

Abstract
Yes‐associated protein (YAP) and myocardin‐related transcription factor (MRTF) play similar roles and exhibit significant crosstalk in directing transcriptional responses to chemical and physical extracellular cues. The mechanism underlying this crosstalk, however, remains unclear. Here, we show MRTF family proteins bind YAP via a conserved PPXY motif that interacts with the YAP WW domain. This interaction allows MRTF to recruit NcoA3 to the TEAD‐YAP transcriptional complex and potentiate its transcriptional activity. We show this interaction of MRTF and YAP is critical for LPA‐induced cancer cell invasion in vitro and breast cancer metastasis to the lung in vivo. We also demonstrate the significance of MRTF‐YAP binding in regulation of YAP activity upon acute actin cytoskeletal damage. Acute actin disruption induces nucleo‐cytoplasmic shuttling of MRTF, and this process underlies the LATS‐independent regulation of YAP activity. Our results provide clear evidence of crosstalk between MRTF and YAP independent of the LATS kinases that normally act upstream of YAP signaling. Our results also suggest a mechanism by which extracellular stimuli can coordinate physiological events downstream of YAP.
Keywords: cytoskeletal damage, metastasis, MRTF, TEAD‐YAP
Subject Categories: Cancer, Signal Transduction, Transcription
Introduction
A cell's fate—its proliferation and differentiation—is influenced by extracellular cues from its microenvironment. Growth factors, contact with neighboring cells, and the physical stiffness of the extracellular matrix all contribute to cellular morphogenesis, function, and division (Joyce, 2005; Scadden, 2006). This means cellular fate determination requires both extracellular detectors of environmental change and intracellular signaling networks that translate these changes to appropriate physiological responses. As so many of these physiological responses comprise significant shifts in gene expression, transcription factors are central players in the intracellular signaling networks that respond to most extracellular cues.
YAP is an oncogene involved in stem/progenitor cell expansion as well as cancer development and progression (Zhao et al, 2011). YAP is an effector and transcriptional coactivator in the Hippo tumor suppressor pathway. It activates transcriptional programs that control tissue homeostasis by inhibiting differentiation and promoting adult stem/progenitor cell proliferation (Yu & Guan, 2013). As an oncogene, YAP is often over‐expressed in various types of cancers (Overholtzer et al, 2006; Zender et al, 2006). Both transgenic mice over‐expressing wild‐type YAP and Hippo pathway mutant mice with hyperactive YAP show undifferentiated stem/progenitor cell expansions in epithelial organs (e.g., lung, liver, skin, and intestine) (Lee et al, 2008; Schlegelmilch et al, 2011; Zhou et al, 2011; Yimlamai et al, 2014) that subsequently develop into tumors. YAP also induces many of the hallmarks of cancer: proliferation, anchorage‐independent growth, cellular invasion and metastasis, as well as tumor initiation activity (Overholtzer et al, 2006; Cordenonsi et al, 2011; Lamar et al, 2012). Although YAP binds several transcription factors (e.g. RUNX3, p73, AP‐1, SRF, and TEAD; Strano et al, 2001; Vassilev et al, 2001; Zaidi et al, 2004; Shao Diane et al, 2014; Kim et al, 2015), TEAD seems to play the major role in YAP's oncogenic functions (Zhao et al, 2008). Consistent with this, pharmacological or genetic disruptions of TEAD‐YAP binding dramatically reduce YAP‐induced oncogenesis (Liu‐Chittenden et al, 2012).
TEAD‐YAP has recently received considerable attention as a central transcriptional mediator of responses to both chemical and physical extracellular cues (Halder et al, 2012). YAP activity is altered by cell‐to‐cell contacts, actomyosin tension, and RhoA‐activating G‐protein‐coupled receptor (GPCR) ligands like lysophosphatidic acid (LPA) (Dupont et al, 2011; Yu et al, 2012). We and others have shown extracellular cues that affect the actin cytoskeleton can alter LATS activity, which subsequently affects YAP localization and activity (Zhao et al, 2012; Kim et al, 2013). Indeed, actin‐disrupting chemicals do not affect the localization of a mutant form of YAP that is refractory to LATS‐mediated phosphorylation (Zhao et al, 2012; Kim et al, 2013). Using LATS1/2‐knockout mouse embryonic fibroblasts (MEFs), we also confirmed LATS is required for YAP export upon disruption of the actin cytoskeleton (see below). Piccolo and colleagues, however, observed reduced YAP target gene expression upon actin‐disrupting chemical treatment even with a mutant form of YAP that cannot be phosphorylated by LATS (Cordenonsi et al, 2011; Aragona et al, 2013). This suggests the existence of a LATS‐independent mechanism for regulating YAP activity in response to changes in the actin cytoskeleton. The identification of such a LATS‐independent mechanism would be a significant advance in our understanding of YAP and its control of cellular differentiation and proliferation.
We noticed the mechanisms of YAP regulation are similar to those that regulate myocardin‐related transcription factor (MRTF) family proteins, which are SRF transcriptional coactivators (Wang et al, 2002). Similar to YAP, MRTF is translocated to the nucleus upon actin cytoskeletal assembly and RhoA activation by GPCR ligands (Miralles et al, 2003). MRTF and YAP both promote epithelial–mesenchymal transition and cancer metastasis (Morita et al, 2007; Medjkane et al, 2009; Lamar et al, 2012). As various extracellular stimuli induce the nuclear localization and activation of both MRTF and YAP, we speculated there may be crosstalk between YAP and MRTF. Consistent with this hypothesis, MRTF is reportedly enriched at genomic loci containing TEAD‐binding sequences (Esnault et al, 2014). In addition, MRTFA and YAP bind and together promote the expression of RhoA‐regulated genes induced by GPCR activation (Yu et al, 2015). Another recent study showed MRTF and TAZ regulate one another's activity (Speight et al, 2016). Together, these studies suggest SRF‐MRTF and TEAD‐YAP may act together to coordinate transcriptional responses to various extracellular stimuli. Unfortunately, these previous studies have several limitations. For example, these studies neither addressed the molecular mechanisms of MRTF‐YAP binding nor of TEAD‐YAP activity regulation, especially at the level of the transcriptome. The in vivo significance of MRTF‐YAP binding is also unclear.
In this study, we examined the crosstalk between MRTF and YAP. We discovered MRTF binding to YAP recruits the NCOA3 transcriptional coactivator, enhancing TEAD‐YAP target gene expression. We found MRTF‐YAP binding is required in vitro for LPA‐induced cancer cell invasion and in vivo for the metastasis of 4T1 breast cancer cells. We also implicated cytoplasmic MRTF translocation in the LATS‐independent regulation of YAP that occurs upon disruption of the actin cytoskeleton. In summary, this study characterizes the molecular interaction between MRTF and YAP that allows TEAD‐YAP to coordinate transcriptional responses to various extracellular stimuli.
Results
MRTF proteins activate TEAD‐YAP transcriptional activity independent of the Hippo pathway
To determine whether MRTF proteins affect TEAD‐YAP activity, we first co‐expressed MRTFA and MRTFB (hereafter collectively referred to as MRTF) with YAP in the presence of a TEAD luciferase reporter containing eight tandem TEAD‐binding DNA motifs (5′‐CATTCC‐3′, TBS‐luc). We found MRTF over‐expression induces a marked up‐regulation of the TEAD luciferase reporter's activity (Fig 1A). We next monitored genome‐wide changes in YAP target gene expression upon MRTF over‐expression. To do this, we performed RNA sequencing on MCF‐10A cells over‐expressing either a hyperactive YAP (YAP 5SA) or a hyperactive MRTFB. MRTFB ΔN lacks the RPEL domains that induces its cytoplasmic sequestration upon binding to globular actin (Miralles et al, 2003). We found a significant overlap between the gene sets induced by over‐expression of YAP and those induced by over‐expression of MRTFB (Fig 1B). According to a gene set enrichment analysis (GSEA) of MRTF‐induced targets, MRTFB over‐expression induces up‐regulation of known YAP target genes (Fig 1C). We next validated our RNA sequencing results using qPCR. Of the YAP target genes up‐regulated by over‐expression of hyperactive MRTFB, some are known targets of SRF‐MRTF (e.g., CTGF, CYR61) while others are not (e.g., PTGS2, EDN1, TGFB2) (Fig 1D and E). In addition, we found MRTFA and/or MRTFB depletion additively down‐regulate YAP target genes (Fig 1F–H).
Figure 1. MRTF enhances TEAD‐YAP transcriptional activity.
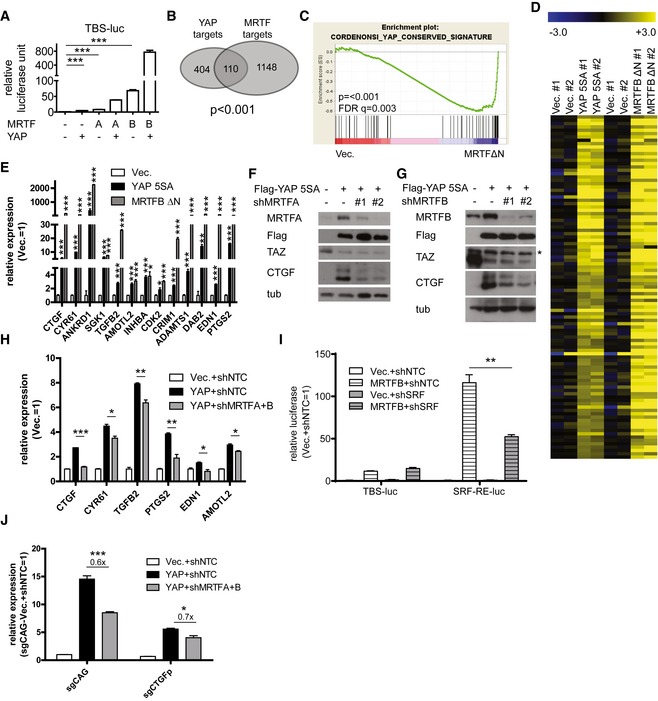
-
ATEAD luciferase reporter assay in 293T cells expressing MRTFA/B and/or YAP (n = 3). Note that the TEAD luciferase reporter construct lacks any putative SRF‐binding DNA motif.
-
BGenes up‐regulated by either YAP or MRTF over‐expression in the RNA sequencing results.
-
CGSEA for MCF‐10A cells over‐expressing a control or constitutively active MRTF.
-
DHeatmap of the RNA sequencing results for MCF‐10A cells over‐expressing either hyperactive YAP or MRTF. Data are presented as fold change relative to average expression level of control cells in Log2.
-
EqPCR analysis of representative TEAD‐YAP target genes in MCF‐10A cells over‐expressing the indicated genes (n = 3).
-
F, GWestern blots from MCF‐10A cells over‐expressing YAP and treated with shRNAs against (F) MRTFA or (G) MRTFB. Asterisk indicates non‐specific band.
-
HqPCR analysis of representative TEAD‐YAP target genes in MCF‐10A cells infected with the indicated viruses (n = 3).
-
ITEAD‐YAP activity reporter (TBS‐luc) and SRF‐MRTF activity reporter (SRF‐RE‐luc) luciferase assay with 293T cells expressing MRTFB and shRNA against SRF (n = 3).
-
JqPCR analysis of CTGF expression in MCF‐10A cells treated with a control guide RNA (sgCAG) or a guide RNA against the SRF‐binding region of the CTGF promoter and infected with the indicated viruses (n = 3).
Since many of the TEAD‐YAP targets, including CTGF and CYR61, are known SRF‐MRTF targets, changes in their expression may merely reflect changes in SRF‐MRTF activity. We first tested whether SRF‐MRTF activity can influence TEAD‐YAP activity by performing TEAD‐activity and SRF‐activity luciferase reporter assays in the presence of MRTFB over‐expression and SRF depletion. We found that while SRF depletion significantly attenuates the activity of the SRF luciferase reporter that is induced by MRTFB, it has no effect on the TEAD‐activity reporter. This suggests MRTF regulates the activity of TEAD‐YAP independent of SRF‐MRTF complex formation (Fig 1I). To confirm MRTF proteins regulate TEAD‐YAP activity, we performed a luciferase reporter assay using a fragment of the CTGF promoter containing its TEAD‐binding sequence but lacking its distal SRF‐binding site (Muehlich et al, 2007). As expected, MRTFB over‐expression potently activates the activity of this TEAD reporter (Fig EV1A). In further confirmation, we used the CRISPR/Cas9 system to make a targeted mutation of the endogenous SRF‐binding site within the CTGF promoter (Fig EV1B). We found CTGF expression in these CTGF promoter mutant cells is insensitive to the SRF‐MRTF activator cytochalasin D (Fig EV1B). YAP induction of CTGF, in contrast, is unaffected in these cells, suggesting normal responsiveness to TEAD‐YAP. Intriguingly, MRTF depletion still reduces CTGF expression in these CTGF promoter mutant cells, presumably due to down‐regulation of TEAD‐YAP activity (Fig 1J). We acknowledge the contribution of SRF‐MRTF activity to the expression of many TEAD‐YAP targets, but these data suggest MRTF can, at least in part, directly regulate TEAD‐YAP transcriptional targets independent of SRF.
Figure EV1. SRF activity‐independent regulation of TEAD‐YAP target genes by MRTF.
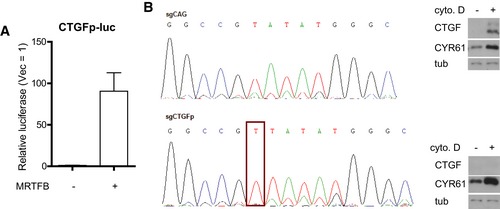
- CTGF luciferase reporter assay in 293T cells expressing MRTFB. Data are presented as means ± s.e.m. (n = 3).
- (left) Genomic sequencing results for MCF‐10A cells treated with CRISPR‐Cas9 against control (sgCAG) or SRF‐binding sequences in the CTGF promoter. A successful mutation in the SRF‐binding motif (CArG box) is outlined in red. (right) Western blot confirming CTGF promoter mutant MCF‐10A cells are unresponsive to SRF by treating them with cytochalasin D (cyto. D, 1 μM, 30 min).
We next investigated the mechanism by which TEAD‐YAP activity is regulated by MRTF. Since YAP is mainly regulated by phosphorylation leading to cytoplasmic sequestration by the Hippo pathway, we asked whether MRTF affects YAP phosphorylation or localization. While MRTF depletion does not alter YAP phosphorylation in MDA‐MB‐231 cells, it reduces both YAP target gene expression and the activity of a TEAD luciferase reporter (Fig 2A). In contrast to recent reports, we were unable to observe any change in TAZ expression upon MRTF depletion (Fig 2A) (Liu et al, 2016a; Speight et al, 2016). Consistent with this, neither MRTF depletion nor MRTFB over‐expression changes the nucleo‐cytoplasmic localization of YAP or TAZ (Figs 2B–D and EV2A). Next, we performed similar experiments in LATS1/2‐knockout MEFs and found MRTF depletion still reduces the expression of TEAD‐YAP target genes (Fig 2E). This confirms MRTF depletion alters TEAD‐YAP activity independent of YAP phosphorylation by LATS. We also used the CRISPR/Cas9 system to knockout LATS1/2 in 293T cells. Similar to the results shown in Fig 2E, MRTF depletion in these cells still reduces TEAD‐YAP target gene expression and TEAD‐YAP luciferase reporter activity (Fig EV2B). These results together suggest MRTF regulates TEAD‐YAP activity independent of the canonical Hippo pathway, especially the LATS‐dependent phosphorylation and translocation of YAP.
Figure 2. MRTF‐mediated regulation of YAP is independent of YAP localization.
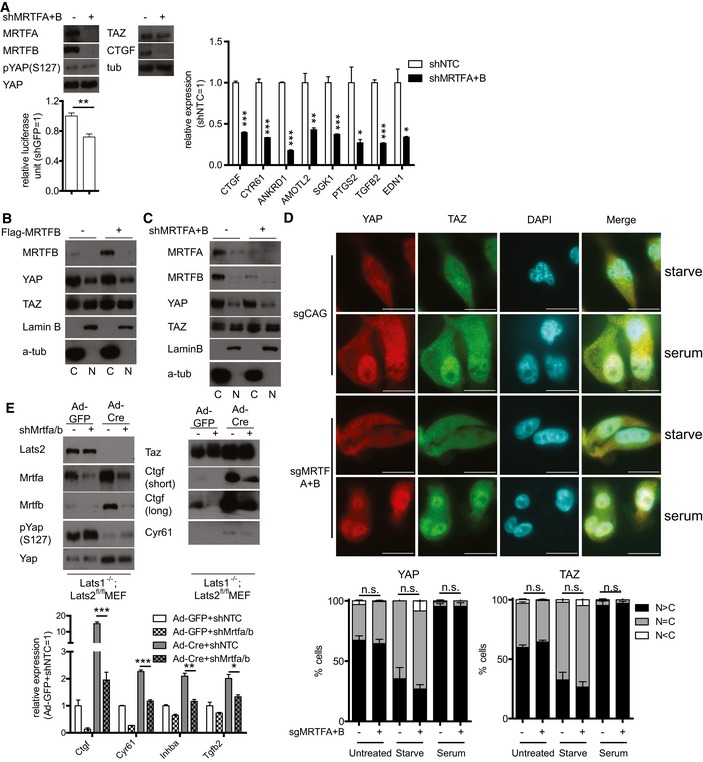
-
A(top left) Western blots, (bottom left) TEAD luciferase reporter assay, and (right) qPCR (n = 3) analysis of representative TEAD‐YAP target genes in MDA‐MB‐231 cells expressing either control shRNAs or shRNAs against MRTFA and MRTFB.
-
B, CNuclear‐cytoplasmic fractionation of MDA‐MB‐231 cells (B) over‐expressing MRTFB or (C) depleted of MRTFA and MRTFB.
-
DRepresentative images of control (sgCAG) or MRTF‐knockout (sgMRTFA+B) MDA‐MB‐231 cells. Note the localization of YAP/TAZ. This localization of YAP (left) and TAZ (right) is quantified below (n = 3, Scale bars: 10 μm).
-
E(top) Western blots and (bottom) qPCR (n = 3) analysis of Lats1/2‐floxed MEFs treated with adenovirus‐expressing Cre and/or lentivirus expressing shRNAs against both Mrtfa and Mrtfb (shMrtfa/b).
Figure EV2. MRTF‐mediated regulation of TEAD‐YAP activity is independent of LATS.
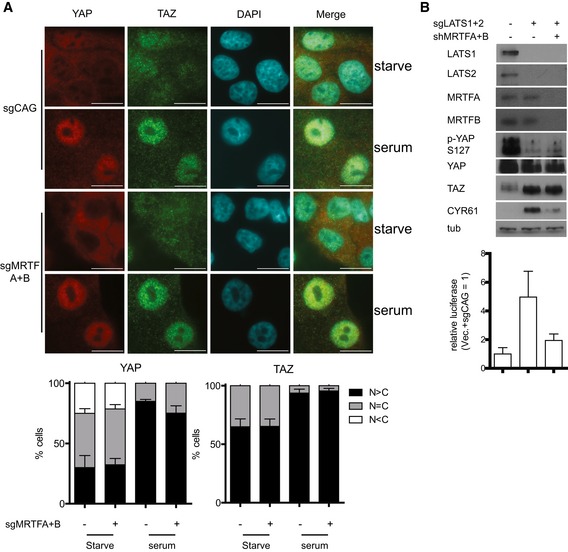
- Control (sgCAG) or MRTF‐knockout (sgMRTFA+B) MCF‐10A cells were serum‐starved for 24 h and stimulated with complete medium for 1 h. Then, YAP and TAZ localization was examined with immunofluorescence. Quantifications for YAP (left) and TAZ (right) are shown below (n = 3, Scale bars: 10 μm).
- Western blots and TEAD‐YAP luciferase reporter assays with control (sgCAG) or LATS1/2‐knockout (sgLATS1 + 2) 293T cells were treated with shRNAs against MRTFA and MRTFB (n = 3).
MRTF proteins physically interact with YAP via a PPXY motif–WW domain interaction
Recent studies suggest the myocardin family of proteins interact with YAP (Xie et al, 2012; Yu et al, 2015). We were able to confirm MRTF and YAP bind one another when expressed at both exogenous and endogenous levels (Fig 3A and B). Since MRTF is enriched at TEAD‐binding genomic loci, we hypothesized TEAD, YAP, and MRTF may form a complex on TEAD‐responsive promoters. Using a DNA pulldown assay, we confirmed binding of all three proteins to the TEAD‐binding DNA motifs of CTGF promoter (Fig 3C). Using a serial co‐immunoprecipitation (IP) assay, we further confirmed all three proteins form a trimeric complex (Fig 3D). We next performed co‐IP experiments with MRTFB and a series of YAP truncation fragments to map the binding domains responsible for their interactions. We found that a WW domain‐containing fragment of YAP binds MRTFB (Fig EV3A). WW domains reportedly bind proline‐proline‐X‐tyrosine (PPXY) motifs (Chen & Sudol, 1995). Strikingly, all members of the myocardin protein family bear a conserved PPXY motif within their C‐terminal transactivation domains (Fig 3E). Targeted deletion of the YAP WW domain completely abolishes MRTFB binding (Fig 3F). Conversely, we confirmed that MRTFB's PPXY motif is required for its binding to YAP (Fig 3G). The MRTFB Y305A mutant, which does not bind SRF (Zaromytidou et al, 2006), and the MRTFB ΔPY mutant, which does not bind YAP, both retain their ability to bind YAP and SRF, respectively. This suggests SRF‐MRTF binding and YAP‐MRTF binding are independent of one another (Fig 3G). TEAD‐YAP binding does not seem to affect YAP‐MRTF binding because the YAP truncation mutant and the TEAD‐binding deficient YAP point mutant both bind MRTFB with affinity comparable to that of wild‐type YAP (Figs 3F and EV3A). We also found consistent differences in the affinity with which MRTFA and MRTFB bind SRF and YAP. While MRTFA shows a preference for SRF binding, MRTFB is biased toward YAP binding (Fig EV3B). This result is consistent with our finding that while MRTFB activates the SRF luciferase reporter twice as much as MRTFA, it activates the TEAD‐YAP luciferase reporter nearly ten times as much as MRTFA (Fig EV3C). Previous studies also reported that MRTFA shows higher affinity for SRF than MRTFB does (Wang et al, 2002). In summary, while MRTFA and MRTFB demonstrate some functional redundancy, MRTFA preferentially activates SRF and its target genes and MRTFB preferentially activate YAP and its target genes.
Figure 3. MRTF and YAP interact via their PPXY and WW domains.
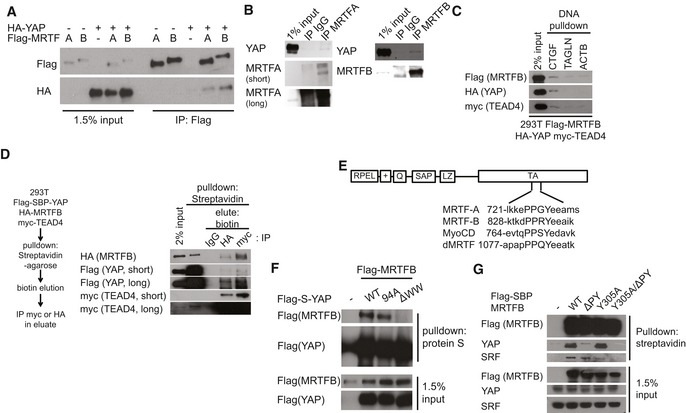
- Co‐immunoprecipitation (IP) analysis with 293T cells expressing the indicated genes.
- Co‐IP analysis with 293T cells examining the interaction of MRTFA or MRTFB with YAP at endogenous levels of expression.
- DNA pulldown analysis with lysates of 293T cells expressing the indicated genes and DNA fragments comprising the promoters of the indicated genes.
- Serial co‐IP analysis with 293T cells expressing the indicated genes.
- Schematics of the functional domains and peptide sequences near the PPXY motif of myocardin family proteins.
- Co‐IP analysis with 293T cells expressing MRTFB and the indicated mutant version of YAP.
- Co‐IP analysis with 293T cells expressing the indicated MRTFB mutants.
Figure EV3. Characterization of MRTF binding to YAP.
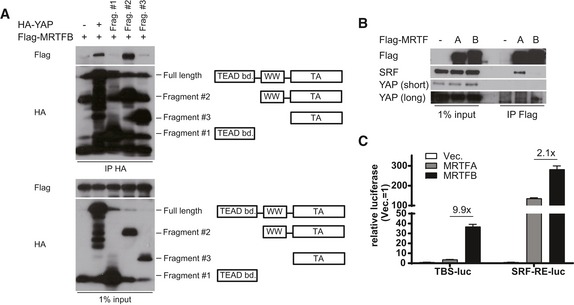
- Co‐IP experiment with MRTFB and series of truncation mutants of YAP.
- Co‐IP experiment with MRTFA or MRTFB with endogenous SRF and YAP. Note that MRTFA and MRTFB have different preferential binding partner.
- TEAD‐activity reporter (TBS‐luc) and SRF‐activity reporter (SRF‐RE‐luc) assay with 293T cells expressing MRTFA or MRTFB (n = 3). Data are presented as means ± s.e.m.
MRTF‐YAP binding is required for TEAD‐YAP target gene activation
We next measured the impact of MRTF binding to its cognate transcriptional regulators by performing transcription reporter assays. Consistent with our observation that SRF‐MRTF binding and YAP‐MRTF binding do not affect one another, the ΔPY and Y305A mutations fail to activate the TEAD‐YAP and SRF luciferase reporters, respectively (Fig 4A). To verify the contribution YAP‐MRTF binding makes to TEAD‐YAP transcriptional activation, we depleted MRTFA and MRTFB and reconstituted MRTFB expression with either wild‐type MRTFB or mutants deficient in binding YAP and/or SRF. Then, we activated TEAD‐YAP target genes by either over‐expressing YAP (Fig 4B) or serum stimulation (Fig 4C) and measured YAP target gene expression. We did not observe any differences in YAP phosphorylation and abundance or TAZ abundance after either MRTF depletion or MRTFB reconstitution (Fig 4B and C). There was one exception, where mutant MRTFB deficient in binding both YAP and SRF (Y305A/ΔPY) decreased YAP phosphorylation in serum‐stimulated condition (Fig EV4A). While the causes of this difference may be of interest for future studies, as decrease in YAP phosphorylation is expected to activate YAP, this change does not explain the failure of this mutant MRTFB to rescue YAP activity. Expectedly, the MRTFB ΔPY mutant produces a far less potent activation of TEAD‐YAP target genes upon YAP over‐expression (Fig 4B) or serum stimulation (Fig 4C). This same mutant does not affect SRF‐MRTF target gene expression upon serum stimulation (Fig EV4B).
Figure 4. MRTF‐YAP binding enhances TEAD‐YAP transcriptional activity.
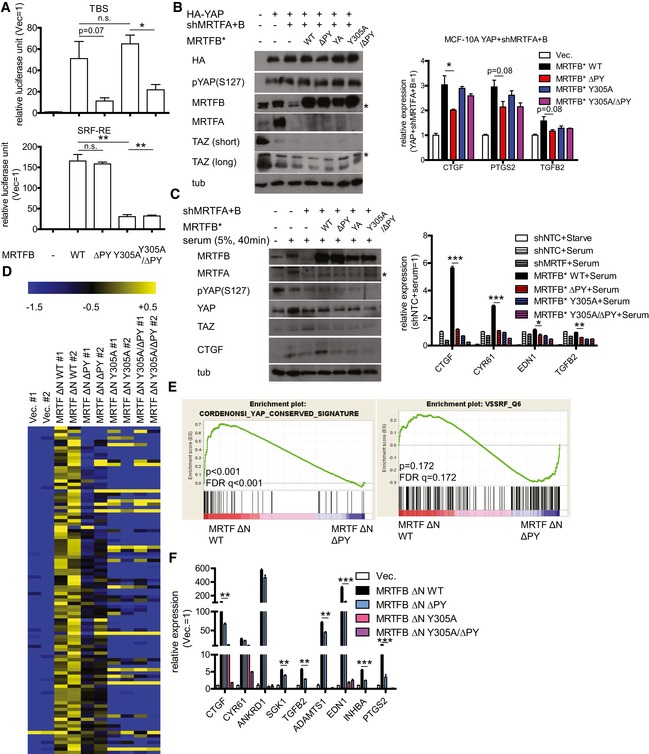
- (top) TEAD luciferase reporter assay and (bottom) SRF luciferase reporter assay in 293T cells expressing the indicated MRTFB mutants (n = 3).
- Western blots (left) and qPCR analysis (n = 3) (right) for MCF‐10A cells expressing the indicated genes. Asterisks indicate non‐specific bands.
- Western blots (left) and qPCR analysis (n = 3) (right) for MCF‐10A cells expressing the indicated genes and serum‐stimulated to activate YAP. Quantification of pYAP(S127)/YAP and TAZ/tubulin ratio is in Fig EV4A. Asterisk indicates non‐specific band.
- Heatmap of selected TEAD‐YAP target genes for MCF‐10A cells expressing the indicated hyperactive MRTFB mutants. Data are presented as fold change relative to average expression level of control cells in Log2.
- GSEA for MCF‐10A cells expressing either wild type (WT) or YAP‐binding deficient (ΔPY) MRTFB.
- qPCR validation of the RNA sequencing data in (D) for representative TEAD‐YAP target genes (n = 3).
Figure EV4. Regulation of TEAD‐YAP and SRF‐MRTF targets by MRTF mutants.
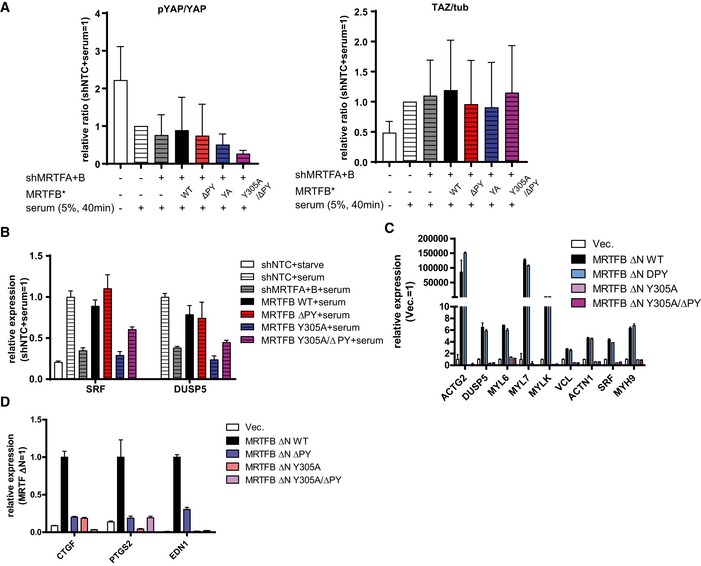
-
AQuantifications of blot in Fig 4C (n = 4).
- B, C
-
DqPCR analysis for selected TEAD‐YAP target genes in MDA‐MB‐231 cells expressing the indicated MRTFB mutant (n = 3).
For a genome‐wide picture of the ways MRTF‐YAP binding affects the transcriptome, we generated MCF‐10A cells over‐expressing mutant forms of MRTFB that are constitutively localized in the nucleus (MRTFB ΔN) but otherwise wild type (WT), YAP‐binding deficient (ΔPY), SRF‐binding deficient (Y305A), or both binding deficient (Y305A/ΔPY). When we performed RNA sequencing on these cells, we found many target genes that were up‐regulated by wild‐type MRTFB and significantly down‐regulated by MRTFB ΔPY (Fig 4D). GSEA indicated these MRTFB ΔPY‐expressing cells show significantly lower expression of YAP signature genes and no change in SRF signature genes (Fig 4E). We also observed consistent reductions in TEAD‐YAP target genes upon MRTFB ΔPY over‐expression and no change in SRF‐MRTF target genes (Figs 4F and EV4C).
It is interesting to note that while the MRTFB Y305A mutant shows no defect in TEAD‐YAP luciferase reporter activation (Fig 4A), it does not properly activate TEAD‐YAP target genes. This suggests that, similar to the TEAD–AP‐1 interactions necessary for TEAD‐YAP target gene expression (Zanconato et al, 2015; Liu et al, 2016b), there are widespread genomic interactions that bring the TEAD and SRF transcription complexes together to activate TEAD‐YAP target gene expression (see the Discussion section). This is supported by recent in vivo ChIP‐seq data showing enrichments for SRF and MRTF at TEAD‐binding DNA motifs (Esnault et al, 2014). Also, since MRTF is expected to depend on nuclear YAP, the relatively modest defect MRTFB ΔPY shows in activating TEAD‐YAP target genes may reflect weak endogenous YAP activity in MCF‐10A cells. Consistent with this interpretation, the MRTFB ΔPY mutant has more pronounced defects in activating TEAD‐YAP target genes in MDA‐MB‐231 cells, which have higher endogenous YAP activity (Fig EV4D) (Lamar et al, 2012). While the exact transcriptional mechanisms of crosstalk between MRTF‐YAP and MRTF‐SRF binding remain unclear, our results suggest MRTF‐YAP binding is required for full TEAD‐YAP target gene activation.
MRTF recruits NcoA3 to the TEAD‐YAP transcription complex
We next wanted to determine how MRTF protein activates the TEAD‐YAP transcription complex. First, we measured the binding affinity of TEAD for YAP in the presence and absence of MRTF, but found no apparent changes (Fig 5A and B). Since MRTF can recruit various transcriptional coactivators and chromatin remodelers (Li et al, 2007; Lockman et al, 2007; Zhang et al, 2007; Kihara et al, 2008), we generated shRNAs against each transcriptional coactivator known to bind MRTF. Then, we asked whether depletion of these candidates alters TEAD‐YAP activity in the presence of either YAP or MRTFB over‐expression. We found shRNAs against NcoA3 (nuclear receptor coactivator 3) consistently reduce the activity of the TEAD‐YAP luciferase reporter in the presence of YAP or MRTFB over‐expression (Fig 5C). NcoA3‐specific shRNAs, but not others, also blunt the YAP over‐expression‐dependent induction of CTGF and ANKRD1 (Fig 5D). We also found NcoA3 depletion reduces the expression of many other genes induced by over‐expression of either wild‐type YAP or a constitutively active MRTFB (Fig 5E and F). NcoA3 depletion also reduces endogenous TEAD‐YAP target gene expression in MDA‐MB‐231 cells (Fig 5G). This indicates NcoA3 is necessary for the activation of target gene expression by the TEAD‐YAP‐MRTF complex. Recently, TEAD and NcoA3 were found to interact for YAP target gene expression (Zhang et al, 2015a; Liu et al, 2016b). Thus, we asked whether MRTF enhances the association of TEAD and YAP with NcoA3. Indeed, we found MRTFB over‐expression increases TEAD2/YAP‐NcoA3 binding (Fig 5H and I), while MRTF depletion decreases TEAD2/YAP‐NcoA3 binding (Fig 5J and K). These results suggest MRTFB activates TEAD‐YAP via recruitment of NcoA3 (Fig 5L).
Figure 5. MRTF recruits NcoA3 to enhance TEAD‐YAP activity.
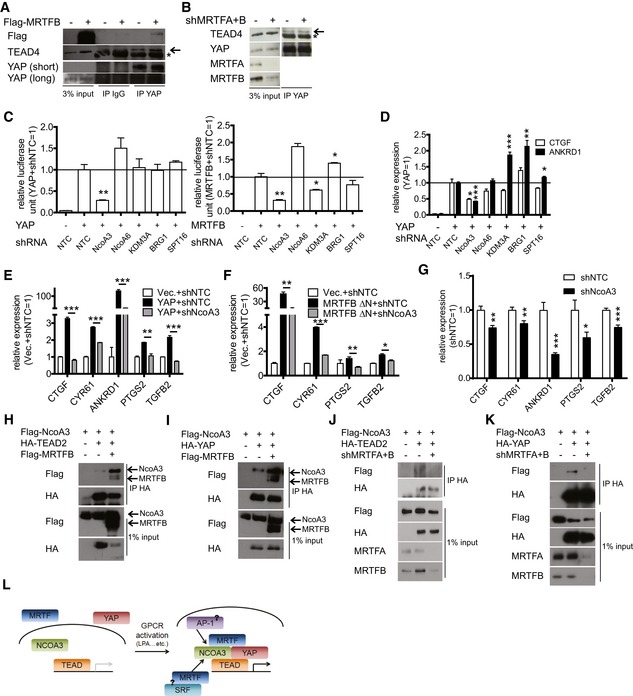
-
A, BCo‐IP analysis of 293T cells either (A) over‐expressing MRTFB or (B) depleted of MRTFA and MRTFB. Asterisk indicates band from heavy chain; arrow indicates the desired specific band.
-
CTEAD luciferase reporter assay with 293T cells expressing either (left) YAP or (right) MRTFB and shRNAs against the indicated genes (n = 3).
-
DqPCR analysis of MCF‐10A cells expressing YAP and shRNAs against the indicated genes (n = 3).
-
E, FqPCR analysis of representative TEAD‐YAP target genes in MCF‐10A cells over‐expressing the indicated genes (n = 3).
-
GqPCR analysis of representative TEAD‐YAP target genes in MDA‐MB‐231 cells infected with lentivirus expressing control (shNTC) or NcoA3 shRNA (n = 3).
-
HCo‐IP analysis for TEAD2 and NcoA3 with 293T cells expressing the indicated genes.
-
ICo‐IP analysis for YAP and NcoA3 with 293T cells expressing the indicated genes.
-
JCo‐IP analysis for TEAD2 and NcoA3 with 293T cells in the presence of shRNAs against MRTFA and MRTFB.
-
KCo‐IP analysis for YAP and NcoA3 with 293T cells in the presence of shRNAs against MRTFA and MRTFB.
-
LSchematic for MRTF‐mediated TEAD‐YAP activation. MRTF recruits NCOA3 to the TEAD‐YAP complex. This may, in turn, recruit other transcription factors for full TEAD‐YAP target gene activation.
MRTF‐YAP binding mediates LPA‐induced cellular invasion and cancer metastasis
The MRTF and YAP coactivators are activated by a common set of extracellular cues, including LPA (lysophosphatidic acid), which induces a GPCR‐dependent activation of RhoA. Thus, we asked whether MRTF and YAP exhibit functional crosstalk upon cellular exposure to LPA. First, we found LPA exposure enhances the binding of YAP with MRTF (Fig 6A). While YAP and MRTFB are located in the cytoplasm of serum‐starved cells, LPA dramatically increases their nuclear co‐localization in various cell lines (Figs 6B and EV5). LPA is also known to induce YAP‐dependent changes in cell migration, invasion, and proliferation (Yu et al, 2012). Therefore, we next attempted to confirm this MRTF/YAP crosstalk upon LPA stimulation by knocking down endogenous MRTF and reconstituting its expression with either wild‐type or mutant MRTFB lacking the ability to bind YAP and/or SRF. We found MRTF knockdown reduces the LPA‐induced invasion of 4T1 mouse breast cancer cells. While wild‐type MRTFB expression effectively restores the metastatic potential of MRTF‐depleted 4T1 cells, mutant MRTFB ΔPY, which cannot bind YAP, does not (Fig 6C and D). To confirm that MRTFB ΔPY's defect in rescuing invasive potential is secondary to a defect in YAP‐MRTF binding, we compared the invasion potential conferred by wild type and ΔPY MRTFB in control and YAP/TAZ‐depleted backgrounds. Intriguingly, not only does YAP/TAZ‐depletion attenuate the invasion induced by MRTFB, it also negates the reduction in invasion induced by expression of MRTFB ΔPY (Fig 6E). This suggests MRTF ΔPY's defect in promoting cell invasion is secondary to a deficit in YAP‐MRTF binding.
Figure 6. MRTF‐YAP binding regulates LPA‐induced cancer cell invasion in vitro and breast cancer metastasis in vivo .
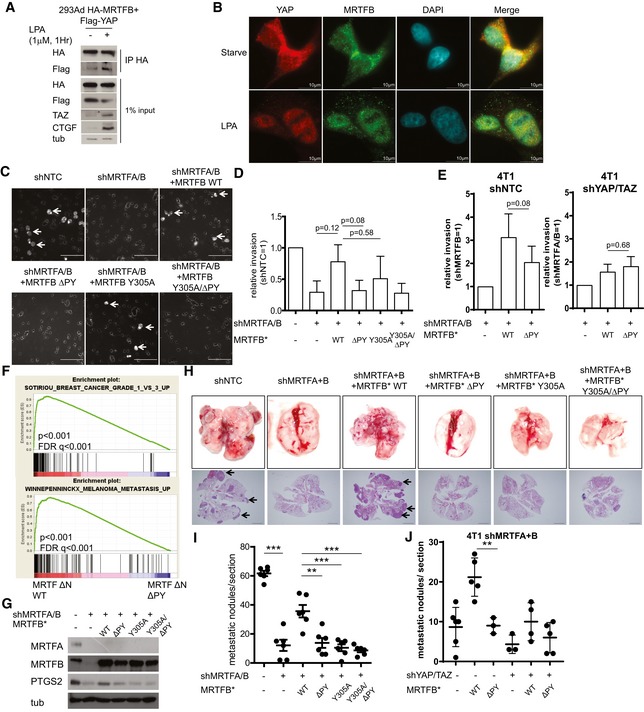
- Co‐IP analysis with 293Ad cells treated with vehicle or LPA for the indicated time.
- Immunofluorescence assay examining the localization of MRTFB and YAP before and after LPA treatment (Scale bars: 10 μm).
- Representative images of the invasion assay with 4T1 cells expressing the indicated genes. Arrows indicated invaded cells (Scale bars: 50 μm).
- Quantification of the invasion assay performed in (C) (n = 3).
- Quantification of the invasion assay with control (shNTC) and YAP/TAZ‐depleted (shYAP/TAZ) 4T1 cells infected with the indicated viruses (n = 3).
- GSEA for the transcriptomes of MCF‐10A cells expressing either wild type (WT) or YAP‐binding deficient (ΔPY) MRTFB.
- Western blots for 4T1 cells expressing the indicated genes.
- Representative images of the in vivo metastasis assay with cells generated in (G). Arrows indicate metastatic nodules (Scale bars: 1 mm).
- Quantification of the metastatic nodules in the metastasis assay performed in (H) (n = 6).
- Quantification of the metastatic nodules in the metastasis assay with 4T1 cells expressing shRNA against MRTFA and MRTFB additionally expressing indicated genes (n = 3–6 for each group). Representative images are shown in Fig EV6.
Figure EV5. LPA induces nuclear co‐localization of YAP and MRTF.
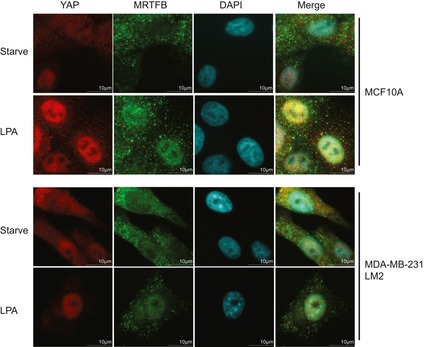
Representative images of the indicated cell lines before and after LPA treatment.
Finally, we examined the physiological significance of MRTF‐YAP binding in vivo. We focused this effort on cancer metastasis because both YAP and MRTF promote metastasis (Medjkane et al, 2009; Lamar et al, 2012). The GSEA for both MRTFB WT and MRTFB ΔPY suggests MRTF‐YAP binding promotes the expression of genes associated with high‐grade metastatic cancers (Fig 6F). In the metastatic breast cancer cell line 4T1, MRTF depletion down‐regulates the expression of PTGS2, which is associated with breast cancer metastases to the lung (Minn et al, 2005; Ma et al, 2006). Although expression of MRTFB WT rescues PTGS2 expression in the same cell line, the rescue induced by the YAP‐binding deficient MRTFB mutant is much less significant (Fig 6G). Therefore, we next performed an in vivo metastasis assay with 4T1 cells expressing the various MRTFB mutants. While wild‐type MRTFB expression increases the metastatic potential of MRTF‐depleted 4T1 cells, MRTFB mutants that do not bind YAP and/or SRF do not (Fig 6H and I). Finally, we examined whether MRTFB ΔPY's deficiency in MRTF‐YAP binding is directly responsible for attenuation in metastatic potential by in vivo metastasis assay with 4T1 cells expressing wild type or ΔPY MRTFB, and compared the promotion of metastatic potential in control and YAP/TAZ‐depleted background (Figs 6J and EV6). Strikingly, not only does YAP/TAZ‐depletion attenuate the metastasis induced by MRTFB, it also negates the attenuation in metastasis induced by expression of MRTFB ΔPY. Together, these results suggest TEAD‐YAP activation by MRTF‐YAP binding enhances the metastatic potential of breast cancer cells.
Figure EV6. Representative images of in vivo metastasis assay in Fig 6J.
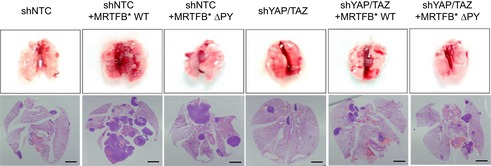
Scale bars: 1 mm.
Nucleo‐cytoplasmic shuttling of MRTF reveals LATS‐independent regulation of TEAD‐YAP activity upon acute cytoskeletal damage
Disruption of the actin cytoskeleton is known to reduce TEAD‐YAP activity even in the presence of mutant YAP that cannot be phosphorylated and that is retained in the nucleus (Dupont et al, 2011; Zhao et al, 2012; Aragona et al, 2013). Consistent with this result, we were able to confirm LATS disruption results in nuclear retention of YAP despite actin disruption using LATS knockout MEFs (Fig 7A). Notably, changes in YAP localization by actin disruption were MRTF independent. MRTF, which we discovered activates nuclear YAP, is also known to respond to changes in the actin cytoskeleton (Miralles et al, 2003). This led us to hypothesize that cytoplasmic sequestration of MRTF upon actin disruption may reduce the activity of nuclear YAP (Fig 7B). While the association of YAP 5SA, which is retained in the nucleus, with wild‐type MRTF is reduced upon actin disruption, its association with the MRTF ΔN mutant, which is also retained in the nucleus, is maintained (Fig 7C). We observed a modest decrease in binding between YAP 5SA and MRTFB ΔN mutant. We believe this is caused when YAP hyperactivity triggers a negative feedback loop (Moroishi et al, 2015; Park et al, 2016) that disrupts YAP‐MRTF binding by an unknown mechanism. Nevertheless, this supports a role for MRTF‐YAP binding in the changes in TEAD‐YAP activity that occur downstream of acute actin disruption. Although YAP 5SA alone does not block the reductions in TEAD‐YAP target gene expression induced by Latrunculin A‐mediated actin disruption, those reductions do not occur when YAP 5SA is expressed in the MRTF‐knockout background (Fig 7D). We observed similar results with actin disruption by other drugs (e.g., blebbistatin and Y27632) (Fig EV7A and B). Knockout of Lats alone does not block the reductions in TEAD‐YAP target gene expression induced by actin disruption. The simultaneous knockout of Mrtf and Lats, however, actually increases TEAD‐YAP target gene expression upon actin disruption (Fig 7E). We found a similar phenomenon when looking at the effects of actin disruption on TEAD‐YAP luciferase reporter with simultaneous expression of both YAP 5SA and MRTF‐specific shRNAs (Fig 7F). Of note, while acute actin disruption does not affect TEAD‐YAP target gene expression in YAP 5SA‐expressing and MRTF‐depleted cells, long‐term actin disruption, as with prolonged dense culture conditions or soft matrix culture conditions, does in the same cells (Fig EV7C and D). These differences between acute and long‐term actin disruption suggests the existence of an alternative, slow‐acting mechanotransducer that attenuates YAP activity upon long‐term disruption of actin cytoskeletal tension. We plan to identify this mechanotransducer in a future study. In conclusion, our results suggest MRTF sequestration represents a largely unexplored mechanism by which acute changes in a cell's external physical environment alter TEAD‐YAP activity.
Figure 7. Cytoskeletal disruption alters TEAD‐YAP activity by inducing nucleo‐cytoplasmic shuttling of MRTF.
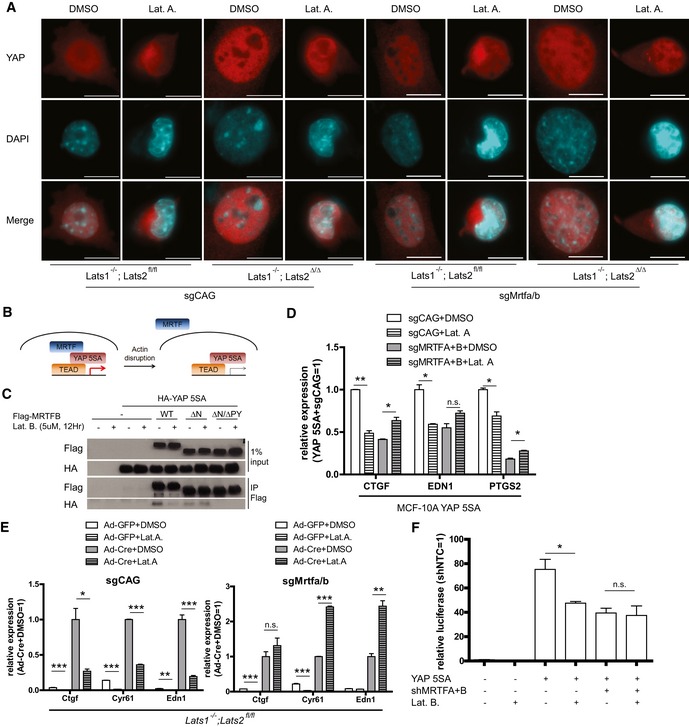
- Lats1−/−; Lats2fl/fl or Lats1−/−; Lats2Δ/Δ cells treated with sgRNAs against control (sgCAG) or Mrtfa and Mrtfb (sgMrtfa/b) were treated with either DMSO or Latrunculin A (Lat. A, 0.5 μM, 1 h). Then, YAP localization was examined using immunofluorescence (scale bars: 10 μm).
- Schematic for the regulation of TEAD‐YAP activity upon actin disruption. Even in the presence of YAP 5SA, which cannot be phosphorylated, actin‐disrupting drugs induce the translocation of MRTF out of the nucleus, reducing TEAD‐YAP activity despite the nuclear retention of YAP.
- Co‐IP analysis with 293T cells transfected with the indicated plasmids and treated with Latrunculin B (Lat. B).
- qPCR analysis of representative TEAD‐YAP target genes in either control (sgCAG) or MRTF‐knockout (sgMRTFA+B) MCF‐10A cells expressing YAP 5SA and treated with Lat. A (n = 3).
- qPCR analysis of representative TEAD‐YAP target genes in either control (sgCAG) or MRTF‐knockout (sgMrtfa/b) Lats1/2 floxed, or deleted cells treated with Lat. A (n = 3).
- TEAD‐YAP luciferase reporter assay with 293T cells transfected with the indicated plasmids and treated with Lat. B (n = 3).
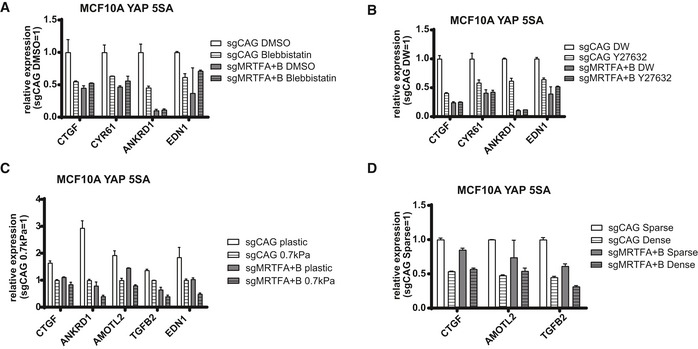
Figure EV7. MRTF mediates LATS‐independent acute mechanical regulation of YAP activity.
-
A–DqPCR analysis of representative TEAD‐YAP target genes for MCF‐10A cells expressing YAP 5SA with sgRNA against control (sgCAG) and MRTFA and MRTFB (sgMRTFA+B) treated with (A) blebbistatin or solvent (DMSO), (B) Y27632 or solvent (distilled water, DW), (C) hard (tissue culture plastic) and soft (0.7 kPa) matrix, (D) sparse/dense cell density. Data are presented as means ± s.e.m. (n = 2 for each figure).
Discussion
In this study, we identified MRTF proteins as key coactivators of the TEAD‐YAP transcriptional complex. Although recent studies have uncovered a functional relationship between MRTF and YAP/TAZ (Yu et al, 2015; Speight et al, 2016), our study substantially improves our understanding of MRTFs and their activation of the TEAD‐YAP transcriptional complex. First, we have demonstrated the significance of MRTF‐YAP binding in the direct regulation of TEAD‐YAP activity independent of its crosstalk with Smad or SRF. Specifically, we have taken a genome‐wide view of the impact of MRTF‐YAP binding on the transcriptome. Second, we have demonstrated the physiological relevance of MRTF‐YAP binding in cancer cell invasion and metastasis. Third, we have identified MRTF as a regulator of nuclear YAP activity. This is particularly interesting because most known YAP regulation occurs via LATS‐mediated YAP phosphorylation and nuclear translocation. Our results add another layer to the regulation of YAP activity, fine‐tuning it in response to GPCR activation (i.e., by LPA) or actin cytoskeletal disruption.
In addition, our results contradict some previously published findings. We failed to observe any change in TAZ expression or localization upon over‐expression or depletion of MRTF. Instead, we discovered MRTF recruits the coactivator NcoA3 to the TEAD‐YAP complex. These differences between our results and those of other studies may reflect different cellular contexts. Future experiments will be necessary to determine the relative contributions of those differences to TEAD‐YAP regulation in each cellular context.
Our study reveals intimate crosstalk between the transcriptional coactivators YAP and MRTF, which both respond to extracellular stimuli. Although MRTF is not required for TEAD‐YAP activity, it does enhance it. While YAP and MRTF are both activated by common extracellular stimuli, they differ in their regulation. MRTF binds globular actin via a RPEL motif, while YAP responds to actomyosin tension in a way that is still poorly characterized (Miralles et al, 2003; Dupont et al, 2011). This suggests MRTF may act to fine‐tune TEAD‐YAP activity. For example, while cell attachment and spreading can induce YAP activation directly, further cytoskeletal maturation characterized by the formation of filamentous actin and the depletion of globular actin may further enhance TEAD‐YAP activity by activating MRTF. We, therefore, suggest MRTF‐mediated enhancement of TEAD‐YAP activity may strengthen a bi‐stable cellular signaling pathway that can be activated either by GPCR activation or by increasing cellular tension. Once activated, this pathway can induce a transcriptional program that reinforces itself. Recently, SRF‐MRTF was found to activate TAZ transcription (Liu et al, 2016a; Speight et al, 2016). This, along with our finding that MRTF potentiates TEAD‐YAP activity, suggests the presence of a positive feedback loop that efficiently modulates cellular phenotypes in response to extracellular stimuli. This bi‐stable switch may contribute to phenomena like tissue morphogenesis, cancer invasion, and cellular differentiation, which are all regulated by cellular tension and GPCR activation (Marinissen & Gutkind, 2001; Paszek et al, 2005). We expect further studies of the physiological relevance of such a multi‐layered regulation of the TEAD‐YAP transcription complex will prove both interesting and important in cancer therapeutics.
Adding to the complexity of TEAD‐YAP activity regulation, two recent studies revealed intimate crosstalk between TEAD‐YAP and other transcriptional machinery including AP‐1 (Zanconato et al, 2015; Liu et al, 2016b). It is thus possible YAP acts as a transcriptional hub relaying various transcription factor complexes to accomplish fine‐tuned regulation of target gene expression. Consistent with this idea, YAP is enriched at several distal enhancers across the genome (Galli Giorgio et al, 2015; Zanconato et al, 2015). We have shown recruitment of NcoA3 to the TEAD‐YAP complex by MRTF is necessary for full TEAD‐YAP activity. Another recent report implicated NcoA3 as a molecular bridge between TEAD‐YAP and AP‐1—and possibly other transcription factors—that is necessary to fully activate target gene expression. Indeed, NcoA3 is already known to act as a molecular hub that brings together the various transcription factors in embryonic stem cells that maintain their pluripotency (Percharde et al, 2012). With our results, these reports suggest the sequential binding of MRTF and NcoA3 to TEAD‐YAP induces the formation of a massive transcriptional complex that maximally activates YAP target genes. Future studies of mutant cells lacking the various components of the full TEAD‐YAP transcriptional complex will reveal the nature of such higher order genomic interactions and how they affect YAP target genes genome‐wide.
We observed that mutant MRTFs lacking the PPXY‐ or SRF‐binding motifs are specifically defective in activating TEAD‐ or SRF‐reporters, respectively. We found it interesting that MRTF that is incapable of binding SRF can neither activate conventional SRF‐MRTF target genes nor TEAD‐YAP target genes. This suggests that, at least in certain genomic contexts, MRTF‐SRF binding is important in regulating TEAD‐YAP activity. Consistent with this idea, SRF and TEAD reportedly bind one another (Gupta et al, 2001). Along with our discovery that MRTF recruits NcoA3 to the TEAD‐YAP transcriptional complex, this suggests MRTF‐SRF binding may coordinate transcriptional responses by facilitating the interaction of SRF with TEAD‐containing transcription complexes through NcoA3.
Although SRF‐MRTF binding plays a pervasive role in regulating all MRTF target genes, YAP‐MRTF binding is important in regulating TEAD‐YAP activity. Finally, in a previous study, we found an association between SRF and YAP for the activation of a specific subset of YAP target genes that endow mammary stem cell‐like properties (Kim et al, 2015). In that study, TEAD and MRTF were not part of the relevant SRF‐YAP transcriptional complex. Thus, we propose the existence of multiple mutually exclusive combinations of transcriptional regulators. The TEAD‐YAP‐MRTF transcriptional complex fully activates most YAP target genes, while the SRF‐YAP transcriptional complex regulates specific subsets of YAP target genes.
Our results suggest a mutual dependence of MRTF and YAP in transcriptional responses to extracellular stimuli. We also propose YAP‐induced tumorigenesis and stem/progenitor cell expansion may depend on MRTF. YAP is a critical oncogenic driver of uveal melanoma, which depends on active RhoA (Feng et al, 2014; Yu et al, 2014). YAP is also responsible for the changes in cell invasion and cell growth in 3D in vitro culture that occur upon manipulation of the stiffness of the extracellular matrix (Aragona et al, 2013). Since these same cellular contexts also activate MRTF, it may also play a role in the onset of uveal melanoma and in the induction of cell invasion upon changes in extracellular matrix stiffness. Our study also suggests a possible role for TEAD‐YAP in cellular responses thought to be induced by SRF‐MRTF. We have shown that MRTF‐mediated cancer metastasis, once thought to be a consequence of SRF‐MRTF, also depends on TEAD‐YAP activity. Previously, MRTFA knockout mice were reported to show defects in mammary myoepithelial cell differentiation (Li et al, 2006; Sun et al, 2006). The fact that TAZ knockout mice show similar defects (Skibinski et al, 2014) strongly suggests the combined action of MRTF‐TAZ induces myoepithelial cell differentiation.
Finally, our study also contributes to a mechanistic understanding of some therapeutics that have been successful against YAP‐associated tumors. Verteporfin is used as an inhibitor of the association between TEAD and YAP (Liu‐Chittenden et al, 2012), but its specificity and mechanism of action remain controversial (Zhang et al, 2015b). Our results suggest CCG‐1423, a Rho/SRF‐MRTF pathway and MICAL2 inhibitor (Evelyn et al, 2007; Lundquist Mark et al, 2014), may be effective as an alternative inhibitor of TEAD‐YAP that blocks its activation by MRTF. NcoA3 is amplified in some breast cancers (Anzick et al, 1997), possibly making it a therapeutically relevant target in YAP‐amplified cancers itself. Chemical NcoA3 inhibitors like bufalin and gossypol (Wang et al, 2011, 2014) may interfere with MRTF's activation of TEAD‐YAP by degrading NcoA3. Despite its initial description as an activator of the estrogen receptor, NcoA3 is frequently over‐expressed in estrogen receptor‐negative breast cancers (Burandt et al, 2013) that usually show higher YAP activity (Kim et al, 2015). NcoA3 inhibitors may therefore be particularly effective against YAP‐active triple‐negative breast cancers. Future experiments should address the in vivo efficacy of candidate inhibitors of the SRF‐MRTF/YAP/NcoA3 pathway for use individually or in combined therapies.
Materials and Methods
Cell culture
293T, 293Ad, 4T1, MDA‐MB‐231, and MDA‐MB‐231‐LM2 cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). MCF‐10A were grown in a 1:1 mixture of DMEM and Ham's F12 medium (DMEM/F12) supplemented with 5% horse serum, 20 ng/ml EGF, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, and 10 μg/ml insulin. All cells except 4T1 and MDA‐MB‐231‐LM2, which was a gift from Mi‐Young Kim, are obtained from ATCC. Cell lines were validated by DNA fingerprinting at TPOX, TH01, vWA, and D5S818 loci. Cells were routinely tested for the presence of mycoplasma with 4,6‐diamidino‐2‐phenylindole staining.
Antibodies
The antibodies used in this study were as follows: anti‐Flag (Sigma M2, 1:500), anti‐HA (Biolegend, 1:1,000), anti‐YAP (raised against 201–450 amino acids of human YAP, 1:1,000), anti‐YAP (Abfrontier 2F12, 1:1,000 for immunofluorescence and immunoprecipitation), TAZ (Cell Signaling, 1:1,000 for Western blot, 1:100 for immunofluorescence), anti‐γ‐tubulin (Santa Cruz, 1:1,000), anti‐MRTFA (Santa Cruz, 1:1,000), anti‐MRTFB (Bethyl Laboratories, 1:1,000 for Western blot, 1:100 for immunofluorescence), anti‐NcoA3 (Santa Cruz, 1:1,000), anti‐CTGF (Santa Cruz, 1:250), anti‐Myc (Santa Cruz 9E10, 1:200), and anti‐phospho‐YAP S127 (Cell Signaling, 1:1,000). Unless otherwise noted, the given dilutions were for Western blots.
shRNA
The shRNA target sequences used for this study were as follows: shMRTFA #1 (human): 5′‐GACTATCTCAAACGGAAGATT‐3′, shMR TFA #2 (human): 5′‐GCTCAAGTACCACCAGTACAT‐3′, shMRTFB #1 (human): 5′‐GCAGACACTTTCACCGAGATT‐3′, shMRTFB #2(human): 5′‐GACACTTTCACCGAGATTATG‐3′, shMrtfa/b (targets both mouse Mrtfa and Mrtfb): 5′‐CATGGAGCTGGTGGAGAAGAA‐3′, shSRF: 5′‐CGATGTTTGCCATGAGTATTA‐3′, shNcoA3: 5′‐GATCAGAAGGCAGGATTATA‐3′, shNcoA6: 5′‐GCCCATTGTTGGTCAACTTAT‐3′, shBRG1: 5′‐CCCGTGGACTTCAAGAAGATA‐3′, shSPT16: 5′‐TGAGCAGATGGAACGAGAAAT‐3′, shKDM3A: 5′‐GCTTTGATTGTGAAGCATTTA‐3′.
CRISPR‐Cas9‐mediated gene editing
Cas9‐mediated gene editing was performed by lentiviral infection of sgRNA and spCas9 using the lentiCRISPR v2 vector (Sanjana et al, 2014). Clones were isolated and screened for successful ablation of genes using Western blot, and sequencing of genomic DNA. The guide RNA sequences used for this study were as follows: CTGF promoter: 5′‐ACTGTCCAGATGCCCATATA‐3′ LATS1: 5′‐GCAACCTAACATACCAGTG‐3′, LATS2: 5′‐GTAGGACGCAAACGAATCG‐3′, MRTFA (human): 5′‐GCTGCGCTGTGACTTCTCAC‐3′, MRTFB (human): 5′‐ATAGACACCGAGGATGAAGT‐3′, Mrtfa/b (mouse): 5′‐TTCTTCTCCACCAGCTCCAT‐3′. Taz (mouse): 5′‐AGAGATACTTCCTTAATCACA‐3′. Yap (mouse): 5′‐TGAGAACAATGACAACCAATA‐3′.
Co‐immunoprecipitation
Cells were lysed in NETN lysis buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.5% NP‐40, 1 mM EDTA). 1 μg of antibody was added to the cleared lysate and incubated overnight. Then, 15 μl of protein A/G agarose was added to the lysate, incubated for 1 h, washed with NETN three times, and boiled in Laemmli's sample buffer. The samples were then subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and Western blot analysis.
DNA pulldown assay
After 48 h, 293T cells transfected with the relevant plasmids were washed once with PBS and lysed in NETN buffer. Next, 1 mg of the cleared lysate was incubated for 1 h with 3 μg of a random 50‐bp competitor probe corresponding to a region of human ACTB (5′‐CGTTGCTATCCAGGCTGTGCTATCCCTGTACGCCTCTGCCGTACCACTG‐3′) to eliminate non‐specific DNA interacting proteins. Meanwhile, 3 μg of biotinylated PCR products containing promoter regions of human CTGF, TAGLN, and ACTB lying 701‐bp downstream of the TSS was incubated with 20 μl of streptavidin agarose (Thermo Scientific). Then, DNA‐bound beads were washed three times in lysis buffer and mixed with the pre−cleared lysate. The beads were again washed three times in lysis buffer and resuspended in Laemmli buffer for resolution by SDS–PAGE.
RT–qPCR
RNA was extracted from cells with easy‐Blue solution (Intron Biotechnology). cDNA was synthesized from total RNA using M‐MLV reverse transcriptase (Enzynomics) for 2 h at 37°C. All gene expression levels were normalized to that of GAPDH. See Table EV1 for the relevant primers.
Immunofluorescence
Cells were seeded on gelatin‐coated coverslips. Cells were fixed in 4% paraformaldehyde (Sigma) in phosphate‐buffered saline (PBS), permeabilized in 0.1% Triton X‐100 in PBS, and blocked with 1% bovine serum albumin (BSA) in PBS. Cells were then incubated in the appropriate primary antibody diluted in 1% BSA in PBS at 4°C overnight. Appropriate secondary antibodies (Jackson Laboratories and Life Technologies) diluted in PBS were then added for 1 h at 37°C. Finally, the coverslips were mounted on slides using Vectashield mounting medium with DAPI (Vector Laboratories).
Luciferase assay
293T cells were seeded in 96‐well plates. Cells were transfected in triplicate with a mixture of DNAs of the desired expression vectors, firefly luciferase vectors driven by promoters of interest, and Renilla luciferase vectors driven by CMV. Twenty‐four hours after transfection, relative luciferase activity was measured with the dual‐Glo luciferase kit (Promega).
RNA sequencing
RNA was extracted as described above, and RNA sequencing was performed with a Truseq RNA library prep kit v2 (Illumina) and Hi‐seq 2000 equipment. The Multi‐experiment Viewer (MeV) program was used to draw heatmaps.
In vitro invasion assay
4T1 cells were trypsinized, resuspended in serum‐free DMEM supplemented with 0.1% BSA, and counted. 1 × 105 cells were seeded in the upper chamber of cell culture inserts with a polyethylene terephthalate (PET) membrane with 8‐μm pores (SPL Life Sciences) coated with 15 μl of 20% matrigel diluted in serum‐free DMEM. Serum‐free DMEM supplemented with 1 μM LPA was added to the lower chamber to serve as a chemoattractant. After 24 h, the membranes of the cell culture inserts were removed and mounted on slides using Vectashield mounting medium with DAPI (Vector Laboratories). Three random fields from each membrane were photographed using a fluorescent microscope for quantification.
In vivo metastasis assay
4T1 cells were trypsinzed, resuspended in PBS supplemented with 0.1% BSA, and counted. 2 × 105 cells were injected into the tail vein of 6‐week‐old BALB/c female mice (Daehan Biolink). Mice were sacrificed 18 days after injection. Then, the lungs were removed and metastatic nodules were counted. This experiment was not performed blind. All animal experiments were performed in accordance with KAIST IACUC. Our work is compliant with ARRIVE guidelines.
Acrylamide hydrogel fabrication
Hydrogels with an elastic modulus of ~0.7 kPa were fabricated according to Cretu et al (2010). Briefly, 18 mm ∅ coverslips were immersed in 0.1N NaOH for 3 min. After aspiration, 3′APES was added for 3 min. Following aspiration and rinse with DW, 0.5% glutaraldehyde was added. After 30 min of incubation, coverslips were rinsed and allowed to dry completely. Next, 4% NHS was added to the acrylamide mix, then poured onto the coverslips. Immediately, coverslips that have been siliconized using DCDMS were carefully placed on the gel mix, and incubated the “sandwich” at 37°C for 2 h for polymerization to occur. Then, the siliconized top coverslips were carefully pushed off, and the bottom coverslip containing the solidified gel was exposed to UV radiation for 30 min. After rinsing with 1× PBS, hydrogels were immersed in 0.1 mg/ml collagen in PBS, then placed on a rocker at 4°C overnight to allow covalent linkage to occur. Next day, remaining collagen was aspirated and rinsed, and then, 1 mg/ml heat‐inactivated BSA in serum‐free media was added for 30 min in 37°C incubator to block any unreacted NHS. Finally, coverslips were washed with PBS, and then, an appropriate number of cells were seeded. Cells were harvested via trypsinization 72 h after plating for assay.
Statistics, sample size, and data analysis
All statistical analysis are done with GraphPad prism 7. Sample sizes were set as default to three samples for in vitro studies, and six samples for in vivo studies. In case sufficient statistical power was not achieved, we performed additional experiments. They usually yielded enough statistical power. There were no excluded data in the analysis. There was no blinding, nor randomization.
Data deposition
The raw and processed RNA sequencing data are uploaded in NCBI gene expression omnibus (GEO) with accession number GSE89182.
Author contributions
TK and DH performed experiments and wrote the manuscript. DL performed tail vein injections of cancer cells. J‐HK and S‐YK performed RNA sequencing. D‐SL supervised the experiments and TK and D‐SL wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We thank Mi‐Young Kim and Joan Massagué for materials. This work was supported by grants from the National Creative Research Program (2012‐0001228) and Science Research Center Program (NRF‐2016R1A5A1010764). DH was supported by the National Research Foundation of Korea (NRF‐2014H1A2A1020575).
The EMBO Journal (2017) 36: 520–535
Contributor Information
Tackhoon Kim, Email: tackhoon.k@kaist.ac.kr.
Dae‐Sik Lim, Email: daesiklim@kaist.ac.kr.
References
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X‐Y, Sauter G, Kallioniemi O‐P, Trent JM, Meltzer PS (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277: 965–968 [DOI] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S (2013) A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin‐processing factors. Cell 154: 1047–1059 [DOI] [PubMed] [Google Scholar]
- Burandt E, Jens G, Holst F, Jänicke F, Müller V, Quaas A, Choschzick M, Wilczak W, Terracciano L, Simon R, Sauter G, Lebeau A (2013) Prognostic relevance of AIB1 (NCoA3) amplification and overexpression in breast cancer. Breast Cancer Res Treat 137: 745–753 [DOI] [PubMed] [Google Scholar]
- Chen HI, Sudol M (1995) The WW domain of Yes‐associated protein binds a proline‐rich ligand that differs from the consensus established for Src homology 3‐binding modules. Proc Natl Acad Sci USA 92: 7819–7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti Anna R, Poletti A, Daidone Maria G, Dupont S, Basso G, Bicciato S, Piccolo S (2011) The hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147: 759–772 [DOI] [PubMed] [Google Scholar]
- Cretu A, Castagnino P, Assoian R (2010) Studying the effects of matrix stiffness on cellular function using acrylamide‐based hydrogels. J Vis Exp 42: e2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183 [DOI] [PubMed] [Google Scholar]
- Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N, Treisman R (2014) Rho‐actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev 28: 943–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evelyn CR, Wade SM, Wang Q, Wu M, Iñiguez‐Lluhí JA, Merajver SD, Neubig RR (2007) CCG‐1423: a small‐molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther 6: 2249–2260 [DOI] [PubMed] [Google Scholar]
- Feng X, Degese Maria S, Iglesias‐Bartolome R, Vaque Jose P, Molinolo Alfredo A, Rodrigues M, Zaidi MR, Ksander Bruce R, Merlino G, Sodhi A, Chen Q, Gutkind JS (2014) Hippo‐independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio‐regulated Rho GTPase signaling circuitry. Cancer Cell 25: 831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli Giorgio G, Carrara M, Yuan W‐C, Valdes‐Quezada C, Gurung B, Pepe‐Mooney B, Zhang T, Geeven G, Gray Nathanael S, de Laat W, Calogero Raffaele A, Camargo Fernando D (2015) YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell 60: 328–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Kogut P, Davis FJ, Belaguli NS, Schwartz RJ, Gupta MP (2001) Physical interaction between the MADS box of serum response factor and the TEA/ATTS DNA‐binding domain of transcription enhancer factor‐1. J Biol Chem 276: 10413–10422 [DOI] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S (2012) Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol 13: 591–600 [DOI] [PubMed] [Google Scholar]
- Joyce JA (2005) Therapeutic targeting of the tumor microenvironment. Cancer Cell 7: 513–520 [DOI] [PubMed] [Google Scholar]
- Kihara T, Kano F, Murata M (2008) Modulation of SRF‐dependent gene expression by association of SPT16 with MKL1. Exp Cell Res 314: 629–637 [DOI] [PubMed] [Google Scholar]
- Kim M, Kim M, Lee S, Kuninaka S, Saya H, Lee H, Lee S, Lim D‐S (2013) cAMP/PKA signalling reinforces the LATS‐YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J 32: 1543–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Yang S‐J, Hwang D, Song J, Kim M, Kyum Kim S, Kang K, Ahn J, Lee D, Kim M‐Y, Kim S, Seung Koo J, Seok Koh S, Kim S‐Y, Lim D‐S (2015) A basal‐like breast cancer‐specific role for SRF‐IL6 in YAP‐induced cancer stemness. Nat Commun 6: 10186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamar JM, Stern P, Liu H, Schindler JW, Jiang Z‐G, Hynes RO (2012) The Hippo pathway target, YAP, promotes metastasis through its TEAD‐interaction domain. Proc Natl Acad Sci USA 109: E2441–E2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J‐H, Kim T‐S, Yang T‐H, Koo B‐K, Oh S‐P, Lee K‐P, Oh H‐J, Lee S‐H, Kong Y‐Y, Kim J‐M, Lim D‐S (2008) A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J 27: 1231–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Chang S, Qi X, Richardson JA, Olson EN (2006) Requirement of a myocardin‐related transcription factor for development of mammary myoepithelial cells. Mol Cell Biol 26: 5797–5808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HJ, Haque Z, Lu Q, Li L, Karas R, Mendelsohn M (2007) Steroid receptor coactivator 3 is a coactivator for myocardin, the regulator of smooth muscle transcription and differentiation. Proc Natl Acad Sci USA 104: 4065–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C‐Y, Chan SW, Guo F, Toloczko A, Cui L, Hong W (2016a) MRTF/SRF dependent transcriptional regulation of TAZ in breast cancer cells. Oncotarget 7: 13706–13716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li H, Rajurkar M, Li Q, Cotton Jennifer L, Ou J, Zhu Lihua J, Goel Hira L, Mercurio Arthur M, Park J‐S, Davis Roger J, Mao J (2016b) Tead and AP1 coordinate transcription and motility. Cell Rep 14: 1169–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu‐Chittenden Y, Huang B, Shim JS, Chen Q, Lee S‐J, Anders RA, Liu JO, Pan D (2012) Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26: 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman K, Taylor JM, Mack CP (2007) The histone demethylase, Jmjd1a, interacts with the myocardin factors to regulate SMC differentiation marker gene expression. Circ Res 101: e115–e123 [DOI] [PubMed] [Google Scholar]
- Lundquist Mark R, Storaska Andrew J, Liu T‐C, Larsen Scott D, Evans T, Neubig Richard R, Jaffrey Samie R (2014) Redox modification of nuclear actin by MICAL‐2 regulates SRF signaling. Cell 156: 563–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Kundu N, Rifat S, Walser T, Fulton AM (2006) Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res 66: 2923–2927 [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS (2001) G‐protein‐coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22: 368–376 [DOI] [PubMed] [Google Scholar]
- Medjkane S, Perez‐Sanchez C, Gaggioli C, Sahai E, Treisman R (2009) Myocardin‐related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol 11: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J (2005) Genes that mediate breast cancer metastasis to lung. Nature 436: 518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou A‐I, Treisman R (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113: 329–342 [DOI] [PubMed] [Google Scholar]
- Morita T, Mayanagi T, Sobue K (2007) Dual roles of myocardin‐related transcription factors in epithelial–mesenchymal transition via slug induction and actin remodeling. J Cell Biol 179: 1027–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu F‐X, Karin M, Pan D, Guan K‐L (2015) A YAP/TAZ‐induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 29: 1271–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muehlich S, Cicha I, Garlichs CD, Krueger B, Posern G, Goppelt‐Struebe M (2007) Actin‐dependent regulation of connective tissue growth factor. Am J Physiol Cell Physiol 292: C1732–C1738 [DOI] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng C‐X, Brugge JS, Haber DA (2006) Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA 103: 12405–12410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park G‐S, Oh H, Kim M, Kim T, Johnson RL, Irvine KD, Lim D‐S (2016) An evolutionarily conserved negative feedback mechanism in the Hippo pathway reflects functional difference between LATS1 and LATS2. Oncotarget 7: 24063–24075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart‐King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM (2005) Tensional homeostasis and the malignant phenotype. Cancer Cell 8: 241–254 [DOI] [PubMed] [Google Scholar]
- Percharde M, Lavial F, Ng JH, Kumar V, Tomaz RA, Martin N, Yeo JC, Gil J, Prabhakar S, Ng HH, Parker MG, Azuara V (2012) Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self‐renewal and reprogramming. Genes Dev 26: 2286–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Meth 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden DT (2006) The stem‐cell niche as an entity of action. Nature 441: 1075–1079 [DOI] [PubMed] [Google Scholar]
- Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger Bridget T, Vasioukhin V, Avruch J, Brummelkamp Thijn R, Camargo Fernando D (2011) Yap1 acts downstream of α‐catenin to control epidermal proliferation. Cell 144: 782–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Diane D, Xue W, Krall Elsa B, Bhutkar A, Piccioni F, Wang X, Schinzel Anna C, Sood S, Rosenbluh J, Kim Jong W, Zwang Y, Roberts Thomas M, Root David E, Jacks T, Hahn William C (2014) KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158: 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski A, Breindel Jerrica L, Prat A, Galván P, Smith E, Rolfs A, Gupta Piyush B, LaBaer J, Kuperwasser C (2014) The hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell Rep 6: 1059–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speight P, Kofler M, Szaszi K, Kapus A (2016) Context‐dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton‐regulated MRTF and TAZ and TGF[beta]‐regulated Smad3. Nat Commun 7: 11642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, Oren M, Sudol M, Cesareni G, Blandino G (2001) Physical interaction with yes‐associated protein enhances p73 transcriptional activity. J Biol Chem 276: 15164–15173 [DOI] [PubMed] [Google Scholar]
- Sun Y, Boyd K, Xu W, Ma J, Jackson CW, Fu A, Shillingford JM, Robinson GW, Hennighausen L, Hitzler JK, Ma Z, Morris SW (2006) Acute myeloid leukemia‐associated Mkl1 (Mrtf‐a) is a key regulator of mammary gland function. Mol Cell Biol 26: 5809–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML (2001) TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes‐associated protein localized in the cytoplasm. Genes Dev 15: 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D‐Z, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, Richardson JA, Nordheim A, Olson EN (2002) Potentiation of serum response factor activity by a family of myocardin‐related transcription factors. Proc Natl Acad Sci USA 99: 14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow D‐C, Palzkill TG, O'Malley BW (2011) Small molecule inhibition of the steroid receptor coactivators, SRC‐3 and SRC‐1. Mol Endocrinol 25: 2041–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow D‐C, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F, Hodder P, Chase P, Griffin PR, Zhou S, Liao L, Xu J, O'Malley BW (2014) Bufalin is a potent small‐molecule inhibitor of the steroid receptor coactivators SRC‐3 and SRC‐1. Cancer Res 74: 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE (2012) Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem 287: 14598–14605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli Giorgio G, Yanger K, Pepe‐Mooney B, Gurung B, Shrestha K, Cahan P, Stanger Ben Z, Camargo Fernando D (2014) Hippo pathway activity influences liver cell fate. Cell 157: 1324–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F‐X, Zhao B, Panupinthu N, Jewell Jenna L, Lian I, Wang Lloyd H, Zhao J, Yuan H, Tumaneng K, Li H, Fu X‐D, Mills Gordon B, Guan K‐L (2012) Regulation of the hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell 150: 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F‐X, Guan K‐L (2013) The Hippo pathway: regulators and regulations. Genes Dev 27: 355–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F‐X, Luo J, Mo J‐S, Liu G, Kim Young C, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, Zhao J, Chen X, Zhang L, Wang C‐Y, Bastian Boris C, Zhang K, Guan K‐L (2014) Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 25: 822–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu O, Miyamoto S, Brown JH (2015) MRTF‐A and YAP exert dual control in GPCR and RhoA‐mediated transcriptional regulation and cell proliferation. Mol Cell Biol 36: 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Sullivan AJ, Medina R, Ito Y, van Wijnen AJ, Stein JL, Lian JB, Stein GS (2004) Tyrosine phosphorylation controls Runx2‐mediated subnuclear targeting of YAP to repress transcription. EMBO J 23: 790–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015) Genome‐wide association between YAP/TAZ/TEAD and AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol 17: 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaromytidou A‐I, Miralles F, Treisman R (2006) MAL and ternary complex factor use different mechanisms to contact a common surface on the serum response factor DNA‐binding domain. Mol Cell Biol 26: 4134–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon‐Cardo C, Silke J, Fan S‐T, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW (2006) Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125: 1253–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Fang H, Zhou J, Herring BP (2007) A novel Role of Brg1 in the regulation of SRF/MRTFA‐dependent smooth muscle‐specific gene expression. J Biol Chem 282: 25708–25716 [DOI] [PubMed] [Google Scholar]
- Zhang C, Robinson Brian S, Xu W, Yang L, Yao B, Zhao H, Byun Phil K, Jin P, Veraksa A, Moberg Kenneth H (2015a) The ecdysone receptor coactivator taiman links yorkie to transcriptional control of germline stem cell factors in somatic tissue. Dev Cell 34: 168–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Győrffy B, Sebolt‐Leopold JS, Dame MK, Varani J, Brenner DE, Fearon ER, Omary MB, Shah YM (2015b) Tumor‐selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal 8: ra98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang C‐Y, Chinnaiyan AM, Lai Z‐C, Guan K‐L (2008) TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22: 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Tumaneng K, Guan K‐L (2011) The Hippo pathway in organ size control, tissue regeneration and stem cell self‐renewal. Nat Cell Biol 13: 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang C‐Y, Yu J, Guan K‐L (2012) Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26: 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, Dawson D, Willis JE, Markowitz SD, Camargo FD, Avruch J (2011) Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes‐associated protein (Yap) overabundance. Proc Natl Acad Sci USA 108: E1312–E1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File