

Abstract
The transport protein particle (TRAPP) was initially identified as a vesicle tethering factor in yeast and as a guanine nucleotide exchange factor (GEF) for Ypt1/Rab1. In mammals, structures and functions of various TRAPP complexes are beginning to be understood. We found that mammalian TRAPPII was a GEF for both Rab18 and Rab1. Inactivation of TRAPPII‐specific subunits by various methods including siRNA depletion and CRISPR–Cas9‐mediated deletion reduced lipolysis and resulted in aberrantly large lipid droplets. Recruitment of Rab18 onto lipid droplet (LD) surface was defective in TRAPPII‐deleted cells, but the localization of Rab1 on Golgi was not affected. COPI regulates LD homeostasis. We found that the previously documented interaction between TRAPPII and COPI was also required for the recruitment of Rab18 to the LD. We hypothesize that the interaction between COPI and TRAPPII helps bring TRAPPII onto LD surface, and TRAPPII, in turn, activates Rab18 and recruits it on the LD surface to facilitate its functions in LD homeostasis.
Keywords: COPI, lipid droplets, Rab18, TRAPPC10, TRAPPC9, TRAPPII
Subject Categories: Membrane & Intracellular Transport
Introduction
The transport protein particle (TRAPP) was originally identified in as a vesicle tethering factor for COPII‐coated vesicles at the Golgi (Sacher et al, 2001). Subsequently, three forms of TRAPP have been identified in yeast, and they have functions in addition to vesicle tethering (Yu & Liang, 2012). The consensus view of the yeast TRAPPI complex is that it contains the subunits Trs20, Trs23, Trs31, Bet3, Bet5, and arguably Trs33 (Liang et al, 2007; Sacher et al, 2008; Tokarev et al, 2009; Barrowman et al, 2010; Lynch‐Day et al, 2010). TRAPPII contains three extra subunits Trs65, Trs120, and Trs130. TRAPPIII contains all the TRAPPI subunits plus Trs85. Recently, other candidate subunits have been reported, but they remain to be fully characterized (Choi et al, 2011). In mammals, the compositions of various forms of TRAPP complex are less clear (Yu & Liang, 2012). Distinct complexes structurally similar to yeast TRAPPII and TRAPPIII were implicated by Zong et al in mammalian cells (Zong et al, 2011) and subsequently were unequivocally demonstrated (Bassik Michael et al, 2013). Mammalian TRAPPII contains TRAPPII‐specific subunits TRAPPC9 and TRAPPC10, orthologs of Trs120 and Trs130, respectively, and TRAPPIII contains TRAPPC8, TRAPPC11, and TRAPPC12 (Scrivens et al, 2011; Bassik Michael et al, 2013). TRAPPC8 is the ortholog of Trs85, but TRAPPC11 and TRAPPC12 are unique in mammals. The functions of mammalian TRAPP could be very different from the yeast counterparts. In yeast, all three forms of TRAPP arguably serve as the guanine nucleotide exchange factor (GEF) for the small GTPase Ypt1 (Barrowman et al, 2010). Mammalian TRAPPII was also shown to have GEF activity toward Rab1, the ortholog of Ypt1 (Yamasaki et al, 2009). TRAPPII‐specific subunits bind to coatomer γ‐COP. Depletion of TRAPPII subunits by siRNA appears to affect Golgi integrity and transport, presumably due to improper tethering of COPI vesicles. TRAPPIII modulates autophagy through the interaction between specific subunit TRAPPC8 and TBC1D14 (Lamb et al, 2016), a GTPase‐activating protein (GAP) for unspecified Rab proteins (Longatti et al, 2012). TRAPPIII also interacts with COPII vesicle coat for ricin trafficking (Bassik Michael et al, 2013).
TRAPPC9 (mammalian ortholog of TRAPPII‐specific subunit Trs120) mutations have been identified in human mentally retarded individuals with postnatal microcephaly (Mir et al, 2009; Mochida et al, 2009; Montpetit & Conibear, 2009). Human skin fibroblasts with mutations in TRAPPC9 have been isolated from one such individual (Philippe et al, 2009). However, these cells are viable and the secretory and endocytic pathways are intact (Results section), even though TRAPPII has been implicated to interact with components of the COPI vesicle coat. COPI was originally discovered as promoting Golgi‐derived protein transport vesicles (Malhotra et al, 1989). RNAi‐based screening and proteomic studies have identified several COPI subunits having regulatory effects on lipid droplet (LD) homeostasis. Depletion of COPI components resulted in lipid over‐storage due to impaired lipolysis (Beller et al, 2008; Guo et al, 2008). LDs are cytosolic organelles that not only store but also regulate synthesis, metabolism, and trafficking of lipids (Murphy, 2001; Martin & Parton, 2006; Farese & Walther, 2009; Brasaemle & Wolins, 2012). Originating from the ER, LDs are composed of a hydrophobic core of neutral lipids, mostly triacylglycerol (TG) and cholesterol esters, surrounded by a phospholipid monolayer, on which a wide range of proteins are present (Blanchette‐Mackie et al, 1995; Tauchi‐Sato et al, 2002; Pol et al, 2004). These proteins include components of COPI coat and Arf (Beller et al, 2008; Guo et al, 2008; Soni et al, 2009). Although it has been reported that retrograde transport mediated by Arf‐COPI indirectly regulates lipid homeostasis (Takashima et al, 2011), both proteomic and cell biological studies suggest a direct role by the Arf‐COPI machinery (Guo et al, 2008; Thiam et al, 2013). Arf1 and COPI components act directly on the LD surfaces to remove phospholipids through budding of nano‐LDs and therefore promote the establishment of membrane bridges between LDs and the ER (Wilfling et al, 2014), allowing rapid targeting of ER‐bound proteins, such as ATGL and GPAT4, to LD surface (Prinz William, 2013; Wilfling et al, 2013). The COPI complex may positively regulate lipolysis through inhibition of ADRP (perilipin‐2) binding to LD surfaces and, in turn, promote ATGL migrating to LDs (Soni et al, 2009).
At least five Rab proteins had been reported to be associated with LDs by proteomic studies and other techniques, indicating their extensive involvement in the intracellular trafficking and/or dynamics of LDs (Tan et al, 2013; Kiss & Nilsson, 2014). Among them, Rab18 is best characterized, but its functions on LD are incompletely understood (Martin et al, 2005; Ozeki et al, 2005). The possible roles for Rab18 may include the regulation of the association of LDs with ER membranes and lipolysis. In adipocytes, both insulin‐stimulated lipogenesis and isoprenaline‐stimulated lipolysis increase localization of Rab18 to LDs (Pulido et al, 2011). Furthermore, only wild‐type and dominant‐active, but not dominant‐negative, forms of Rab18, are localized to LDs when transfected into cells, suggesting that the localization of Rab18 to LDs is guanine nucleotide‐dependent (Ozeki et al, 2005). Rab18 mediates the apposition of the LD phospholipid monolayer and the ER membrane (Ozeki et al, 2005). The GTP form of Rab18 has been reported to interact with the NAG‐RINT1‐ZW10 (NRZ) complex, an ER tethering complex which facilitates the fusion of vesicles from the Golgi (Gillingham et al, 2014). It is possible that a Rab18‐NRZ interaction could mediate interaction between ER and LDs. However, the mechanism leading to its activation and, hence, its recruitment onto LDs remain elusive.
Rab18 has also been localized to other membrane structures including ER, Golgi, endosomes, and peroxisomes (Dejgaard et al, 2008; Gronemeyer et al, 2013). The Rab3GAP complex, besides acting as the GAP for Rab3 (as well as other Rabs), was reported to serve as the GEF for Rab18 and regulate its function on ER morphology (Gerondopoulos et al, 2014). Rab18, Rab3GAP, and TBC1D20 are mutated in cases of Warburg micro syndrome and a clinically related Martsolf syndrome (Aligianis et al, 2005; Bem et al, 2011; Liegel Ryan et al, 2013). This class of genetic diseases is mainly characterized by abnormalities in eye, brain, and sexual organ development. Clinical presentations include congenital bilateral cataracts, intellectual disability, and postnatal microcephaly. Fibroblasts isolated from the affected individuals all have a common feature at the subcellular level: formation of aberrantly large LDs (Liegel Ryan et al, 2013).
In this paper, we investigated the functions of mammalian TRAPPII and unexpectedly found that it served as a GEF for Rab18 and regulated its recruitment onto LD surfaces. Based on the well‐established interaction between TRAPPII and COPI coatomers, we investigated the functional relationship between COPI, TRAPPII, and Rab18 in LD homeostasis and found that COPI helped recruit TRAPPII onto LD, so that TRAPPII could activate Rab18 and allow the small GTPase to regulate LD homeostasis.
Results
TRAPPII is a GEF for Rab18
A weak signal of Rab18 in a mass spectrometry analysis of TRAPPC9‐interacting proteins prompted us to investigate the possibility of an interaction between Rab18 and TRAPPII (data not shown). We co‐transfected HA‐tagged Rab18 with Myc‐tagged TRAPPC9 and immunoprecipitated lysates of the transfected cells with anti‐Myc antibody and found Rab18 bound to TRAPPC9. This interaction was much stronger in the presence of EDTA (Fig 1A), although longer exposure of the same immunoblot also detected weak binding in conditions in which Rab18 was bound to GTPγS or GDP (data not shown). EDTA chelates the magnesium ions from Rab18 and prevents it from binding guanine nucleotide. We confirmed that Rab18 bound to TRAPPII most strongly when it was nucleotide‐free since a nucleotide‐free mutant, Rab18[N221], but not wild‐type Rab18, was able to co‐precipitate with TRAPPC9 in the presence of Mg2+ ions (Fig 1B). HA‐tagged Rab18 and Rab1, but not Rab2, were able to co‐precipitate endogenous TRAPPII‐specific subunits (top two panels, Fig 1C). In these co‐IP experiments, TRAPPIII‐specific subunit, TRAPPC12, was not present in Rab1 and Rab18 immunoprecipitates (fourth panel, Fig 1C), suggesting the mammalian TRAPPIII complex may not activate Rab1 (later in the results section). GST‐Rab18 purified from bacteria could pull down endogenous TRAPPII‐specific subunits from 293T cell lysates (Fig 1D). In both cases, we could detect endogenous TRAPPC2 and TRAPPC3, common subunits of TRAPP complexes, suggesting that Rab18 interacted with the TRAPPII complex. Again, the interaction appeared to be the strongest when Rab18 was nucleotide‐free (i.e., pre‐treated with EDTA). Since most nucleotide exchange factors bind most strongly to small GTPases in a nucleotide‐free state, we suspected TRAPPII to be a GEF for Rab18.
Figure 1. The TRAPPII complex binds preferentially to the nucleotide‐free form of Rab18.
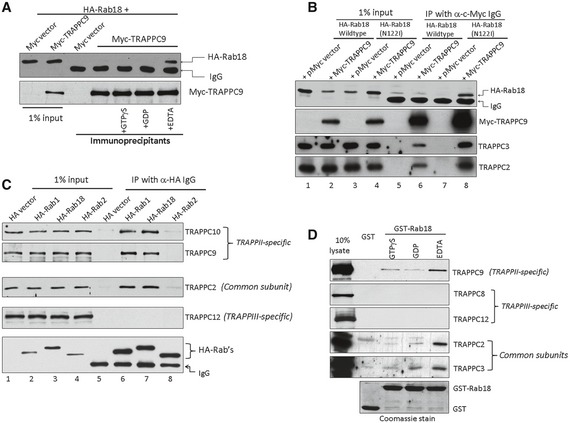
- Lysates from 293T cells transfected with the indicated DNA constructs were incubated with 1 mM Mg2+ plus 0.1 mM GTPγS, GDP, or 2 mM EDTA and immunoprecipitated with anti‐Myc antibody. Rab18‐HA co‐immunoprecipitated with Myc‐TRAPPC9 when it was nucleotide‐free (+EDTA).
- A mutant of Rab18 unable to bind nucleotide, N122I, was co‐immunoprecipitated with Myc‐TRAPPC9 by anti‐Myc antibody.
- HA‐tagged Rab1 and Rab18, but not Rab2, could co‐immunoprecipitate endogenous TRAPPII complex but not TRAPPIII complex.
- GST‐Rab18 could pull down endogenous TRAPPII complex in an in vitro binding assay. GST‐Rab18 was pre‐loaded with GTPγS or GDP or stripped of nucleotide by EDTA. Lysates from 293T cells were subjected to GST‐Rab18 pull‐down. The presence of various TRAPP subunits was detected by immunoblotting. TRAPPII complex, but not TRAPPIII complex, was co‐isolated with GST‐Rab18, most strongly with Rab18 in nucleotide‐free state.
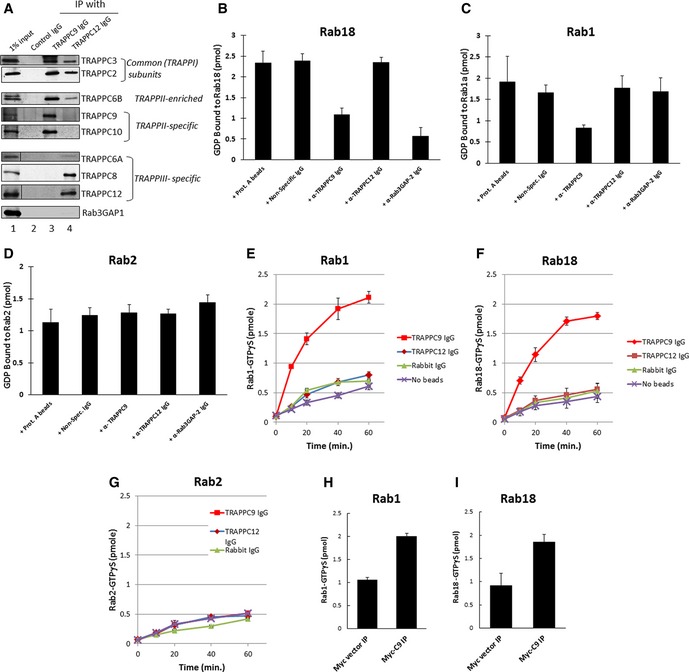
We performed a nucleotide exchange assay to determine whether TRAPPII was a bona fide GEF for Rab18. To do this, we immunoprecipitated TRAPPII from mammalian cell lysates using an antibody against TRAPPC9 (Fig 2A, lane 3). TRAPPIII, which served as a control, was also precipitated using antibody against TRAPPC12 (Fig 2A, lane 4). In these immunoprecipitates, both TRAPP complexes contained common subunits TRAPPC3 and TRAPPC2. TRAPPII also contained TRAPPC10 and TRAPPC6B was enriched in this complex. TRAPPIII also contained TRAPPC8 and TRAPPC6A. Rab3GAP1 precipitated with neither TRAPP complex (Fig 2A, bottom panel), eliminating the possibility that TRAPP might interact with a known GEF for Rab18. Nucleotide exchange assays were performed on GST‐Rab18, GST‐Rab1, and GST‐Rab2 pre‐loaded with [3H]GDP (Fig 2B–D). Immunoprecipitates using the indicated antibodies were added to the reaction to enzymatically promote exchange of non‐radioactive GTP for radioactive GDP on the indicated Rab proteins, causing a reduction in the radiolabel signal when the Rab proteins were captured on nitrocellulose filters in a subsequent step. From these experiments, we observed that TRAPPII promoted guanine nucleotide exchange for Rab18 and Rab1 but not Rab2. TRAPPIII could not promote exchange for any of the Rab proteins tested, whereas Rab3GAP could promote exchange for Rab18, but not Rab1, as previously reported. To measure the uptake of GTP as an additional experiment for nucleotide exchange, we performed uptake of [35S]GTPγS for the Rab proteins that were pre‐loaded with non‐radioactive GDP (Fig 2E–G). Again, the nucleotide uptake experiments demonstrated that TRAPPII promoted nucleotide exchange for Rab1 and Rab18, but not Rab2, in a time‐dependent manner, whereas TRAPPIII did not serve as a GEF for the Rabs tested. To eliminate the possibility that the TRAPPC9 antibody non‐specifically pulled down a factor that had GEF activity for Rab18, we changed the antibody for immunoprecipitation by transfecting HEK293T cells with Myc‐TRAPPC9, immunoprecipitated TRAPPII complex with anti‐Myc antibody, and then performed nucleotide exchange experiments with the immunoprecipitates. Myc‐TRAPPC9 immunoprecipitate, but not Myc vector‐transfected immunoprecipitate, was able to promote nucleotide exchange on Rab1 and Rab18 (Fig 2H and I). These results unambiguously demonstrated that mammalian TRAPPII served as a GEF for Rab18, in addition to Rab1.
Figure 2. TRAPPII complex stimulates guanine nucleotide exchange on Rab1 and Rab18.
-
ATRAPPII and TRAPPIII complexes were immunoprecipitated by antibodies specifically recognizing TRAPPC9 or TRAPPC12, and the presence of various co‐precipitating TRAPP subunits was detected by immunoblotting.
-
B–DTRAPP‐stimulated guanine nucleotide exchange on purified Rab18, Rab1, and Rab2 GST fusion proteins measured by release of [3H]GDP that was pre‐loaded onto the Rab proteins.
-
E–GNucleotide exchange reactions measured by time‐dependent binding of [35S]GTPγS. Immunoprecipitates using the indicated antibodies were included in the assay as the GEF to be tested. In the [3H]GDP‐release experiments, the reactions were incubated for 30 min at 37°C before the Rab proteins were subjected to nitrocellulose filter binding. In the [35S]GTPγS binding experiments, the reactions were incubated for the indicated amount of time before filter binding.
-
H, IAnti‐Myc antibody immunoprecipitates from lysates of HEK293T cells transfected with Myc‐TRAPPC9 were used as the GEF for GST‐Rab1 or GST‐Rab18 in 30‐min incubations at 37°C with [35S]GTPγS.
TRAPPII regulates LD size and the association of Rab18 on LD surface
Rab18 regulates lipid homeostasis. Therefore, we investigated whether TRAPPII also regulated this process as an upstream activator of Rab18. We depleted TRAPP subunits by siRNA in HEK293T cells and then determined whether the size of lipid droplets was larger after the cells were incubated with oleic acid to accumulate lipid droplets (Fig 3A). Immunoblotting showed that siRNA specific to TRAPPC9 and TRAPPC8 could specifically deplete the respective proteins in HEK293T cells. TRAPPC9 depletion, but not TRAPPC8 or control firefly luciferase depletion, caused drastic increase in intensity of fluorescence labeling of LDs (Fig 3A). Because of the small volume of cytoplasm in HEK293T cells, it was difficult for us to discern whether the increase in LD staining was due to size increase or clustering of small LDs. We repeated the siRNA depletion experiment in HeLa cells (Appendix Fig S1) and observed an increase in LD size in TRAPPC9‐depleted cells after careful measurement of the diameters of individual lipid droplets. This result further suggested that the large LD phenotype described here was not cell line‐specific. Furthermore, skin fibroblasts isolated from a human individual with a mutation in TRAPPC9 also contained aberrantly large LDs after lipid uptake compared to similarly isolated fibroblasts from wild‐type individual (Fig 3B). These results demonstrated that defective TRAPPII complex caused the same aberrantly large LDs as reduced activity of Rab18, suggesting TRAPPII and Rab18 could be functionally related in the regulation of LD homeostasis.
Figure 3. Loss of TRAPPC9 function resulted in aberrantly large lipid droplets.
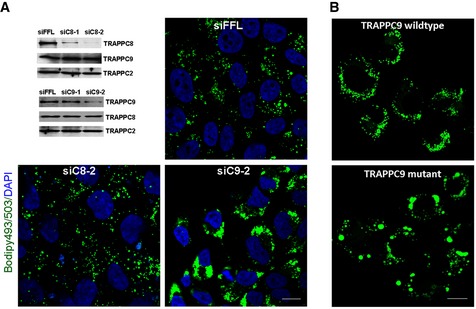
- Depleting TRAPPC9, but not TRAPPC8, increased lipid droplet size in HEK293T cells. The efficiency of siRNA depletion of TRAPPC8 and TRAPPC9 in HeLa cells is shown by immunoblotting in the top left panel. Representative fluorescence images show the status of lipid droplets in these siRNA‐treated cells after oleic acid incubation. Scale bar = 10 μm.
- After loading with oleic acid, human skin fibroblasts containing a R475* mutation in TRAPPC9 accumulated lipid droplets of greater sizes than skin fibroblasts similarly isolated from a human individual with TRAPPC9 wild type. Scale bar = 10 μm.
Markers for the early secretory pathway are morphologically normal in the TRAPPC9 mutant cells (Appendix Fig S2). Since the siRNA depletion of TRAPPC9 and the mutant TRAPPC9 fibroblasts did not reveal any observable defect in the early secretory pathway, we were concerned that such lack of phenotype was due to incomplete inactivation of this protein or TRAPPII complex. To inactivate the function of TRAPPII more efficiently, we carried out CRISPR–Cas9‐mediated deletion of the TRAPPC9 and/or TRAPPC10, major subunits of TRAPPII complex. The strategy of deletion and the identification of knockout cell clones are presented in Appendix Fig S3. TRAPPC9 deletion did not result in an observable defect in the early secretory pathway, but accumulation of large LDs was very obvious (data not shown), though not as drastic as in cells treated with TRAPPC9‐specific siRNA. Contrary to our previous conclusion that TRAPPC9 mediated the interaction between TRAPPC10 and common subunits (Zong et al, 2011), we found that partially functional TRAPPII might still be present in the TRAPPC9‐deleted cells. Therefore, we further deleted TRAPPC10 in cells with wild‐type and TRAPPC9‐deletion backgrounds and investigated whether the secretory pathway and LD formation were changed in these cells. The TRAPPC9 and TRAPPC10 singly or doubly deleted cells appeared to be normal in the early secretory pathway (Fig EV1A–C). Internalization of fluorescent‐transferrin in these cells was also not different from wild‐type HEK293T cells (Fig EV1D and E). ER‐to‐Golgi traffic of FM‐CD8 and VSV‐G was no different from wild‐type control (Fig EV2 and Appendix Fig S4, respectively). However, accumulation of large LDs was obvious in cells with TRAPPC10 knockout and those with double knockout of TRAPPC9/C10 (double knockout will now be called TRAPPII‐deleted cells) (Fig 4A). To a lesser extent, the TRAPPC9‐knockout cells also accumulated aberrantly large LDs. We observed that TRAPPC10 deletion caused partial loss of TRAPPC9 protein by degradation, and hence, TRAPPC10 deletion was almost equivalent to TRAPPC9 and TRAPPC10 double deletion (data not shown).
Figure EV1. Deletion of TRAPPII subunits does not affect the morphologies of the indicated cellular organelle markers or inhibit the internalization of transferrin.
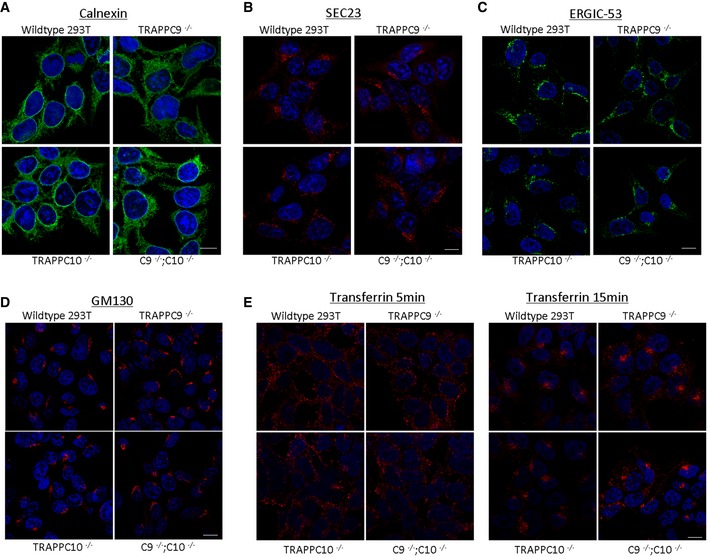
- ER marker calnexin.
- ER exit site marker Sec23.
- ERGIC marker ERGIC‐53.
- Golgi marker GM130.
- Internalization of rhodamine‐transferrin (red) for 5 min (left panels) and 15 min (right panels).
Figure EV2. Deletion of TRAPPII subunits does not affect ER‐to‐Golgi traffic as monitored by the trafficking of GFP‐FM4‐CD8 protein.
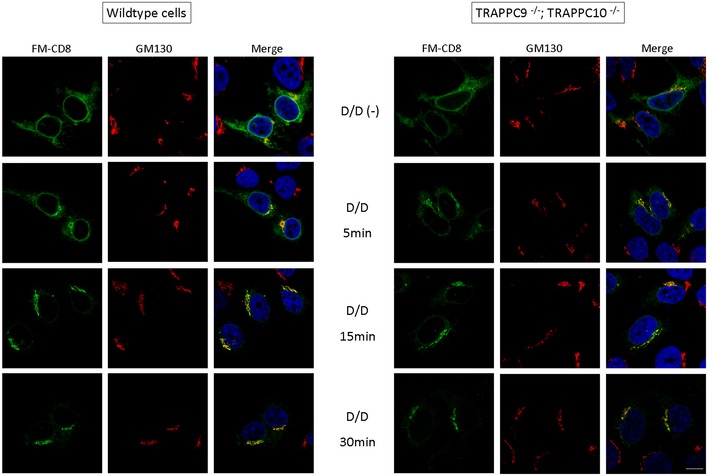
Transport of FM4‐CD8 out of the ER was initiated by applying the disaggregating drug AP21998 (D/D) to a final concentration of 2 μM for the indicated time (Lavieu et al 2013). Transport of GFP‐FM4‐CD8 to the Golgi (GM130) was monitored for up to 30 min in wild‐type and TRAPPII‐deleted (TRAPPC9−/−;TRAPPC10−/−) HEK293T cells. Scale bar = 10 μm.
Figure 4. Recruitment of Rab18 onto small lipid droplets was defective in TRAPPII‐deleted cells.
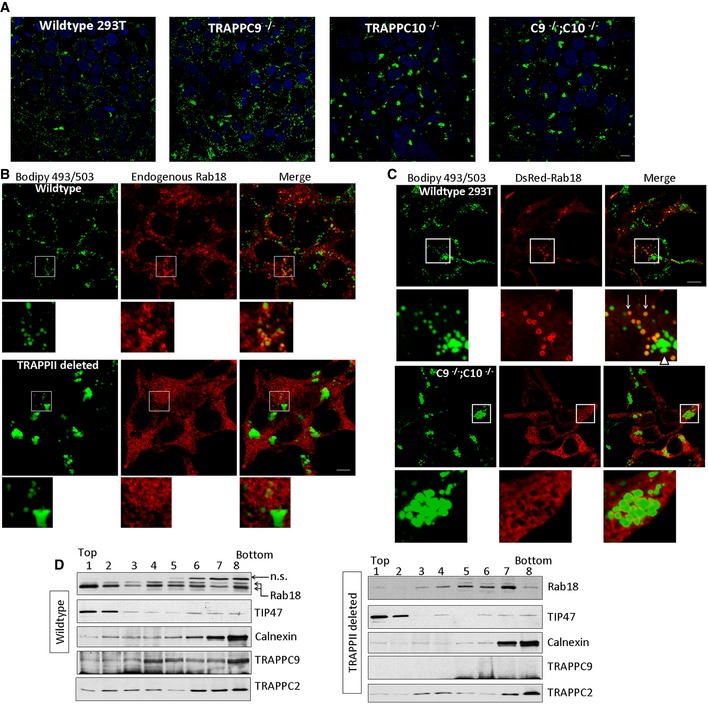
- HEK293T cells with the indicated genotypes were loaded with 400 μM oleic acid for 24 h before staining with Bodipy 493/503. Scale bar = 10 μm.
- Wild‐type or TRAPPII‐deleted cells were loaded with oleic acid and stained with Bodipy 493/503 (green) and endogenous Rab18 by immunofluorescence (red). Scale bar = 10 μm.
- DsRed‐Rab18 was transfected into wild‐type (top panels and insets) or TRAPPII‐deleted HEK293T cells (bottom panels and insets) that were loaded with oleic acid and stained with Bodipy 493/503 (green). Scale bar = 10 μm.
- Homogenates of wild‐type or TRAPPII‐deleted cells were subjected to sucrose gradient fractionation. Fractions 1 and 2 were enriched with LDs. The presence of Rab18, TRAPPC9, TRAPPC2, LD marker TIP47, and ER marker calnexin was detected by SDS–PAGE followed by immunoblotting.
Many GEFs determine the subcellular localizations of their target Rab proteins to specify the Rab functions. Rab18 had been reported to function at the ER, Golgi, and LDs. We found endogenous Rab18 largely colocalized with Golgi marker GM130 in the indicated cells, although ER‐localized signal was also observed (Fig EV3A). In HEK293T, Rab18 signal was found mainly cytosolic and perhaps on the ER (data not shown). After oleic acid loading, Rab18 was redistributed onto the surface of a percentage of the LDs, but the efficiency of LD association varied among several commonly used cell lines (Figs EV3B–D). We chose to investigate the Rab18‐LD association in HEK293T cells and Huh‐7 hepatocytes because Rab18‐LD associations were the strongest. Intense Rab18 signals were observed to decorate a subset of small‐size LDs in wild‐type HEK293T cells (Fig 4B, top panels). However, no such intense Rab18 coating was observed on LDs from TRAPPII‐deleted cells (Fig 4B, bottom panels). The Rab18 signals were, instead, in a diffused staining pattern, likely representing the ER or in the cytosol. The LDs were aberrantly large in size in these TRAPPII‐deleted cells, similar to that shown in Fig 4A. Since overexpressing Rab18 could increase its association with LD, we wondered whether such manipulation in TRAPPII‐deleted cells could increase its LD association or reduce the size of LD to normal. When DsRed‐Rab18 was transfected into wild‐type HEK293T, it was found to surround a subset of small LDs (arrows, Fig 4C, top panels and insets), but not the larger‐sized LDs (arrowhead, Fig 4C). However, in the TRAPPII‐deleted cells, DsRed‐Rab18 signals remained in the ER but not on LD surface (Fig 4C, bottom panels and insets). When we performed the same experiment with overexpression of DsRed‐Rab1, also a substrate of TRAPPII, we observed that DsRed‐Rab1 signal was mainly enriched in the cis‐Golgi in both wild‐type and TRAPPII‐deleted cells (Appendix Fig S5). Therefore, unlike Rab18, the localization of Rab1 was not affected by TRAPPII deletion. To confirm whether defective TRAPPII caused reduced LD association of Rab18, we performed sucrose density gradient fractionation and determined whether the LD‐enriched fractions isolated from TRAPPII‐deleted cells contained Rab18 (Fig 4D). LD‐enriched fractions were detected at the top of the gradient in fractions 1 and 2 due to the light density of LD, as indicated by the well‐documented LD marker TIP47. Enrichment of Rab18 in these fractions was observed from wild‐type homogenate. In the gradient of TRAPPII‐deleted cell homogenate, majority of the Rab18 signal was found in fractions other than the LD‐enriched fractions. We investigated whether TRAPPC9 was also concentrated in the LD‐enriched fractions and found that only a minor pool of it was present in these fractions. TRAPPC9 signal peaked at fraction 4 in the sucrose gradient fractionation of wild‐type homogenate (Fig 4D). As expected, no TRAPPC9 signal was detected in TRAPPII‐deleted cells. Combining the microscopic and biochemical results, we demonstrated that TRAPPII was required for the recruitment of Rab18 onto LD surfaces.
Figure EV3. The ability of Rab18 to be associated with LD surface varies among different cell lines.
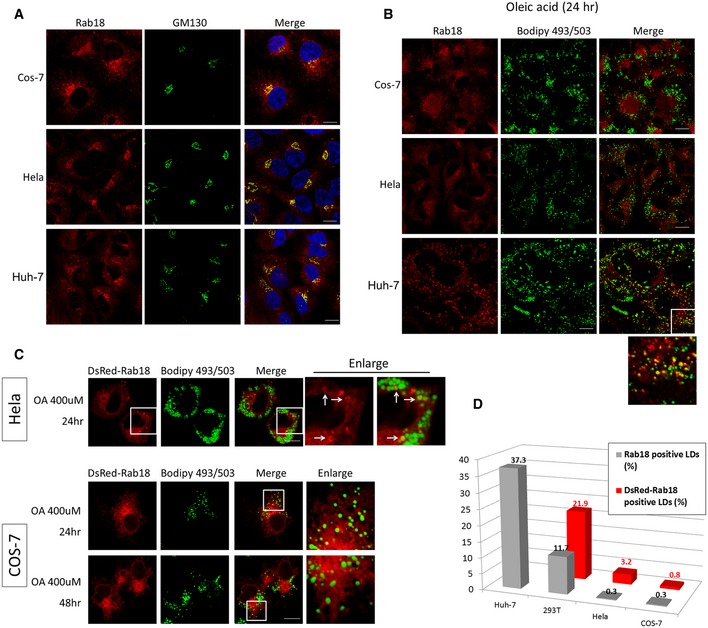
- Endogenous Rab18 is enriched in the perinuclear region and colocalized largely with Golgi marker GM130 in cells grown in growth medium. Scale bars = 10 μm.
- The cells were incubated with 400 μM of oleic acid for 24 h before staining with Rab18. Rab18 signals on the Golgi were slightly reduced and became more dispersed throughout the cytoplasm in COS and HeLa cells, but were very poorly colocalized with LDs. In Huh‐7 cells, Rab18 signals redistributed to highly punctate structures which partially decorated the LDs. Scale bars = 10 μm.
- Overexpression of DsRed‐Rab18 slightly increased its association with LDs in HeLa but not in COS cells even after 48 h of oleic acid loading. Scale bars = 10 μm.
- Statistical quantification of the percentage of LDs decorated with Rab18 in each of the indicated cell lines. Gray bars = endogenous Rab18; red bars = overexpressed DsRed‐Rab18. Total number of LD fluorescence dots counted: n = 834, 1041, 670, and 383, for Huh‐7, COS, HeLa, and HEK293T, respectively, in the endogenous Rab18 group (gray bars). N = 860, 1119, and 469 for COS, HeLa, and HEK293T, respectively, in the DsRed‐Rab18‐transfected group (red bars). In each cell sample, the LD fluorescence dots were derived from at least 10 different cells.
Rab18, TRAPPII, and COPI regulate lipolysis
Since Rab18 and COPI had been implicated in lipolysis of LDs and TRAPPII interacted with both molecules, we determined whether lipolysis was defective in TRAPPII‐deleted cells by measuring the rate of secretion of 3H‐labeled non‐esterified free fatty acids (NEFA) that had been pre‐incubated with the cells. Release of fatty acids in TRAPPII‐deleted cells at various time points was only 50–60% of wild‐type HEK293T cells (Fig 5). The extent of reduction was similar to depletion of Rab18 by siRNA in the same experiment. Release of COPI from Golgi membranes by the drug brefeldin A (BFA) or by siRNA depletion of β‐COP (siCOPB) caused a similar extent of lipolysis reduction as depletion of Rab18 or deletion of TRAPPII (Lippincott‐Schwartz et al, 1989; Donaldson et al, 1992; Scheel et al, 1997), suggesting potential relationship between COPI, TRAPPII, and Rab18 in LD lipolysis.
Figure 5. Release of NEFA at various time points by 293T cells with the indicated genotypes or treatments.
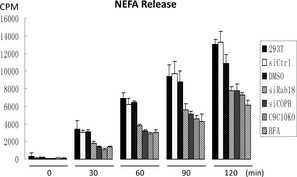
Control siRNA depletion (siCtrl) was conducted with siRNA oligos specific to a firefly luciferase sequence; siCOPB = depletion of β‐COP; siRab18 = depletion of Rab18. C9C10KO = HEK293T cells with TRAPPC9 and TRAPPC10 genes deleted. N = 3, error bars = SD.
COPI, TRAPPII, and Rab18 are in complex
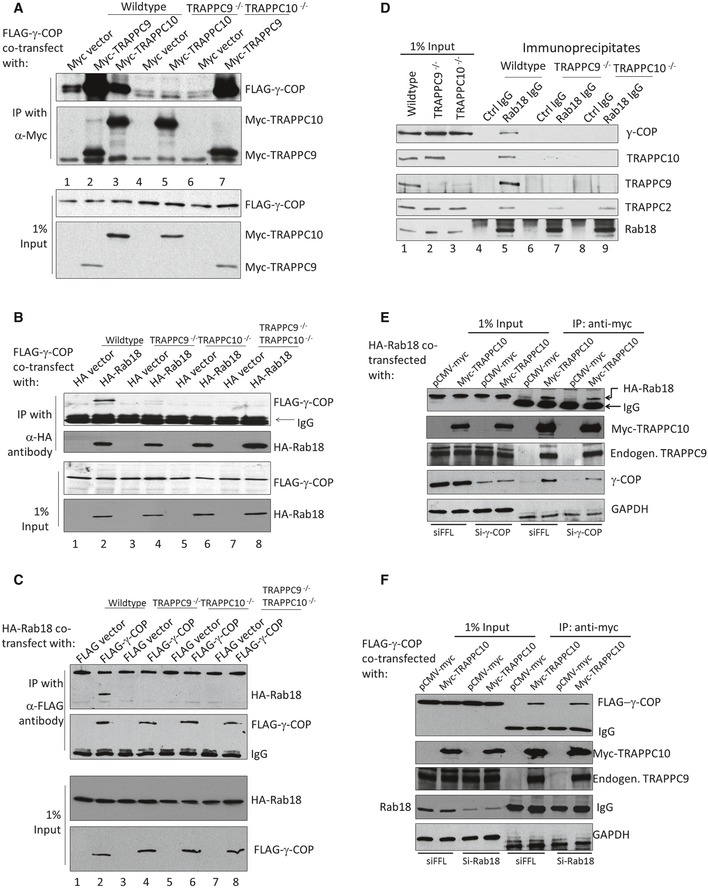
Next, we began to investigate the functional relationship between COPI, TRAPPII, and Rab18 in LD homeostasis. TRAPPC9 and γ‐COP have been suggested to mediate the interaction between TRAPPII and COPI (Yamasaki et al, 2009). We investigated this by interaction studies carried out in cell lines with various TRAPPII deletions. In an experiment in which FLAG‐γ‐COP and Myc‐TRAPPC9 or Myc‐TRAPPC10 were co‐transfected and their interactions were tested with co‐IP, we observed that Myc‐TRAPPC9 and, to a lesser extent, Myc‐TRAPPC10 were both capable of co‐precipitating FLAG‐γ‐COP in HEK293T cells of wild‐type background (Fig 6A, lanes 1–3). However, Myc‐TRAPPC10 could not co‐precipitate FLAG‐γ‐COP in TRAPPC9‐deleted cells (lane 5, Fig 6A), whereas Myc‐TRAPPC9 was able to co‐precipitate FLAG‐γ‐COP in TRAPPC10‐deleted cells (lane 7, Fig 6A). TRAPPC9 could bind to FLAG‐γ‐COP regardless of the presence of TRAPPC10, strongly suggesting TRAPPC9 and γ‐COP directly interacted with each other. To determine whether Rab18 could interact with γ‐COP in a TRAPPII‐dependent manner, we performed a co‐IP experiment in wild‐type and various TRAPPII‐deletion backgrounds. As shown in Fig 6B, FLAG‐γ‐COP co‐precipitated with HA‐Rab18 in a TRAPPII wild‐type background but not in various TRAPPII‐deletion backgrounds, suggesting the interaction between γ‐COP and Rab18 requires intact TRAPPII. Likewise, when FLAG‐γ‐COP was precipitated, HA‐Rab18 came down only in a wild‐type 293T background but not in a TRAPPII‐deletion background (Fig 6C). Based on these overexpression studies, we investigated whether endogenous COPI–TRAPPII–Rab18 complex existed in cells. Co‐IP experiment using anti‐Rab18 antibody, but not control rabbit IgG, was able to co‐precipitate COPI and TRAPPII complexes as indicated by the presence of their representative subunits in the immunoblots (Fig 6D). In TRAPPC9−/− or TRAPPC10−/− backgrounds, the interaction between COPI and Rab18 was not observed, even though TRAPPC2, a subunit common to all TRAPP complexes, co‐precipitated with Rab18 (second bottom panel, Fig 6D). Such result implicated that the core TRAPPI complex might be responsible for binding to Rab18. COPI was not required for the interaction between TRAPPII and Rab18 because siRNA depletion of γ‐COP did not weaken the interaction between TRAPPII and Rab18 (Fig 6E), even though the amount of co‐precipitated γ‐COP was significantly reduced in a Myc‐TRAPPC10 immunoprecipitation. Rab18 was also not required for the interaction between TRAPPII and γ‐COP because siRNA depletion of Rab18 did not weaken that interaction (Fig 6F). Taken together, these results demonstrated that TRAPPII mediated the interaction between COPI and Rab18. This raised the possibility that functional COPI was required to recruit TRAPPII complex to the LD surface, and then TRAPPII activated Rab18, allowing the small GTPase to be recruited onto the LD.
Figure 6. Interaction between COPI and Rab18 is TRAPPII‐dependent.
-
AMyc‐TRAPPC9 directly interacted with FLAG‐γ‐COP. The presence of FLAG‐γ‐COP in immunoprecipitations of Myc‐TRAPPC9 and Myc‐TRAPPC10 from cell lysates of the indicated genetic backgrounds was investigated by immunoblotting.
-
B, CCo‐immunoprecipitation between FLAG‐γ‐COP and HA‐Rab18 in HEK293T cells with indicated genetic backgrounds. The presence of FLAG‐γ‐COP in immunoprecipitates of HA‐Rab18 (and reciprocal IP in C) was investigated by immunoblotting.
-
DThe interaction between endogenous COPI and Rab18 was TRAPPII‐dependent. Endogenous Rab18 was immunoprecipitated by anti‐Rab18 antibody from lysates of indicated genetic background. The presence of γ‐COP and various TRAPP subunits was detected by their specific antibodies as indicated.
-
ECo‐immunoprecipitation between TRAPPII and HA‐Rab18 in control (siFFL) and γ‐COP (si‐γ‐COP) siRNA‐depleted cells. TRAPPII was precipitated with transfected Myc‐TRAPPC10, and the presence of HA‐Rab18 was determined by immunoblotting.
-
FCo‐immunoprecipitation between FLAG‐γ‐COP and TRAPPII in control (siFFL) and Rab18 (si‐Rab18) siRNA‐depleted cells. TRAPPII was precipitated with Myc‐TRAPPC10, and the presence of FLAG‐γ‐COP was determined by immunoblotting. The molecular weight of endogenous Rab18 overlaps with IgG light chain, and therefore, the amount of endogenous Rab18 precipitated with TRAPPII cannot be determined in this experiment.
COPI is required for Rab18 to be recruited onto LD
To determine whether COPI was required for LD association of Rab18, we depleted γ‐COP and investigated whether Rab18 was LD‐associated. Rab18 signals were no longer associated with LD surface in γ‐COP‐depleted Huh‐7 cells (Fig 7A). However, when we tried to confirm such observation by inhibiting COPI membrane binding with BFA, we found that BFA could not stop Rab18 from being recruited to LD surface (right panel and inset, Fig 7B). Further, TRAPPC9 signal was found largely in the cytosol in Huh‐7 cells (Appendix Fig S6A). Even if we depleted the cytosolic pool with digitonin prior to fixation and staining, TRAPPC9 was mainly found on Golgi membrane but not on LD surface (Fig 7C and Appendix Fig S6B). Puzzled by the apparently contradictory findings, we suspected COPI activity was required only briefly after oleic acid loading. To determine how γ‐COP, TRAPPII, and Rab18 localizations changed when cells were loaded with oleic acid, we monitored the subcellular localizations of these proteins at various time points after the cells were incubated with oleic acid. Because of the high capacity of Huh‐7 cells to take up lipid, we needed to serum‐starve the cells in order to lower the level of LD. In the absence of any source of fatty acid, TRAPPC9 was found on the Golgi (Figs 8A and EV4). Eight hours after oleic acid was applied, TRAPPC9 was relocated to LD surface with high fluorescence intensity, colocalized with LD marker ADRP. By 20 h, TRAPPC9 once again returned to the Golgi. In serum‐starved Huh‐7 cells, Rab18 and γ‐COP were found in the perinuclear structures and in tubular networks (top panels, Fig 8B), consistent with the reported subcellular localization at the Golgi and ER tubules for Rab18 and ERGIC and cis‐Golgi for COPI. After 4–8 h of incubation with oleic acid, relocation of γ‐COP and Rab18 to cytosolic space surrounding the LDs became obvious (Figs 8B and EV5). Frequently, γ‐COP signals appeared as intense fluorescence puncta juxtaposed the LDs. This feature was most obvious at 12 h as the Rab18 signal started to consolidate onto LD surface, in agreement with the observation by others (Wilfling et al, 2014). By 20–24 h after oleic acid loading, γ‐COP signal had largely returned to the Golgi, behaving like TRAPPC9, while Rab18 remained on LD surface (bottom panels and insets, Fig 8B). During the period between 4 and 16 h of oleic acid incubation, the γ‐COP signal became highly fragmented, possibly due to heavy traffic and metabolism of the absorbed oleic acid. Golgi marker GM130 and Rab3GAP1 were not observed to undergo such drastic redistribution (Fig EV6A). Rab3GAP1 localization was also not changed in TRAPPII‐deleted cells (Fig EV6B).
Figure 7. Differential effect of COPI inhibition by siRNA depletion and BFA on Rab18 recruitment onto LD surface.
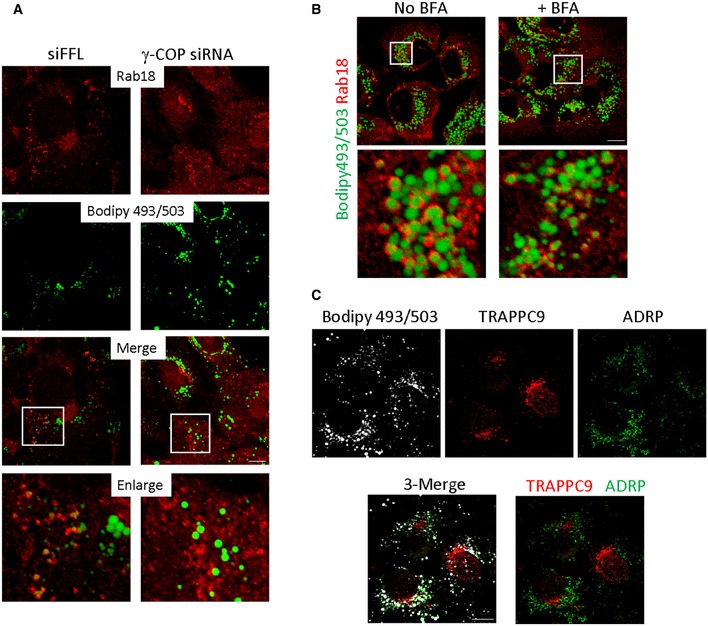
- Huh‐7 cells were depleted with control siRNA specific to firefly luciferase sequence (top panels) or with siRNA specific to γ‐COP (bottom panels). Endogenous Rab18 (red) and LD (green) were stained and visualized. Scale bar = 10 μm.
- Huh‐7 cells were treated with or without BFA for 6 h before staining for Rab18 (red) and LD (green). Scale bar = 10 μm.
- TRAPPC9 were detected on Golgi but not LD surface. Huh‐7 cells were permeabilized with digitonin before fixation and staining. LDs were labeled with Bodipy 493/503 and pseudocolored in white. TRAPPC9 (red) and LD surface marker ADRP (green) were detected by their respective antibodies. Scale bar = 10 μm.
Figure 8. Time‐dependent relocations of COPI, TRAPPII, and Rab18 onto LD surface upon oleic acid incubation.
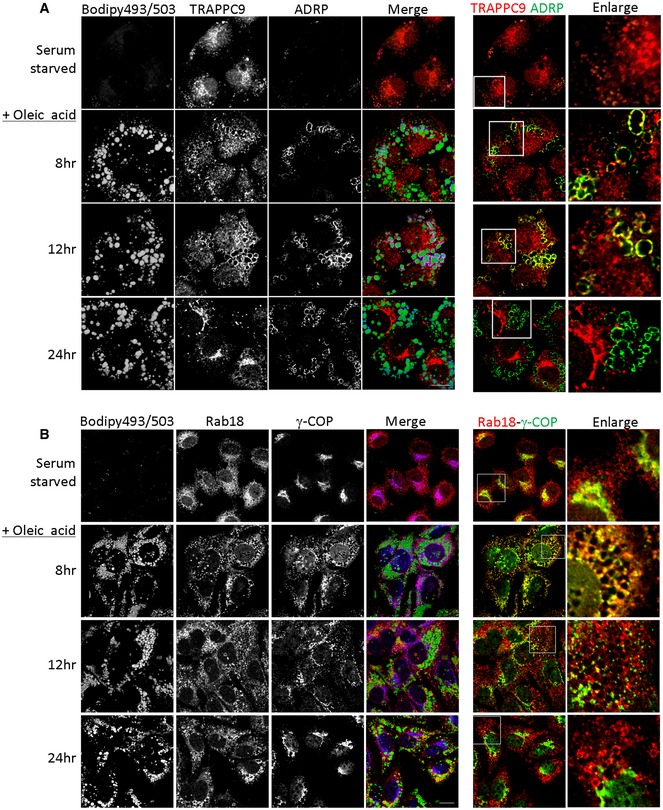
- In the merged images, Bodipy 493/503 was pseudocolored in green, TRAPPC9 in red, and ADRP in blue. In the colocalization between TRAPPC9 and ADRP on the right side, the ADRP signal was re‐colored to green for easy visualization. Scale bar = 10 μm.
- Bodipy 493/503 was pseudocolored in green, Rab18 in red, and γ‐COP in blue. In the colocalization between Rab18 and γ‐COP on the right side, the γ‐COP signal was re‐colored to green for easy visualization. Scale bar = 10 μm.
Figure EV4. TRAPPC9 localization on LD surface was investigated as a function of time after oleic acid incubation.
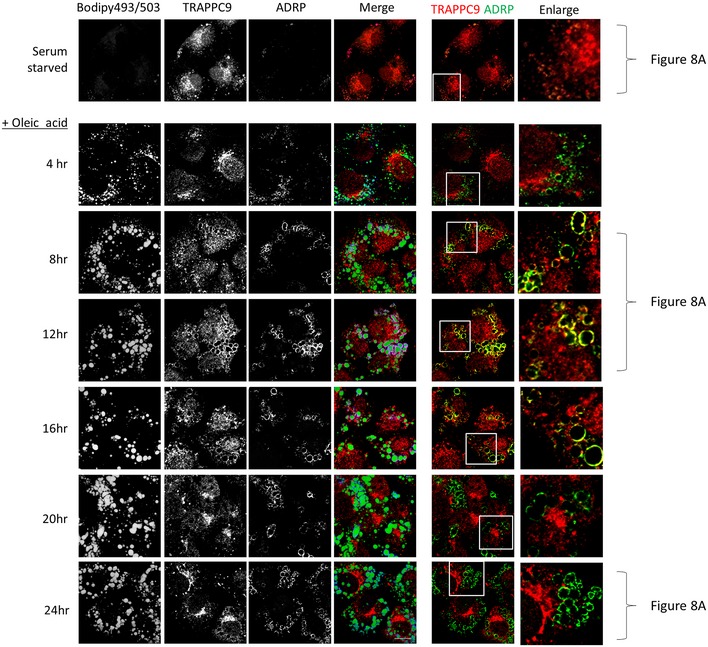
Huh‐7 cells were first serum‐starved and then incubated with oleic acid for the indicated time before staining with Bodipy 493/503, TRAPPC9, and ADRP. In the merge images, Bodipy 493/503 was pseudocolored in green, TRAPPC9 in red, and ADRP in blue. In the colocalization between TRAPPC9 and ADRP on the right side, the ADRP signal was re‐colored to green for easy visualization and overlapped signals around the LDs became yellow. Scale bar = 10 μm.
Figure EV5. COPI and Rab18 localizations were investigated as a function of time after oleic acid incubation.
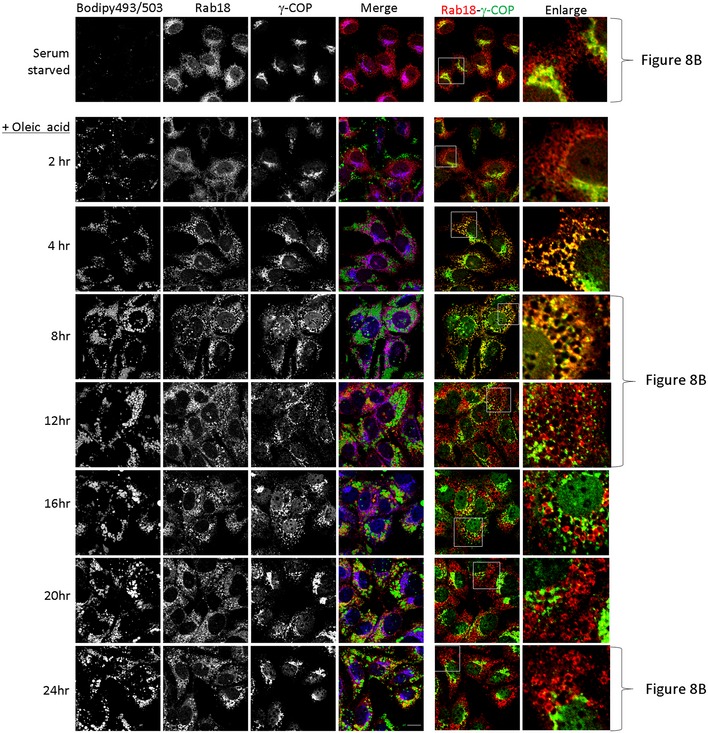
Huh‐7 cells were first serum‐starved and then incubated with oleic acid for the indicated time before staining with Bodipy 493/503, Rab18, and γ‐COP. In the merged images, Bodipy 493/503 was pseudocolored in green, Rab18 in red, and γ‐COP in blue. In the colocalization between Rab18 and γ‐COP on the right side, the γ‐COP signal was re‐colored to green for easy visualization. Scale bar = 10 μm.
Figure EV6. Subcellular localization of Rab3GAP1 and GM130 in response to oleic acid incubation and TRAPPII deletion.

- Rab3GAP1 and GM130 were stained after Huh‐7 cells were serum‐starved (top panels), and then incubated with oleic acid for 8 and 24 h. Bodipy 493/503 was pseudocolored in green, Rab3GAP1 in red, and GM130 in blue. Rab3GAP1 signals remained perinuclear and significantly colocalized with Golgi marker GM130 regardless of oleic acid incubation. Scale bar = 10 μm.
- Rab3GAP1 and GM130 were stained after HEK293T cells of indicated genetic background were incubated with oleic acid for 24 h. TRAPPII deletion did not change the perinuclear signal of Rab3GAP1. Rab3GAP1 was not associated with LDs in any of the cells shown. Scale bar = 10 μm.
Based on such information, we tested again whether disrupting COPI from membranes with BFA during this period after oleic acid incubation would affect LD association of Rab18. As shown in Fig 9, Rab18 failed to be recruited onto LDs when BFA was applied to the cells together with oleic acid in the first 6 h. However, when BFA was added 10 h after oleic acid incubation, it no longer prevented Rab18 from being recruited to LDs. We investigated how TRAPPC9 responded to BFA treatment in the first 6 h of oleic acid loading and found that TRAPPC9 signal was drastically reduced. This was probably due to its release from Golgi for LD metabolism, and the protein was depleted by digitonin treatment. Nonetheless, any remaining TRAPPC9 signal was not associated with LD in this condition (Appendix Fig S7). A similar change in subcellular localization of Rab18 and γ‐COP was also observed when we investigated HEK293T cells (Appendix Fig S8), except that γ‐COP was weakly localized to LD surface and it took 16 h of oleic acid incubation for Rab18 to be recruited onto LDs. These results suggested that, with respect to the recruitment of Rab18 onto LDs, the function of COPI and TRAPPII appeared to be most critical in the first 4–8 h (for Huh‐7) and 12–16 h (for HEK293T) after oleic acid loading. When the HEK293T cells were treated with BFA 12 h after oleic acid loading, Rab18 was unable to associate with LDs (Appendix Fig S9). Addition of BFA after 18 h of oleic acid loading no longer prevented association of Rab18 with LDs. Overall, these results suggested that the recruitment of TRAPPII and Rab18 on the LD surface was COPI‐dependent, but once the role of COPI was fulfilled, Rab18 did not need COPI to maintain its LD association, and therefore, BFA treatment could not remove Rab18 from the LD surface.
Figure 9. Recruitment of Rab18 onto LD is dependent on COPI.
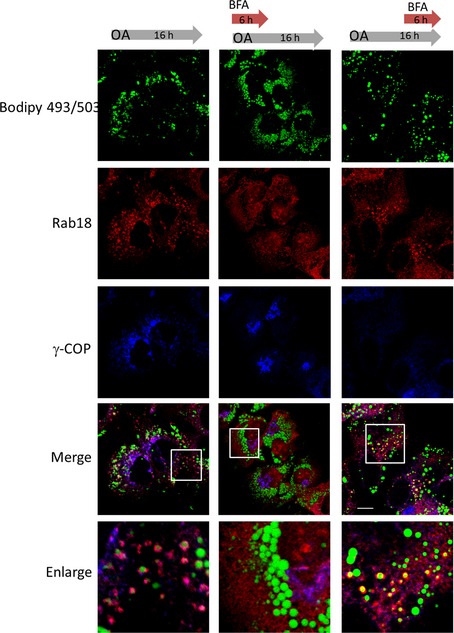
Huh‐7 cells were first starved with serum‐free medium and then incubated with oleic acid for 12 h. Condition 1 (control): The cells were not treated with BFA at any time point (left column). Condition 2: The cells were incubated with both 5 μg/ml of BFA and oleic acid incubation for 6 h and then in oleic acid alone for 10 h (middle column). Condition 3: The cells were treated with 5 μg/ml of BFA starting at 12 h after oleic acid incubation (right column). Scale bar = 10 μm.
Discussion
In this study, we have discovered a COPI–TRAPPII–Rab18 signaling mechanism in LD homeostasis. The association of Rab18 with LDs is regulated by COPI coatomers and TRAPPII in a spatiotemporally critical manner. COPI recruits TRAPPII via the interaction between γ‐COP and TRAPPC9. Then, TRAPPII serves as a GEF for Rab18 and recruits the small GTPase onto the LD surface (Fig 10). Arf1‐COPI machinery causes increased surface tension of LDs and promotes the budding of nano‐LDs. Here, we suggest that the COPI–TRAPPII interaction activates Rab18 so that LDs and the ER are in proximity to facilitate the lipogenic and/or lipolytic activities of Rab18.
Figure 10. Hypothesis of how COPI and TRAPPII activate Rab18 and promote the association of Rab18 on LD surface.
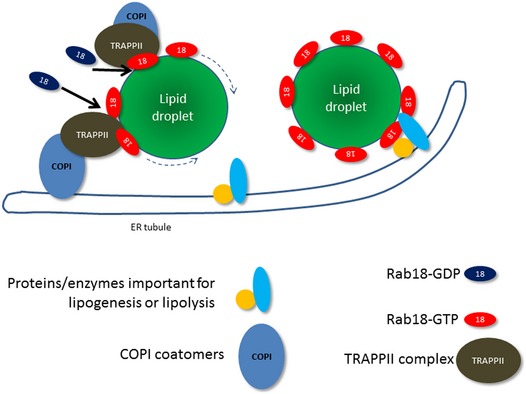
COPI relocates from Golgi to LD surface or relocates to the surface of ER tubules via retrograde transport. The interaction between COPI and TRAPPII allows the GEF activity of TRAPPII to activate Rab18. Rab18‐GTP becomes LD‐associated and promotes the formation of ER–LD bridge so that lipid metabolizing enzymes can access the content of LD.
Due to the high degree of sequence similarity between Rab1 and Rab18, we suspect that the GEF catalytic center for both Rabs lies in the TRAPPI core as suggested by structural studies (Cai et al, 2008). However, the inability to isolate TRAPPI prevented us from directly addressing whether the TRAPPI core was capable of activating both Rabs. Unlike yeast TRAPPIII, we did not observe GEF activity of mammalian TRAPPIII toward Rab1. The reason that mammalian TRAPPIII fails to activate either Rab1 or Rab18 is not clear at present, but we suspect the TRAPPIII‐specific subunits may have blocked the access of Rab1 or Rab18 for nucleotide exchange. More importantly, since siRNA depletion of the TRAPPC8 and CRISPR–Cas9 deletion of TRAPPC12 (both are TRAPPIII‐specific subunits) did not cause formation of aberrantly large LDs (Fig 3A and data not shown), it was very unlikely that TRAPPIII was involved in Rab18‐mediated LD homeostasis. Rab3GAP complex also functions as a GEF localizing Rab18 at the ER or stabilizing it at the cis‐Golgi (Gerondopoulos et al, 2014; Handley et al, 2015). TRAPPII and Rab3GAP may activate Rab18 at specific locations to regulate different cellular events. In agreement with this idea, depleting either subunit of the Rab3GAP complex redistributed Rab18 from the ER to the cytosol, whereas depletion of TRAPPII subunits specifically prevented Rab18 from locating on LDs but left Rab18 still associated with ER or Golgi membranes. Endogenous Rab18 relocated from a perinuclear pattern (i.e., ER and Golgi) to encircle small LDs when cells were loaded with oleic acid (Fig 8), but oleic acid loading, as well as TRAPPII deletion, did not change the ER and Golgi localization of Rab3GAP (Fig EV6). The efficiency of Rab18 association with LDs varied among the cell lines we investigated. Since TRAPPII and COPI are functional in these cell lines and the protein expression levels for TRAPPII and COPI subunits were very similar (data not shown), we think factor(s) other than the components of the COPI–TRAPPII–Rab18 pathway must also regulate the ability of Rab18 to be recruited to LDs. Such factor(s) may be very limiting or missing in COS cells, making regulation of ER morphology by Rab18 a prominent function in this cell line (Gerondopoulos et al, 2014).
siRNA depletion of TRAPPII‐specific subunits does not affect ER‐to‐Golgi traffic in COS and HEK293T cells (Yamasaki et al, 2009; this study). TRAPPII may thus not play an essential role in the early secretory pathway in mammals as implicated by yeast genetic studies. TRAPPII activates Rab1, which plays important roles in ER‐to‐Golgi traffic. However, the subcellular localization of Rab1 is normal in TRAPPII‐depleted cells (Appendix Fig S5), suggesting that TRAPPI is sufficient in maintaining the Rab1 function in ER‐to‐Golgi traffic in these cells. Only the function of Rab18 activation is not compensated in TRAPPII‐depleted cells, leading hence to the aberrantly large LD phenotype.
Human individuals suffering from Warburg micro syndrome contain mutations in Rab18, Rab3GAP, or TBC1D20 genes, and fibroblasts isolated from these individuals all contain aberrantly large LDs. TRAPPC9 mutations have been reported to cause microcephaly and intellectual disability. At the cellular level, we have found that TRAPPC9 mutant skin fibroblasts also contain aberrantly large LDs, like the TRAPPII‐deleted cells. Here again, human genetic evidence suggests Rab18 and TRAPPII may function in the same pathway in LD metabolism. Recently, Rab18 has been reported to be a retinoic acid‐responsive, LD‐associated protein in hepatic stellate cells, which are involved in pathological progression of liver fibrosis when these cells are activated (O'Mahony et al, 2015). The protein expression and LD insertion of Rab18 increase during stellate cell activation, whereas knockdown or reduction of its LD‐membrane insertion inhibits the activation. In this context, the regulation of Rab18‐LD association by TRAPPII makes this protein complex a potential target for anti‐liver fibrosis treatment.
Materials and Methods
Reagents, plasmids, and antibodies
Transfection reagent Lipofectamine 2000 was from Invitrogen. Glutathione agarose 4B was from Macherey‐Nagel. Unless otherwise indicated, all other chemicals and reagents were purchased from Sigma‐Aldrich. Construction of Myc‐tagged mammalian TRAPP subunits has been previously described (Zong et al, 2011, 2012). FLAG‐γ‐COP mammalian expression plasmid was obtained from Dr. Tagaya (Tokyo University of Pharmacy and Life Sciences, Japan). Rab18 was subcloned in frame into a N‐terminal 3×HA‐tagged pcDNA3.1(−), pDsRed‐monomer‐C1, and pGEX‐4T1 vector using the EcoRI and BamHI restriction sites. Rab18 mutants were generated using QuikChange site‐directed mutagenesis (Agilent Technologies). Rabbit polyclonal antibodies against TRAPP subunits have been described previously (Zong et al, 2011, 2012). Rabbit anti‐Rab18 antibody (SAB4200173) and monoclonal ANTI‐FLAG® M2 antibody (F3165) were purchased from Sigma‐Aldrich. Mouse monoclonal antibodies against γ‐COP (sc‐271362), TRAPPC10 (clone RR‐18, sc‐101259), ADRP (sc‐377229), and LAMP2 (sc‐18822) were purchased from Santa Cruz Biotechnology. Mouse monoclonal antibodies against GM130 (clone 35, 610822) and Sec31A (clone 32, 612350) were purchased from BD Biosciences. Monoclonal antibody against golgin‐97 (CDF4, A‐21270) was obtained from Invitrogen. Polyclonal antibody against calnexin was purchased from Sigma (C4731). Rabbit polyclonal antibodies against TIP47 (10694‐1‐AP) and Rab3GAP1 (21663‐1‐AP) were from Proteintech. Mouse monoclonal antibody against ERGIC‐53 was obtained from Hans‐Peter Hauri (Schweizer et al, 1993). Anti‐GAPDH antibody (ab37168) was from Abcam. Monoclonal antibodies against HA were from Roche (catalog no. 11583816001) or from Trans Inc. (catalog no. HT‐301; Shenzhen, China).
Cell culture, transfection, and RNA interference
HEK293T, HeLa, COS‐7, and Huh‐7 cells and human fibroblasts were cultured in DMEM (Invitrogen), supplemented with 10% (v/v) fetal bovine serum (FBS; Invitrogen) at 37°C in a 5% CO2 incubator. For plasmid transfection, GenJet™ transfection reagent Ver.II (SignaGen) and polyethylenimine (PEI; Sigma) were used for Huh‐7 cells or other cell lines, respectively, according to the manufacturer's instructions. For siRNA transfection, Lipofectamine 2000 (Invitrogen) was used. For microscope imaging, cells were cultured on 24‐well plates to reach 70% confluence on the day of transfection. Two microlitre of transfection reagent and 1 μl of 20 μM siRNA were added separately into 50 μl of Opti‐MEM medium and incubated for 5 min. Both solutions were mixed and incubated for an additional 20 min. The transfection mixture was added to culture wells in complete DMEM without antibiotics. After incubation at 37°C for 6 h, the cells were changed to fresh medium and cultured for an additional 24 h. The cells were then transfected for the second time with the same condition. After 6‐h incubation, cells were trypsinized and seeded onto 12‐mm poly‐L‐lysine‐coated coverslips. For Western blot analysis, cells were cultured in 10‐cm dishes, and 40 μl of transfection reagent and 20 μl of 20 μM siRNA were used for each transfection. Unless otherwise indicated, all of the experiments were performed at 72 h after the first siRNA transfection. Stealth™ siRNAs targeting to human TRAPPC9 (catalog # 129003) were purchased from Invitrogen. The sequences of each siRNA Oligonucleotides (Genepharm, Shanghai, China) target to human Rab18, human β‐COP, and human γ‐COP are as follows: Rab18 siRNA(1): 5′‐GUCACAAGAAGAGAUACAU‐3′; Rab18 siRNA(2): 5′‐GCCUGAAAUUUGCACGAAA‐3′; β‐COP siRNA: 5′‐GGAUCACACUAUCAAGAAA‐3′; γ‐COP siRNA(1): 5′‐GAGAUGUGUUACCCAGUAUCU‐3′; γ‐COP siRNA(2): 5′‐CUUGUGAGAGGUCAGACAA‐3′.
The formation of lipid droplets in 293T, HeLa, and COS7 cells was induced with complete medium supplemented with 400 μM oleic acid (OA) for the indicated time. For Huh‐7 cells, the cells were first cultured with DMEM plus 1% fatty acid‐free BSA for 16 h prior to stimulation with 400 μM OA. For experiments involving brefeldin A (BFA) treatment, 293T cells were either treated with 400 μM OA and 5 μg/ml BFA for 6 h followed by addition of 400 μM OA and incubation for 18 h or incubated with 400 μM OA first for 18 h and then with medium containing 400 μM OA and 5 μg/ml BFA for 6 h. For Huh‐7 cells, after starvation for 16 h, 400 μM OA and 5 μg/ml BFA were added for 6 h followed by an additional 10 h of OA incubation. Another condition involved an incubation with 400 μM OA for 10 h, followed by 400 μM OA and 5 μg/ml BFA for 6 h.
CRISPR–Cas9 deletion of TRAPPC9 and TRAPPC10 in HEK293T cells
A cluster of guide RNAs that encompassed about 500 bp of DNA within the gene loci of TRAPPC9 or TRAPPC10 were first cloned into the vector lentiCRISPR version 2 (Addgene plasmid # 52961) (Sanjana et al, 2014). These plasmids were transfected into HEK293T cells in a 12‐well plate. The transfected cells were expanded into a 10‐cm plate and selected with growth medium containing 1.5 μg/ml puromycin. After 2 weeks, the puromycin‐resistant clones were isolated for PCR genotyping and further cell expansions. Double deletion of TRAPPC9 and TRAPPC10 was carried out on TRAPPC9‐deleted cells. Detailed information on TRAPPC9 and TRAPPC10 gene loci and strategy of generating indels with guide RNAs can be found in Fig EV3.
Nucleotide exchange assays
For the expression and purification of GST‐tagged Rab1a, Rab2, and Rab18, E. coli strain BL21‐Ai, transformed with a plasmid (pGEX4T‐2) that contained individual Rab protein coding sequences, was grown at 37°C to an OD600 of 1.0. Expression of the recombinant proteins was induced at 18°C for 16 h in the presence of 0.1 mM IPTG and 0.2% l‐arabinose. The cells were harvested and resuspended in lysis buffer (PBS with 5 mM MgCl2 and a protease inhibitor cocktail), sonicated, and centrifuged at 12,000 g for 30 min. The supernatants were incubated with glutathione–Sepharose beads for 1 h at 4°C. After that, the beads were washed extensively with lysis buffer. For GTP‐binding assays, the beads were incubated in 50 mM HEPES–NaOH, pH 8.0, 1 mg/ml BSA, and 1 mM EDTA for 30 min. 10 μM GDP and 5 mM MgCl2 were added and incubated for an additional 45 min. Finally, the beads were transferred to a column and eluted with lysis buffer containing 10 mM glutathione. The purified proteins were stored at −80°C until use.
The nucleotide exchange reaction was carried out in a final volume of 100 μl containing GEF buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 2 mM MgCl2, and 50 μg/ml BSA), 4 pmol Rab‐GDP, and 50 pmol guanosine 5′‐O‐(3‐thio)triphosphate (GTPγS) ([35S], 104 cpm/pmol). Immunoprecipitates containing the TRAPP complex or a control were transferred into the reaction mixture and incubated for the indicated times at 37°C. The reaction was stopped by adding 2 ml of ice‐cold stop buffer (25 mM Tris, pH 8.0, and 20 mM MgCl2) and then was filtered through a 0.4‐μm nitrocellulose membrane (Whatman) and washed with stop buffer extensively. The membrane was measured for the Rab proteins labeled with [35S]GTPγS by scintillation counting. For GDP‐release assays, 5 μg of purified GST‐tagged Rab protein was incubated in 100 μl assay buffer containing 50 mM HEPES–NaOH, pH 6.8, 1 mg/ml BSA, 125 μM EDTA, 10 μM Mg‐GDP, and 5 μCi [3H]GDP for 15 min at 30°C. At the end of incubation, 10 mM Mg‐GTP and 10 μl of immunoprecipitate of TRAPP complex or control were added and further incubated for 30 min at 37°C. The reaction was stopped by adding 2 ml of ice‐cold stop buffer and filtered through a 0.4‐nitrocellulose membrane (Millipore) with extensive wash using the stop buffer. The membrane was taken for scintillation measurement of the captured [3H]GDP.
Immunoprecipitation and in vitro binding assay
Pellets of 293T cells harvested from each 15‐cm plate were lysed with 1 ml of lysis buffer consisting of 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.1% NP‐40, and protease inhibitor cocktails. The lysates were centrifuged at 12,000 g for 30 min at 4°C. The supernatants could be subjected to the in vitro binding assay described below or to isolation of TRAPP complex by immunoprecipitation. For immunoprecipitation, lysates were incubated with 1 μg of corresponding antibodies or non‐specific IgG as control at 4°C overnight. The immunoprecipitates were washed five times with lysis buffer and resuspended in 50 μl of PBS for nucleotide exchange assays or eluted by boiling the beads in 2× SDS loading buffer and analyzed by immunoblot.
For co‐immunoprecipitation detecting interaction between endogenous Rab18 and γ‐COP, HEK293T cells harvested from a 15‐cm plate were lysed with 1 ml of lysis buffer consisting of 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 10 mM EDTA, 0.1% NP‐40, and protease inhibitor cocktails. The lysates were centrifuged at 12,000 g for 30 min at 4°C, and then, the supernatants were incubated overnight at 4°C with 2 μg of rabbit anti‐Rab18 antibody or 2 μg non‐specific rabbit IgG as control. The next day, 50 μl of protein A–Sepharose 4B slurry was added to the mixture for further incubation for 4 h. The immunoprecipitates were washed five times with lysis buffer and eluted by boiling the beads in 2× SDS loading buffer and analyzed by immunoblotting.
Expression and purification of GST or GST‐Rab fusion proteins were as described above. For an in vitro binding assay, 50 μg of purified GST‐Rab or GST (control) was incubated with 50 μl glutathione–Sepharose in 1 ml NE100 buffer (20 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.1% Triton X‐100) for 60 min at 4°C. The beads were washed with 500 μl NE100 buffer four times and resuspended in 200 μl of NE100 buffer (the nucleotide‐free form) or in 200 μl of NL100 buffer (20 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 5 mM MgCl2, and 0.1% Triton X‐100), and then, 20 μl of 10 mM GDP or GTPγS was added. Then, the beads were incubated with 200 μl of 293T cell extracts for 60 min at 4°C on a roller. After incubation, the beads were centrifuged and washed with NL100 buffer four times. Finally, 40 μl of 2×SDS loading buffer was added and the beads were boiled for 5 min for SDS–PAGE and immunoblotting.
LD fractionation
LDs were isolated using a modified protocol of Martin et al (2005). Briefly, four 15‐cm dishes of 293T cells grown to 80% of confluence were incubated in growth medium supplemented with 400 μM oleic acid for 24 h. Then, cells were scraped and collected in 2 ml of disruption buffer (25 mM Tris–HCl, pH 7.4, 100 mM KCl, 1 mM EDTA, 5 mM EGTA, and protease inhibitor cocktail), kept on ice for 30 min, and disrupted by passing through a needle (27‐gauge). Afterward, the cell lysates were centrifuged at 1500 g for 5 min. The supernatants were adjusted to 3 ml final volume containing 0.33 M sucrose and transferred into a 13‐ml polycarbonate ultracentrifuge tube, and then, 3 ml of 0.25 M, 3 ml of 0.125 M sucrose solution, and 3 ml of disruption buffer were overlaid sequentially. After centrifugation at 150,000 g for 2 h, 1.5‐ml fractions were collected from the top of the tube and the protein contents of these fractions were analyzed by immunoblotting.
Lipolysis measurements
Lipolytic activity was assessed through measurements of NEFA released from lipid droplets as previously described, with minor modifications (Beller et al, 2008). Briefly, cells grown on 24‐well plates at a density of 1 × 105/well were incubated for 24 h with F12 medium supplemented with 400 μM oleic acid complexed to 0.4% BSA. [3H]‐labeled oleic acid was also added at 5 × 105 dpm/well. After incubation, the cells were washed three times with 2% defatted BSA in F12 medium to remove unincorporated oleic acid, and then, 500 μl of F12 medium containing 10 μM triacsin C was added to inhibit intracellular synthesis of fatty acids. The medium was collected and replaced at indicated time. NEFA released was measured by the radioactivity released to the cell culture medium by scintillation counting.
Fluorescence microscopy
Cells were grown on poly‐L‐lysine‐coated coverslips, washed three times with PBS, and fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. After fixation, the cells were permeabilized with 0.1% saponin in PBS for 20 min, blocked in blocking buffer (PBS containing 1% BSA and 0.05% saponin) for 30 min. The coverslips were sequentially incubated with primary antibodies and fluorophore‐conjugated secondary antibodies diluted in blocking buffer. For staining of TRAPPC9, Huh‐7 cells were first permeabilized with 40 μg/ml digitonin in PBS for 5 min on ice. After three washes with PBS, the samples were fixed with 4% PFA for 15 min at room temperature and blocked with PBS containing 1% BSA and 0.05% saponin for 30 min. Then, the samples were subjected to antibody incubations as described above. For lipid droplet staining, Bodipy 493/503 was added after fixation or combined with the secondary antibody at a final concentration of 2 μg/ml for 30 min. Nuclei were stained with DAPI (5 ng/ml in PBS) for an additional 5 min. The coverslips were mounted on glass slides with Fluoromount‐G (Southern BioTech, USA). Images were captured using an FV1200 Olympus confocal microscope equipped with a 60× objective (NA = 1.4). When two or three markers were imaged in the same cells, each fluorophore was excited and detected sequentially.
Author contributions
CL performed the experiments with assistance from XL, SZ, GKYS and SSBY. CL, AS, YL, HCC and SSBY analyzed and interpreted data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank the Genome Centre of University of Hong Kong for mass spectrometry identification of TRAPPC9 co‐purified proteins. The study was supported by the General Research Fund numbers 479410 and 477912 of Hong Kong RGC, and the one‐line budget of CUHK. We thank Dr. Michael G. Roth (U.T. Southwestern) for critical comments on and language editing the manuscript.
See also: F Zappa et al (February 2017)
References
- Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K, Maina EN, Morgan NV, Tee L, Morton J, Ainsworth JR, Horn D, Rosser E, Cole TRP, Stolte‐Dijkstra I, Fieggen K, Clayton‐Smith J, Megarbane A, Shield JP, Newbury‐Ecob R et al (2005) Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet 37: 221–224 [DOI] [PubMed] [Google Scholar]
- Barrowman J, Bhandari D, Reinisch K, Ferro‐Novick S (2010) TRAPP complexes in membrane traffic: convergence through a common Rab. Nat Rev Mol Cell Biol 11: 759–763 [DOI] [PubMed] [Google Scholar]
- Bassik Michael C, Kampmann M, Lebbink Robert J, Wang S, Hein Marco Y, Poser I, Weibezahn J, Horlbeck Max A, Chen S, Mann M, Hyman Anthony A, LeProust Emily M, McManus Michael T, Weissman Jonathan S (2013) A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 152: 909–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beller M, Sztalryd C, Southall N, Bell M, Jackle H, Auld DS, Oliver B (2008) COPI complex is a regulator of lipid homeostasis. PLoS Biol 6: e292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bem D, Yoshimura S‐I, Nunes‐Bastos R, Bond Frances F, Kurian Manju A, Rahman F, Handley Mark TW, Hadzhiev Y, Masood I, Straatman‐Iwanowska Ania A, Cullinane Andrew R, McNeill A, Pasha Shanaz S, Kirby Gail A, Foster K, Ahmed Z, Morton Jenny E, Williams D, Graham John M, Dobyns William B et al (2011) Loss‐of‐function mutations in RAB18 cause Warburg micro syndrome. Am J Hum Genet 88: 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette‐Mackie EJ, Dwyer NK, Barber T, Coxey RA, Takeda T, Rondinone CM, Theodorakis JL, Greenberg AS, Londos C (1995) Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J Lipid Res 36: 1211–1226 [PubMed] [Google Scholar]
- Brasaemle DL, Wolins NE (2012) Packaging of fat: an evolving model of lipid droplet assembly and expansion. J Biol Chem 287: 2273–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Chin HF, Lazarova D, Menon S, Fu C, Cai H, Sclafani A, Rodgers DW, De La Cruz EM, Ferro‐Novick S, Reinisch KM (2008) The structural basis for activation of the Rab Ypt1p by the TRAPP membrane‐tethering complexes. Cell 133: 1202–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Davey M, Schluter C, Pandher P, Fang Y, Foster LJ, Conibear E (2011) Organization and assembly of the TRAPPII complex. Traffic 12: 715–725 [DOI] [PubMed] [Google Scholar]
- Dejgaard SY, Murshid A, Erman A, Kizilay O, Verbich D, Lodge R, Dejgaard K, Ly‐Hartig TB, Pepperkok R, Simpson JC, Presley JF (2008) Rab18 and Rab43 have key roles in ER‐Golgi trafficking. J Cell Sci 121: 2768–2781 [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD (1992) Brefeldin A inhibits Golgi membrane‐catalysed exchange of guanine nucleotide onto ARF protein. Nature 360: 350–352 [DOI] [PubMed] [Google Scholar]
- Farese RV Jr, Walther TC (2009) Lipid droplets finally get a little R‐E‐S‐P‐E‐C‐T. Cell 139: 855–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerondopoulos A, Bastos RN, Yoshimura S, Anderson R, Carpanini S, Aligianis I, Handley MT, Barr FA (2014) Rab18 and a Rab18 GEF complex are required for normal ER structure. J Cell Biol 205: 707–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham AK, Sinka R, Torres IL, Lilley KS, Munro S (2014) Toward a comprehensive map of the effectors of rab GTPases. Dev Cell 31: 358–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronemeyer T, Wiese S, Grinhagens S, Schollenberger L, Satyagraha A, Huber LA, Meyer HE, Warscheid B, Just WW (2013) Localization of Rab proteins to peroxisomes: a proteomics and immunofluorescence study. FEBS Lett 587: 328–338 [DOI] [PubMed] [Google Scholar]
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, Farese RV (2008) Functional genomic screen reveals genes involved in lipid‐droplet formation and utilization. Nature 453: 657–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley MT, Carpanini SM, Mali GR, Sidjanin DJ, Aligianis IA, Jackson IJ, FitzPatrick DR (2015) Warburg micro syndrome is caused by RAB18 deficiency or dysregulation. Open Biol 5: 150047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss RS, Nilsson T (2014) Rab proteins implicated in lipid storage and mobilization. J Biomed Res 28: 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb CA, Nuhlen S, Judith D, Frith D, Snijders AP, Behrends C, Tooze SA (2016) TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J 35: 281–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieu G, Zheng H, Rothman JE (2013) Stapled Golgi cisternae remain in place as cargo passes through the stack. Elife 2: e00558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Morozova N, Tokarev AA, Mulholland JW, Segev N (2007) The role of Trs65 in the Ypt/Rab guanine nucleotide exchange factor function of the TRAPP II complex. Mol Biol Cell 18: 2533–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liegel Ryan P, Handley Mark T, Ronchetti A, Brown S, Langemeyer L, Linford A, Chang B, Morris‐Rosendahl Deborah J, Carpanini S, Posmyk R, Harthill V, Sheridan E, Abdel‐Salam Ghada MH, Terhal Paulien A, Faravelli F, Accorsi P, Giordano L, Pinelli L, Hartmann B, Ebert Allison D et al (2013) Loss‐of‐function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am J Hum Genet 93: 1001–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott‐Schwartz J, Yuan LC, Bonifacino JS, Klausner RD (1989) Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longatti A, Lamb CA, Razi M, Yoshimura S‐I, Barr FA, Tooze SA (2012) TBC1D14 regulates autophagosome formation via Rab11‐ and ULK1‐positive recycling endosomes. J Cell Biol 197: 659–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch‐Day MA, Bhandari D, Menon S, Huang J, Cai H, Bartholomew CR, Brumell JH, Ferro‐Novick S, Klionsky DJ (2010) Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci USA 107: 7811–7816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra V, Serafini T, Orci L, Shepherd JC, Rothman JE (1989) Purification of a novel class of coated vesicles mediating biosynthetic protein transport through the Golgi stack. Cell 58: 329–336 [DOI] [PubMed] [Google Scholar]
- Martin S, Driessen K, Nixon SJ, Zerial M, Parton RG (2005) Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J Biol Chem 280: 42325–42335 [DOI] [PubMed] [Google Scholar]
- Martin S, Parton RG (2006) Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol 7: 373–378 [DOI] [PubMed] [Google Scholar]
- Mir A, Kaufman L, Noor A, Motazacker MM, Jamil T, Azam M, Kahrizi K, Rafiq MA, Weksberg R, Nasr T, Naeem F, Tzschach A, Kuss AW, Ishak GE, Doherty D, Ropers HH, Barkovich AJ, Najmabadi H, Ayub M, Vincent JB (2009) Identification of mutations in TRAPPC9, which encodes the NIK‐ and IKK‐[beta]‐binding protein, in nonsyndromic autosomal‐recessive mental retardation. Am J Hum Genet 85: 909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH, Mahajnah M, Hill AD, Basel‐Vanagaite L, Gleason D, Hill RS, Bodell A, Crosier M, Straussberg R, Walsh CA (2009) A truncating mutation of TRAPPC9 is associated with autosomal‐recessive intellectual disability and postnatal microcephaly. Am J Hum Genet 85: 897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit B, Conibear E (2009) Identification of the novel TRAPP associated protein Tca17. Traffic 10: 713–723 [DOI] [PubMed] [Google Scholar]
- Murphy DJ (2001) The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res 40: 325–438 [DOI] [PubMed] [Google Scholar]
- O'Mahony F, Wroblewski K, O'Byrne SM, Jiang H, Clerkin K, Benhammou J, Blaner WS, Beaven SW (2015) Liver X receptors balance lipid stores in hepatic stellate cells through Rab18, a retinoid responsive lipid droplet protein. Hepatology 62: 615–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozeki S, Cheng J, Tauchi‐Sato K, Hatano N, Taniguchi H, Fujimoto T (2005) Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum‐derived membrane. J Cell Sci 118: 2601–2611 [DOI] [PubMed] [Google Scholar]
- Philippe O, Rio M, Carioux A, Plaza J‐M, Guigue P, Molinari F, Boddaert N, Bole‐Feysot C, Nitschke P, Smahi A, Munnich A, Colleaux L (2009) Combination of linkage mapping and microarray‐expression analysis identifies NF‐[kappa]B signaling defect as a cause of autosomal‐recessive mental retardation. Am J Hum Genet 85: 903–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol A, Martin S, Fernandez MA, Ferguson C, Carozzi A, Luetterforst R, Enrich C, Parton RG (2004) Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol Biol Cell 15: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz William A (2013) A bridge to understanding lipid droplet growth. Dev Cell 24: 335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido MR, Diaz‐Ruiz A, Jimenez‐Gomez Y, Garcia‐Navarro S, Gracia‐Navarro F, Tinahones F, Lopez‐Miranda J, Fruhbeck G, Vazquez‐Martinez R, Malagon MM (2011) Rab18 dynamics in adipocytes in relation to lipogenesis, lipolysis and obesity. PLoS ONE 6: e22931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, Ferro‐Novick S (2001) TRAPP I implicated in the specificity of tethering in ER‐to‐Golgi transport. Mol Cell 7: 433–442 [DOI] [PubMed] [Google Scholar]
- Sacher M, Kim YG, Lavie A, Oh BH, Segev N (2008) The TRAPP complex: insights into its architecture and function. Traffic 9: 2032–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Meth 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel J, Pepperkok R, Lowe M, Griffiths G, Kreis TE (1997) Dissociation of coatomer from membranes is required for brefeldin A‐induced transfer of Golgi enzymes to the endoplasmic reticulum. J Cell Biol 137: 319–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A, Ericsson M, Bachi T, Griffiths G, Hauri H (1993) Characterization of a novel 63 kDa membrane protein. Implications for the organization of the ER‐to‐Golgi pathway. J Cell Sci 104: 671–683 [DOI] [PubMed] [Google Scholar]
- Scrivens PJ, Noueihed B, Shahrzad N, Hul S, Brunet S, Sacher M (2011) C4orf41 and TTC‐15 are mammalian TRAPP components with a role at an early stage in ER‐to‐Golgi trafficking. Mol Biol Cell 22: 2083–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni KG, Mardones GA, Sougrat R, Smirnova E, Jackson CL, Bonifacino JS (2009) Coatomer‐dependent protein delivery to lipid droplets. J Cell Sci 122: 1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima K, Saitoh A, Hirose S, Nakai W, Kondo Y, Takasu Y, Kakeya H, Shin HW, Nakayama K (2011) GBF1‐Arf‐COPI‐ArfGAP‐mediated Golgi‐to‐ER transport involved in regulation of lipid homeostasis. Cell Struct Funct 36: 223–235 [DOI] [PubMed] [Google Scholar]
- Tan R, Wang W, Wang S, Wang Z, Sun L, He W, Fan R, Zhou Y, Xu X, Hong W, Wang T (2013) Small GTPase Rab40c associates with lipid droplets and modulates the biogenesis of lipid droplets. PLoS ONE 8: e63213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi‐Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T (2002) The surface of lipid droplets is a phospholipid monolayer with a unique Fatty Acid composition. J Biol Chem 277: 44507–44512 [DOI] [PubMed] [Google Scholar]
- Thiam AR, Antonny B, Wang J, Delacotte J, Wilfling F, Walther TC, Beck R, Rothman JE, Pincet F (2013) COPI buds 60‐nm lipid droplets from reconstituted water‐phospholipid‐triacylglyceride interfaces, suggesting a tension clamp function. Proc Natl Acad Sci USA 110: 13244–13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarev AA, Taussig D, Sundaram G, Lipatova Z, Liang Y, Mulholland JW, Segev N (2009) TRAPP II complex assembly requires Trs33 or Trs65. Traffic 10: 1831–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas Joel T, Krahmer N, Gould Travis J, Uchida A, Cheng J‐X, Graham M, Christiano R, Fröhlich F, Liu X, Buhman Kimberly K, Coleman Rosalind A, Bewersdorf J, Farese Jr Robert V, Walther Tobias C (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 24: 384–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Thiam AR, Olarte MJ, Wang J, Beck R, Gould TJ, Allgeyer ES, Pincet F, Bewersdorf J, Farese RV Jr, Walther TC (2014) Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife 3: e01607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki A, Menon S, Yu S, Barrowman J, Meerloo T, Oorschot V, Klumperman J, Satoh A, Ferro‐Novick S (2009) mTrs130 is a component of a mammalian TRAPPII complex, a Rab1 GEF that binds to COPI coated vesicles. Mol Biol Cell 20: 4205–4215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Liang Y (2012) A trapper keeper for TRAPP, its structures and functions. Cell Mol Life Sci 69: 3933–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong M, Wu XG, Chan CW, Choi MY, Chan HC, Tanner JA, Yu S (2011) The adaptor function of TRAPPC2 in mammalian TRAPPs explains TRAPPC2‐associated SEDT and TRAPPC9‐associated congenital intellectual disability. PLoS ONE 6: e23350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong M, Satoh A, Yu MK, Siu KY, Ng WY, Chan HC, Tanner JA, Yu S (2012) TRAPPC9 mediates the interaction between p150 and COPII vesicles at the target membrane. PLoS ONE 7: e29995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File