

Abstract
Timely resolution of sister chromatid cohesion in G2/M is essential for genome integrity. Resolution at telomeres requires the poly(ADP‐ribose) polymerase tankyrase 1, but the mechanism that times its action is unknown. Here, we show that tankyrase 1 activity at telomeres is controlled by a ubiquitination/deubiquitination cycle depending on opposing ubiquitin ligase and deubiquitinase activities. In late S/G2 phase, the DNA damage‐responsive E3 ligase RNF8 conjugates K63‐linked ubiquitin chains to tankyrase 1, while in G1 phase such ubiquitin chains are removed by BRISC, an ABRO1/BRCC36‐containing deubiquitinase complex. We show that K63‐linked ubiquitin chains accumulate on tankyrase 1 in late S/G2 to promote its stabilization, association with telomeres, and resolution of cohesion. Timing of this posttranslational modification coincides with the ATM‐mediated DNA damage response that occurs on functional telomeres following replication in G2. Removal of ubiquitin chains is controlled by ABRO1/BRCC36 and occurs as cells exit mitosis and enter G1, ensuring that telomere cohesion is not resolved prematurely in S phase. Our studies suggest that a cell cycle‐regulated posttranslational mechanism couples resolution of telomere cohesion with completion of telomere replication to ensure genome integrity.
Keywords: cohesion, poly(ADP‐ribose), RNF8, tankyrase, telomere
Subject Categories: DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Mammalian chromosome ends are protected from eliciting a DNA damage response through the actions of shelterin, the multisubunit complex that coats the telomeric TTAGGG repeats (Palm & de Lange, 2008). Shelterin promotes formation of a protective t‐loop structure, where the single‐stranded G‐overhang is buried (and thereby protected) in the double‐stranded TTAGGG repeats (Griffith et al, 1999). Replication of telomeric DNA is a multistep process that involves passage of the replication fork, nuclease C‐strand digestion, G‐strand elongation by telomerase, and C‐strand fill‐in by Polα/primase (Martinez & Blasco, 2015). The DNA damage response machinery plays a central role ensuring that the protective structure is maintained during (and regenerated after) telomere replication (O'Sullivan & Karlseder, 2010). Indeed, many proteins that function in detection and signaling of DNA damage such as the MRN complex and ATM localize to functional telomeres during and after replication (Zhu et al, 2000; Verdun et al, 2005).
Sister chromatids are associated (cohered) from the moment they are replicated in S phase until their separation at mitosis (Peters & Nishiyama, 2012). Cohesion between sister telomeres is mediated by the cohesin subunit SA1 along with the shelterin subunits TRF1 and TIN2 (Canudas et al, 2007; Canudas & Smith, 2009). Resolution of telomere cohesion requires the PARP, tankyrase 1 (Dynek & Smith, 2004), which PARsylates itself and TRF1 (Smith et al, 1998) and localizes to telomeres in late S/G2 to resolve telomere cohesion (Bisht et al, 2012, 2013). The timing of resolution of telomere cohesion is important for telomere integrity. Premature loss of cohesion between sister telomeres results in an inability to repair breaks following DNA replication and in sister telomere loss (Canudas & Smith, 2009) and leads to fragile telomeres (Remeseiro et al, 2012). In the human inherited disease dyskeratosis congenita, patient cells that harbor TIN2 mutations exhibit premature loss of sister telomere cohesion (Canudas et al, 2011) and have extremely short telomeres (Savage et al, 2008; Walne et al, 2008), suggesting that telomere cohesion may be important for telomere lengthening by telomerase or by recombination (Canudas et al, 2011; Houghtaling et al, 2012). Conversely, persistent cohesion between sister telomeres (induced by depletion of tankyrase 1) results in deprotection of chromosome ends; sister chromatids are inappropriately fused by nonhomologous end joining (Hsiao & Smith, 2009), suggesting that timely removal of sister telomere cohesion is essential for reformation of a protective structure.
Ubiquitination is a critical posttranslational regulator of protein activity and function (Ciechanover & Schwartz, 2004). Ubiquitin molecules can be linked through one of seven lysine residues in a polyubiquitin chain. K48‐linked polyubiquitin chains are the most abundant in vivo and act as a signal for degradation by the proteasome (Komander & Rape, 2012). This pathway plays a major role in tankyrase 1 stability. The E3 ligase RNF146 adds K48‐linked polyubiquitin chains to autoPARsylated tankyrase 1, targeting it for proteasomal degradation (Callow et al, 2011; Zhang et al, 2011). K63‐linked polyubiquitin, on the other hand, has nonproteolytic roles (Chen & Sun, 2009) in processes as diverse as the interferon response (Fuchs, 2012) and double‐strand break repair (Bartocci & Denchi, 2013). The K63‐ubiquitination response to double‐strand breaks is initiated by ATM, which accumulates at breaks, dependent on MRN. ATM phosphorylates H2AX, leading to recruitment of MDC1 and the E3 ubiquitin ligase RNF8 (Huen et al, 2007; Kolas et al, 2007; Mailand et al, 2007). RNF8 then (along with a second E3 ligase RNF168) promotes K63‐linked ubiquitination of histones, providing a platform for DNA damage signaling and recruitment of repair factors (Al‐Hakim et al, 2010; Bekker‐Jensen & Mailand, 2010; Thorslund et al, 2015).
The ubiquitination status of a protein is controlled by the counteracting activities of E3 ligases that promote ubiquitin ligation and deubiquitinating enzymes (DUBs) that remove ubiquitin (Clague et al, 2013; Eletr & Wilkinson, 2014). The K63‐linked chains assembled at sites of DNA damage can be dismantled through the action of BRCC36, a K63‐specific DUB to control the DNA damage response. BRCC36 is part of a nuclear complex (BRCA1‐A) containing ABRAXAS, MERIT40, BRCC45, and the K63‐ubiquitin binding protein RAP80, where ABRAXAS serves as a scaffold binding all four subunits with its N‐terminus and BRCA1 with its C‐terminus (Dong et al, 2003; Kim et al, 2007a,b; Sobhian et al, 2007; Wang & Elledge, 2007; Wang et al, 2007; Shao et al, 2009). The BRCC36 DUB is also part of a similar cytoplasmic complex BRISC, which has (in place of the ABRAXAS scaffold) the related ABRO1 (Abraxas brother 1); ABRO1 contains N‐terminal binding sites for BRCC36, MERIT40, and BRCC45, but not for RAP80, and it lacks the C‐terminal binding site for BRCA1 (Cooper et al, 2009, 2010; Feng et al, 2010; Patterson‐Fortin et al, 2010; Hu et al, 2011).
In an effort to elucidate mechanisms of tankyrase 1 function, we performed a proteomic screen to identify binding proteins. We identified ABRO1 (the distinguishing scaffold subunit of the BRISC complex) as a significant partner, leading us to investigate the role of K63‐ubiquitin in tankyrase 1 function. Here, we elucidate the mechanism of the cell cycle‐regulated K63‐ubiquitination of tankyrase 1 and determine its impact on the timing of resolution of sister chromatid cohesion at telomeres.
Results
Tankyrase 1 binds the BRISC DUB
Tankyrase 1 contains a large ankyrin repeat domain that serves as a scaffold for multiple binding partners including TRF1 (Smith et al, 1998; Smith, 2015). To identify novel tankyrase‐binding partners, we isolated Flag‐epitope‐tagged tankyrase 1‐associated protein complexes from HeLa cells using tandem affinity purification (Bisht et al, 2012) followed by multidimensional protein identification technology liquid chromatography–tandem mass spectrometry (MudPIT LC‐MS/MS) (Giannone et al, 2010). We identified (in addition to several known tankyrase‐binding proteins) ABRO1 (Fig 1A), the scaffold subunit of the BRISC DUB (Fig 1B). The detection of ABRO1 along with a second subunit of BRISC, MERIT40, a validated tankyrase‐binding protein (Guettler et al, 2011), prompted us to investigate the interaction of tankyrase 1 with BRISC.
Figure 1. Tankyrase 1 is a target of ABRO1 and the BRISC DUB.
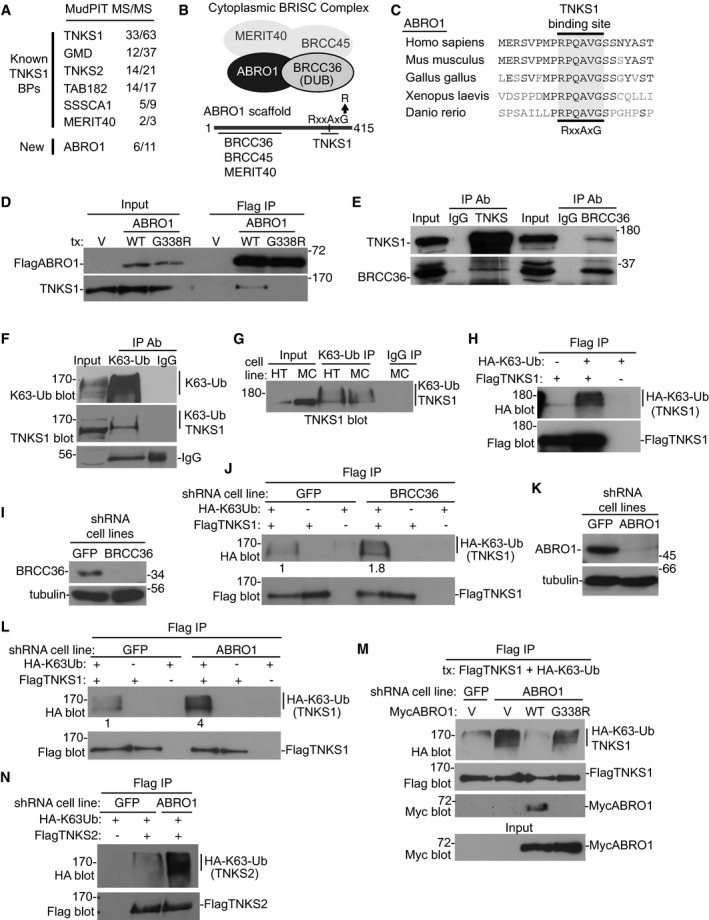
-
AIdentification of tankyrase 1 binding proteins by MudPIT MS/MS. Unique peptides/total peptide numbers are indicated.
-
BSchematic diagram showing the four‐subunit BRISC complex with a line diagram below indicating ABRO1 containing a consensus tankyrase‐binding motif.
-
CAlignment of the tankyrase‐binding site in ABRO1 across multiple species. Identical amino acids are in black.
-
DFlagABRO1 WT, but not the tankyrase‐binding site mutant G338R, binds tankyrase 1. Lysates from HTC75 cells transfected with vector (V), FlagABRO1.WT, or FlagABRO1.G338R were immunoprecipitated with anti‐Flag antibody and immunoblotted with anti‐Flag or TNKS1 antibodies.
-
EReciprocal coimmunoprecipitation of endogenous tankyrase 1 and BRCC36. HTC75 cell lysates were immunoprecipitated with anti‐TNKS1, BRCC36, or IgG antibodies and immunoblotted with anti‐TNKS1 or BRCC36 antibodies.
-
FEndogenous tankyrase 1 is K63‐linked polyubiquitinated. HTC75 cell extracts were immunoprecipitated with anti‐K63‐Ub antibody and immunoblotted with antibodies against K63‐Ub or tankyrase 1.
-
GEndogenous tankyrase 1 is K63‐linked polyubiquitinated in multiple cell lines. Whole‐cell extracts from HTC75 (HT) or MCF7 (MC) cells were immunoprecipitated with anti‐K63‐Ub antibody and immunoblotted with anti‐TNKS1 antibody.
-
HFlagTNKS1 is K63‐linked polyubiquitinated. HTC75 cells were cotransfected with FlagTNKS1 and HA‐K63‐Ub, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐Flag or HA antibodies.
-
I, JK63‐Ub chains are increased on tankyrase 1 in BRCC36‐depleted cells. (I) Immunoblot analysis of HTC75 cell lines stably expressing GFP or BRCC36 shRNA. (J) GFP or BRCC36 HTC75 shRNA cell lines were cotransfected with FlagTNKS1 and HA‐K63‐Ub, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐Flag or HA antibodies. Protein levels relative to FlagTNKS1 and normalized to the GFP control are indicated below the blot.
-
K, LK63‐Ub chains are increased on tankyrase 1 in ABRO1‐depleted cells. (K) Immunoblot analysis of HTC75 cell lines stably expressing GFP or ABRO1 shRNA. (L) GFP or ABRO1 HTC75 shRNA cell lines were cotransfected with FlagTNKS1 and HA‐K63‐Ub, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐Flag or HA antibodies. Protein levels relative to FlagTNKS1 and normalized to the GFP control are indicated below the blot.
-
MThe increase in K63‐Ub chains in ABRO1‐depleted cells is rescued by ABRO1 WT, but not by the G338R tankyrase‐binding mutant. GFP or ABRO1 HTC75 shRNA cell lines were cotransfected with FlagTNKS1, HA‐K63‐Ub, and a vector control or MycABRO1 WT or G338R, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐HA, Flag, or Myc antibodies.
-
NFlagTNKS2 is modified by K63‐Ub chains that are increased in ABRO1‐depleted cells. GFP or ABRO1 HTC75 shRNA cell lines were cotransfected with FlagTNKS2 and HA‐K63‐Ub, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐Flag or HA antibodies.
Source data are available online for this figure.
Examination of the amino acid sequence of ABRO1 revealed a canonical tankyrase‐binding motif (RxxAxG) at amino acids 333–338 (Fig 1B) that was highly conserved across species (Fig 1C). We generated Flag‐epitope‐tagged ABRO1 wild‐type (WT) and mutant (G338R; the glycine at position 338 was mutated to arginine) alleles, transfected them into HTC75 cells, and performed immunoprecipitation analysis. As shown in Fig 1D, Flag ABRO1 WT, but not G338R, bound endogenous tankyrase 1. Additional immunoprecipitation analysis showed that endogenous BRCC36 and tankyrase 1 each coimmunoprecipitated the other (Fig 1E), confirming tankyrase association with the DUB.
Tankyrase 1 is modified by K63‐Ub chains that are cleaved by BRISC
Given that tankyrase 1 binds ABRO1 and BRCC36, we wondered whether it was a target of the K63‐specific DUB. To address this, we first determined whether tankyrase 1 was modified by K63‐linked polyubiquitin chains. To measure endogenous tankyrase 1, HTC75 cell extracts were immunoprecipitated with anti‐K63‐Ub antibody, which detects polyubiquitin chains formed by the K63 linkage, followed by immunoblotting with antibodies against K63‐Ub or tankyrase 1 (Fig 1F). Detection with anti‐K63‐Ub antibody revealed a smear indicating all proteins that are modified by K63‐Ub, whereas detection with anti‐TNKS1 antibody revealed just a few slower migrating bands indicating K63‐Ub TNKS1. We reproducibly detected slower migrating forms of tankyrase 1 following immunoprecipitation with anti‐K63‐Ub and detection with anti‐TNKS1 antibodies in HTC75 and other (MCF7) cell lines (Fig 1G).
As an alternative way to measure K63‐Ub chains on tankyrase 1, we utilized a HA‐tagged ubiquitin in which all lysines except K63 were mutated to arginine (HA‐K63‐Ub), thus enabling in vivo synthesis of polyubiquitin chains of the K63‐specific linkage. HTC75 cells were transfected with FlagTNKS1 and HA‐K63‐Ub, subjected to Flag immunoprecipitation, and analyzed by immunoblotting with anti‐Flag to detect tankyrase 1 and anti‐HA antibodies to detect K63‐Ub. As shown in Fig 1H, FlagTNKS1 was detected as a K63‐linked polyubiquitinated protein.
To determine whether BRISC cleaves the K63‐Ub chains off tankyrase 1, we used shRNA lentiviral infection to generate BRCC36‐depleted or GFP control HTC75 cell lines (Fig 1I) and then analyzed K63‐ubiquitination of tankyrase 1. Cells were transfected with FlagTNKS1 and HA‐K63‐Ub, subjected to Flag immunoprecipitation, and analyzed by immunoblot with anti‐Flag or HA antibodies. As shown in Fig 1J, we observed a 1.8‐fold increase in K63‐Ub chains on tankyrase 1 in BRCC36‐depleted cells. To determine whether K63‐Ub chain removal was ABRO1‐dependent, ABRO1‐depleted cells (generated by lentiviral shRNA infection) (Fig 1K) were transfected with FlagTNKS1 and HA‐K63‐Ub, subjected to Flag immunoprecipitation, and analyzed by immunoblot with anti‐Flag or HA antibodies. As shown in Fig 1L, we observed a fourfold increase in K63‐Ub chains on tankyrase 1 in ABRO1‐depleted cells. To confirm that ABRO1 was responsible for chain removal, we introduced shRNA‐resistant ABRO1 alleles into ABRO1‐depleted cells and measured K63‐Ub TNKS1. As shown in Fig 1M, ABRO1 WT, but not the G338R tankyrase 1‐binding site mutant, rescued the increase in K63‐Ub TNKS1. Finally, we show that the closely related isoform tankyrase 2 is also K63‐ubiquitinated and the K63‐Ub chains are increased in ABRO1‐depleted cells (Fig 1N).
The E3 ligase RNF8 is required for K63‐ubiquitination of tankyrase 1
We next sought to determine the E3 ligase that was responsible for K63‐ubiquitination of tankyrase 1. While many E3 ligases that promote K48‐Ub chains have been identified, only a few have been shown to promote K63‐Ub chains. The well‐characterized RNF8 is recruited to sites of double‐strand breaks, where it promotes K63‐ubiquitination of H1‐type‐linker histones as part of a DNA damage‐signaling cascade (Mailand et al, 2007; Thorslund et al, 2015). Before considering RNF8 (a nuclear E3 ligase) as a candidate, we determined the subcellular localization of K63‐Ub TNKS1. First, cells were fractionated into nuclear and cytosolic fractions and analyzed by immunoblot. Tankyrase 1 was detected in both the nuclear and cytosolic compartments of the cell, whereas the bulk of the K63‐Ub proteins in the tankyrase size range fractionated to the nuclear compartment along with RNF8 (Fig 2A). To confirm that K63‐Ub TNKS1 associated with the nuclear compartment, nuclear and cytosolic extracts were prepared from cells cotransfected with FlagTNKS1 and HA‐K63‐Ub and subjected to Flag immunoprecipitation, followed by immunoblotting with anti‐Flag or HA antibodies. As shown in Fig 2B, K63‐Ub TNKS1 was detected in the nuclear fraction.
Figure 2. Tankyrase is K63‐linked polyubiquitinated by the E3 ligase RNF8.
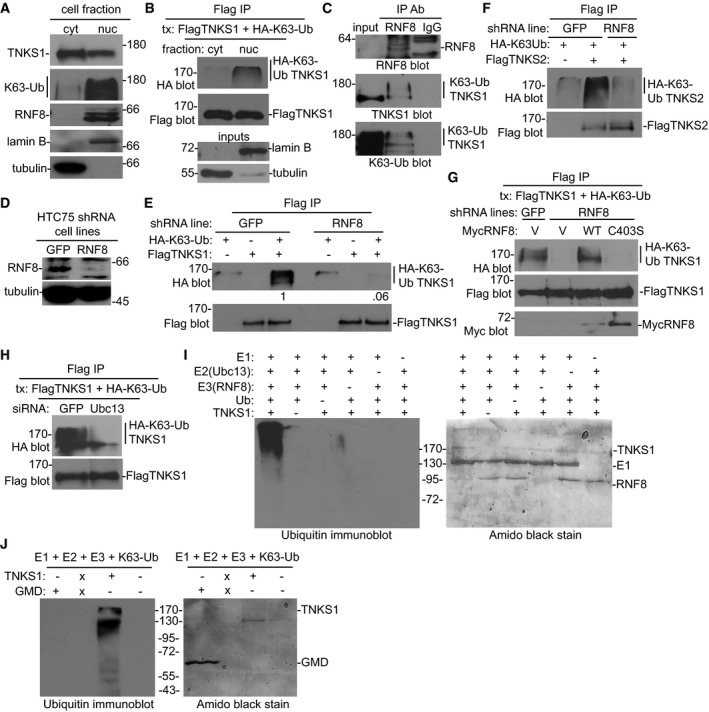
-
AK63‐Ub is enriched in the nuclear fraction. Immunoblot analysis of nuclear and cytosolic extracts with the indicated antibodies.
-
BK63‐Ub TNKS1 is in the nuclear fraction. Cytosolic and nuclear extracts from HTC75 cells cotransfected with FlagTNKS1 and HA‐K63‐Ub were immunoprecipitated with anti‐Flag antibody and immunoblotted with anti‐Flag or HA antibodies.
-
CEndogenous RNF8 is bound to endogenous K63‐linked polyubiquitinated tankyrase 1. HTC75 cell extracts were immunoprecipitated with anti‐RNF8 antibody and immunoblotted with anti‐RNF8, TNKS1, or K63‐Ub antibodies.
-
D, EK63‐Ub chains are diminished on tankyrase 1 in RNF8‐depleted cells. (D) Immunoblot analysis of HTC75 cell lines stably expressing GFP or RNF8 shRNA. (E) GFP or RNF8 shRNA cell lines were cotransfected with FlagTNKS1 and HA‐K63‐Ub, immunoprecipitated with anti‐Flag antibody, and immunoblotted with anti‐Flag or HA antibodies. Protein levels relative to FlagTNKS1 and normalized to the GFP control are indicated below the blot.
-
FFlagTNKS2 is modified by K63‐Ub chains that are decreased in RNF8‐depleted cells. GFP or RNF8 HTC75 shRNA cell lines were cotransfected with FlagTNKS2 and HA‐K63‐Ub, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐Flag or HA antibodies.
-
GThe decrease in K63‐Ub chains in RNF8‐depleted cells is rescued by RNF8 WT, but not by the C403S RING‐deficient mutant. GFP or RNF8 HTC75 shRNA cell lines were cotransfected with FlagTNKS1, HA‐K63‐Ub, and a vector control (V) or MycRNF8 WT or C403S, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐HA, Flag, or Myc antibodies.
-
HK63‐Ub chains are diminished on tankyrase 1 in Ubc13‐depleted cells. HTC75 cells were transfected with GFP or Ubc13 siRNA for 24 h, followed by cotransfection with FlagTNKS1 and HA‐K63‐Ub for an additional 24 h. Cell extracts were immunoprecipitated with anti‐Flag and immunoblotted with anti‐HA and anti‐Flag antibodies.
-
IRNF8 ubiquitinates tankyrase 1 in vitro. Ubiquitination reactions containing the indicated purified proteins were fractionated on SDS–PAGE, visualized by staining with amido black (right panel) and by immunoblotting with anti‐ubiquitin antibody (left panel).
-
JRNF8 K63‐ubiquitinates tankyrase 1 (but not an unrelated protein GMD) in vitro. Ubiquitination reactions containing the E1, E2, E3, and K63‐Ub and GMD or TNKS1 were fractionated on SDS–PAGE, visualized by staining with amido black (right panel) and by immunoblotting with anti‐ubiquitin antibody (left panel). X indicates a blank lane.
Source data are available online for this figure.
To determine whether RNF8 binds to tankyrase in cells, endogenous RNF8 was immunoprecipitated from whole‐cell extracts and analyzed by immunoblotting with antibodies against RNF8, tankyrase 1, and K63‐Ub. As shown in Fig 2C, endogenous K63‐Ub TNKS1 coimmunoprecipitated with endogenous RNF8. To determine whether RNF8 is required for K63‐ubiquitination of tankyrase 1, we analyzed the K63‐Ub status of tankyrase 1 in RNF8‐depleted or GFP control HTC75 cell lines (generated by shRNA lentiviral infection) (Fig 2D). Cells were transfected with FlagTNKS1 and HA‐K63‐Ub, subjected to Flag immunoprecipitation, and analyzed by immunoblot with anti‐Flag or HA antibodies. As shown in Fig 2E, K63‐Ub TNKS1 was dramatically reduced in RNF8‐depleted cells. A similar analysis using FlagTNKS2 shows that RNF8 is required for K63‐ubiquitination of tankyrase 2 (Fig 2F); the K63‐Ub chains on tankyrase 2 are greatly diminished in RNF8‐depleted cells. To confirm that RNF8 was responsible for K63‐Ub chain addition to tankyrase 1, we introduced shRNA‐resistant RNF8 alleles into RNF8‐depleted cells and measured K63‐Ub TNKS1. As shown in Fig 2G, RNF8 WT, but not the C403S catalytically inactive RING mutant (Mailand et al, 2007), rescued the loss in K63‐Ub TNKS1. To determine whether K63‐ubiquitination of tankyrase 1 was catalyzed by Ubc13, the E2‐ubiquitin‐conjugating enzyme that collaborates with RNF8 to generate specifically K63‐linked ubiquitin chains (Hofmann & Pickart, 1999; Kolas et al, 2007; Wang & Elledge, 2007), cells were transfected with GFP or Ubc13 siRNA, followed by transfection with FlagTNKS1 and HA‐K63‐Ub, then subjected to Flag immunoprecipitation, and analyzed by immunoblot with anti‐Flag or HA antibodies. As shown in Fig 2H, K63‐Ub TNKS1 was dramatically reduced in Ubc13 siRNA treated cells.
Finally, to demonstrate that RNF8 ubiquitinates tankyrase 1 directly, we performed ubiquitination in vitro with the following purified components: tankyrase 1, E3 (GST‐RNF8), E1, E2 (Ubc13), and ubiquitin. The reaction products were fractionated by SDS–PAGE and visualized by staining with amido black and by immunoblotting with anti‐ubiquitin antibody. As shown in Fig 2I, left panel, a ladder of ubiquitinated tankyrase 1 was detected. Importantly, ubiquitination depended on each of the five reagents; omission of any one led to loss of the ubiquitinated tankyrase 1 products. We further show that RNF8 adds K63‐Ub chains to tankyrase 1, but not to an unrelated protein, the tankyrase 1‐binding protein GMD (Bisht et al, 2012) (Fig 2J).
K63‐Ub chains increase tankyrase 1 stability
Tankyrase 1 protein is rapidly turned over due to RNF146‐mediated K48‐linked polyubiquitination and degradation by the proteasome (Callow et al, 2011; Zhang et al, 2011). We reasoned that K63‐linked polyubiquitination of tankyrase 1 might influence its stability by preventing degradation by the proteasome. First, we overexpressed HA‐K63‐Ub in cells by transient transfection and measured levels of endogenous tankyrase 1 by immunoblot. As shown in Fig 3A (Input), we observed a dramatic stabilization of endogenous tankyrase 1 in HA‐K63‐Ub‐ versus vector‐transfected cells. Immunoprecipitation with anti‐HA antibody followed by immunoblotting with anti‐TNKS1 confirmed that endogenous tankyrase 1 was modified by K63‐Ub chains (Fig 3A; HA IP). Transfection with K48‐Ub (Fig 3B) or with a K63R‐Ub mutant (Fig 3C) did not induce stabilization of tankyrase 1, indicating that stabilization was due to K63‐Ub chains. To determine whether K63‐Ub TNKS1 is more stable than K48‐Ub TNKS1, we measured their half‐lives in vivo. HTC75 cells were transfected with FlagTNKS1 and HA‐K63‐Ub or HA‐K48‐Ub, in the presence or absence of cycloheximide, subjected to Flag immunoprecipitation, and analyzed by immunoblotting with anti‐Flag antibody to detect tankyrase 1 and anti‐HA antibody to detect K48‐Ub and K63‐Ub. As shown in Fig 3D and E, we observed a greater than twofold increase in K63‐Ub TNKS1 compared to K48‐Ub TNKS1 following 3 h of cycloheximide treatment.
Figure 3. K63‐Ub chains increase tankyrase 1 stability.
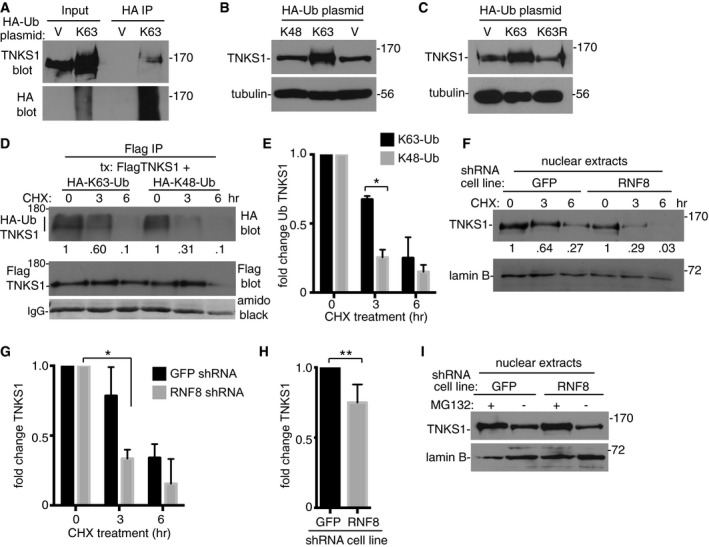
-
AEndogenous tankyrase 1 is modified and stabilized by K63‐Ub chains. HTC75 cells were transfected with vector (V) or HA‐K63‐Ub, immunoprecipitated with anti‐HA, and immunoblotted with anti‐HA or TNKS1 antibodies.
-
B, CNeither HA‐K48‐Ub nor HA‐K63R‐Ub induced stabilization of tankyrase 1. HTC75 cells were transfected with V, HA‐K63‐Ub, and (B) HA‐K48‐Ub or (C) HA‐K63R‐Ub and analyzed by immunoblot with anti‐TNKS1 or α‐tubulin antibodies.
-
D, EHA‐K63‐Ub TNKS1 has a longer half‐life than HA‐K48‐Ub TNKS1. (D) HTC75 cells were cotransfected with FlagTNKS1 and HA‐K63‐Ub or HA‐K48‐Ub, treated with cycloheximide for 0, 3, or 6 h prior to harvest, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐Flag or HA antibodies. Protein levels of HA‐K63‐Ub or HA‐K48‐Ub TNKS1 relative to FlagTNKS1 and normalized to the 0‐h time point are indicated below the blot. (E) Graphical representation of the fold change. Average of two independent experiments ± SEM. *P ≤ 0.05; Student's unpaired t‐test.
-
F, GThe half‐life of nuclear tankyrase 1 is decreased in RNF8‐depleted cells. (F) Nuclear extracts were prepared from GFP or RNF8 shRNA HTC75 cells following treatment with cycloheximide for 0, 3, or 6 h and analyzed by immunoblotting with anti‐TNKS1 or lamin B antibodies. Protein levels of tankyrase 1 in GFP shRNA‐ or RNF8 shRNA‐depleted cells relative to lamin B and normalized to the 0‐h time point are indicated below the blot. (G) Graphical representation of the fold change. Average of two independent experiments ± SEM. *P ≤ 0.05; Student's unpaired t‐test.
-
HTankyrase 1 protein levels are decreased in nuclear extracts from RNF8‐depleted cells. Graphical representation of the fold change in tankyrase 1 relative to lamin B measured by immunoblot. Average of four independent experiments ± SD. **P ≤ 0.01; Student's unpaired t‐test.
-
IRNF8 protects tankyrase 1 from proteasomal degradation. Immunoblot analysis of nuclear extracts from HTC75 cell lines stably expressing RNF8 or GFP shRNA treated with and without MG132 for 4 h prior to harvest.
Source data are available online for this figure.
The experiments described above predict that the stability and half‐life of tankyrase 1 protein should be decreased in RNF8‐depleted cells. We analyzed tankyrase 1 in nuclear extracts from RNF8‐depleted versus control cells following cycloheximide treatment. As shown in Fig 3F and G, we observed a threefold decrease in tankyrase 1 levels in RNF8‐depleted cells after 3 h of cycloheximide treatment, indicating that RNF8 increases the half‐life of tankyrase 1. Immunoblot analysis indicates that we can detect a relatively small but statistically significant decrease in tankyrase 1 levels in nuclear extracts even without cycloheximide treatment (Fig 3H). Lastly, we measured the impact of RNF8‐depletion on tankyrase 1 stability in the presence or absence of the proteasome inhibitor MG132. As shown in Fig 3I, treatment with MG132 increased endogenous levels of tankyrase 1 in RNF8‐depleted cells to levels in control cells, indicating that RNF8 protects tankyrase 1 from proteasomal degradation.
Tankyrase 1 K63‐ubiquitination is regulated by the cell cycle
We showed thus far that RNF8 is required to generate K63‐Ub chains on tankyrase 1 and these chains can be readily detected in undamaged cells. However, since RNF8 is known as a DNA damage‐responsive E3 ligase, we asked whether the modification could be stimulated further by exogenous DNA damage. Treatment of cells with phleomycin to induce double‐strand breaks led to a marked increase in K63‐Ub chains on tankyrase 1 (Fig 4A), indicating that the modification is responsive to DNA damage. Nonetheless, the high basal levels of K63‐Ub TNKS1 observed in untreated cells were striking, and we considered that it might result from endogenous DNA damage, perhaps generated by cell cycle‐dependent processes. To determine whether the K63‐Ub modification of tankyrase 1 occurred during cell cycle progression, HeLaI.2.11 cells were synchronized at the G1/S border by a double thymidine block, released, and harvested every 2 h. Cell extracts were immunoprecipitated with anti‐K63‐Ub antibody followed by immunoblotting with anti‐TNKS1 antibody. As shown in Fig 4B, K63‐Ub TNKS1 was detected as early as the 4‐h time point (mid‐S phase) and slower migrating forms were detected at 6, 8, and 10 h, indicating that tankyrase 1 was modified by K63‐Ub in late S/G2/M phase of the cell cycle.
Figure 4. K63‐linked polyubiquitination of tankyrase 1 is cell cycle‐regulated.
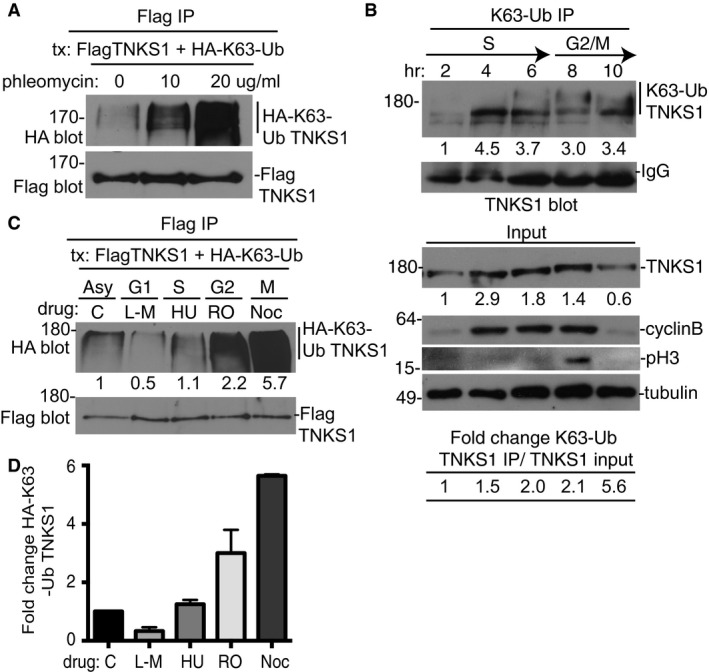
-
AK63‐ubiquitination of tankyrase 1 occurs without exogenous damage, but can be stimulated by double‐strand breaks. HTC75 cells were cotransfected with FlagTNKS1 and HA‐K63‐Ub, treated with 0, 10, or 20 μg/ml phleomycin for 4 h prior to harvest, immunoprecipitated with anti‐Flag antibody, and immunoblotted with anti‐Flag or HA antibodies.
-
BK63‐ubiquitination of tankyrase 1 is dependent upon cell cycle progression. HeLa cells were synchronized by a double thymidine block, released, and collected every 2 h. Whole‐cell extracts were immunoprecipitated with anti‐K63‐Ub antibody and analyzed by immunoblotting with anti‐TNKS1 antibody. Protein levels of K63‐Ub TNKS1 relative to IgG and normalized to the 2‐h time point are indicated below the blot. Immunoblot analysis of input samples with the indicated antibodies is shown below. Protein levels of TNKS1 relative to tubulin and normalized to the 2‐h time point are indicated below the blot. The fold change of immunoprecipitated K63‐Ub TNKS1 relative to input TNKS1 is indicated at the bottom.
-
C, DHA‐K63‐linked polyubiquitinated FlagTNKS1 is high in S/G2/M and diminished in G1 phase. (C) HTC75 cells were cotransfected with FlagTNKS1 and HA‐K63‐Ub and synchronized by addition of L‐mimosine (G1), hydroxyurea (S) R03306 (G2), or nocodazole (M). Cell extracts were immunoprecipitated with anti‐Flag and immunoblotted with anti‐Flag or HA antibodies. Protein levels of HA‐K63‐Ub TNKS1 relative to FlagTNKS1 and normalized to the asynchronized (asy) control are indicated below the blot. (D) Graphical representation of the fold change. Average of two independent experiments ± SEM.
Source data are available online for this figure.
We used an alternative method of synchronization and detection to confirm the cell cycle regulation. HTC75 cells were transfected with FlagTNKS1 and HA‐K63‐Ub and arrested at different stages of the cell cycle by treatment with the inhibitors L‐mimosine (G1), hydroxyurea (S phase), R03306 (G2), and nocodazole (M), subjected to Flag immunoprecipitation, and analyzed by immunoblotting with anti‐Flag antibody to detect tankyrase 1 and anti‐HA antibody to detect K63‐Ub. As shown in Fig 4C and D, K63‐Ub TNKS1 was low in G1 and high in S/G2/M phases of the cell cycle. Together these data demonstrate that the K63‐Ub modification of tankyrase 1 is restricted to the late S/G2/M phases of the cell cycle.
RNF8 promotes association of tankyrase with telomeres and resolution of sister telomere cohesion
Previous studies showed that tankyrase 1 localized to telomeres in late S/G2/M phases of the cell cycle (measured by telomere chromatin immunoprecipitation; ChIP) (Bisht et al, 2012). To determine whether RNF8 influenced this association, we generated RNF8‐depleted or GFP control HeLaI.2.11 cell lines by shRNA lentiviral infection (Fig 5A) and performed telomere ChIP. As shown in Fig 5B and C, tankyrase 1 binding to telomeres was reduced in RNF8‐depleted cells. To determine whether the reduction in tankyrase 1 at telomeres impacted resolution of telomere cohesion, cells were isolated by mitotic shake‐off and analyzed using chromosome‐specific FISH with a 16p telomere probe. As shown in Fig 5D and E, we observed a threefold decrease in resolution of sister telomere cohesion in RNF8‐depleted cells. Previous studies showed that unresolved telomere cohesion (induced by tankyrase 1 depletion) led to DNA ligase IV‐dependent fusion of sister telomeres (Hsiao & Smith, 2009). To determine whether unresolved telomere cohesion induced by RNF8 depletion led to a similar dysfunction, we examined metaphase spreads by PNA FISH using a telomere repeat probe. As shown in Fig 5F and G, we observed a fourfold increase in sister telomere fusions in RNF8‐ versus GFP‐depleted cells. We then generated ligase IV‐depleted cells by lentiviral infection of RNF8‐shRNA cells with two different DNA ligase 4 shRNA lentiviruses (Hsiao & Smith, 2009). As shown in Fig 5H, sister telomere fusions in RNF8‐depleted cells were suppressed by ligase IV depletion.
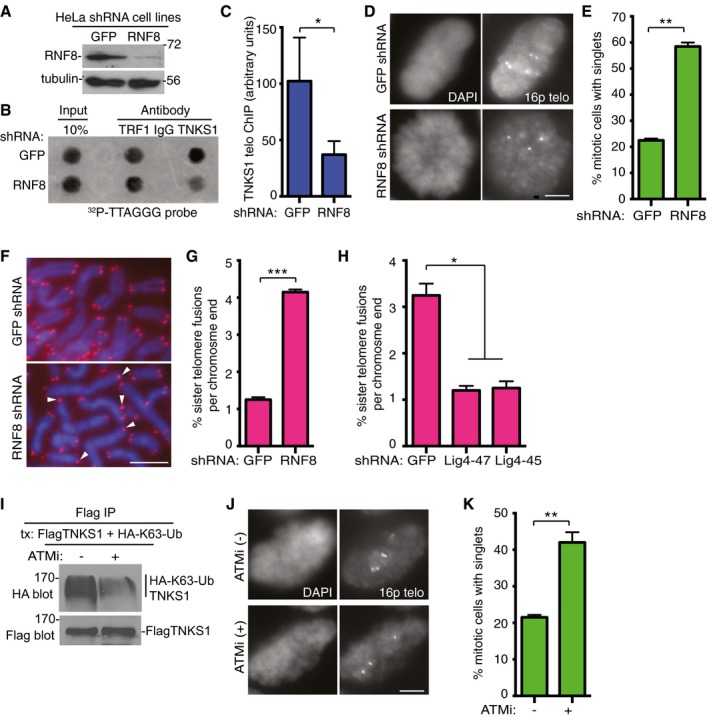
Figure 5. RNF8 is required for binding of tankyrase 1 to telomeres and for timely resolution of telomere cohesion.
-
A–CTankyrase 1 association with telomeres is reduced in RNF8‐depleted cells. (A) Immunoblot analysis of HeLaI.2.11 cell lines stably expressing GFP or RNF8 shRNA. (B) Telomeric DNA ChIP analysis of GFP or RNF8 shRNA HeLaI.2.11 cell lines using the indicated antibodies. (C) Quantification of the signal intensity of telomeric DNA immunoprecipitated by anti‐TNKS1 antibody. Average of three independent experiments ± SD. *P ≤ 0.05; Student's unpaired t‐test.
-
D, EResolution of telomere cohesion depends on RNF8. (D) Mitotic cells were isolated from HeLaI.2.11 cell lines stably expressing GFP or RNF8 shRNA by mitotic shake‐off and subjected to FISH analysis with a 16ptelo probe. Scale bar, 5 μm. (E) Graphical representation of the frequency of mitotic cells with cohered telomeres. Average of two independent experiments (n = 60 cells each) ± SEM. **P ≤ 0.01; Student's unpaired t‐test.
-
F, GRNF8 depletion leads to sister telomere fusions. (F) Telomere FISH analysis with a (CCCTAA)3 repeat probe (red) of metaphase spreads from GFP or RNF8 shRNA HTC75 cell lines. DNA was stained with DAPI (blue). White arrowheads indicate sister telomere fusions. Scale bar, 5 μm. (G) Graphical representation of the frequency of sister telomere fusions per chromosome end. Average of two independent experiments (n = 756–1,012 chromosome ends each) ± SEM. ***P ≤ 0.001; Student's unpaired t‐test.
-
HDNA ligase IV depletion rescues sister telomere fusions in RNF8 shRNA cells following infection with GFP or Lig4 (47 or 45) shRNA lentivirus. Graphical representation of the frequency of sister telomere fusions per chromosome end. Average of two independent experiments (n = 702–1,072 chromosome ends each) ± SEM. *P ≤ 0.05; Student's unpaired t‐test.
-
IInhibition of ATM reduces K63‐Ub chains on tankyrase 1. HeLaI.2.11 cells were cotransfected with FlagTNKS1 and HA‐K63‐Ub, treated with or without the ATM inhibitor (ATMi) KU‐55933 for 4 h prior to harvest, immunoprecipitated with anti‐Flag, and immunoblotted with anti‐Flag or HA antibodies.
-
J, KInhibition of ATM reduces resolution of telomere cohesion. (J) HeLaI.2.11 cells were isolated by mitotic shake‐off following 4‐h treatment with or without ATMi and subjected to FISH analysis with a 16ptelo probe. Scale bar, 5 μm. (K) Graphical representation of the frequency of mitotic cells with cohered telomeres. Average of two independent experiments (n = 60 cells each) ± SEM. **P ≤ 0.01; Student's unpaired t‐test.
Source data are available online for this figure.
Finally, previous studies showed that the timing of tankyrase 1 association with telomeres (Bisht et al, 2012) and resolution of telomere cohesion (Bisht et al, 2013) coincided with the timing of the ATM‐mediated DNA damage response that occurs on functional telomeres in G2 phase of the cell cycle (Verdun et al, 2005). Our demonstration that appearance of K63‐Ub chains coincides with this timing (Fig 4B–D), depends on the DNA damage‐responsive E3 ligase RNF8 (Fig 2E), and occurs independent of (but stimulated by) exogenous DNA damage (Fig 4A) suggested a potential link to the ATM‐dependent DNA damage response that occurs on functional telomeres. Consistent with this notion, treatment of cells with the ATM inhibitor KU‐55933 led to a decrease in K63‐Ub chains on tankyrase 1 (Fig 5I) and to reduced resolution of telomere cohesion (Fig 5J and K), similar to RNF8 depletion.
ABRO1 removes K63‐Ub chains from tankyrase 1 in G1 to prevent premature resolution of sister telomere cohesion
As described above, K63‐Ub chains appear on tankyrase 1 in late S/G2/M phase of the cell cycle. We noticed that the chains were barely detectable in G1 phase of the cell cycle; they were detected on tankyrase 1 neither following a 2‐hour release from a double thymidine block (Fig 4B) nor following arrest in G1 by L‐mimosine (Fig 4C and D), suggesting that chain removal might occur in G1. To determine whether chain removal by ABRO1 occurred in G1, ABRO1 or GFP shRNA HTC75 cell lines were transfected with FlagTNKS1 and HA‐K63‐Ub, arrested in mitosis by nocodazole, isolated by mitotic shake‐off, returned to culture, and harvested at 0, 90, and 120 min. Cell extracts were subjected to Flag immunoprecipitation and analyzed by immunoblotting with anti‐Flag or anti‐HA antibodies. As shown in Fig 6A, in control GFP shRNA HTC75 cells K63‐HA‐Ub TNKS1 was detected at time 0, but was then reduced at 90 min and not detected at 120 min following release from mitotic arrest. By contrast, in ABRO1‐depleted cells K63‐Ub TNKS1 was present at much higher levels than in GFP control cells at time 0 and did not diminish as cells were released into G1. These data indicate that ABRO1 removes the K63‐Ub chains from tankyrase 1 as cells exit mitosis and enter G1. Similar results were obtained in BRCC36‐depleted cells (Fig 6B).
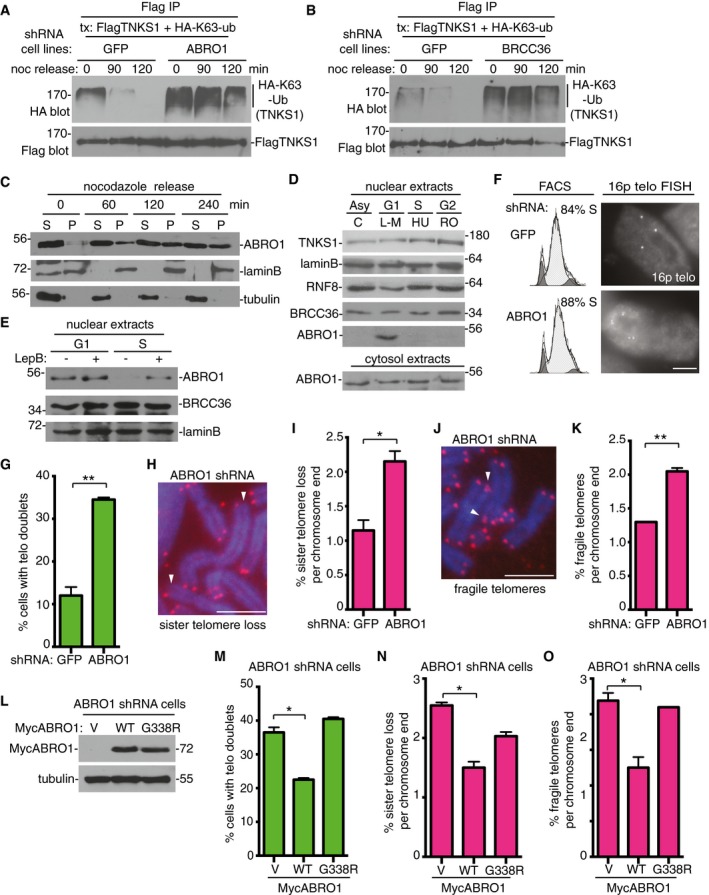
Figure 6. ABRO1 is required to remove K63‐Ub chains from tankyrase 1 in G1 to prevent premature resolution of sister telomere cohesion.
-
AABRO1 is required for removal of K63‐Ub chains from tankyrase 1 as cells exit mitosis and enter G1. GFP or ABRO1 shRNA HTC75 cell lines were transfected with FlagTNKS1 and HA‐K63‐Ub and treated with nocodazole for 20 h. Cells were isolated by mitotic shake‐off, replated, and harvested at 0, 90, and 120 min. Cell extracts were immunoprecipitated with anti‐Flag antibody and analyzed by immunoblotting with anti‐HA or Flag antibodies.
-
BBRCC36 is required for removal of K63‐Ub chains from tankyrase 1 as cells exit mitosis and enter G1. GFP or BRCC36 shRNA HTC75 cell lines were processed as described in (A).
-
CABRO1 enters the nuclear fraction as cells exit mitosis and enter G1. Following 20‐h nocodazole treatment, HTC75 cells were isolated by mitotic shake‐off, replated, harvested at 0, 60, 120, and 240 min, separated into nuclear pellet (P) and cytosolic supernatant (S) fractions, and analyzed by immunoblotting with the indicated antibodies.
-
DABRO1 localizes to the nuclear fraction in G1 phase of the cell cycle. HTC75 cells were synchronized by addition of L‐mimosine (G1), hydroxyurea (S), or R03306 (G2) for 18–20 h, harvested, separated into nuclear and cytosolic fractions, and analyzed by immunoblotting with the indicated antibodies.
-
EABRO1 is exported from the nucleus in S phase. HTC75 cells were synchronized in G1 or S (as above) and leptomycin B was added 3 h prior to harvest. The nuclear fractions were analyzed by immunoblotting with the indicated antibodies.
-
F, GTelomere cohesion is resolved prematurely in ABRO1‐depleted cells. (F) GFP or ABRO1 HTC75 shRNA cell lines were synchronized by a double thymidine block and analyzed by FACS and telomere FISH with a telomere 16ptelo probe 4 h after release from the second thymidine block. Scale bar, 5 μm. (G) Graphical representation of the frequency of cells with telomere doublets in S phase. Average of two independent experiments (n = 60 cells each) ± SEM. **P ≤ 0.01; Student's unpaired t‐test.
-
H–KABRO1 depletion leads to (H and I) sister telomere loss and (J and K) fragile telomeres. (H and J) Telomere FISH analysis with a (CCCTAA)3 repeat probe (red) of metaphase spreads from GFP or ABRO1 shRNA HTC75 cell lines. DNA was stained with DAPI (blue). Scale bars, 5 μm. Arrows indicate (H) sister telomere loss or (J) fragile telomeres. (I and K) Graphical representation of the frequency of (I) sister telomere loss or (K) or fragile telomeres per chromosome end. Average of two independent experiments (n = 798–846 chromosome ends each) ± SEM. *P ≤ 0.05, **P ≤ 0.01; Student's unpaired t‐test.
-
L–OThe telomere dysfunction in ABRO1‐depleted cells is rescued by ABRO1 WT, but not by the ABRO1 G338R tankyrase‐binding mutant. ABRO1 HTC75 shRNA cells were transfected with vector (V) or MycABRO1 WT or G338R and analyzed by (L) immunoblot with anti‐Myc or tubulin antibodies, (M) FISH with a telomere 16ptelo probe 4 h after release from a double thymidine block, or (N and O) telomere FISH with a (CCCTAA)3 repeat probe of metaphase spreads. (M) Graphical representation of the frequency of cells with telomere doublets in S phase. Average of two independent experiments (n = 40–58 cells each) ± SEM. *P ≤ 0.05; Student's unpaired t‐test. (N, O) Graphical representation of the frequency of (N) sister telomere loss or (O) fragile telomeres. Average of two independent experiments (n = 632–932 chromosome ends) ± SEM. *P ≤ 0.05; Student's unpaired t‐test.
Source data are available online for this figure.
To determine whether ABRO1 associates with the nuclear compartment upon G1 entry, HTC75 cells were arrested in mitosis by nocodazole, isolated by mitotic shake‐off, returned to culture, harvested at 0, 1, 2, and 4 h, fractionated into nuclear (pellet) and cytosolic (sup) extracts, and analyzed by immunoblot (Fig 6C). At time 0 when cells were in mitosis and the nuclear envelope dissolved, ABRO1 was detected in the soluble fraction, as were the controls: tubulin and lamin B. Some lamin B was already detected in the pellet fraction, likely due to the insoluble lamina or chromatin binding. Within 1 h of release from mitosis (and increasing at the 2‐ and 4‐h time points), ABRO1 distributed between the nuclear (pellet; marked by lamin B) and cytosolic (supernatant; marked by tubulin) fractions, indicating a nuclear localization for a portion of ABRO1 as the nuclear envelope reforms in G1 (Fig 6C).
As an alternative way to measure localization across the cell cycle, HTC75 cells were arrested at different stages of the cell cycle by treatment with the inhibitors, fractionated into nuclear and cytosolic extracts, and analyzed by immunoblot. As shown in Fig 6D, ABRO1, which was detected in the cytosol at all stages of the cell cycle (as expected), could be detected in the nuclear compartment, but only in G1. To determine whether ABRO1 was actively exported out of the nucleus following G1, cells were synchronized in G1 or S phase. Leptomycin B (an inhibitor of nuclear export) was added 3 h prior to harvest, and nuclear extracts were prepared and analyzed by immunoblot. As shown in Fig 6E, upon inhibition of nuclear export, ABRO1 could be detected in the nuclear compartment in S phase. Together these data indicate that ABRO1 distributes between the nuclear and cytoplasmic compartments as cells exit mitosis and enter G1 and is then exported out of the nucleus to the cytoplasm as cells enter S phase.
We next queried the consequences of not removing K63‐Ub chains from tankyrase 1. We considered that the G1‐specific removal might be required to prevent early association of tankyrase 1 with telomeres and premature resolution of telomere cohesion. To address this question, we synchronized GFP and ABRO1 HTC75 shRNA cell lines with a double thymidine block, released for 4 h, and performed FACS and FISH analysis (Fig 6F and G). FACS analysis showed that GFP control and ABRO1‐depleted cells were similarly synchronized in mid‐S phase. FISH analysis revealed a greater than threefold increase in doublets in ABRO1‐depleted cells compared to control, indicating premature resolution of sister telomere cohesion in S phase. Previous studies showed that premature resolution of cohesion led to sister telomere loss (Canudas & Smith, 2009) and to fragile telomeres (Remeseiro et al, 2012). Consistent with this, we found an increase in sister telomere loss (Fig 6H and I) and fragile telomeres (Fig 6J and K) in ABRO1‐depleted cells. Finally, to confirm that ABRO1 and its association with tankyrase 1 were responsible for the telomere dysfunction, we introduced shRNA‐resistant ABRO1 alleles into ABRO1‐depleted cells (Fig 6L) and measured telomere functions. We found that ABRO1 WT, but not the G338R tankyrase 1‐binding site mutant, led to a reduction in premature resolution of sister telomere cohesion (Fig 6M), sister telomere loss (Fig 6N), and fragile telomeres (Fig 6O).
Discussion
The timely resolution of sister chromatid cohesion is essential for genome integrity. Holding sisters together through S and G2 phase of the cell cycle ensures that chromatids have a copy template to use for recombination and repair during and after DNA replication (Sjogren & Nasmyth, 2001). This timing may be particularly important for telomeres, which, due to their repetitive G‐rich sequence, encounter replication problems (Sfeir et al, 2009). In addition, telomeres undergo multiple processing steps following DNA replication, including elongation by telomerase, nucleolytic processing, and fill‐in (Martinez & Blasco, 2015), which may require coordination with resolution of cohesion. Premature resolution of telomere cohesion results in sister telomere loss (Canudas & Smith, 2009) and fragile telomeres (Remeseiro et al, 2012). Here, we show that the timing of sister chromatid resolution at telomeres is ensured through the cell cycle‐regulated activities of the RNF8 E3 ligase and the BRISC DUB. Perturbation of this regulation, either by depletion of RNF8 and consequent reduction in K63‐Ub chains or by depletion of ABRO1 and consequent excessive K63‐Ub chains, results in persistent unresolved cohesion in mitosis or premature loss of cohesion in S phase, respectively.
Tankyrase localizes to telomeres through its interaction with TRF1. However, although tankyrase can be detected in nuclear extracts throughout the cell cycle, it accumulates on telomeres only in late S/G2/M (Bisht et al, 2012). We show that this accumulation is coincident with the appearance of RNF8‐dependent K63‐Ub chains on tankyrase 1. Although RNF8 is generally associated with the DNA damage response, the K63‐Ub chains on tankyrase 1 are detected in the absence of exogenous DNA damage. The timing of the modification, however, suggests a connection to the ATM‐dependent DNA damage response that occurs naturally on functional telomeres in G2 (Verdun et al, 2005). In support of this notion, inhibition of ATM prevents resolution of sister telomere cohesion. The coupling of these two events (the damage response on functional telomeres and association of tankyrase 1 with telomeres) would ensure that cohesion is resolved only after telomeres are replicated and could offer an opportunity to coordinate processing events between sister telomeres.
Previous studies showed that RNF8 promoted K63‐linked ubiquitination of histones at sites of DS breaks, providing a platform for DNA damage signaling and recruitment of repair factors to promote genome stability (Al‐Hakim et al, 2010; Bekker‐Jensen & Mailand, 2010; Thorslund et al, 2015). Subsequent analyses showed that RNF8 had a similar activity at deprotected telomeres (Peuscher & Jacobs, 2011; Rai et al, 2011; Okamoto et al, 2013; Orthwein et al, 2014). However, in contrast to a DS break, when a deprotected telomere is the target, the outcome of DNA repair (telomere fusion by NHEJ) is detrimental to genome stability. In our study, we show that RNF8 promotes K63‐ubiquitination of tankyrase 1 at functional telomeres to prevent telomere fusions and thereby safeguard genome integrity. These observations raise the question of what dictates RNF8 target choice. Perhaps having intact shelterin on a functional telomere distinguishes it from a DS break or a deprotected telomere and thereby favors a nonhistone target. Interestingly, the shelterin subunit TPP1 was found to be K63‐ubiquitinated at functional telomeres by RNF8 (Rai et al, 2011). In this case, the modification promoted TPP1 association with telomeres and prevented telomere fusions, consistent with a role for RNF8 in promoting telomere integrity.
How do K63‐Ub chains stabilize tankyrase 1? Previous studies showed that tankyrase 1 was rapidly turned over due to RNF146‐mediated K48 ubiquitination and degradation by the proteasome (Callow et al, 2011; Zhang et al, 2011). We show here that the K63‐Ub chains increase the half‐life of tankyrase 1 and that RNF8 prevents proteasome‐mediated degradation of tankyrase 1. Thus, K63‐ubiquitination shunts tankyrase 1 out of the K48 ubiquitination pathway, thereby preventing its rapid turnover and leading to its stabilization. K63‐linked ubiquitination of tankyrase 1 might prevent K48‐linked ubiquitination by reducing the interaction between tankyrase 1 and RNF146, or K63‐ubiquitination could compete directly for the same lysine residues on tankyrase 1 and thereby diminish the K48 modification. We further demonstrate that RNF8 promotes increased association of tankyrase 1 with telomeres. This increase is likely due (at least in part) to the increased stability of tankyrase 1, but it is additionally possible that the K63‐Ub chains on tankyrase 1 promote (yet to be identified) protein interactions at telomeres that retain it.
Removal of K63‐Ub chains from tankyrase 1 is subject to cell cycle regulation; the chains are removed as cells exit mitosis and transit into G1, dependent on ABRO1. This removal ensures that resolution of telomere cohesion does not occur prematurely in S phase. Indeed, ABRO1‐depletion led to premature resolution of telomere cohesion. However, it was not initially clear how ABRO1 could access the nuclear K63‐Ub TNKS1 since ABRO1 is primarily cytoplasmic. We found that ABRO1 localized to the nuclear compartment, specifically in G1 phase of the cell cycle. Previous studies show that ABRO1 can translocate to the nucleus during oxidative stress (Ambivero et al, 2012) or upon DNA damage (Zhang et al, 2014). Here, we show that the cell cycle controls ABRO1 localization to the nucleus; a fraction of ABRO1 localizes to the nucleus in G1 following mitosis and is then actively exported out of the nucleus in S phase.
We present our results in terms of a model of cell cycle‐regulated addition and removal of K63‐Ub chains (Fig 7). Tankyrase 1 is present in the nucleus throughout the cell cycle, but it localizes to telomeres and resolves cohesion only in G2/M (Bisht et al, 2012, 2013). We speculate that following telomere replication in late S/G2 in response to the ATM‐mediated endogenous DNA damage signal that occurs on functional telomeres, RNF8‐mediated K63‐linked polyubiquitination of tankyrase 1 promotes its stability and association with telomeres. In mitosis, once telomere cohesion is resolved, ABRO1 gains access to K63‐Ub TNKS1 upon nuclear envelope breakdown when nuclear and cytoplasmic components mix. ABRO1 (and BRCC36) remains associated with tankyrase 1 in the nucleus to keep the K63‐Ub chains off tankyrase 1 until S phase (when ABRO1 is exported from the nucleus), thereby ensuring that telomere cohesion is not resolved prematurely.
Figure 7. Model showing cell cycle‐regulated control of resolution of telomere cohesion by K63‐ubiquitination of tankyrase 1.
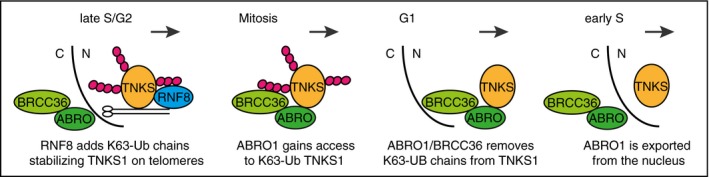
In late S/G2 following DNA replication (and in conjunction with the ATM‐mediated DNA damage response at functional telomeres), RNF8 puts K63‐Ub chains on tankyrase 1, promoting its association with telomeres and sister telomere resolution. Following nuclear envelope breakdown, ABRO1 (and BRCC36) gains access to K63‐Ub TNKS1 and removes the chains. ABRO1 remains in the nucleus through G1 keeping the K63‐Ub chains off tankyrase 1 and preventing premature resolution of telomere cohesion in S phase. ABRO1 is exported from the nucleus in S phase to permit K63‐ubiquitination of tankyrase 1. C, cytoplasm; N, nucleus.
Materials and Methods
Plasmids
FlagTNKS1, full‐length TNKS1 cloned into a lentiviral vector, was described previously (pLSJH.TNKS1WT) (Kim & Smith, 2014). For FlagTNKS2, full‐length TNKS2 was generated by PCR and cloned into the SalI and PacI sites of pLSJH (a modified pLKO.1ps lentiviral vector) to generate pLSJH.TNKS2WT. HA‐Ub K63 and HA‐Ub K48 contain HA‐tagged ubiquitin with either K63 or K48 only, other lysines mutated to arginines, in the pRK5‐HA vector. HA‐Ub K63R contains lysine 63 mutated to arginine; the other lysines are wild‐type. FlagABRO1 contains N‐terminal Flag/HA epitope‐tagged ABRO1 cloned into the pOZ‐N retroviral vector (Patterson‐Fortin et al, 2010) (Addgene plasmid 27499; deposited by Roger Greenberg). The FlagABRO1 G338R mutation was created by substituting the glycine (G) at position 338, with arginine (R) by site‐directed mutagenesis of FlagABRO1.WT using the oligonucleotide 5′‐cgacctcaagctgtgcgctcttccaattatgctt‐3′. MycABRO1 contains N‐terminal 3xMyc epitope‐tagged ABRO1 cloned into pCMV‐3Tag‐2A vector. To generate shRNA‐resistant MycABRO1, silent mutations were introduced into the shAbro1 target sequence (CAGAGCCTTCTAATAGTGAAT) using the oligonucleotide 5′‐gattgaccctacagagcctagtaacagtgaatactcacatt‐3′. MycRNF8 contains N‐terminal 3xMyc epitope‐tagged RNF8 cloned into pCMV‐3Tag‐2A vector. To generate shRNA‐resistant Myc‐RNF8, silent mutations were introduced into the shRNF8 target sequence (CCAAAGAATGACCAAATGATA) by using the oligonucleotide 5′‐ctttccccaaagaacgatcaaatgatagaaaaaaataag‐3′. The RING C403S mutation was generated using the oligonucleotide 5′‐gctagagaatgagctccaaagtattatttgttcagaatac‐3′.
Mutagenesis was performed using the Stratagene QuikChange site‐directed mutagenesis kit according to the manufacturer's instructions. shRNA lentiviral vectors contained sequences targeting GFP, RNF8 (TRCN0000003441), ABRO1 (TRCN0000133846), BRCC36 (TRCN0000073970), and DNA ligase IV (LigIV‐45 TRCN0000009847 and LigIV‐47 TRCN0000040003) from Thermo Scientific.
Cell lines
HeLaI.2.11 (van Steensel et al, 1998), HTC75 (van Steensel & de Lange, 1997), and MCF7 cells (ATCC) were grown under standard conditions.
Cell synchronization and inhibitor treatments
For release across the cell cycle, cells were grown in the presence of 2 mM thymidine for 16 h, washed three times with phosphate‐buffered saline (PBS), released into fresh medium for 10 h, treated again with 2 mM thymidine for 16 h, washed three times with PBS, released into fresh medium, and harvested by trypsinization at 2‐h intervals. For transfection of synchronized cells, cells were transfected prior to the second thymidine treatment.
For cell cycle arrest, cells were synchronized in G1 by incubation with 400 μM L‐mimosine (Sigma) for 18 h, in S phase by incubation in 2 mM hydroxyurea (HU) (Sigma) for 20 h, in G2 by incubation in 9 μM R03306 (Cal Biochem) for 18 h, or in mitosis by incubation in 30 ng/ml nocodazole (Sigma) for 20 h. For nocodazole release, cells were arrested in mitosis by incubation in 30 ng/ml nocodazole for 20 h, isolated by mitotic shake‐off, replated, and harvested at 0, 90, and 120 min.
To inhibit protein synthesis cells were treated with cycloheximide (Sigma) (100 μg/ml) and harvested after 0, 3, and 6 h prior to harvesting. For ATM inhibition, cells were treated with KU‐55933 (Tocris Bioscience) (10 μM) for 4 h prior to harvesting. To inhibit the proteasome, cells were treated with proteasome inhibitor MG132 (Boston Biochem) (10 μM) for 4 h prior to harvesting. For inhibition of nuclear export, cells were treated with leptomycin B (Sigma) (10 ng/ml) for 3 h prior to harvest. To generate DNA damage cells were treated with phleomycin (Invivogen) (10 or 20 μg/ml) for 4 h prior to harvesting.
Lentiviral infection
Lentiviruses were produced by transfection of 293FT (Invitrogen) packaging cells with a three‐plasmid system as described previously (Naldini et al, 1996; Zufferey et al, 1997). 293FT cells were seeded in a 6‐cm dish at 1.2 × 106 cells and 24 h later were transfected with 1 μg lentiviral vector, 1 μg pCMVΔR.89 packaging plasmid, and 100 ng pMD.G envelope plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Lentiviral supernatants were collected at 48 h after transfection, filtered with a 0.45‐μm filter (Millipore), and frozen at −80°C. Twenty‐four hours before infection, target cells were seeded at a density of 2.2 × 105. Target cells were infected for 48–72 h with lentiviral supernatants supplemented with 8 μg/ml polybrene (Sigma‐Aldrich). Cells were subcultured 1:2 into medium containing 2 μg/ml puromycin.
Plasmid and siRNA transfection
For plasmid transfection, cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol for 18–20 h. When inhibitor treatment was employed, cells were transfected for 26 h and inhibitors were added 20 h prior to harvest. For siRNA transfection, cells were transfected with Ubc13 siRNA (final concentration 200 nM) (Santa Cruz, sc‐43551) (Wang et al, 2006) or GFP Duplex I (Dharmacon Research Inc.) with Oligofectamine (Invitrogen) according to the manufacturer's protocol for 24 h, followed by transfection with FlagTNKS1 and HA‐K63‐Ub for an additional 24 h.
Cell fractionation
For whole‐cell extracts, cells were resuspended in 4 volumes of TNE buffer [10 mM Tris (pH 7.8), 1% Nonidet P‐40, 0.15 M NaCl, 1 mM EDTA, and 2.5% protease inhibitor cocktail (PIC) (Sigma)] and incubated for 1 h on ice. Suspensions were pelleted at 8,000 × g for 15 min. Equal amounts of supernatant proteins (determined by Bio‐Rad protein assay) were fractionated by SDS–PAGE and analyzed by immunoblotting.
For nuclear and cytosol extracts, PBS‐washed cell pellets were washed with 5 mM MgCl2–PBS and with buffer A [10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 20% glycerol, 1 mM dithiothreitol (DTT), and PIC]. The pellet was resuspended in buffer A and homogenized on ice with a Dounce homogenizer. After centrifugation at 3,220 × g for 10 min at 4°C, the supernatant was collected as the cytoplasmic fraction. The resulting nuclear pellet was then washed in buffer A, resuspended in TNE buffer, and incubated for 30 min on ice. After centrifugation at 12,000 × g for 10 min at 4°C, the supernatant was collected as the nuclear fraction.
When extracts were to be used for analysis of ubiquitinated proteins, 4 mM N‐ethylmaleimide (Sigma) and 4 mM 1,10‐phenanthroline (Sigma) were included in the buffers.
Immunoblot analysis
Immunoblots were incubated separately with the following primary antibodies: rabbit anti‐tankyrase 1 762 (1 μg/ml) (Scherthan et al, 2000); rabbit anti‐Flag (1 μg/ml) (Sigma‐Aldrich, F7425); rabbit c‐Myc (0.2 μg/ml) (Santa Cruz, sc‐789); mouse anti‐α‐tubulin ascites (1:10,000) (Sigma‐Aldrich, T5768); mouse cyclin B1 (0.2 μg/ml) (Santa Cruz, sc‐245); rabbit phospho‐H3 (1 μg/ml) (Millipore, 06‐570); rabbit HA (0.5 μg/ml) (Abcam, ab9110); rabbit anti‐RNF8 (1 μg/ml) (Abcam, ab4183); rabbit lamin B serum (1:1,000) (Chaudhary & Courvalin, 1993); rabbit anti‐BRCC36 (0.5 μg/ml) (Bethyl, A302‐517A); rabbit anti‐ABRO1 (1 μg/ml) (Bethyl, A301‐255A); and rabbit anti‐ubiquitin Lys63‐specific clone Apu3 (1:1,000) (Millipore, 05‐1308), followed by horseradish peroxidase‐conjugated donkey anti‐rabbit or anti‐mouse IgG (Amersham) (1:2,500). Bound antibody was detected with SuperSignal West Pico (Thermo Scientific).
In vitro ubiquitination
In vitro ubiquitination reactions were performed in 20‐μl reaction mixtures containing ubiquitination buffer (50 mM Tris [pH 8], 5 mM MgCl2, 1 mM dithiothreitol [DTT], and 2 mM ATP), 100 ng human recombinant E1 UBE1 (Boston Biochem), 300 ng human recombinant E2 (UBC13) UBE2N/UBE2V2 (Boston Biochem), 300 ng of human recombinant GST‐RNF8 (Abnova), 10 μg of ubiquitin or K63‐ubiquitin (Boston Biochem), and 250 ng tankyrase 1 (Trevigen) or GMD (Bisht et al, 2012). Reactions were incubated at 37°C for 3 h, terminated by addition of 2× SDS loading buffer, and processed for immunoblot analysis with rabbit anti‐Ub antibody (Dako, Z0458) (1:300).
Immunoprecipitation
Cells were lysed as above and supernatants precleared with protein G–Sepharose rotating at 4°C for 30 min. Nonspecific protein aggregates were removed by centrifugation, and the supernatant was used for immunoprecipitation analysis or fractionated directly on SDS–PAGE (indicated as input, ∼5% of the amount used in the immunoprecipitation).
For immunoprecipitation of Flag‐ or HA epitope‐tagged proteins, supernatants were incubated with 15 μl of Flag M2 agarose (Sigma) and anti‐HA affinity matrix (Roche) for 3 h. For immunoprecipitation of endogenous proteins, supernatants were incubated with the following primary antibodies: 3 μl rabbit anti‐ubiquitin Lys63‐specific clone Apu3 (Millipore, 05‐1308); 1.5 μg/ml rabbit anti‐RNF8 (Abcam, ab4183); 1 μg/ml rabbit anti‐tankyrase 1 762 (Scherthan et al, 2000); or 1 μg/ml rabbit anti‐BRCC36 (Bethyl, A302‐517A), followed by protein G–Sepharose. For all immunoprecipitations, beads were washed three times with 1 ml of TNE buffer, fractionated by SDS–PAGE, and processed for immunoblotting as described above.
Chromatin immunoprecipitation
Cells were processed for ChIP as described previously (Bisht et al, 2012). Following preparation of cell lysates, a fraction was saved as input and immunoprecipitation was performed by addition of the following antibodies: 2 μg of rabbit IgG; 2 μg of rabbit anti‐tankyrase 1 762 (Scherthan et al, 2000); and 5 μl of rabbit TRF1 415 crude sera (Cook et al, 2002). Hybridization with a 32P‐TTAGGG probe was performed in Church buffer [0.5 M sodium phosphate buffer (pH 7.2), 1% BSA, 1 mM EDTA, 7% SDS] as described previously (de Lange, 1992). Washed membranes were imaged using PhosphorImager Storm860. ImageJ software was used to quantify the percentage of telomeric DNA immunoprecipitated by anti‐TNKS1 antibody.
Chromosome‐specific FISH
Cells were fixed and processed as described previously (Dynek & Smith, 2004). Briefly, cells were fixed twice in methanol:acetic acid (3:1) for 15 min, cytospun (Shandon Cytospin) at 900× g for 2 min onto slides, rehydrated in 2× SSC at 37°C for 2 min, and dehydrated in an ethanol series of 70, 80, and 95% for 2 min each. Cells were denatured at 75°C for 2 min and hybridized overnight at 37°C with a subtelomeric FITC‐conjugated probe 16ptelo from Cytocell. Cells were washed in 0.4× SSC at 72°C for 2 min, and in 2× SSC with 0.05% Tween‐20 at RT for 30 s. DNA was stained with 0.2 μg/ml DAPI.
PNA FISH of metaphase spreads
Cells were treated with 0.5 μg/ml colcemid for 4 h, harvested by trypsinization, hypotonically swollen in 10 mM Tris, pH 7.4, 10 mM NaCl, and 5 mM MgCl2 for 10 min at 37°C, and fixed twice for 15 min in methanol:acetic acid (3:1). Metaphase spreads were prepared by dropping fixed cells on coverslips. Cells on coverslips were rehydrated in PBS for 5 min and fixed in 3.7% formaldehyde for 2 min. Cells were washed three times in PBS, dehydrated in an ethanol series of 70, 95, and 100%, and denatured at 80°C for 3 min in hybridization mix [0.5 μM of a Cy3‐conjugated (CCCTAA)3 PNA probe (PNAbio) in 10 mM Tris, pH 7.2, 70% formamide, 0.5% blocking reagent (Roche)]. Cells were then hybridized for 2 h at room temperature. Cells were washed twice for 15 min in 70% formamide, 10 mM Tris, pH 7.2, and 0.1% BSA and were washed three times for 5 min in 0.1 M Tris, pH 7.2, 0.15 M NaCl, and 0.08% Tween‐20. After the washes, cells were dehydrated in an ethanol series of 70, 95, and 100%, and DNA was counterstained with 0.5 μg/ml DAPI.
Image acquisition
Images were acquired using a microscope (Axioplan 2; Carl Zeiss, Inc.) with a Plan‐Apochromat 63× NA 1.4 oil immersion lens (Carl Zeiss, Inc.) and a digital camera (C4742‐95; Hamamatsu Photonics). Images were acquired and processed using Openlab software (Perkin Elmer).
Statistical analysis
Statistical analysis was performed using Prism 6 software. Student's unpaired t‐test was applied. Data are shown as mean ± SEM (standard error of the mean) or as mean ± SD (standard deviation); P < 0.05 values were considered significant.
Author contributions
ET and SS contributed to the conception and design of the study. ET performed the experiments. ET and SS contributed to data analysis and interpretation. SS drafted the manuscript. ET and SS contributed to editing of the manuscript. SS obtained funding and supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Review Process File
Acknowledgements
We thank the Smith Lab for critical reading of the manuscript, Kameron Azarm for technical assistance, Roger Greenberg, Adolfo Sastre, and Timothy Thomson for providing plasmids, and Richard Giannone and Yie Liu for MudPIT analysis. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number R01CA116352.
The EMBO Journal (2017) 36: 503–519
References
- Al‐Hakim A, Escribano‐Diaz C, Landry MC, O'Donnell L, Panier S, Szilard RK, Durocher D (2010) The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair 9: 1229–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambivero CT, Cilenti L, Zervos AS (2012) ATF4 interacts with Abro1/KIAA0157 scaffold protein and participates in a cytoprotective pathway. Biochim Biophys Acta 1823: 2149–2156 [DOI] [PubMed] [Google Scholar]
- Bartocci C, Denchi EL (2013) Put a RING on it: regulation and inhibition of RNF8 and RNF168 RING finger E3 ligases at DNA damage sites. Front Genet 4: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker‐Jensen S, Mailand N (2010) Assembly and function of DNA double‐strand break repair foci in mammalian cells. DNA Repair 9: 1219–1228 [DOI] [PubMed] [Google Scholar]
- Bisht KK, Dudognon C, Chang WG, Sokol ES, Ramirez A, Smith S (2012) GDP‐mannose‐4,6‐dehydratase is a cytosolic partner of tankyrase 1 that inhibits its poly(ADP‐ribose) polymerase activity. Mol Cell Biol 32: 3044–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisht KK, Daniloski Z, Smith S (2013) SA1 binds directly to DNA through its unique AT‐hook to promote sister chromatid cohesion at telomeres. J Cell Sci 126: 3493–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN, Liu PS, Bheddah S, Tao J, Lill JR, Hongo JA, Davis D, Kirpatrick DS, Polakis P, Costa M (2011) Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS One 6: e22595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Houghtaling BR, Kim JY, Dynek JN, Chang WG, Smith S (2007) Protein requirements for sister telomere association in human cells. EMBO J 26: 4867–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Smith S (2009) Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J Cell Biol 187: 165–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Houghtaling BR, Bhanot M, Sasa G, Savage SA, Bertuch AA, Smith S (2011) A role for heterochromatin protein 1{gamma} at human telomeres. Genes Dev 25: 1807–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary N, Courvalin JC (1993) Stepwise reassembly of the nuclear envelope at the end of mitosis. J Cell Biol 122: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33: 275–286 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Schwartz AL (2004) The ubiquitin system: pathogenesis of human diseases and drug targeting. Biochim Biophys Acta 1695: 3–17 [DOI] [PubMed] [Google Scholar]
- Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbe S (2013) Deubiquitylases from genes to organism. Physiol Rev 93: 1289–1315 [DOI] [PubMed] [Google Scholar]
- Cook BD, Dynek JN, Chang W, Shostak G, Smith S (2002) Role for the related poly(ADP‐Ribose) polymerases tankyrase 1 and 2 at human telomeres. Mol Cell Biol 22: 332–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, Cohen RE (2009) K63‐specific deubiquitination by two JAMM/MPN+ complexes: BRISC‐associated Brcc36 and proteasomal Poh1. EMBO J 28: 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EM, Boeke JD, Cohen RE (2010) Specificity of the BRISC deubiquitinating enzyme is not due to selective binding to Lys63‐linked polyubiquitin. J Biol Chem 285: 10344–10352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Hakimi MA, Chen X, Kumaraswamy E, Cooch NS, Godwin AK, Shiekhattar R (2003) Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome‐like subunit and its role in DNA repair. Mol Cell 12: 1087–1099 [DOI] [PubMed] [Google Scholar]
- Dynek JN, Smith S (2004) Resolution of sister telomere association is required for progression through mitosis. Science 304: 97–100 [DOI] [PubMed] [Google Scholar]
- Eletr ZM, Wilkinson KD (2014) Regulation of proteolysis by human deubiquitinating enzymes. Biochim Biophys Acta 1843: 114–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wang J, Chen J (2010) The Lys63‐specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J Biol Chem 285: 30982–30988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SY (2012) Ubiquitination‐mediated regulation of interferon responses. Growth Factors 30: 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone RJ, McDonald HW, Hurst GB, Shen RF, Wang Y, Liu Y (2010) The protein network surrounding the human telomere repeat binding factors TRF1, TRF2, and POT1. PLoS One 5: e12407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999) Mammalian telomeres end in a large duplex loop. Cell 97: 503–514 [DOI] [PubMed] [Google Scholar]
- Guettler S, LaRose J, Petsalaki E, Gish G, Scotter A, Pawson T, Rottapel R, Sicheri F (2011) Structural basis and sequence rules for substrate recognition by Tankyrase explain the basis for cherubism disease. Cell 147: 1340–1354 [DOI] [PubMed] [Google Scholar]
- Hofmann RM, Pickart CM (1999) Noncanonical MMS2‐encoded ubiquitin‐conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 96: 645–653 [DOI] [PubMed] [Google Scholar]
- Houghtaling BR, Canudas S, Smith S (2012) A role for sister telomere cohesion in telomere elongation by telomerase. Cell Cycle 11: 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao SJ, Smith S (2009) Sister telomeres rendered dysfunctional by persistent cohesion are fused by NHEJ. J Cell Biol 184: 515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Kim JA, Castillo A, Huang M, Liu J, Wang B (2011) NBA1/MERIT40 and BRE interaction is required for the integrity of two distinct deubiquitinating enzyme BRCC36‐containing complexes. J Biol Chem 286: 11734–11745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J (2007) RNF8 transduces the DNA‐damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131: 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X (2007a) Ubiquitin‐binding protein RAP80 mediates BRCA1‐dependent DNA damage response. Science 316: 1202–1205 [DOI] [PubMed] [Google Scholar]
- Kim H, Huang J, Chen J (2007b) CCDC98 is a BRCA1‐BRCT domain‐binding protein involved in the DNA damage response. Nat Struct Mol Biol 14: 710–715 [DOI] [PubMed] [Google Scholar]
- Kim MK, Smith S (2014) Persistent telomere cohesion triggers a prolonged anaphase. Mol Biol Cell 25: 30–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D (2007) Orchestration of the DNA‐damage response by the RNF8 ubiquitin ligase. Science 318: 1637–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81: 203–229 [DOI] [PubMed] [Google Scholar]
- de Lange T (1992) Human telomeres are attached to the nuclear matrix. EMBO J 11: 717–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Bekker‐Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (2007) RNF8 ubiquitylates histones at DNA double‐strand breaks and promotes assembly of repair proteins. Cell 131: 887–900 [DOI] [PubMed] [Google Scholar]
- Martinez P, Blasco MA (2015) Replicating through telomeres: a means to an end. Trends Biochem Sci 40: 504–515 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Bartocci C, Ouzounov I, Diedrich JK, Yates JR III, Denchi EL (2013) A two‐step mechanism for TRF2‐mediated chromosome‐end protection. Nature 494: 502–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Fradet‐Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano‐Diaz C, Durocher D (2014) Mitosis inhibits DNA double‐strand break repair to guard against telomere fusions. Science 344: 189–193 [DOI] [PubMed] [Google Scholar]
- O'Sullivan RJ, Karlseder J (2010) Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 11: 171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T (2008) How shelterin protects mammalian telomeres. Annu Rev Genet 42: 301–334 [DOI] [PubMed] [Google Scholar]
- Patterson‐Fortin J, Shao G, Bretscher H, Messick TE, Greenberg RA (2010) Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J Biol Chem 285: 30971–30981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Nishiyama T (2012) Sister chromatid cohesion. Cold Spring Harb Perspect Biol 4: a011130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peuscher MH, Jacobs JJ (2011) DNA‐damage response and repair activities at uncapped telomeres depend on RNF8. Nat Cell Biol 13: 1139–1145 [DOI] [PubMed] [Google Scholar]
- Rai R, Li JM, Zheng H, Lok GT, Deng Y, Huen MS, Chen J, Jin J, Chang S (2011) The E3 ubiquitin ligase Rnf8 stabilizes Tpp1 to promote telomere end protection. Nat Struct Mol Biol 18: 1400–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S, Cuadrado A, Carretero M, Martinez P, Drosopoulos WC, Canamero M, Schildkraut CL, Blasco MA, Losada A (2012) Cohesin‐SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J 31: 2076–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP (2008) TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet 82: 501–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherthan H, Jerratsch M, Li B, Smith S, Hulten M, Lock T, de Lange T (2000) Mammalian meiotic telomeres: protein composition and redistribution in relation to nuclear pores. Mol Biol Cell 11: 4189–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138: 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao G, Lilli DR, Patterson‐Fortin J, Coleman KA, Morrissey DE, Greenberg RA (2009) The Rap80‐BRCC36 de‐ubiquitinating enzyme complex antagonizes RNF8‐Ubc13‐dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci USA 106: 3166–3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren C, Nasmyth K (2001) Sister chromatid cohesion is required for postreplicative double‐strand break repair in Saccharomyces cerevisiae. Curr Biol 11: 991–995 [DOI] [PubMed] [Google Scholar]
- Smith S, Giriat I, Schmitt A, de Lange T (1998) Tankyrase, a poly(ADP‐ribose) polymerase at human telomeres. Science 282: 1484–1487 [DOI] [PubMed] [Google Scholar]
- Smith S (2015) TIPs: tankyrase Interacting Proteins In PARP inhibitors for cancer therapy, Curtin N, Sharma R. (eds), pp 79–98. New York: Springer; [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA (2007) RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316: 1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, de Lange T (1997) Control of telomere length by the human telomeric protein TRF1. Nature 385: 740–743 [DOI] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T (1998) TRF2 protects human telomeres from end‐to‐end fusions. Cell 92: 401–413 [DOI] [PubMed] [Google Scholar]
- Thorslund T., Ripplinger A., Hoffmann S., Wild T., Uckelmann M., Villumsen B., Narita T., Sixma T.K., Choudhary C., Bekker‐Jensen S., Mailand N (2015) Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527: 389–393 [DOI] [PubMed] [Google Scholar]
- Verdun RE, Crabbe L, Haggblom C, Karlseder J (2005) Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell 20: 551–561 [DOI] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I (2008) TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 112: 3594–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, Hung MC (2006) Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol 8: 1359–1368 [DOI] [PubMed] [Google Scholar]
- Wang B, Elledge SJ (2007) Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA 104: 20759–20763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ (2007) Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316: 1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, Myer VE, Finan PM, Porter JA, Huang SM, Cong F (2011) RNF146 is a poly(ADP‐ribose)‐directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13: 623–629 [DOI] [PubMed] [Google Scholar]
- Zhang J, Cao M, Dong J, Li C, Xu W, Zhan Y, Wang X, Yu M, Ge C, Ge Z, Yang X (2014) ABRO1 suppresses tumourigenesis and regulates the DNA damage response by stabilizing p53. Nat Commun 5: 5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XD, Kuster B, Mann M, Petrini JH, Lange T (2000) Cell‐cycle‐regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres [In Process Citation]. Nat Genet 25: 347–352 [DOI] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo . Nat Biotechnol 15: 871–875 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Review Process File