the rats in PM2.5 group were exposed to PM2.5 suspension. The rats in atorvastatin group were given atorvastatin by gavage with 10 mg·kg-1·per day for 12 weeks until PM2.5 injection. After 24 h, all rats in each group were sacrificed. Pathological analysis, immunohistochemistry (IHC) and electrophoretic mobility shift assays (EMSA) were carried out.
Results
PM2.5 exposure significantly reduced the levels of triglyceride (TG), high density lipoprotein (HDL) and superoxide dismutase (SOD), but promoted the levels of total cholesterol (TC), low density lipoprotein (LDL), atherosclerosis index (AI), malondialdehyde (MDA), tumor necrosis factor-α (TNF-α) and high-sensitivity C-reactive protein (hs-CRP) in the rats of PM2.5 group than MC group (p < 0.05). PM2.5 group showed activated nuclear factor-kappa B (NF-κB), seriously damaged myocardial coronary branches and the highest nuclear translocation rate. Atorvastatin significantly improved the levels of TG, HDL, SOD, interleukin-6 (IL-6), and reduced the levels of TC, LDL, AI, MDA, TNF-α, hs-CRP, oxidized low-density lipoprotein (ox-LDL) and blood pressure, even the nuclear translocation rate.
Conclusions
PM2.5 exposure contributes to atherosclerosis in rats, which correlate with the levels of cholesterol, oxidative stress and inflammatory response. Atorvastatin could attenuate myocardial inflammation caused by PM2.5 exposure in rats.
Keywords: Atorvastatin, Coronary atherosclerosis, Inflammatory reaction, Oxidative stress, PM2.5
INTRODUCTION
As the largest developing country in the world, China is now facing a severe air pollution problem.1 Increasing epidemiologic evidences suggests that particulate air pollution is associated with an increased risk of human mortality and morbidity, especially that caused by cardiopulmonary diseases in the urban environment.2-5 Particulate matter (PM) with a mean aerodynamic diameter of < 10 um (PM10) has been associated with a range of effects on human health due to cardiovascular and respiratory illnesses.3 Increasing evidence has also shown that smaller particles may cause more severe adverse health effects, in particular fine particles such as PM2.5.2 PM2.5 infiltration efficiency is defined as the fraction of outdoor concentration that penetrates indoors and remains suspended, and it has been reported to vary between communities and homes.6
Although increasing epidemiological evidence indicates that ambient PM can promote cardiovascular injury and atherosclerosis, the mechanisms of the cardiovascular injury and proatherogenic effects are unclear. Atherosclerosis is a lifelong process involving a range of mechanisms including lipid peroxidation and inflammation affecting the vascular wall.7 In China, the incidence of the disease is increasing every year. In atherosclerosis, fatty streaks develop on the inner arterial wall and cause obstruction of blood flow.8 The progression of atherosclerotic plaque may be caused by uncontrolled cholesterol and lipid accumulation in macrophages and smooth muscle cells during atherosclerosis,9 and exposure to ambient air pollution may contribute to the acceleration of the development of atherosclerosis.7,10 For example, a prospective cohort study reported a close correlation between long-term residential exposure to high traffic levels of PM2.5 and coronary atherosclerosis.11
Epidemiological studies have also shown that even under concentrations below the air quality standards, an increasing concentration of PM2.5 is correlated with increases in the incidence of heart and lung disease and mortality, especially in individuals suffering from respiratory and cardiovascular diseases and the elderly.12,13 Daily mortality has also been reported to be significantly correlated with PM2.5.14,15 Short-term exposure to PM2.5 can cause a reduction in healthy heart rate variability, suggesting that PM2.5 is potentially toxic to the cardiovascular system.16 The incidence of acute myocardial infarction has also been positively correlated with ambient air pollution 2-24 hours before the onset.3 In China, PM2.5 reportedly caused 1.2 million premature deaths in 2010, accounting for almost 40% of the total global death.17
Numerous animal models have been established to study disease susceptibility to PM pollution, including models of lung-disease,18 hypertension,19 bronchitis,20 emphysema,21 and asthma,22 and they have been shown to be a useful research method to further explore the pathogenesis and risk factors of atherosclerosis. In the present study, a rat coronary atherosclerosis model was established. Rats were exposed to PM2.5 suspension through tail intravenous injections, and atorvastatin was then applied to explore the effect on atherosclerosis. Lipid metabolism, inflammatory cytokines, and enzyme activities of malondialdehyde (MDA) and superoxide dismutase (SOD) were analyzed. Histological examinations were carried out by analyzing coronary arteries. Immunohistochemistry and electrophoretic mobility shift assay (EMSA) were used to analyze nuclear transcription factor-kappa B (NF-κB) nuclear translocation rates.
MATERIALS AND METHODS
Subjects
Thirty-two healthy male Wistar rats weighing 180-220 g (Animals Certificate Number scxy 2015-001) were provided by the animal center of Shanxi Medical University. All of the rats were raised in a laboratory with a room temperature of 25 ± 2 °C and relative humidity of 50 ± 10%. The rats were fed with standard laboratory chow (wheat flour 30%, 15% soybean meal, corn 40%, wheat bran 8%, 5% fish meal, bone meal 1%, salt 1%) and water ad libitum. All of the animal procedures conformed to the National Institutes of Health (NIH) guidelines and were approved by the Local Ethics Committee of Shanxi Medical University Animal Center.
PM2.5 samples were provided by Dr. Wang of the National Institute of Environmental Health Sciences (The label #2783, NIST, US), and consisted of a PM2.5 suspension with a concentration of 40 mg/ml and used immediately when injection. Atorvastatin was purchased from Pfizer Company (Lot Number 65837008, New York, US). VD3 was purchased from Shanghai General Pharmaceutical Co., Ltd. (Lot Number 050506, Shanghai, China).
Grouping and establishment of the atherosclerosis model
All the rats were fed adaptively for 1 week, and then used to establish the atherosclerosis model. The rats were evenly divided into four groups, including a normal control group (NC), model control group (MC), model PM2.5 group (PM2.5) and model Atorvastatin group (Atorvastatin).
To establish the atherosclerosis model, all of the rats were raised for another 12 weeks. The NC group were fed with standard laboratory chow as before, and given total saline 2 m/kg body weight per day by intraperitoneal injection for the first three days. Twelve weeks later, the rats were given saline 1 ml/kg body weight by intravenous injection. The rats in the MC and PM2.5 groups were given new fodder based on the standard laboratory chow, containing 4% cholesterol, 10% lard, 5% sucrose, 0.5% sodium cholate, 0.2% propylthiouracil and 80.3% basal diet. The rats in both groups were treated with VD3 2 ml/kg (60,000 IU/kg) (Lot Number 050506, Shanghai General Pharmaceutical Co., Ltd., Shanghai, China) body weight per day by intraperitoneal injection for the first three days. Twelve weeks later, the rats in the MC group were given saline 1 ml/kg body weight by intravenous injection, and the rats in the PM2.5 group were given PM2.5 injections. As with the rats in the MC and PM2.5 groups, the rats in the Atorvastatin group were given new fodder and VD3 and also atorvastatin 10 mg/kg per day by gavage for 12 weeks until PM2.5 injection.
Exposure to PM2.5 was achieved through suspension injections in this study. Compared with aerosol inhalation, suspension injections are lossless and time-saving, and through this method PM2.5 can directly enter the blood. Thus, the effect of PM2.5 on atherosclerosis can be induced within a short time. In aerosol inhalation, the PM2.5 particles first enter pulmonary alveoli though the respiratory tract and then enter the blood. Some PM2.5 particles may remain on the pulmonary alveoli, thereby decreasing the concentration of PM2.5 in the blood making it difficult to quantify the amount of PM2.5 particles. Moreover, serious air pollution via respiratory tract inhalation in the real world may last for several days or even several weeks, increasing the potential for the destructive effects of PM2.5. The degree of PM2.5 exposure with suspension injections may be 100 times higher than in the real world, or dozens of times higher than aerosol inhalation, thereby allowing for the investigation of the effect of PM2.5 on atherosclerosis in a rat model.
Sampling and biochemical variables
Twenty-four hours after the PM2.5 injection, the rats in each group were anaesthetized by intraperitoneal injection of 25% urethane per 4 ml/kg body weight. Venous blood was collected from the orbital venous plexus of each rat. The rats were then placed in the supine position with the head and limbs fixedon the operating table, and cut from the middle of the abdomen. The abdominal aorta was separated and arterial blood was collected by a syringe. The heart was then exposed via thoracotomy, and the heart and aorta were quickly removed and washed in pre-chilled HEPES buffer. Portions of the heart and aorta were immediately frozen in liquid nitrogen, and the rest were fixed in 10% neutral formalin solution for further testing. After 30 minutes, the blood samples were centrifuged at 3000 rpm for 20 minutes. The supernatant was preserved at -20 °C for further testing.
Total cholesterol (TC), triglyceride (TG), high density lipoprotein (HDL), and low density lipoprotein (LDL) were measured using kits (BIOSINO Biotechnology Company, Beijing, China). MDA content was measured by TBA, and SOD content was measured using the xanthine oxidase method. Interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) were measured using an RIA Kit (Technology Development Center of PLA General Hospital, Beijing, China). High-sensitivity C-reactive protein (hs-CRP) and oxidized low-density lipoprotein (ox-LDL) were measured by enzyme linked immunosorbent assay (ELISA) (Dalian Fanbang Reagent Company, Dalian, China). Blood pressure (BP) was recorded using a multi-channel polygraph (CR Bard Inc., NJ, US), and atherosclerosis index (AI) was calculated using the formula: AI = TC – HDL / HDL
Histological examination and immunohistochemistry (IHC)
As described previously, the heart and aorta of the rats in each group were separated. Myocardial tissue blocks were cut into 5-um sections, fixed with formalin and embedded in paraffin. For pathological analysis, the paraffin sections were stained with hematoxylin and eosin (Jimei, Beijing, China) and observed under an ordinary optical microscope (OLYMPUS BX51, OLYMPUS, Tokyo, Japan). For IHC, samples were first incubated with NF-κB p65 monoclonal antibody (Santa Cruz, California, US), and then incubated with a biotinylated secondary antibody using a two-step Immunohistochemical Detection Kit (Zhongshan Golden Bridge Biotech Co., Ltd., Beijing, China). All stained sections were examined under a light microscope at 400 × magnification.
EMSA
NF-κB nucleoproteins were extracted from the myocardial tissue of the rats using a nuclear protein extraction kit (KEYGEN Biotech Co., Ltd., Nanjing, China). The concentration of nucleoprotein was measured using a Coomassie brilliant blue protein assay kit (Lot number 20070111, Jiancheng Institute of Biotechnology, Nanjing, China). The probes were labeled using annealing biotin-labeled oligonucleotides, and the oligonucleotides used for EMSA were amplified using the primers 5’-AGTTGAGGGGACTTTCCCAGGC-3’ and 5’-GCCTGGG AAAGTCCCCTCAACT-3’. Of the templates, 100 ng were 5’ end-labeled with T4 polynucleotide kinase (Fermentas, Hanover, MD, US) and [γ-32P]-ATP (GE Healthcare, Piscataway, PA, US). The labeled fragments were purified from the unincorporated nucleotides using Sephadex G-50 columns (GE Healthcare), and the final DNA concentration assuming 100% recovery was 22 nM in the stock solution. The labeled and purified DNA was then incubated for 30’ at 30 °C with different amounts of the protein in a BB buffer containing 12 mM HEPES, 12% glycerol, 100 mM Tris-HCl, pH 7.9, 0.5 mM NaCl, 10 mM DTT, 10 mM EDTA, 0.001 mg/ml dI/dC and 0.06 mg/ml BSA in a volume of 25 ml, where the final DNA concentration was 1 nM. Approximately 10 ug of purified NF-κB protein was used for each EMSA reaction. Activated NF-κB was quantitatively analyzed using a BI 2000 medical image analysis system (Thai Union Technology Co., Ltd., Chengdu, China).
Statistical analysis
Statistical analysis was conducted using SPSS 17.0. Data are expressed as means ± SD. Comparisons among groups were conducted using one-way analysis of variance (ANOVA) followed by the LSD test. A p value of < 0.05 or < 0.01 was taken to indicate a significant difference.
RESULTS
Biochemical variable analysis
Figure 1 shows that the levels of biochemical variables including TC, TG, LDL, AI, MDA, SOD, TNF-α, hs-CRP, ox-LDL and BP were significantly increased in the MC group compared to the NC group (p < 0.05), whereas the levels of HDL and IL-6 were significantly decreased in the MC groups compared to the NC group (p < 0.05). After PM2.5 injection, the levels of TG, HDL and SOD were significantly decreased, and the levels of TC, LDL, AI, MDA, TNF-α and hs-CRP were significantly increased in the PM2.5 group compared to the MC group (p < 0.05). In the atorvastatin group, atorvastatin significantly increased levels of TG, HDL, SOD and IL-6, and decreased levels of TC, LDL, AI, MDA, TNF-α, hs-CRP, ox-LDL and BP compared to the PM2.5 group (p < 0.05).
Figure 1.

Comparison of biochemical variables among four groups, including normal control group (NC), model control group (MC), model PM2.5 group (PM2.5) and model atorvastatin group (Atorvastatin). Error bars correspond to the standard deviation of the mean ± SD of three replicates. The letters above the bars indicate a statistically significant difference (p < 0.05) between groups with different letters and no difference between groups with the same letters.
Morphology analysis
Morphology analysis illustrated that the smooth muscle cells were spindle-shaped and arranged regularly, and that the lumen were regular and intact in the myocardial coronary branch of the NC group (Figure 2). In the MC group, the myocardial coronary branch showed missing and damaged endothelial cells, a large number of lipid droplets, thinned tunica media, and irregular smooth muscle cells and lumen. In addition, the lumen in the PM2.5 group was narrower and more irregular, and the inner membrane was damaged with missing endothelial cells. Furthermore, the inner elastic lamina was broken, the smooth muscle cells were arranged disorderly, and smooth muscle hyperplasia was obvious. In the Atorvastatin group, no fat cells were observed in the myocardial coronary branch, although the endothelial cells were damage and the internal elastic lamina structure was unclear to some extent.
Figure 2.
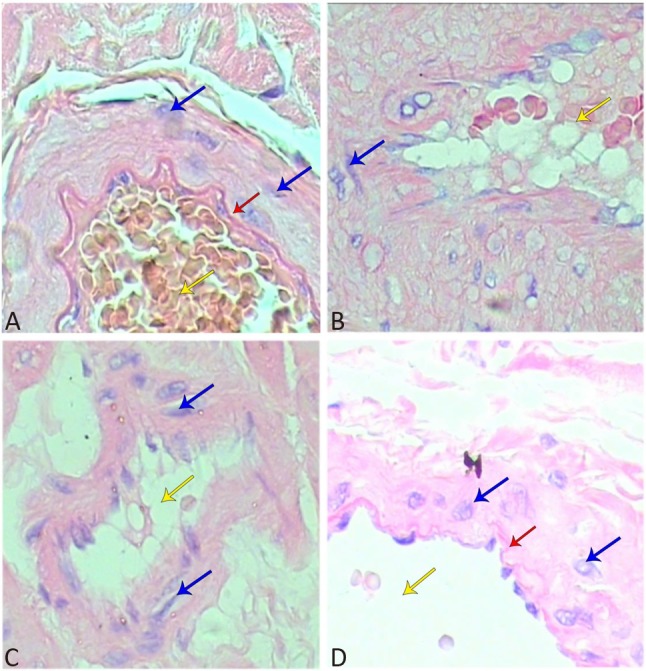
Histopathological analysis of coronary branch (400×). HE staining was carried out. (A) NC group, (B) MC group, (C) PM2.5 group, (D) Atorvastatin group. Red arrows indicate the elastic plate; yellow arrows indicate lumen, and blue arrows indicate smooth muscle cells.
NF-κB transcriptional activity analysis in the rats
IHC was performed to elucidate the NF-κB transcriptional activity in the rats (Figure 3). NF-κB was found to exist in the cytoplasm in an inactivated state under normal conditions, and blue stained nuclei and yellow particles were distributed in the cytoplasm in the NC group. When NF-κB is activated, it enters the nucleus from the cytoplasm (i.e. nuclear translocation). The nuclei of the NF-κB-positive cells showed light to dark brown on the basis of blue, and the number of yellow particles in the cytoplasm was obviously decreased in the PM2.5 group. The nuclear translocation rate was consistent with the IHC results. The PM2.5 group had the highest nuclear translocation rate. Compared to the NC group, the nuclear translocation rate was significantly higher in the rats with atherosclerosis (p < 0.05). Moreover, atorvastatin markedly reduced the nuclear translocation rate in the Atorvastatin group compared to the PM2.5 group (p < 0.05).
Figure 3.
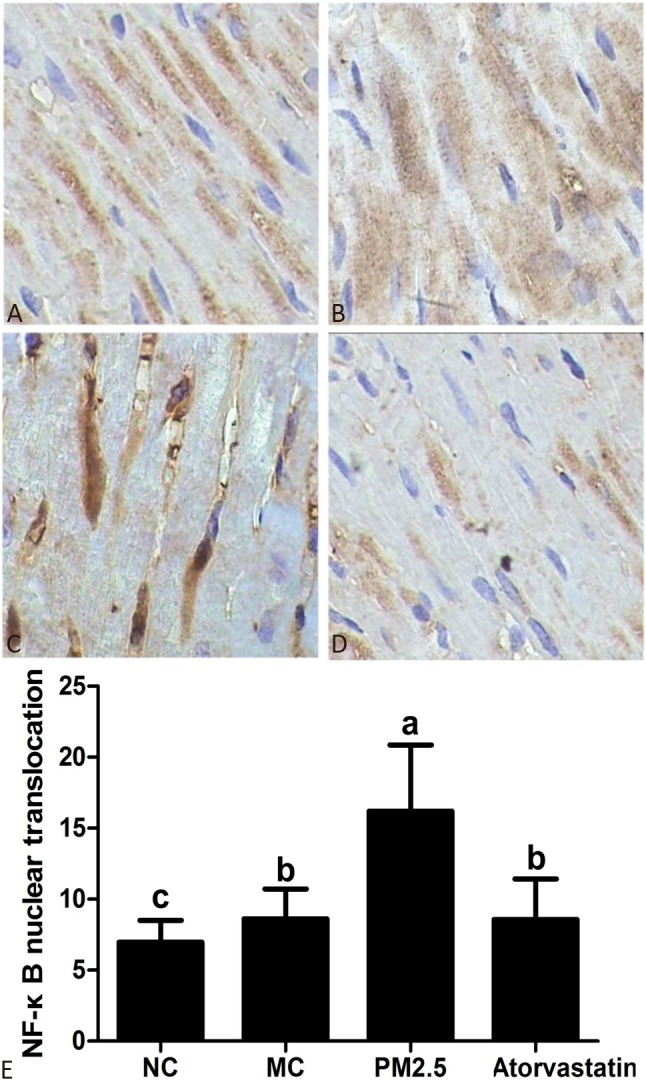
Immunohistochemistry analysis of NF-κB (400×). (A) NC group, (B) MC group, (C) PM2.5 group, (D) Atorvastatin group, (E) The rate of NF-κB nuclear translocation. The letters above the bars indicate a statistically significant difference (p < 0.05) between groups with different letters and no difference between groups with the same letters.
The EMSA results demonstrated that the PM2.5 group had the highest NF-κB concentration and that NC group had the lowest NF-κB concentration (Figure 4). NF-κB could be activated in this atherosclerosis model. Atorvastatin markedly reduced the NF-κB concentration in the Atorvastatin group compared to the PM2.5 group, which was consistent with IHC results.
Figure 4.
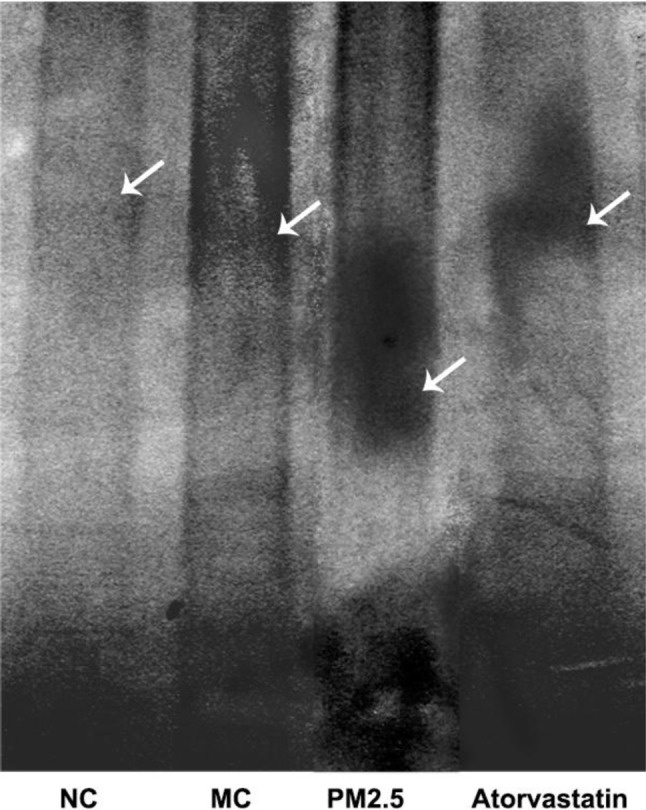
Electrophoretic Mobility Shift Assay (EMSA). White arrows indicate the locations of the bands.
DISCUSSION
PM2.5 is a mixture of various substances. The spatial and temporal variation in the chemical composition of PM2.5 has been widely studied, and has been shown to vary according to district and climate.1,6,23 Major contributors to PM2.5 mass include elemental carbon, organic carbon,24 sulfate (SO42-), nitrate (NO3-), and several metal ions such as iron, nickel, and zinc.25,26 Although epidemiological evidence has demonstrated an association between PM2.5 and cardiovascular morbidity, the mechanism between exposure to PM2.5 and atherosclerosis is still unclear. In the present study, we studied the effect of PM2.5 on atherosclerosis in rats exposed to PM2.5 suspension by tail intravenous injections. The related parameters of cholesterol, oxidative stress and inflammatory response were measured.
Hyperlipidemia has been shown to promote thrombosis through increasing superoxide anion (O2-) production.27 In acute coronary syndrome (ACS), LDL is oxidized to ox-LDL28 which has been associated with vulnerable plaque, which can both induce apoptosis of smooth muscle cells and also promote levels of cytokines such as IL-6 and TNF-α.29 Elevated levels of ox-LDL have been reported to be positively associated with the severity of ACS.30 HDL is an antioxidant that can inhibit the expression of adhesion molecules and protease.28 In the present study, the levels of TC, LDL and ox-LDL showed a similar pattern to the level of TNF-α but a contrasting pattern to the level of IL-6. Atherosclerosis significantly increased the levels of cholesterol in rats, such as TC, TG and LDL. In contrast, HDL significantly decreased the level of LDL in the MC group compared to the NC group. Meanwhile, PM2.5 exposure significantly reduced the level of HDL but promoted the accumulation of the LDL. In addition, the levels of ox-LDL were higher levels in the MC and PM2.5 groups compared to the NC group, which is consistent with a previous study.30 The AI was also highest in the PM2.5 group. Interestingly, morphology analysis illustrated that the myocardium of the rats in the MC group showed damaged endothelial cells, a large number of lipid droplets, thinned tunica media, irregular smooth muscle cells and lumen, and that PM2.5 exposure exacerbated the severity of damage to the myocardium. Our findings provide evidence that PM2.5 exposure can affect cholesterol levels and thereby regulate atherosclerosis.31
IL-6 and TNF-α are known to promote atherosclerosis disease, which is also closely correlated to an increased risk of coronary heart disease.32,33 TNF-α can induce endothelial cell procoagulant activity, increase its permeability, promote plaque formation and the expression of metal matrix proteinase in monocytes and macrophages, and activate T lymphocytes, all of which contribute to atherosclerosis.34-36 As a sensitive marker of nonspecific inflammatory responses, hs-CRP can be promoted during infection and inflammatory diseases.37 In the current study, levels of hs-CRP showed a similar pattern to TNF-α, but a contrasting pattern to IL-6 and HDL, which is consistent with previous studies. Some studies have reported that hs-CRP is negatively correlated with increased levels of HDL during the acute phase of atherosclerosis.38 A positive linear correlation has also been reported between peak levels of plasma IL-6 and hs-CRP in patients with acute myocardial infarction.39 Inflammatory cytokines have been reported to stimulate hs-CRP production by human coronary artery smooth muscle cells.40 In the current study, PM2.5 exposure significantly increased levels of TNF-α and hs-CRP and reduced levels of IL-6 in the PM2.5 group compared to the MC group (p < 0.05). The severity of damage to the myocardium was also correlated with the levels of inflammatory cytokines. A previous study suggested that the PM2.5-mediated enhancement of atherosclerosis is likely due to its pro-oxidant and pro-inflammatory effects, involving multiple organs, different cell types, and various molecular mediators.41 Our results demonstrated that PM2.5 exposure may affect inflammatory cytokines and thereby regulate atherosclerosis.
NF-κB has been shown to be a vital transcription factor involved in inflammatory responses and oxidative stress in atherosclerosis.42 PM2.5 exposure can activate NF-κB and inducethe expression of cytokines such as IL-6 and IL-8.43 In the current study, NF-κB was activated in our atherosclerosis model, and the nuclear translocation rate was highest in the PM2.5 group, which was consistent with the pattern of TC, LDL, ox-LDL, AI, TNF-α and hs-CRP. Moreover, the levels of MDA and SOD were significantly promoted by PM2.5 exposure. Morphology analysis and EMSA both showed damage to the myocardial coronary branch in the PM2.5 group. Epidemiological studies, animal experiments and cellular data support the association between air pollutants and systemic oxidative stress, inflammation, and atherosclerosis.44 Further, our findings illustrated that PM2.5 exposure may affect oxidative stress and the regulation of atherosclerosis.41,44
Atorvastatin has been widely used to regulate cholesterol.45 Our findings demonstrated that atorvastatin significantly reduced levels of TC, LDL and ox-LDL, and promoted levels of TG and HDL. In addition, BP was significantly decreased after atorvastatin treatment. Moreover, the levels of TNF-α and hs-CRP, the concentration of MDA and NF-κB nuclear translocation rate were also significantly decreased in the rats in the Atorvastatin group. These results demonstrated consistencies between the histological, IHC and EMSA results, and that atorvastatin could effectively reduce inflammatory responses and oxidative stress.
CONCLUSIONS
PM2.5 exposure has potential cardiac toxicity, which may induce the occurrence and aggravate the severity of atherosclerosis. PM2.5 exposure may activate NF-κB, alter the levels of cholesterol, oxidative stress and inflammatory responses in the regulation of atherosclerosis in rats. Atorvastatin could effectively attenuate the myocardial inflammation induced by PM2.5 in the rats in this study.
Acknowledgments
PM2.5 sample was kindly provided by Prof. De-Suo Wang, National Institute of Environmental Health Sciences, Carolina state, US. We also thank Dr. Wang for his help during the process of our experiment.
FUNDING
This research was supported by a grant from National Natural Science Foundation (No. 81070225) and Dr. Foundation of Shanxi Medical University (No. 03200819).
DECLARATION OF INTEREST
The authors report no conflicts of interest.
ETHICAL APPROVAL
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
REFERENCES
- 1.Kan H, Chen R, Tong S. Ambient air pollution, climate change, and population health in China. Environ Int. 2012;42:10–19. doi: 10.1016/j.envint.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Araujo JA, Barajas B, Kleinman M, et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102:589–596. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peters A, Dockery DW, Muller JE, et al. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103:2810–2815. doi: 10.1161/01.cir.103.23.2810. [DOI] [PubMed] [Google Scholar]
- 4.Pope CA, III, Burnett RT, Thun MJ, et al. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA. 2002;287:1132–1141. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pope CA, Burnett RT, Thurston GD, et al. Cardiovascular mortality and long-term exposure to particulate air pollution epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Zhao B. Review of relationship between indoor and outdoor particles: I/O ratio, infiltration factor and penetration factor. Atmos Environ. 2011;45:275–288. [Google Scholar]
- 7.Künzli N, Jerrett M, Garcia-Esteban R, et al. Ambient air pollution and the progression of atherosclerosis in adults. PloS One. 2010;5:e9096. doi: 10.1371/journal.pone.0009096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Ishibashi S, Perrey S, et al. Troglitazone inhibits atherosclerosis in apolipoprotein E–knockout mice pleiotropic effects on CD36 expression and HDL. Arterioscl Throm Va. 2001;21:372–377. doi: 10.1161/01.atv.21.3.372. [DOI] [PubMed] [Google Scholar]
- 9.Zingg JM, Ricciarelli R, Azzi A. Scavenger receptor regulation and atherosclerosis. Biofactors. 2000;11:189–200. doi: 10.1002/biof.5520110305. [DOI] [PubMed] [Google Scholar]
- 10.Künzli N, Jerrett M, Mack WJ, et al. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Pers. 2005:201–206. doi: 10.1289/ehp.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navab M, Hama SY, Hough GP, et al. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J Lipid Res. 2001;42:1308–1317. [PubMed] [Google Scholar]
- 12.Saldiva PH, Pope CA, III, Schwartz J, et al. Air pollution and mortality in elderly people: a time-series study in Sao Paulo, Brazil. Arch Environ Health. 1995;50:159–163. doi: 10.1080/00039896.1995.9940893. [DOI] [PubMed] [Google Scholar]
- 13.Ballester F, Tenias J, Perez-Hoyos S. Air pollution and emergency hospital admissions for cardiovascular diseases in Valencia, Spain. J Epidemiol Commun H. 2001;55:57–65. doi: 10.1136/jech.55.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz J, Dockery DW, Neas LM. Is daily mortality associated specifically with fine particles? J Air Waste Manage. 1996;46:927–939. [PubMed] [Google Scholar]
- 15.Klemm RJ, Mason RM, Jr., Heilig CM, et al. Is daily mortality associated specifically with fine particles? Data reconstruction and replication of analyses. J Air Waste Manage. 2000;50:1215–1222. doi: 10.1080/10473289.2000.10464149. [DOI] [PubMed] [Google Scholar]
- 16.Gold DR, Litonjua A, Schwartz J, et al. Ambient pollution and heart rate variability. Circulation. 2000;101:1267–1273. doi: 10.1161/01.cir.101.11.1267. [DOI] [PubMed] [Google Scholar]
- 17.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2013;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costa DL, Kodavanti UP. Toxic responses of the lung to inhaled pollutants: benefits and limitations of lung-disease models. Toxicol Lett. 2003;140:195–203. doi: 10.1016/s0378-4274(02)00515-5. [DOI] [PubMed] [Google Scholar]
- 19.Kodavanti UP, Schladweiler MC, Ledbetter AD, et al. The spontaneously hypertensive rat as a model of human cardiovascular disease: evidence of exacerbated cardiopulmonary injury and oxidative stress from inhaled emission particulate matter. Toxicol Appl Pharm. 2000;164:250–263. doi: 10.1006/taap.2000.8899. [DOI] [PubMed] [Google Scholar]
- 20.Kodavanti UP, Mebane R, Ledbetter A, et al. Variable pulmonary responses from exposure to concentrated ambient air particles in a rat model of bronchitis. Toxicol Sci. 2000;54:441–451. doi: 10.1093/toxsci/54.2.441. [DOI] [PubMed] [Google Scholar]
- 21.Sweeney T, Brain J, Leavitt S, et al. Emphysema alters the deposition pattern of inhaled particles in hamsters. Am J Pathol. 1987;128:19. [PMC free article] [PubMed] [Google Scholar]
- 22.Gavett SH, Madison SL, Stevens MA, et al. Residual oil fly ash amplifies allergic cytokines, airway responsiveness, and inflammation in mice. Am J Resp Crit Care. 1999;160:1897–1904. doi: 10.1164/ajrccm.160.6.9901053. [DOI] [PubMed] [Google Scholar]
- 23.Bell ML, Dominici F, Ebisu K, et al. Spatial and temporal variation in PM2.5 chemical composition in the United States for health effects studies. Environ Health Perspect. 2007:989–995. doi: 10.1289/ehp.9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostro B, Feng WY, Broadwin R, et al. The effects of components of fine particulate air pollution on mortality in California: results from CALFINE. Environ Health Perspect. 2007:13–19. doi: 10.1289/ehp.9281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fairley D. Daily mortality and air pollution in Santa Clara County, California: 1989-1996. Environ Health Perspect. 1999;107:637. doi: 10.1289/ehp.99107637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao J, Xu H, Xu Q, et al. Fine particulate matter constituents and cardiopulmonary mortality in a heavily polluted Chinese city. Environ Health Perspect. 2012;120:373. doi: 10.1289/ehp.1103671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eitzman DT, Westrick RJ, Xu Z, et al. Hyperlipidemia promotes thrombosis after injury to atherosclerotic vessels in apolipoprotein E–deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:1831–1834. doi: 10.1161/01.atv.20.7.1831. [DOI] [PubMed] [Google Scholar]
- 28.Forrester JS. Role of plaque rupture in acute coronary syndromes. Am J Cardiol. 2000;86:15–23. doi: 10.1016/s0002-9149(00)01195-4. [DOI] [PubMed] [Google Scholar]
- 29.Okura Y, Brink M, Itabe H, et al. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation. 2000;102:2680–2686. doi: 10.1161/01.cir.102.22.2680. [DOI] [PubMed] [Google Scholar]
- 30.Ehara S, Ueda M, Naruko T, et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation. 2001;103:1955–1960. doi: 10.1161/01.cir.103.15.1955. [DOI] [PubMed] [Google Scholar]
- 31.Rao X, Zhong J, Maiseyeu A, et al. CD36-dependent 7-ketocholesterol accumulation in macrophages mediates progression of atherosclerosis in response to chronic air pollution exposure. Circ Res. 2014;115:770–780. doi: 10.1161/CIRCRESAHA.115.304666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haddy N, Sass C, Droesch S, et al. IL-6, TNF-α and atherosclerosis risk indicators in a healthy family population: the Stanislas cohort. Atherosclerosis. 2003;170:277–283. doi: 10.1016/s0021-9150(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 33.Mazurek T, Zhang L, Zalewski A, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. 2003;108:2460–2466. doi: 10.1161/01.CIR.0000099542.57313.C5. [DOI] [PubMed] [Google Scholar]
- 34.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis the good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 35.Saren P, Welgus HG, Kovanen PT. TNF-alpha and IL-1 beta selectively induce expression of 92-kDa gelatinase by human macrophages. J Immunol. 1996;157:4159–4165. [PubMed] [Google Scholar]
- 36.Libby P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 37.Torzewski J, Torzewski M, Bowyer DE, et al. C-reactive protein frequently colocalizes with the terminal complement complex in the intima of early atherosclerotic lesions of human coronary arteries. Arterioscler Thromb Vasc Biol. 1998;18:1386–1392. doi: 10.1161/01.atv.18.9.1386. [DOI] [PubMed] [Google Scholar]
- 38.Grønholdt ML, Sillesen H, Wiebe B, et al. Increased acute phase reactants are associated with levels of lipoproteins and increased carotid plaque volume. Eur J Vasc Endovasc Surg. 2001;21:227–234. doi: 10.1053/ejvs.2001.1321. [DOI] [PubMed] [Google Scholar]
- 39.Miyao Y, Yasue H, Ogawa H, et al. Elevated plasma interleukin-6 levels in patients with acute myocardial infarction. Am Heart J. 1993;126:1299–1304. doi: 10.1016/0002-8703(93)90526-f. [DOI] [PubMed] [Google Scholar]
- 40.Calabró P, Willerson JT, Yeh ET. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation. 2003;108:1930–1932. doi: 10.1161/01.CIR.0000096055.62724.C5. [DOI] [PubMed] [Google Scholar]
- 41.Bai Y, Sun Q. Fine particulate matter air pollution and atherosclerosis: mechanistic insights. Biochim Biophys Acta. 2016;1860:2863–2868. doi: 10.1016/j.bbagen.2016.04.030. [DOI] [PubMed] [Google Scholar]
- 42.Pueyo ME, Gonzalez W, Nicoletti A, et al. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-κB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–651. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 43.Dye JA, Adler KB, Richards JH, et al. Role of soluble metals in oil fly ash-induced airway epithelial injury and cytokine gene expression. Am J Physiol-Lung C. 1999;277:L498–L510. doi: 10.1152/ajplung.1999.277.3.L498. [DOI] [PubMed] [Google Scholar]
- 44.Araujo JA. Particulate air pollution, systemic oxidative stress, inflammation, and atherosclerosis. Air Qual Atmos Hlth. 2011;4:79–93. doi: 10.1007/s11869-010-0101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pedersen T. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Atheroscler Suppl. 2004;5:81–87. doi: 10.1016/j.atherosclerosissup.2004.08.027. [DOI] [PubMed] [Google Scholar]