

Abstract
Background
KCa3.1 ion channels play an important role during atherosclerosis. We aimed to investigate the effect of exenatide on KCa3.1 expression in aortic vascular smooth muscle cells (VSMCs) of diabetic rats.
Methods
Sprague-Dawley rats were randomly divided into normal control (NC), diabetes model (DM), and exenatide treatment (ET) groups. Hematoxylin and eosin and α-actin immunohistochemical staining were used to detect changes in rat aortic vascular smooth muscle. Quantitative RT-PCR and Western blot analysis were used to detect changes in KCa3.1 mRNA and protein levels, respectively.
Results
Aortic tissue staining in the DM group revealed an absence of smooth or integrated endothelium, increased smooth muscle cell proliferation in the media, smooth muscle hyperplasia, disorganized smooth muscle cells, and an increased number of collagen fibers, relative to the NC and ET groups. KCa3.1 mRNA expression was higher in the DM group than in the NC and ET groups. Similarly, the KCa3.1 protein level was higher in the DM group than in the NC and ET groups. The KCa3.1 protein level did not significantly differ between the ET and NC groups.
Conclusions
Exenatide could inhibit the expression of the KCa3.1 channel in VSMCs of diabetic rats.
Keywords: Atherosclerosis, Ion channel, Vascular smooth muscle cells
INTRODUCTION
Diabetes is a common chronic condition frequently complicated by vascular lesions. Diabetic vascular lesions can cause cardio-cerebral vascular events, and result in serious damage, since hyperglycemia can accelerate atherosclerosis.1 Vascular smooth muscle cells (VSMCs) are vital during atherosclerosis, and proliferation of VSMCs is an essential step in the pathogenesis of atherosclerosis.2
KCa3.1 calcium-activated potassium ion channels play an important role during atherosclerosis.3 The KCa3.1 ion channel regulates transcription factors and cell cycle proteins by controlling the intracellular calcium concentration and proliferation of VSMCs.4 The contractile phenotype of VSMCs changes to the proliferative phenotype during arteriosclerosis in humans and rats, and the KCa1.1 and KCa3.1 ion channels of VSMCs are downregulated and upregulated, respectively.5 Insulin and advanced glycosylation end products upregulate the KCa3.1 channel through their receptors, promote the proliferation and migration of VSMCs, and result in the development and progression of arteriosclerosis.6,7 Drug or gene knockdown interventions for KCa3.1 may slow the development of atherosclerosis.8,9
Drugs that can lower blood sugar and serum cholesterol levels are of clinical significance in diabetes treatment. The glucagon-like peptide-1 (GLP-1) receptor is present in arterial endothelial cells and arterial VSMCs.10,11 Active GLP-1 in the blood has been correlated with atherosclerotic plaques, as it has been found to stabilize such plaques.12 Exenatide is a GLP-1 analogue that has been widely used to treat hyperglycemia in type 2 diabetes patients, and it has been shown to protect the arterial endothelium directly by activating the adenosine monophosphate-activated protein kinase/phosphatidylinositol-3-kinase/protein kinase B/endothelial nitric oxide synthase (AMPK/PI3K/Akt/eNOS) pathway.13 Studies have shown that exenatide reduces proliferation of impaired artery intima14,15 and relieves cell aging induced by an angiotensin II receptor blocker.16 Exenatide has also been shown to affect macrophagocytes, exert an anti-inflammatory effect, attenuate proliferation of VSMCs, and delay atherosclerosis.17
VSMC proliferation results in upregulation of KCa3.1 during atherosclerosis; however, it remains to be determined if exenatide could prevent atherosclerosis formation by altering KCa3.1 expression. Here, we established a rat model of diabetes to observe the effect of exenatide on KCa3.1 expression in aortic VSMCs using pathology, immunohistochemistry, and molecular biology.
MATERIALS AND METHODS
Experimental animals
A total of 24 healthy, specific pathogen-free, 8-week-old Sprague-Dawley (SD) rats (average weight, 200-250 g) were purchased from the Laboratory Animal Center of the Fourth Military Medical University. All protocols were approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University.
Main equipment and reagents
Streptozotocin (STZ) (02100557.1; MP Biomedicals, Santa Ana, CA, USA), exenatide injection (AstraZeneca, London, UK), glucose testing kit (F006; Nanjing Jiancheng Bioengineering Institute, Nanjing, China), total cholesterol test kit (F002-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, China), KCa3.1 antibodies (60276-1-Ig; Proteintech, Wuhan, China), DAB reagent kit (SP-9000; ZSGB-BIO, Beijing, China), PrimeScript RT reagent kit with gDNA Eraser (Perfect Real Time; TaKaRa, Dalian, China), SYBR® Premix ExTaqTM II (Tli RNase H Plus; TaKaRa, Dalian, China), TRIZOLTM Reagent (Life Technologies, Carlsbad, CA, USA), Goat Anti-Rabbit IgG (Thermo Fisher Scientific, Rockford, IL, USA), Goat Anti-Mouse IgG (Thermo Fisher Scientific, Rockford, IL, USA), and α-actin antibodies (55135-1-AP; Proteintech, Wuhan, China).
SD rat diabetic model
Six 8-week-old SD rats were randomly classified into the normal control (NC) group and given standard chow. Eighteen 8-week-old SD rats were fed a high sugar and high fat diet for 8 weeks (feed formula: 10% lard, 20% sucrose, 2.5% cholesterol, 1% bile salts, and 66.5% normal feed)18 to initiate the development of the DM. At 16 weeks of age, the 18 diabetic rats were given intraperitoneal injections of STZ (30 mg/kg) dissolved in citric acid buffer (0.1 mol/L, pH 4.4) administered following a 12-hour fast. Rats in the NC group were injected with the same volume of citrate buffer at 16 weeks of age. Blood samples were extracted from the tail tips of all rats at 17 and 23 weeks of age, and used to test fasting blood glucose and total cholesterol levels. A total of 17 diabetic rats were successfully produced (fasting blood glucose > 11.1 mmol/L).
Grouping and drug administration
The diabetic rat model was successfully established, and then the rats were divided into two groups: nine rats were placed in the diabetes model (DM) group, and eight in the exenatide treatment (ET) group. Rats in both these groups were fed a high sugar and high fat diet for 4 weeks, followed by a normal diet for 2 weeks, while those in the NC group were fed a normal diet for 6 weeks. Six rats died during the experiment (three each from the DM and ET groups group). The rats in the ET group were administered 5 μg exenatide once daily via a subcutaneous injection into the abdomen. The animals in the DM and NC groups received the same volume of saline daily via subcutaneous injection into the abdomen.
Collection and storage of tissue and blood samples
All the rats were anesthetized with intraperitoneal injections of chloral hydrate (5%; 0.8 mL/100 g). Blood samples were extracted from the tail tip of rats in all three groups and used to test fasting blood glucose and total cholesterol levels at 23 weeks of age. The thoracic aortas were removed, weighed, snap-frozen in liquid nitrogen, and stored at -80 °C.
Staining of the thoracic aortas
After the rats had been anesthetized, the thoracic aortas were removed and fixed in 4% paraformaldehyde. They were then embedded in paraffin and sectioned continuously. Sections were stained with hematoxylin and eosin (H&E), Masson’s trichrome, or with labeled antibodies for the immunohistochemical identification of α-actin. Changes in the aortic structure were observed under a microscope and photographed.
Changes in the KCa3.1 mRNA expression in the thoracic aortas
Total RNA was extracted from arterial smooth muscles using TRIzol, and the mRNA levels were quantified by NanoDrop 2000c (Thermo Scientific, Wilmington, DE, USA). To confirm the quality of the RNA, RNA extract was subjected to electrophoresis (20 minutes, 1% agarose gel). The electrophoresis images were observed by gel imaging analysis system (Shanghai Peiqing Technology Co., Ltd., Shanghai, China) and clear 28S and 18S bands and a blurry 5S band appeared. The RNA was then used for reverse transcription (RT) to cDNA. Quantitative real-time RT-PCR was then performed in triplicate with a SYBR® Premix Ex Taq kit (25 μL reaction volume; TaKaRa, Dalian, China). The reaction included initial denaturation at 95 °C (30 seconds), followed by 40 cycles of denaturation at 95 °C (5 seconds), annealing at 56 °C (30 seconds), and extension at 60 °C (40 seconds). Absolute gene transcription was normalized to the level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Table 1 lists the primer sequences used for each gene. The relative fold change of each group was calculated using the 2–ΔΔCT method.
Table 1. Oligonucleotide sequences of primers used for RT-PCR.
| Gene | Accession No | Forward primer | Reverse primer |
| H-GAPDH | NM_017008 | CCATGTTCGTCATGGGTGTGAACCA | GCCAGTAGAGGCAGGGATGATGTTC |
| KCNN4 | NM_023021 | AACTGGCATCGGACTCAT | AGACAAAGGAGGAAGGCAG |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KCNN4, gene encoding KCa3.1 protein.
Change in KCa3.1 protein expression in the thoracic aortas
Smooth muscle cells of each group were lysed with RIPA lysis buffer and used for total protein extraction. The protein extract was then quantified. Protein (25 μg) was separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% (w/v) skimmed milk powder in Tris-buffered saline with Tween for 1 hour, incubated with primary antibodies to KCa3.1 (1:300 dilution) for 2 hours at room temperature, and then overnight at 4 °C. After washing the membranes, they were incubated with secondary antibodies (1:10,000 dilution; horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG) for 1 hour at room temperature. Signals were detected using enhanced chemiluminescence.
Statistical analysis
Data were analyzed using SPSS v19.0 software. Data were presented as mean ± standard deviation. Normally distributed data were analyzed using one-way analysis of variance (ANOVA). When variance was uneven, data were analyzed using one-way ANOVA by reciprocal transformation. Comparisons among groups were performed using Fisher’s least-significant difference test. Differences were considered statistically significant at a p value < 0.05.
RESULTS
Characteristics of the SD diabetic rat model
The diabetic SD rats exhibited significant weight loss, increased blood sugar levels, and blood lipid levels. Table 2 presents the body weight at 16, 17, and 23 weeks of age, and the blood sugar (glucose) and the blood lipid (cholesterol) levels of the three groups at 17 and 23 weeks of age. The results revealed that the average weight did not significantly differ among the three groups at 16 weeks of age. However, at 17 weeks, the average weight of the NC group was significantly higher compared to the DM and ET groups (p < 0.01). The average body weight did not significantly differ between the ET and DM groups at 17 weeks of age (p > 0.05), however it was significantly higher in the NC group compared to the DM and ET groups at 23 weeks of age (p < 0.01). The average body weight did not significantly differ between the ET and DM groups at 23 weeks of age (p > 0.05).
Table 2. Weight and biochemical characteristics of Sprague-Dawley rats.
| Group | Weight of rat (g) | Glucose (mmol/L) | Cholesterol (mmol/L) | ||||
| 16 w | 17 w | 23 w | 17 w | 23 w | 17 w | 23 w | |
| Normal control (NC) | 572.11 ± 60.84 (n = 6) | 575.38 ± 65.85 (n = 6) | 606.06 ± 64.90 (n = 6) | 6.34 ± 0.35 (n = 6) | 6.68 ± 0.31 (n = 6) | 1.26 ± 0.03 (n = 6) | 1.32 ± 0.05 (n = 6) |
| Diabetes model (DM) | 507.34 ± 57.78 (n = 9) | 462.79 ± 62.38* (n = 9) | 422.79 ± 45.37* (n = 6) | 19.10 ± 3.82* (n = 9) | 21.81 ± 2.66* (n = 6) | 1.92 ± 0.06* (n = 9) | 1.96 ± 0.07* (n = 6) |
| Exenatide treatment (ET) | 504.56 ± 77.34 (n = 9) | 451.10 ± 73.62* (n = 8) | 468.66 ± 70.05* (n = 6) | 18.75 ± 3.09* (n = 8) | 9.90 ± 0.55*# (n = 6) | 1.95 ± 0.07* (n = 8) | 1.61 ± 0.05*# (n = 6) |
* Indicates p < 0.01 when compared to the normal control (NC). # Indicates p < 0.01 when compared to the diabetes model (DM). Data are expressed as mean ± standard deviation (SD). w, weeks of age.
The blood glucose levels of the DM and ET rats met the diagnostic criterion for diabetes. The average blood glucose levels of the DM and ET groups were significantly higher than the NC group at 17 weeks of age. Importantly, the average blood glucose level of the ET group was significantly lower than that of the DM group, but still higher than that of the NC group at 23 weeks of age (p < 0.01). Similarly, the average cholesterol level was significantly higher in the ET and DM groups compared to the NC groups, and there was no significant difference between the ET and DM groups at 17 weeks of age. At 23 weeks of age, the average cholesterol was the highest in the DM group and lowest in the NC group (p < 0.01; Table 2).
Microscopic structure of rat thoracic aortas
Rat thoracic aorta tissue was examined through staining and histological analyses to identify potential vascular lesions. The results of the H&E staining showed intact and smooth intima, intact media and adventitia, and no obvious proliferation of smooth muscle cells in the media of the NC group (Figure 1). In the DM group, the intima was not intact or smooth, and the smooth muscles in the media showed obvious proliferation. In the ET group, some smooth muscle proliferation was visible.
Figure 1.
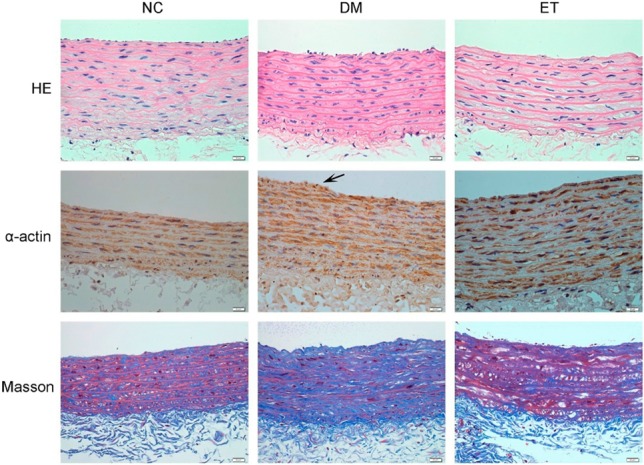
Representative photomicrographs of rat aorta (Original magnification: ×400). DM, diabetes model; ET, exenatide treatment; HE, hematoxylin and eosinstaining; Masson, Masson’s trichrome staining; NC, normal control; α-actin, immunohistochemistry for alpha-actin.
The immunohistochemical staining for α-smooth muscle actin revealed well-distributed smooth muscle and intact intima in the NC group. In the DM group, smooth muscle cells formed intima hyperplasia that was relatively dense in some areas, and parts of the vascular walls protruded into the artery lumen. Aggregates of smooth muscle under the intima were observed in the ET group.
As shown in Figure 1, following Masson’s trichrome staining, the collagen fibers were stained blue, the muscle fibers were stained red, and the nuclei were stained bluish-purple. The results of the staining revealed that the media of the NC group had regular smooth muscle fibers, and the tissues of the DM group had an obvious increase in collagen fibers, derangement of collagen fibers and smooth muscles, and a lack of a smooth and intact intima. In addition, an increase in collagen fibers was observed in the ET group.
Changes in the KCa3.1 mRNA level in thoracic aorta smooth muscles
Fluorescent quantitative RT-PCR results indicated that the average KCa3.1 mRNA level was higher in the DM group compared to the NC and ET groups (p < 0.01; Figure 2). There was no significant difference in average KCa3.1 mRNA level between the NC and ET groups (p > 0.05; Figure 2).
Figure 2.
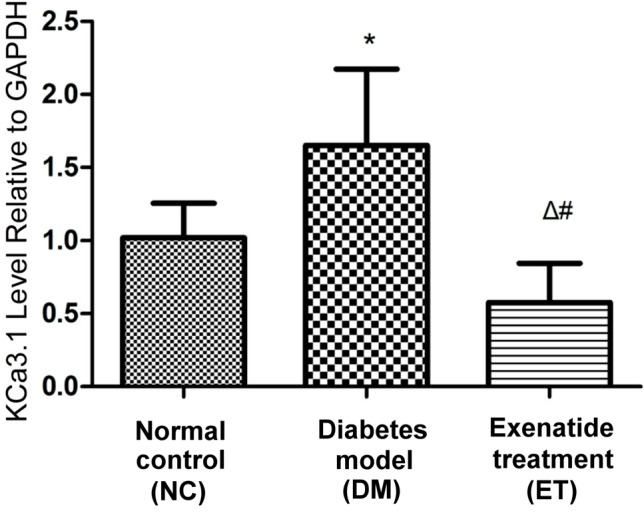
KCa3.1 channel mRNA expression in smooth muscles of rat aorta detected by quantitative RT-PCR. Statistics: * p < 0.01, vs. normal control group; # p < 0.01, vs. diabetes model group; Δ p > 0.05, vs. normal control group.
Changes in the KCa3.1 protein level in thoracic aorta smooth muscles
The KCa3.1 protein level in the thoracic aorta smooth muscles in the diabetic rats was observed by Western blot analysis, which showed that the average KCa3.1 protein level was significantly different among the three groups (p < 0.01; Figure 3). The average KCa3.1 protein level was significantly higher in the DM group than in the NC and ET groups, but it did not significantly differ between the ET and NC groups (p > 0.05; Figure 3).
Figure 3.
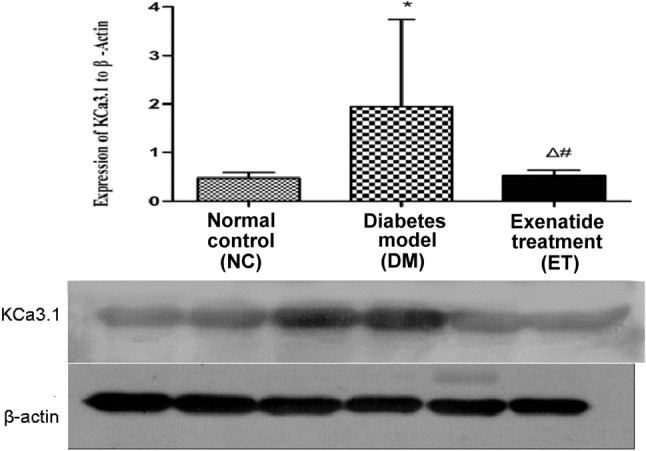
KCa3.1 channel protein expression in the smooth muscle of rat aorta detected by western blotting. Statistics: * p < 0.01, vs. normal control group; # p < 0.01, vs. diabetes model group; Δ p > 0.05, vs. normal control group.
DISCUSSION
Diabetes is one of the major causes of atherosclerosis and accelerated atherosclerosis.19 Smooth muscle proliferation is an essential step in atherosclerosis, and it has been closely correlated with KCa3.1.20 The KCa3.1 level in smooth muscle cells has been found to be significantly elevated during atherosclerosis.21 In addition, previous studies have reported that treatment of aortic smooth muscle cells in normal rats with serum from diabetic rats resulted in upregulation of KCa3.1 expression in these cells.5,22 In the present study, we also demonstrated that KCa3.1 expression was upregulated in the aortic smooth muscle cells of diabetic rats.
The GLP-1 receptor is present in arterial smooth muscle cells. Exenatide can affect arterial smooth muscle cells by inhibiting the proliferation of smooth muscle cells, and inhibiting vascular remodeling by dynamically regulating mitochondria.23 Exenatide can also prevent endothelial vessel dysfunction by opening the K-ATP channel, illustrating that exenatide can regulate the ion channels of cells.24 In the present study, we showed that exenatide can decrease KCa3.1 mRNA and protein expression levels in aortic smooth muscles in diabetic rats.
After establishing DM at 17 weeks of age, the weight of the rats decreased and the blood glucose levels met the criteria for diabetes. These features are similar to dia-betic models used in previous studies.25,26 At 23 weeks of age, the average blood glucose and serum cholesterol levels were lower in the ET group compared to the DM group (p < 0.01). These findings indicate that exenatide could effectively lower blood glucose and serum cholesterol levels.
Micrographs of aortic smooth muscles of the DM and ET groups revealed vascular smooth muscle proliferation of the artery wall, which is a feature of early-stage atherosclerosis. These micrographs suggested that the thoracic aortas of the rats could develop atherosclerosis under hyperglycemia and high blood lipid levels, which is consistent with other reports.22 At 23 weeks of age, the average blood glucose and serum cholesterol levels were lower in the ET group than in the DM group. Moreover, the average KCa3.1 mRNA and protein levels in the 23-week-old rats were lower in the ET group compared to the DM group, and the average blood glucose and serum cholesterol levels in the 23-week-old rats were higher in the ET group than in the NC group. High blood glucose can increase the KCa3.1 expression in VSMCs, and if exenatide did not participate in inhibiting KCa3.1 in VSMCs, the level of KCa3.1 in VSMCs would have been higher in the ET group than in the NC group. However, the KCa3.1 mRNA and protein levels did not significantly differ between the ET and NC groups, suggesting that exenatide itself participated in inhibiting KCa3.1 ex-pression of the aortic VSMCs in the diabetic rats.
Atherosclerosis is the result of long-term inflammatory proliferative vascular lesions. It is associated with endothelial cells, VSMCs, fibroblasts, macrophages, T cells, B cells, and platelets. KCa3.1 is expressed in these cells during atherosclerosis, and it has been shown to participate in VSMC proliferation, macrophage migration, fibroblast proliferation, and aggregation of T cells and B cells.27-32 KCa3.1 also plays a role in the migration and transformation of VSMCs in the process of atherosclerosis. High blood glucose promotes the upregulation of KCa3.1, and KCa3.1 upregulation is essential in the pathogenesis of atherosclerosis.
Many studies have shown that GLP-1 receptor agonists affect the endothelium, macrophages, and smooth muscle cells33-35 due to their anti-atherosclerosis effect. A previous study suggested that GLP-1 receptor agonists could attenuate high blood glucose levels that induced the proliferation and migration of smooth muscle cells by inhibiting the extracellular signal-regulated kinases 1 and 2 (ERK1/2) and phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) signaling pathways.36 In addition, GLP-1 receptor agonists have been reported to decrease the intima media thickness of the carotid artery in patients with type 2 diabetes.37 Another study also showed that exenatide improved coronary endothelial function in human umbilical vein endothelial cells, and that exenatide increased nitric oxide (NO) production and eNOS activation via the glucagon-like peptide-1 receptor/cyclic adenosine monophosphate (GLP-1R/cAMP) signaling pathways. Furthermore, exenatide has been shown to promote endothelial nitricoxide synthase (eNOS) activation in the adenosine monophosphate-activated protein kinase (AMPK) and PI3K/Akt signaling pathways.13 eNOS is a major enzyme that helps control the rate of NO biosynthesis in vascular endothelial cells, when increased in vascular endothelial cells, NO can inhibit the proliferation and migration of VSMCs.38,39 Therefore, exenatide could indirectly inhibit VSMC proliferation and migration. These effects of exenatide depend on GLP-1R, and they can thus be eliminated by using a GLP-1R antagonist.13 Proliferation and migration of VSMCs has been found to be increased in atherosclerosis, and the proliferation of smooth muscle cells is associated with KCa3.1 upregulation at the transcriptional level. VSMCs switch from the contractile phenotype to the proliferative phenotype, and KCa3.1 expression is rapidly upregulated in atherosclerosis.31 Exenatide can inhibit the proliferation and migration of VSMCs, resulting in the downregulation of KCa3.1. Angiotensin II (Ang II) can also induce the proliferation and migration of VSMCs.40 A previous study showed that ERK1/2 and c-Jun N-terminal kinase (JNK) played important roles in Ang II-induced rat aortic smooth muscle cell (RASMC) proliferation and migration, where Ang II caused a phenotypic switch of RASMCs from the contractile type to the synthetic proliferative type, which is a key step in atherosclerosis. Furthermore, exenatide inhibited Ang II-induced phosphorylation of ERK1/2 and JNK, which prevented Ang II-induced RASMC proliferation and migration.12 These findings suggest that exenatide, which activates GLP-1R in the endothelium and in VSMCs, can prevent the proliferation and migration of VSMCs via different cellar pathways, resulting in the downregulation of KCa3.1.
CONCLUSIONS
In summary, the GLP-1 receptor agonist exenatide inhibited smooth muscle proliferation and migration by activating GLP-1R via different cellular pathways, resulting in inhibition of KCa3.1 upregulation in aortic vascular smooth muscles of diabetic rats.
Acknowledgments
The study was supported by the project of youth fund of the second affiliated hospital of Xi’an Jiaotong University (NO.YJ(QN)201413). Part of the exenatide injections were gifted by MS Miao Li (AstraZeneca Corporation).
CONFLICT OF INTEREST
There were no conflicts of interest in this study.
REFERENCES
- 1.Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–585. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39:86–93. [PubMed] [Google Scholar]
- 3.Jackson WF. Potassium channels and proliferation of vascular smooth muscle cells. Circ Res. 2005;97:1211–1212. doi: 10.1161/01.RES.0000196742.65848.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bi D, Lemaitre V, Takai J, et al. The intermediate-conductance Ca2+-activated K+ channel KCa3.1 regulates proliferation of human coronary smooth muscle cells. Circulation. 2012;126(Suppl):A16586. [Google Scholar]
- 5.Su XL, Zhang H, Yu W, et al. Role of KCa3.1 channels in proliferation and migration of vascular smooth muscle cells by diabetic rat serum. Chin J Physiol. 2013;56:155–162. doi: 10.4077/CJP.2013.BAB104. [DOI] [PubMed] [Google Scholar]
- 6.Su X, Wang Y, Zhang W, et al. Insulin-mediated upregulation of K3.1 channels promotes cell migration and proliferation in rat vascular smooth muscle. J Mol Cell Cardiol. 2011;51:51–57. doi: 10.1016/j.yjmcc.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Zhao LM, Su XL, Wang Y, et al. KCa3.1 channels mediate the increase of cell migration and proliferation by advanced glycation end products in cultured rat vascular smooth muscle cells. Lab Invest. 2012;93:159–167. doi: 10.1038/labinvest.2012.163. [DOI] [PubMed] [Google Scholar]
- 8.Toyama K, Wulff H, Chandy KG, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025–3037. doi: 10.1172/JCI30836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tharp DL, Wamhoff BR, Wulff H, et al. Local delivery of the KCa3.1 blocker, TRAM-34, prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis. Arterioscler Thromb Vasc Biol. 2008;28:1084–1089. doi: 10.1161/ATVBAHA.107.155796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaspari T, Liu H, Welungoda I, et al. Dear AE. A GLP-1 receptor agonist liraglutide inhibits endothelial cell dysfunction and vascular adhesion molecule expression in an ApoE-/- mouse model. Diab Vasc Dis Res. 2011;8:117–124. doi: 10.1177/1479164111404257. [DOI] [PubMed] [Google Scholar]
- 11.Koska J, Sands M, Burciu C, et al. Exenatide protects against glucose and lipid-induced endothelial dysfunction: evidence for direct vasodilation effect of GLP-1 receptor agonists in humans. Diabetes. 2015;64:2624–2635. doi: 10.2337/db14-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akiyama E, Sugiyama S, Matsubara J, et al. Decreased plasma levels of active glucagon-like peptide-1 in coronary artery disease. J Am Coll Cardiol. 2015;65:754–755. doi: 10.1016/j.jacc.2014.11.043. [DOI] [PubMed] [Google Scholar]
- 13.Wei R, Ma S, Wang C, et al. Exenatide exerts direct protective effects on endothelial cells through AMPK/Akt/eNOS pathway in a GLP-1 receptor dependent manner. Am J Physiol Endocrinol Metab. 2016;10:E947–E957. doi: 10.1152/ajpendo.00400.2015. [DOI] [PubMed] [Google Scholar]
- 14.Hirata Y, Kurobe H, Nishio C, et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, attenuates neointimal hyperplasia after vascular injury. Eur J Pharmacol. 2013;699:106–111. doi: 10.1016/j.ejphar.2012.11.057. [DOI] [PubMed] [Google Scholar]
- 15.Goto H, Nomiyama T, Mita T, et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces intimal thickening after vascular injury. Biochem Biophys Res Commun. 2011;405:79–84. doi: 10.1016/j.bbrc.2010.12.131. [DOI] [PubMed] [Google Scholar]
- 16.Zhao L, Li AQ, Zhou TF, et al. Exendin-4 alleviates angiotensin II-induced senescence in vascular smooth muscle cells by inhibiting Rac1 activation via a cAMP/PKA-dependent pathway. Am J Physiol Cell Physiol. 2014;307:1130–1141. doi: 10.1152/ajpcell.00151.2014. [DOI] [PubMed] [Google Scholar]
- 17.Bułdak Ł, Machnik G, Bułdak RJ, et al. Exenatide and metformin express their anti-inflammatory effects on human monocytes/macrophages by the attenuation of MAPKs and NFκB signaling. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:1103–1115. doi: 10.1007/s00210-016-1277-8. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Zhang HT, Su XL, et al. Experimental diabetes mellitus down-regulates large-conductance Ca2+-activated K+ channels in cerebral artery smooth muscle and alters functional conductance. Curr Neurovasc Res. 2010;7:75–84. doi: 10.2174/156720210791184925. [DOI] [PubMed] [Google Scholar]
- 19.Gray SP, Di Marco E, Okabe J, et al. NADPH oxidase 1 plays a key role in diabetes mellitus–accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 20.Zhao LM, Su XL, Wang Y, et al. KCa3.1 channels mediate the increase of cell migration and proliferation by advanced glycation endproducts in cultured rat vascular smooth muscle cells. Lab Invest. 2013;93:159–167. doi: 10.1038/labinvest.2012.163. [DOI] [PubMed] [Google Scholar]
- 21.Tharp D, Bowles D. KCa3.1 inhibition decreases size and alters composition of atherosclerotic lesions induced by low, oscillatory flow. FASEB J. 2015;29(Suppl):803.8. doi: 10.2991/artres.k.210202.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Zhao L, Xingli SU, et al. Ca~(2+)-activated K~+ channel switching in smooth muscle participates in atherosclerosis development in diabetic rats. Nan Fang Yi Ke Da Xue Bao. 2014;34:188–192. [PubMed] [Google Scholar]
- 23.Torres G, Morales PE, García-Miguel M, et al. Glucagon-like peptide-1 inhibits vascular smooth muscle cell dedifferentiation through mitochondrial dynamics regulation. Biochem Pharmacol. 2016;104:52–61. doi: 10.1016/j.bcp.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ha SJ, Kim W, Woo JS, et al. Preventive effects of exenatide on endothelial dysfunction induced by ischemia-reperfusion injury via KATP channels. Arterioscler Thromb Vasc Biol. 2012;32:474–480. doi: 10.1161/ATVBAHA.110.222653. [DOI] [PubMed] [Google Scholar]
- 25.Reed MJ, Meszaros K, Entes LJ, et al. A new rat model of type 2 diabetes: the fat-fed, streptozotocin-treated rat. Metabolism. 2000;49:1390–1394. doi: 10.1053/meta.2000.17721. [DOI] [PubMed] [Google Scholar]
- 26.Srinivasan K, Viswanad B, Asrat L, et al. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52:313–320. doi: 10.1016/j.phrs.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 27.Gerlach AC, Gangopadhyay NN, Devor DC. Kinase-dependent regulation of the intermediate conductance, calcium-dependent potassium channel, hIK1. J Biol Chem. 2000;275:585–598. doi: 10.1074/jbc.275.1.585. [DOI] [PubMed] [Google Scholar]
- 28.Srivastava S, Li Z, Ko K, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol Cell. 2006;24:665–675. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Neylon CB, Lang RJ, Fu Y, et al. Molecular cloning, characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle relationship between KCa channel diversity and smooth muscle cell function. Circ Res. 1999;85:e33–e43. doi: 10.1161/01.res.85.9.e33. [DOI] [PubMed] [Google Scholar]
- 30.Chung I, Zelivyanskaya M, Gendelman HE. Mononuclear phagocyte biophysiology influences brain transendothelial and tissue migration: implication for HIV-1-associated dementia. J Neuroimmunol. 2002;122:40–54. doi: 10.1016/s0165-5728(01)00462-3. [DOI] [PubMed] [Google Scholar]
- 31.Tharp DL, Wamhoff BR, Turk JR, et al. Upregulation of intermediate-conductance Ca2+-activated K+ channel (IKCa1) mediates phenotypic modulation of coronary smooth muscle. Am J Physiol Heart Circ Physiol. 2006;291:H2493–H2503. doi: 10.1152/ajpheart.01254.2005. [DOI] [PubMed] [Google Scholar]
- 32.Wolfs JL, Wielders SJ, Comfurius P, et al. Reversible inhibition of the platelet procoagulant response through manipulation of the Gardos channel. Blood. 2006;108:2223–2228. doi: 10.1182/blood-2006-01-009613. [DOI] [PubMed] [Google Scholar]
- 33.Tashiro Y, Sato K, Watanabe T, et al. A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides. 2014;54:19–26. doi: 10.1016/j.peptides.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 34.Dai Y, Mehta JL, Chen M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa B activation. Cardiovasc Drugs Ther. 2013;27:371–380. doi: 10.1007/s10557-013-6463-z. [DOI] [PubMed] [Google Scholar]
- 35.Gaspari T, Welungoda I, Widdop RE, et al. The GLP-1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an ApoE-/- mouse model. Diab Vasc Dis Res. 2013;10:353–360. doi: 10.1177/1479164113481817. [DOI] [PubMed] [Google Scholar]
- 36.Shi L, Ji Y, Jiang X, et al. Liraglutide attenuates high glucose-induced abnormal cell migration, proliferation, and apoptosis of vascular smooth muscle cells by activating the GLP-1 receptor, and inhibiting ERK1/2 and PI3K/Akt signaling pathways. Cardiovasc Diabetol. 2015;14:18. doi: 10.1186/s12933-015-0177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rizzo M, Chandalia M, Patti AM, et al. Liraglutide decreases carotid intima-media thickness in patients with type 2 diabetes: 8-month prospective pilot study. Cardiovasc Diabetol. 2014;13:49. doi: 10.1186/1475-2840-13-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsihlis ND, Oustwani CS, Vavra AK, et al. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochem Biophys. 2011;60:89–97. doi: 10.1007/s12013-011-9179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarkar R, Meinberg EG, Stanley JC, et al. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res. 1996;78:225–230. doi: 10.1161/01.res.78.2.225. [DOI] [PubMed] [Google Scholar]
- 40.Berk BC, Haendeler J, Sottile J. Angiotensin II, atherosclerosis, and aortic aneurysms. J Clin Invest. 2000;105:1525–1526. doi: 10.1172/JCI9820. [DOI] [PMC free article] [PubMed] [Google Scholar]