

Abstract
In B lymphocytes, immunoglobulin class switch recombination (CSR) is induced by activation induced cytidine deaminase (AID) which initiates a cascade of events leading to DNA double stand break (DSB) formation in switch (S) regions. Resolution of DSBs proceeds through formation of S-S synaptic complexes. S-S synapsis is mediated by a chromatin loop that spans the constant region domain of the Igh locus. S-S junctions are joined via a nonhomologous end joining DNA repair process. CSR occurs via an intra-chromosomal looping out and deletion mechanism that is 53BP1 dependent. However, the mechanism by which 53BP1 facilitates deletional CSR and inhibits inversional switching events remains unknown. We report a novel architectural role for 53BP1 in Igh chromatin looping in mouse B cells. Long range interactions between the Eμ and 3’Eα enhancers are significantly diminished in the absence of 53BP1. In contrast, germline transcript promoter:3’Eα looping interactions interactions are unaffected by 53BP1 deficiency. Furthermore, 53BP1 chromatin occupancy at sites in the Igh locus is B cell specific, correlated with histone H4 lysine 20 marks is subject to chromatin spreading. Thus, 53BP1 is required for three dimensional organization of the Igh locus and provides a plausible explanation for the link between 53BP1 enforcement of deletional CSR.
INTRODUCTION
Immunoglobulin class switch recombination (CSR) is induced by activation-induced cytidine deaminase (AID) which initiates the formation of DNA double strand breaks (DSB) in switch (S) regions that flank Cμ and downstream CH regions (reviewed in (1, 2)). DSB intermediates are resolved via intrachromosomal deletion using a distinct nonhomologous end joining (NHEJ) pathway (3). A DNA damage response is activated upon introduction of AID initiated DSBs and leads to formation of repair foci at the Igh locus which accumulate Mre11/Rad50/Nbs1 (MRN), γH2AX and 53BP1 (reviewed in (4)). In cells free of DNA lesions, 53BP1 constitutively binds the chromatin mark histone 4 dimethyl lysine 20 (H4K20me2), via tandem Tudor domains (5-7). In response to DSBs, 53BP1 is phosphorylated by ATM and then accumulates at sites of damage in a manner dependent on phosphorylated H2AX (γH2AX), MDC1 and RNF8 histone ubiquitylation (8-12) and on histone 4 lysine 20 methyl 2 (H4K20me2) (13). It has been proposed that DNA damage promotes high affinity binding of 53BP1 to chromatin which in turn amplifies the efficiency of long range synapsis of broken DNA ends (14). However, deficiencies of ATM, H2AX, MDC1, MRN or Suv4-20h1,h2, the histone methyltransferases responsible for H4K20 modifications, only modestly reduce CSR frequency as compared to the profound reduction found in the absence of 53BP1 (15-21). This dichotomy suggests that 53BP1 performs a function in CSR outside its canonical role in DSB repair.
In mature B lymphocytes, Ig heavy chain effector function is determined by CH genes and is diversified through CSR, while maintaining the original antigen binding specificity arising from V(D)J recombination. The mouse Igh locus contains a series of eight CH genes (μ, δ, γ3, γ1, γ2b, γ2ba, ɛ, and α) that are located downstream of the V, D and JH segments. Each CH gene (except Cδ) is paired with a repetitive S region that is a target for CSR. A transcriptional promoter coupled with an I (intervening) exon is located upstream of each S region (22). Germline transcription initiates at the I exon, proceeds through the S region and terminates downstream of the corresponding CH gene. CSR involves the introduction and joining of DSBs in Sμ and a downstream S region with the concomitant deletion of the intervening genomic material and generation of Sμ-Sx hybrid junctions.
CSR is dependent on three-dimensional (3D) chromatin architecture mediated by long range intra-chromosomal interactions between distantly located transcriptional elements (23-25). In resting splenic B cells, the enhancers Eμ and 3’Eα that are separated by 220 kb interact and form a chromatin loop (23). B cell activation leads to cytokine dependent recruitment of the germline transcript (GLT) promoters to the Eμ:3’Eα complex and enables transcription of S regions targeted for CSR (23, 26). This looped structure facilitates S-S synapsis since Sμ is proximal to Eμ and a downstream S region is co-recruited with the targeted GLT promoter to Eμ:3’Eα complex.
53BP1 deficiency leads to nearly complete abrogation of long range CSR, and a concomitant increase of short range intra-S region deletion, degradation of unrepaired DNA ends and to elevated inversional CSR (14, 17, 27-29). Several models have been proposed to explain how 53BP1 supports genome integrity and facilitates CSR. 53BP1 may facilitate joining of distant DSBs by altering local chromatin structure or by increasing chromatin mobility (30, 31). 53BP1 may favor CSR by protecting broken DNA ends in Ig S regions from resection (28). However, alternative functions are also possible since 53BP1 is a complex domain structure (32). Based on the observation that ablation of 53BP1 leads to elevated inversional CSR, we considered the possibility that 53BP1 plays a unique structural role in the 3D spatial organization of the Igh locus in response to AID induced DSBs.
We used complementing strategies to explore the participation of 53BP1 in CSR and its functional association with Igh chromatin. We mapped chromatin interactions across the Igh locus using chromosome conformation capture (3C) methods and found that the 3’RR, in accord with it’s palindromic structure (33), assumes a unique topological configuration and that Eμ preferentially contacts 3’Eα in activated B cells. Strikingly, induction of Eμ:3’Eα looping was significantly reduced in 53BP1 deficient B cells, modestly impaired in the absence of AID and essentially abolished in 53BP1-/-AID-/- B cells highlighting the dependency of Eμ:3’Eα contacts on AID induced DSBs and 53BP1. In contrast, GLT promoter:3’Eα contacts are only modestly diminished in activated 53BP1-/-AID-/- B cells. Chromatin immunoprecipitation (ChIP) studies demonstrate that 53BP1 re-distributes across the Igh locus in an AID dependent fashion upon B cell activation in accord with a propensity to spread in chromatin. Our studies have revealed a novel architectural function for 53BP1 in supporting the 3D structure of the locus that is integrally linked to facilitating S-S synapsis and promoting completion of the CSR reaction.
MATERIALS AND METHODS
Cell Culture and Statistics
Mice, C57BL/6 (WT) were purchased from Jackson Laboratories, 53BP1-/- and 53BP1-/-AID-/- mice were gifts from Dr. F. Alt (Harvard), AID-/- mice (34) were a gift from Dr. T. Honjo (Kyoto University). Mice were bred under specific pathogen-free conditions in a fully accredited animal facility at the University of Illinois College of Medicine. All procedures involving mice were approved by the Institutional Animal Care Committee of the University of Illinois College of Medicine or the National Institute of Aging. Splenic B cells were sorted for CD43- resting B cells using CD43 magnetic microbeads (MACS, Miltenyi) or were enriched for T cells using the mouse T cell enrichment column kit (MTCC-5, R&D) and cultured as previously described (23). Splenic T cells were cultured at a density of 5 × 105 to 1 × 106 and stimulated in RPMI and glutamine (4 μM), penicillin-streptomycin supplemented with FCS (10% vol/vol) and activated with ConcanavalinA (ConA) (5ng/ml) (15324505, MP Biomedical). To prepare for flow cytometry, B cells activated for 4 days were washed in HBSS plus 2% FCS and stained with antibodies conjugated with fluorescein isothiocyanate (FITC-IgG3 -553403, FITC-IgG1 -553443; Pharmingen). The flow cytometry analyses represented 5000-10,000 events and were gated for live lymphoid cells determined by forward and side scatter with CyanADP and Summit software (Becton Dickenson). P values were calculated by using two-tailed Student’s t test.
Chromosome conformation capture (3C) and analysis
Optimized 3C assays for the Igh locus was performed as described (26). Briefly, cells were crosslinked with 1% formaldehyde for 8 minutes at room temperature and the reaction was quenched with glycine. Cleavage with a restriction enzyme is followed by intramolecular ligation of interacting crosslinked DNA fragments under low concentration conditions. After ligation, crosslinks were reversed and 3C ligation product concentration determined by qPCR with primer pairs located at the ends of the relevant restriction fragments. Controls were performed to confirm the efficacy of the 3C procedure. 3C primers and probes were designed to avoid strain polymorphisms and primers were designed using Primer Express software (ABI) (Table S2). Template concentration using the mb1 primers (Table S2) was determined as previously described (23). The efficiency of HindIII restriction site digestion in chromatin was monitored by real-time PCR analysis using primers spanning specific restriction sites as described previously (Table S1, Table S2) (26). To control for differences in primer efficiencies a control template was constructed in which all possible 3C ligation products are present in equimolar concentration (Table S2). DNA fragments spanning restriction sites of interest were PCR amplified, mixed in equimolar concentrations, then digested with Hind III and ligated as previously described (26). The ligated mix was added to genomic DNA that had been digested and ligated, to serve as the 3C control template and which was used in a standard curve in 3C qPCR analyses. Dilutions of control mix were assayed under optimized conditions to determine the linear range of amplification (64 to 0.03 pg of ligated mix per 50 ng of genomic DNA per μL). For all qPCR 3C reactions, 100 ng of chromatin was used. The data were normalized using the interaction frequency between two fragments within the non-expressed Amylase I (Amy) gene (Table S1). The relative crosslinking frequency between two Igh restriction fragments is calculated: XIgh = [SIgh/SAmy] Cell Type/[SIgh/SAmy] Control mix. SIgh is obtained using primer pairs for two different Igh restriction fragments and SAmy is the signal obtained with primer pairs for the Amy I locus fragments. The crosslinking frequency for the two Amy1 fragments wass arbitrarily set to a value of 1 to permit sample to sample comparisons. Quantitative PCR (qPCR) was used in combination with 5’FAM and 3’BHQ1 modified probes (IDT) to detect of 3C products (Table S2). Primer and probe optimization were carried out according to the manufacturer’s recommendations, (http://www3.appliedbiosystems.com/cms/groups/mcb_support/documents/generaldocuments/cms_042996.pdf). A complete laboratory protocol for 3C is available upon request.
Chromatin Immunoprecipitation and quantitative PCR
Chromatin immunoprecipitation (ChIP) assays were performed according to the Millipore protocol (http://www.millipore.com/techpublications/tech1/mcproto407) as in previous work (35) with slight modifications. Chromatin was sonicated (SonicDismembrator 550, Fisher Scientific Microtip) to an average length of 500 bp. The sonicated cell suspension was diluted 10-fold with a buffer containing 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl and incubated with 80 μl salmon sperm DNA/protein A Agarose 50% Slurry (Upstate Biotechnology Inc.) for 3 h with rotation at 4°C to preclear. The one third of the precleared chromatin was incubated with 2 μg of polyclonal or control antibodies. One tenth of the precleared chromatin was saved as input. ChIP DNA pellets were resuspended in 60 μl of TE. Samples were analyzed by qPCR analysis using SYBR Green PCR Mix (Applied Biosystems) and an ABI7900HT system according to the manufacturer’s instructions. Primers were designed using Primer Express software (ABI) and all the primers have approximately equal efficiency of amplification (Table S2). To ensure that a single PCR product was amplified from each primer pair, the dissociation curves were examined and the PCR products were run on agarose gels. All samples were analyzed in duplicate and averaged. The amount of PCR product amplified was calculated relative to a standard curve (50 ng to 0.04 ng of genomic DNA). The signal from the control anti-IgG or anti-GFP immunoprecipitation was subtracted from the specific IP prior to final calculations. The ChIP Index = bound/input. SEMs were calculated using data from independent experiments. P values were calculated by using two-tailed Student’s t test. ChIP assays were performed with anti-H4K20Me1 and anti-H4K20Me2 (gifts from T. Jenuwein), anti-H4K5,8,12,16Ac (Millipore Upstate 06-866) anti-H4K20Me3 (Abcam ab9053), anti-H4K16Ac (Millipore, 07-329), anti-H3K9Ac (Millipore, 06-942), anti-H3K9me3 (Abcam, ab8898) and anti-HP1γ (Millipore, 05-690), anti-NBS1 (Abcam, ab32074 clone Y112) and control antibodies (Millipore, 12-370). 53BP1 antisera were anti-53BP1 (N) (Novus, NB100-304) a rabbit antibody against human 53BP1 residues 350 and 400, and anti-53BP1 (PC) is a rabbit antibody raised against a GST fusion between human 53BP1 protein N- terminal residues 1-524 as was previously described (36). Each anti-53BP1 or anti-NBS1 sample was derived from three independent ChIPs that were pooled to achieve a six-fold concentration. Backgrounds were determined using the anti-GFP control antibody (Abcam ab290) and were subtracted. Primer sequences used in ChIP assays were forward and reverse SμU, SμD.2, SμD, CμU, Sγ3U, Sγ3D, Sγ1U, Sγ1D, SɛU, SɛD, Hs4, β-globin (37), Sγ1 (38), and Hs4 (39).
RESULTS
CSR is an intra-chromosomal deletion event that is dependent on AID induction and GLT expression at S regions (2) (fig. 1A). In resting splenic B cells Eμ and the 3’RR are separated by 220 kb in the linear genome and are anchors for a B cell specific chromatin loop (23) (fig. 1B). Upon induction of GLT expression and prior to the onset of recombination, the spatial organization of the Igh locus at the 3’ end is configured by three-way interactions of Eμ:3’Eα:GLT promoter that facilitates GLT expression and S/S synapsis during CSR (23, 26) (fig. 1B). This 3D conformation may contribute to locus stability when AID induced DSBs are formed in S regions and within 3’Eα(40-42) (fig. 1B). To determine whether 53BP1 acts as a bridging factor that tethers distantly located broken S regions to support deletional CSR we assessed locus conformation in resting and activated B cells from WT, AID-/-, 53BP1-/-, 53BP1-/-AID-/- mice. To begin, we generated a detailed map of WT looping contacts between the Eμ and 3’RR and within the 3’RR. This information is leveraged to assess the impact of 53BP1 deficiency in the presence and absence of AID induced DSBs on the spatial organization of the Igh locus in activated B cells.
Figure 1. The 3’RR forms a chromatin hub in activated B cells.
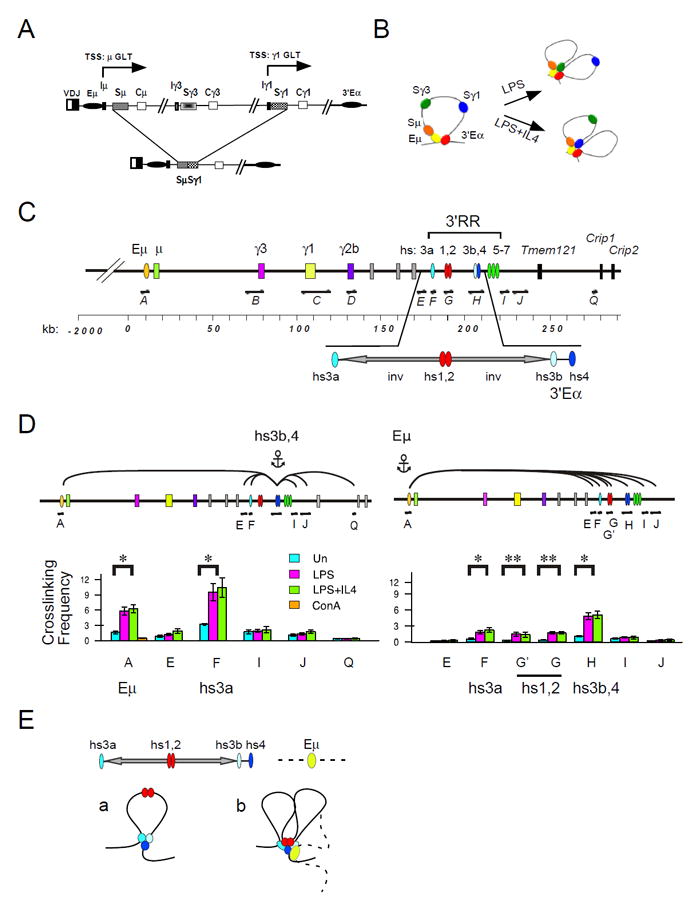
A) CSR is diagrammatically shown. Transcription start sites (TSS) downstream of the μ and γ1 GLT promoters are indicated. LPS+IL4 induces the expression of the γ1 GLT and the intra-chromosomal deletional recombination between Sμ and Sγ1. B) The Igh locus is configured as a chromatin loop tethered by association of the Eμ:3’Eα enhancers in resting B cells. Following B cell activation with LPS or LPS+IL4 the appropriate GLT promoters interact with the 3’Eα in a cytokine dependent fashion. C) A map of the 3’ end of the Igh locus is drawn to scale. The Igh locus contains eight CH region genes each paired with a S region and a GLT promoter (with the exception of Cδ) and are bracketed by the intronic Eμ and the 3’Eα regulatory region located at the 5’- and 3’-ends, respectively. The 3’Eα contains hypersensitive sites (hs) 3a, hs1,2, hs3b,4 that function together as an LCR (reviewed in (69)) and are adjacent to islands of CTCF binding associated with hs 5-7 (39). Inverted (inv) repeat sequences separate hs3a and hs3b and form a >25 kb palindrome with hs1,2 at the center. HindIII restriction fragments used in the 3C analysis are indicated (fragments A-Q). D) 3C chromatin templates were derived from splenic B cells that were resting or activated with LPS or LPS+IL4 for 40 hours or splenic T cells activated with ConA 3 days. 3C assays were anchored at Eμ (fragment A) or hs3b,4 (fragment H) were analyzed for interaction with elements within 3’RR (fragments E-Q). P values of 0.05 and 0.002 or less are indicated by * and **, respectively, and were derived using two-tailed Student’s t tests. E) The 3’RR resides within a palindrome that is composed of two inverted repeats (arrows) flanked by hs3a and hs3b. a) The 3’RR is tethered by hs3a and hs3b,4 enhancers in activated B cells. b) The looping structure of the Eμ:3’RR. Hs1,2 is speculatively shown engaged in chromatin association with hs3a and hs3b,4.
3C chromatin templates were prepared from splenic B cells that were unstimulated and activated for 40 hours with LPS and LPS+IL4 (Suppl. Table I). At this activation time point GLTs and AID are well expressed, S region specific AID dependent DSBs have begun to form (35, 40, 41), however CSR is not yet detectable (23). We confirmed that GLT expression was appropriately induced and essentially identical in activated B cells from WT and genetically deficient mice (Suppl. fig. 1A). AID transcripts were highly expressed in WT and 53BP1-/- but not in AID-/- or 53BP1-/-AID-/- derived B cells (Suppl. fig. 1A). FACS analyses confirmed that IgG3 and IgG1 switching is correlated with GLT expression in activated WT B cells and absent in AID-deficient B cells (Suppl. fig.1B). We note a complete loss of μ→γ3 and an 8-fold reduction of μ→γ1 CSR in 53BP1 deficient B cells (Suppl. fig.1B) consistent with previous reports (17). Thus, the chromatin templates used in our 3C assays derive from appropriately activated B cells.
Eμ preferentially interacts with 3’Eα in B cells
The 3’RR is comprised of five DNase hypersensitivity sites (DHSS) (hs3a, hs1,2, hs3b and hs4) (43)(fig. 1C). Hs3a and hs3b flank inverted repeat sequences that form a >25 kb palindrome with hs1,2 at the center (fig. 1C). Hs4 lies downstream of the palindrome and adjacent to hs3b. Hs3b,4 confer transcriptional enhancer activity and are also referred to as 3’Eα. The palindromic structure of the 3’RR raises the possibility that this region is configured in a distinct spatial organization that may be functionally important.
We interrogated the 3’RR to identify elements that interact with anchor Eμ. Although Eμ looping with hs3a (A:F), and hs1,2 (A:G; A:G’) were significantly induced, Eμ:hs3b,4 (A:H) interactions are preferentially increased (5-fold; p<0.004) in activated- relative to resting- B cells (fig. 1D, right panel). Eμ:hs3b,4 interactions are specific since Eμ looping with the 3’Eα flanking fragments E, I, and J were not induced in activated B cells. These studies have identified hs3b,4 (3’Eα) as the preferred interaction partner with Eμ. Our earlier studies indicated that hs3b,4 preferentially associates with GLT promoters and is critical for GLT expression (23, 26).
Next, we scanned multiple sites within the 3’ regulatory region (3’RR) for interaction with anchor hs3b,4 (fragment H) (fig. 1D, left panel). Hs3b and hs4 reside on a single Hind III fragment and were not separable in 3C assays. It was not feasible to analyze the interactions between hs3b,4 and hs1,2 (fragments H and G) due to proximity effects. Chromatin interactions in activated B cells were induced between the anchor hs3b,4 and hs3a (H:F) (3-fold; p<0.02) and attain the exceptionally high crosslinking frequency of 10 relative to resting B cells (fig. 1D, left panel). Hs3b,4:hs3a (H:F) interactions were not due to spatial proximity since the flanking fragments E, I and J were only modestly interactive with hs3b,4 and were not induced in activated B cells (fig. 1D). Hs3b,4 mediated interactions fall off drastically between fragments J and Q, located ~80 kb downstream of hs3b,4 and 5’-proximal to Crip 1 (fig. 1D, left panel). These findings indicate that the 3’RR attains a unique 3D chromatin architecture in which hs3a is induced to associate with hs3b,4 in activated B cells (fig. 1Ea). We speculate that hs1,2 is engaged in similar interactions. Taken together, the integrity of the palindromic structure (33) and the presence of the topological fold demonstrated here strongly suggests that the 3’RR forms a chromatin hub that mediates looping interactions with Eμ and GLT transcriptional elements (fig. 1Eb).
3’RR chromatin topology and Eμ:3’Eα looping are 53BP1 dependent
It has been suggested that 53BP1 acts as a bridging factor involved in tethering distantly located broken S regions at Igh to support CSR (14, 44). To determine whether 53BP1 mediates chromatin interactions that facilitate S/S synapsis in response to AID initiated DNA damage we performed 3C assays in resting and activated B cells from AID- and 53BP1-deficient mice. Hs3b,4:Eμ (H:A), hs3b,4:hs3a (H:F) and hs3b,4:γ1(H:C) chromatin interactions are similar in resting B cells for all genotypes studied (fig. 2A). Although hs3b,4:γ1 GLT promoter (H:C) interactions are induced in all activated B cells tested their frequency is only somewhat diminished in 53BP1-/-AID-/- relative to WT B cells (fig. 2A). In contrast, induced intra-3’RR (hs3b,4:hs3a (H:F)) looping interactions are abolished for 53BP1-/-AID-/- B cells relative to all other gentoypes indicating that AID and 53BP1 contribute to the induction of this topological fold (fig. 2A). In accord with earlier findings, hs3b,4:Eμ (H:A) contacts are induced upon B cell activation and trend lower in AID deficient- as compared to WT B cells (26). Strikingly, hs3b,4:Eμ (H:A) looping is significantly impaired in activated 53BP1-/- B cells (1.9-fold; p < 0.0002) and is essentially abolished in 53BP1-/-AID-/- (3.0-fold; p < 0.00001) relative to WT (fig. 2A,B). We conclude that Eμ:3’Eα (H:A) looping in activated B cells 1) is 53BP1 dependent, 2) that 53BP1 and AID appear to act additively, and 3) that 53BP1 may have an architectural function that is independent of the DNA damage response since looping is severely reduced when both 53BP1 and AID are absent. Furthermore, intra-3’RR interactions are dependent on both 53BP1 and AID. Our findings provide a possible explanation for 53BP1 enforcement of deletional CSR.
Figure 2. 53BP1 facilitates Eμ:3’Eα looping and the 3’RR topological fold.
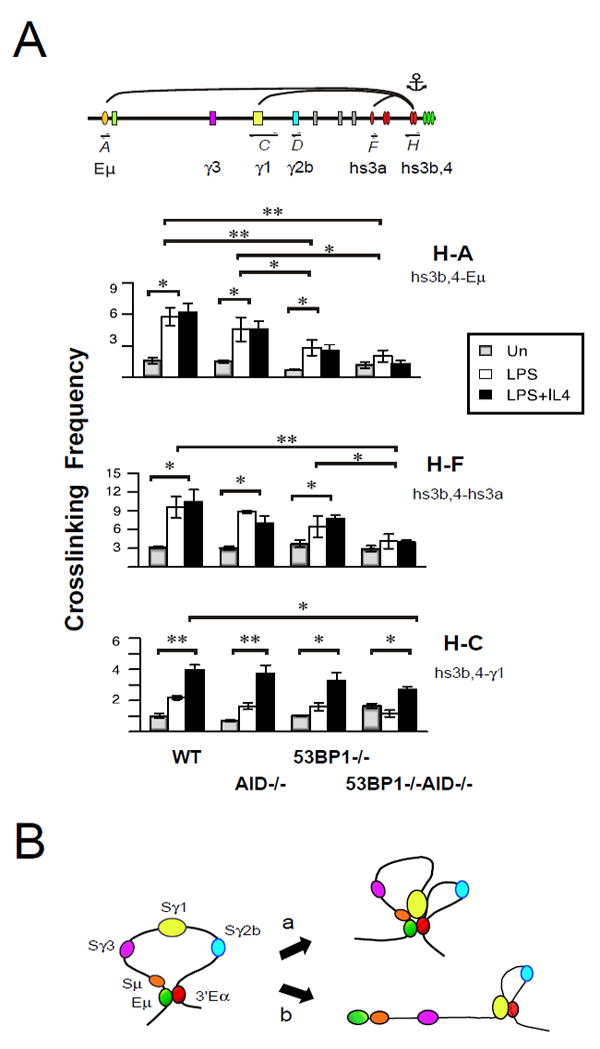
A) 3C chromatin templates were prepared from WT, AID-/-, 53BP1-/-, 53BP1-/-xAID-/- B cells that were resting or activated with LPS or LPS+IL4 for 40 hours. A) 3C assays that were anchored at hs3b,4 (fragment H) and were analyzed for interaction with Eu (fragment A) hs3a (fragment F) and the g1 locus (fragment C). P values of 0.03 and 0.001 or less are indicated by * and **, respectively using a two-tailed Students t test. B) The Igh locus is configured as a chromatin loop tethered by Eμ:3’Eα enhancers in resting B cells. Following B cell activation the appropriate GLT promoters interact with the 3’Eα in a cytokine dependent fashion when cells are 53BP1- sufficient (a) and 53BP1 deficient (b).
53BP1 chromatin association is B cell specific
Given the contribution of 53BP1 to Igh looping it was of interest to characterize the distribution of 53BP1 in B cell chromatin and determine its relationship to the presence of AID dependent DSBs in S regions and in 3’Eα (40, 41). There are three mechanisms that modulate 53BP1 chromatin occupancy: 1) DNA damage, 2) chromatin spreading and 3) the histone mark H4K20me2. In response to DNA damage 53BP1 is phosphorylated by ATM, accumulates at sites of damage in a manner contingent on the presence of γH2AX, MDC1 and RNF8 histone ubiquitylation (8-12) and on H4K20me2 (13). 53BP1 chromatin association is subject to spreading across substantial distances over time (45) and implying that focal enrichment may not occur directly at sites of AID induced DNA damage. In cells free of DNA lesions 53BP1 binds the chromatin modification H4K20me2, via tandem Tudor domains (5-7). We postulated that 53BP1 would accumulate in S regions, which are the sites of AID induced DSBs in activated B cells. However, it may be difficult to predict the level of 53BP1 binding at any given site since multiple variables influence 53BP1 accumulation in chromatin. In the sections that follow we examine AID expression, chromatin spreading and H4K20me2 as mediators of 53BP1 chromatin distribution.
Sites SμU, SμD.2, SμD, Cμ, Sγ1, hs4 (3’Eα), β-globin were chosen for analysis to determine whether 53BP1 chromatin association is regulated by transcription and/or AID expression (fig.1C, 3A). The Sμ-Cμ interval is constitutively transcribed in resting B cells and transcription is augmented upon B cell activation. Although S and C regions are subject to germline transcription, only S regions are targets for AID attack. Downstream S regions such as Sγ1, are transcriptionally silent in resting B cells and are subject to cytokine induced germline transcription in activated B cells (with the exception of the γ3 locus). Hs4 (3’Eα is transcriptionally active and acquires AID dependent DSBs in activated B cells (42). In contrast, the β-globin locus is transcriptionally silent in B cells. To begin, histone H3 lysine 9 acetylation (H3K9Ac) marks were present in resting- and elevated in activated B cells at the downstream end of Sμ (SμD), as previously observed, confirming chromatin quality (fig. 3A, B, right panel)(37). 53BP1 chromatin enrichment was detected at SμD in resting splenic B cells relative to activated B- and T cells using two independent anti-53BP1 antibodies (fig. 3B left panel). Furthermore, 53BP1 binding was significantly elevated in 53BP1 sufficient- but not in deficient- resting B cells validating the specificity of the ChIP assay (fig. 3B, middle panel). Elevated 53BP1 chromatin occupancy in resting B cells was detected at multiple sites within the Sμ-Cμ region, hs4 (3’Eα) and at the β globin gene (fig. 3C). Only the transcriptionally silent Sγ1 region lacked significant 53BP1 binding (fig. 3C). We conclude that 53BP1 chromatin association in resting B cells is not dependent on transcription per se and is not limited to the Igh locus since binding is detected at the β globin gene (fig. 3C). Moreover, significant 53BP1 chromatin occupancy at Cμ which is not targeted by AID suggests that binding may not directly reflect DNA damage (fig. 3C). Indeed, the preferential enrichment of chromatin bound 53BP1 in resting B cells was unanticipated since maximal AID induced DNA damage is associated with activated B cells. Elevated 53BP1 binding at multiple sites in resting B cells may be mediated by a mechanism(s) unrelated to AID expression.
Figure 3. 53BP1 chromatin association is B cell specific.
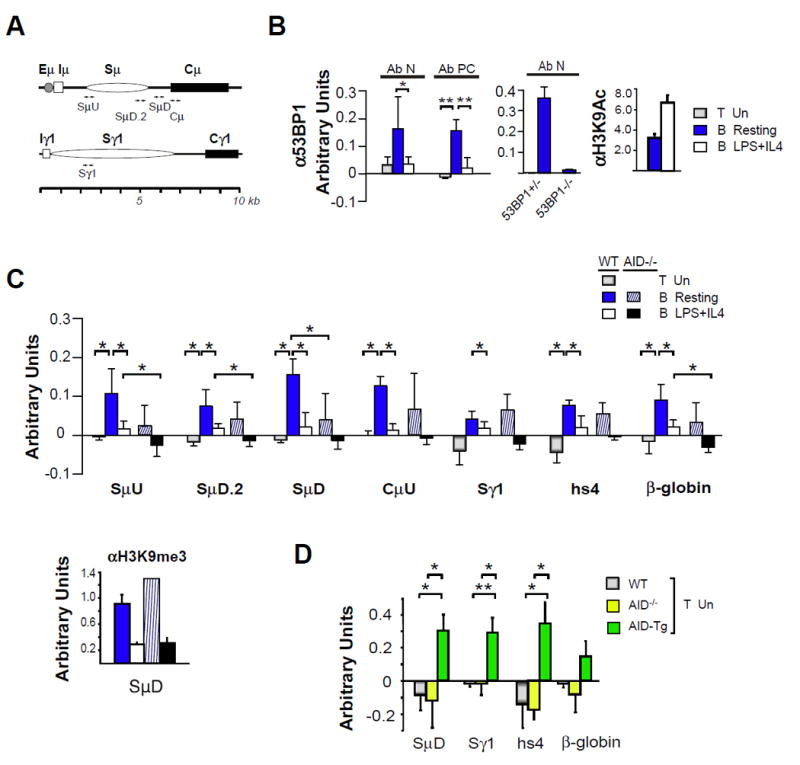
A) Schematics for the μ•and γ1 I-S-CH loci with primer pairs indicated (arrowheads). B,C) ChIP assays for 53BP1 used unstimulated splenic T cells, resting B cells or B cells activated by LPS+IL4 for 48 hours and anti-53BP1 antisera were Ab N and Ab PC, as indicated. Each anti-53BP1 sample was composed of three independent ChIPs that were pooled and concentrated six-fold. Each sample was analyzed in duplicate. Backgrounds were determined using the anti-GFP control antibody (Abcam ab290). Backgrounds and were subtracted from values determined using anti-53BP1. Negative values occurred when the anti-53BP1 value was lower than background. Anti-H3K9Ac or anti-H3K9me3 antisera were used in ChIP assays as controls for chromatin integrity, as indicated. B) ChIP assays were performed using WT, 53BP1 +/- or 53BP1 -/- B cells with SμD primers. Assays used two or three samples from two independent experiments. C) ChIP assays using anti-53BP1 Ab PC (upper panel) are from three to five samples from three independent experiments, respectively. P < 0.05 or 0.005 are indicated by * or **, respectively. ChIP assays using anti-H3K9me3 are shown as loading controls (lower panel). D) ChIP assays using anti-53BP1 and unstimulated splenic T cells from WT, AID-/- or AID-/-AIDTg+/- and Ab N. P < 0.05 and 0.01 indicated by * and **, respectively.
The 53BP1 chromatin distribution pattern undergoes a major shift in response to B cell activation. Because 53BP1 chromatin occupancy was reduced in activated B cells it was difficult to determine whether binding was specific and regulated (fig. 3B, C). We more directly tested the proposition that AID modulates 53BP1 chromatin association by comparing 53BP1 chromatin binding in WT and AID deficient mice (fig. 3C). The overall 53BP1 distribution pattern in AID deficient B cells paralleled that found in WT. Although 53BP1 chromatin binding generally trended higher in WT- as compared to AID- deficient resting B cells, this difference achieved statistical significance only at SμD (fig. 3C). Similarly, 53BP1 chromatin occupancy trended higher in WT- as compared to AID- deficient activated B cells and was significantly enriched at SμU, SμD.2 and β-globin sites (fig. 3C). These findings suggest that 53BP1 binding may be influenced by AID expression but is not entirely determined by it in B cells.
To further explore whether AID expression influences 53BP1 chromatin binding we analyzed T cell chromatin from AID deficient, sufficient and transgenic mice. AID transgenic (Tg) mice were bred onto the AID-/- background and are referred to as AID-Tg. AID-Tg mice are devoid of endogenous AID and express high levels of transgenic AID in all tissues tested including T cells (46, 47). 53BP1 chromatin binding is significantly enriched at Igh sites, SμD, Sγ1, hs4 and is modestly higher at the β-globin gene in AID-Tg- relative to WT and AID deficient T cells (fig. 3D). Thus, AID expression clearly contributes to 53BP1 chromatin occupancy in unstimulated T cells and is likely to influence 53BP1 binding in B cells.
Active redistribution of 53BP1 in chromatin of activated B cells
Accumulation of 53BP1 at AID induced DSBs may be difficult to detect in ChIP assays since DNA damage is distributed across the S region (48), present in only a subset of activated B cells and is subject to spreading long distances from the initial site of DNA damage, a feature shared with γH2AX and MDC1 (45). To investigate the possibility that 53BP1 undergoes chromatin spreading in activated B cells, we evaluated 53BP1 and NBS1 chromatin binding in AID sufficient- and deficient- B cells. NBS1 is a component of the MRN complex that also contains MRE11 and RAD50 and is a sensor for DNA damage required for efficient CSR (16). Consistent with earlier studies, H3K9Ac modifications peak at SμU and diminish sharply at SμD.2 and Cμ, confirming the integrity of the chromatin samples (fig. 4). 53BP1 accumulated in an AID dependent fashion at SμU and not at SμD.2 or CμU at 48 hours of activation (fig. 4). This 53BP1 distribution shifted at 72 hours of activation where 53BP1 chromatin enrichment was highest at CμU in AID sufficient- but not in deficient- B cells (fig. 4). At the same time point NBS1 accumulated at SμU and was undetectable at downstream SμD.2 and CμU in AID sufficient- but not in deficient- activated B cells indicating focused enrichment in the upstream Sμ region, an area most prone to AID induced DNA damage (fig. 4) (48). Thus, 53BP1 dynamically redistributes across the Sμ-Cμ interval over time, consistent with its propensity to undergo chromatin spreading (45). Thus, detection of focused accumulation of 53BP1 at sites of AID induced DNA damaged is complicated by dynamic chromatin spreading.
Figure 4. 53BP1 chromatin association dynamically spreads from S into C regions.
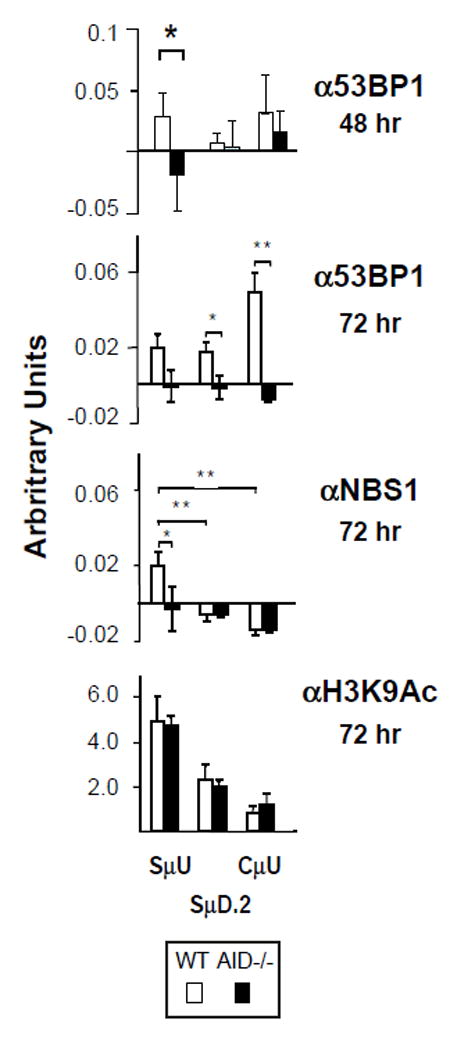
Chromatin for ChIP was derived from WT or AID-/- B cells activated with LPS+IL4 for 48 (A) or 72 hours (B) and used anti-53BP1 (Ab N) or anti-NBS1 (three to six samples) or anti-H3K9Ac (two samples) from two independent experiments. Data for the 48 hour time point was extracted from Figure 3D. P < 0.04 or 0.005 are indicated by * or **, respectively.
53BP1 chromatin binding in resting B cells is correlated with elevated H4K20me2
H4K20me2 marks are deposited by the SUV4-20H1/H2 enzymes and SUV4-20H1/H2 deficiency leads to conversion of H4K20me2 and H4K20me3 to mono-methyl marks (20). Reduced CSR results from abolition of H4K20me1,2 modifications in Suv4-20h1,h2 conditionally deficient B cells (20). On this basis we hypothesized that 53BP1 might be tethered to chromatin via H4K20me2. We profiled several histone H4 modifications to assess the potential for 53BP1 association with Igh locus chromatin in resting and activated B cells.
H4K16Ac is capable of altering higher order chromatin structure (49), and can co-exist with all H4K20me states (50). We found constitutive H4K16Ac enrichment in S regions that was unrelated to B cell activation state or transcriptional activity and functions as a control for the integrity of the chromatin samples (fig. 5). In contrast, acquisition of H4K5,8,12,16Ac (H4Ac) modifications in S regions was associated with B cell activation in accord with previous findings (51-53) (fig. 5). Notably, H4K20me2,3 modifications were markedly enriched in S regions of resting B cells and were depleted following B cell activation, irrespective of transcriptional status (fig. 5). H4K20me1 uniquely marked transcriptionally active Sμ and Sγ3 regions in resting B cells and was reduced in activated B cells (fig. 5). Our findings demonstrate H4K20me2 chromatin enrichment at multiple S regions in resting B cells without regard to transcriptional status and prior to initiation of AID induced DNA damage. Thus, the 53BP1 chromatin association is tightly correlated with the presence of H4K20me2 marks in resting B cells. However, relatively little is known regarding the epigenetic landscape of Igh locus in resting B cells.
Figure 5. Elevated H4K20me2 modifications in resting B cells.
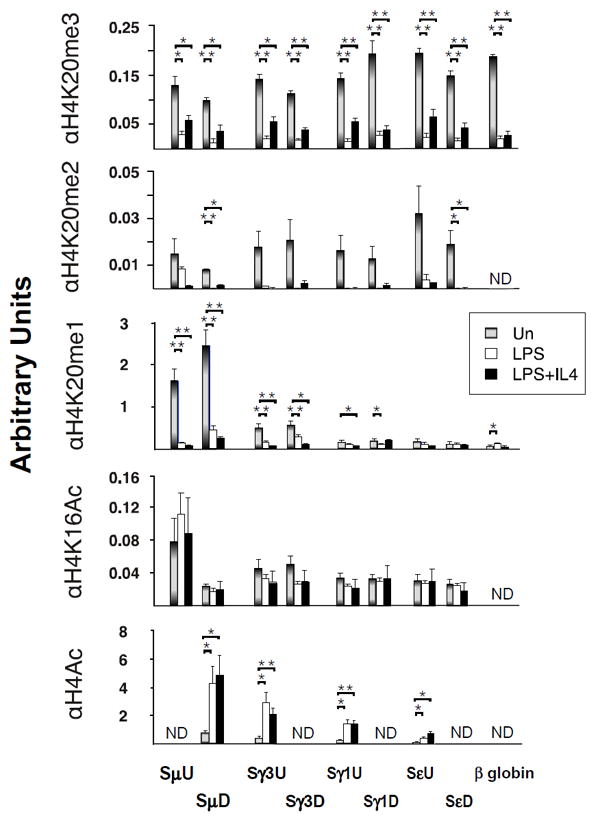
ChIP assays on B cells that were resting or activated with LPS or LPS+IL4 for 48 hours using four to six samples derived from two independent experiments. Primer pairs are indicated. P < 0.05, and 0.001 are indicated by *, and ** respectively and derived using two-tailed Student’s t tests.
Distinct chromatin landscape in resting and activated B cells
H4K20me3 is a repressive histone mark that is both evolutionarily conserved and associated with heterochromatin (54) that is preferentially detected in resting B cells (fig. 5). In contrast, activating histone modifications play a role in targeting CSR machinery to S regions by providing binding motifs for CSR co-factors and by creating chromatin accessibility (35, 37, 38, 55, 56). To more fully characterize the epigenetic landscape of S-C regions in resting and activated B cells we evaluated H3K9Ac an activating mark, and H3K9me3 and HP1γ binding, classical markers for heterochromatin (reviewed in (57)) that have been reported at transcribed S regions (38, 55, 56). H3K9Ac was focused to actively transcribed Sμ and diminished in Cμ in resting B cells in accord with previous work (fig. 6A) (35, 37). H3K9Ac levels increased Sγ1 regions upon LPS+IL4 stimulation and reflect induced γ1 GLT expression (fig. 6A). We and others (55) note that H3K9me3 was present at multiple S-C regions in resting B cells and was not induced by B cell stimulation in transcriptionally active Sμ or in downstream S regions (fig. 6A,B) (55). Furthermore, H3K9me3 was highly enriched within the Sμ region (SμU, SμD.2, SμD), was depleted in Cμ in resting B cells and was essentially absent in activated B cells (fig. 6A,B). Previously H3K9me3 enrichment was reported to increase at transcribed S regions in activated B cells (38, 56). This difference may be related to different protocols used for B cell isolation. The HP1γ binding pattern bore striking similarities H4K20me1 in that it was focused to the μ locus, peaked at 5’Sμ (SμU) in resting cells and was retained at reduced levels in activated B cells (fig.4, 5A,B). HP1γ chromatin occupancy was similar in AID sufficient and deficient B cells and was unlike H3K9,14Ac which we previously found to be reduced in AID deficient B cells (fig. 6B) (35). Thus, HP1γ uniquely marks transcriptionally active donor Sμ regions in resting B cells. Collectively, our findings indicate that repressive histone marks decorate the Igh locus landscape in resting B cells and are consistent with enriched deposition of the 53BP1 ligand, H4K20me2.
Figure 6. S regions are decorated with repressive histone marks in resting B cells.
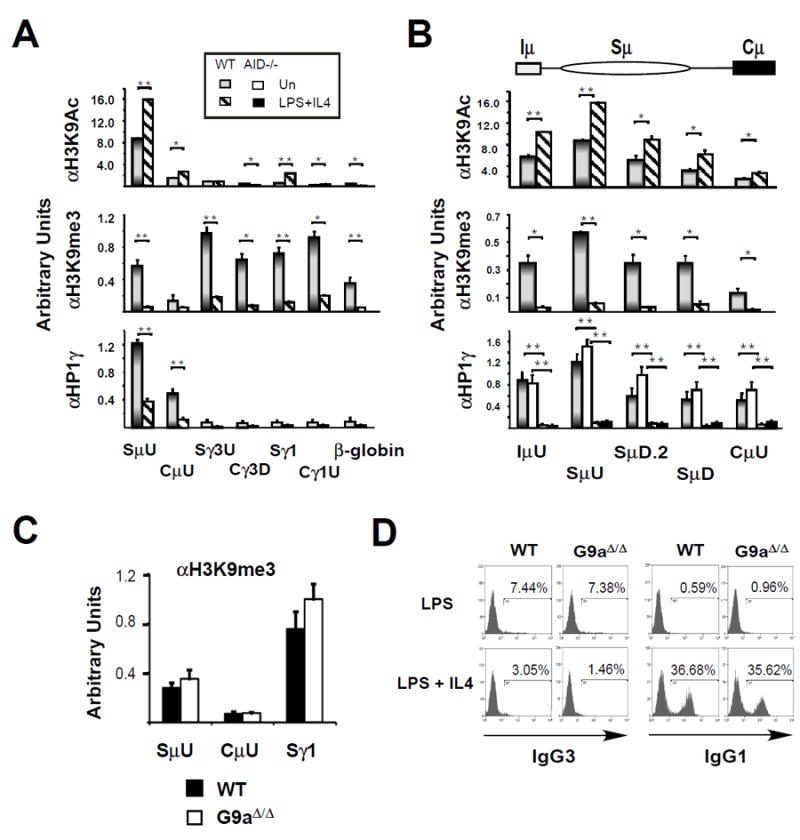
ChIP assays on WT, AID-/- or G9a∆/∆ splenic B cells that were resting or activated with LPS+ IL4 for 48 hours and anti-H3K9Ac or anti-H3K9me3 or anti-HP1γ using two to eight samples derived from two to three independent experiments. P < 0.05, and 0.001 are indicated by *, and ** respectively. A) ChIP analyses for μ, γ3, and γ1 I-S-CH loci and the β-globin gene are shown. B) ChIP analyses for the μ I-S-CH locus are shown. C,D) WT and G9a∆/∆ splenic B cells were activated with LPS or LPS+ IL4 for 48 hours or 4 days as indicated. C) ChIP analyses using and anti-H3K9Ac for SμD, CμU, Sγ1 sites in WT and G9a∆/∆ splenic B following 48 hours of LPS+IL4 activation. D) FACS analysis of IgG3 and IgG1 surface expression on WT and G9a∆/∆ splenic B cells following 4 days of activation as indicated.
Because G9a serves as the major H3K9me2 histone methyltransferases in B lymphocytes and G9a deficiency exhibits significantly reduced global levels of H3K9me2, the precursor of H3K9me3 (53)(58) we asked whether the H3K9me3 modifications in the Sμ region are G9a dependent and influence CSR. There was no change in the abundance of H3K9me3 marks at Sμ, Cμ and Sγ1 regions in WT and conditionally deleted G9a (G9aΔ/Δ) resting B cells (fig. 6C). Furthermore, FACS analyses indicated that G9a deficiency had no impact on the frequency of IgG3 or IgG1 switching in activated B cells (fig. 6D). We conclude that H3K9me3 modifications at the μ locus arose independent of G9a.
DISCUSSION
Our studies indicate that 53BP1 facilitates Eμ:3’Eα looping and is largely dispensable for 3’Eα:GLT promoter interactions in activated B cells. 53BP1 dependent Eμ:3’Eα looping in the presence or absence of AID suggests a structural role for 53BP1 that may explain its function in mediating deletional CSR (29). Two lines of evidence suggest that 53BP1 has functions that are independent of the ATM/γH2AX/MDC1/RNF8 DNA damage response. First, the localization of 53BP1 to DNA repair foci in B cells undergoing CSR is dependent on H2AX phosphorylation (γH2AX), MDC1, and RNF8 (19, 59-61). However, loss of MDC1 or γH2AX produces a milder deficit on CSR than does deletion of 53BP1 (17, 18, 21, 27, 60, 62). 53BP1 association with repair foci analyzed during metaphase may not be fully representative of its function during CSR because AID initiated DSBs are induced and repaired in G1 of the cell cycle (19, 63). Thus, Igh repair foci detected during metaphase may represent difficult to resolve DNA breaks that persist through the cell cycle and may not reflect the distribution and dynamics of 53BP1 chromatin binding in B cells. Second, 53BP1 is a large and complex protein with multiple domains that are differentially involved in chromatin association, repair focus formation, DNA end protection and CSR (64). These separation of function studies provide support for the notion that 53BP1 mediated chromatin looping may operate independently of the DNA damage response.
We identified 3’Eα (hs3b,4) as the major anchor for looping interactions with Eμ (fig. 1) and with GLT promoters (26). Deletion of 3’Eα (hs3b,4) in mice leads to loss of Eμ:3’Eα looping (23), severely impaired CSR and most GLT expression, albeit with little impact on SHM (65, 66). Here we report that the 3’RR is encompassed within an inducible topological fold in which 3’Eα (hs3b,4) and hs3a preferentially interact and anchor the palindromic unit. Disruption of 3’RR palindromic architecture by removal of the hs3a/hs1,2 interval or by deletion of the inverted repeats leads to reduction of CSR and near abolition of SHM (33). These observations suggest that the 3’RR topological fold is dictated by integrity of the palindromic unit and that perturbation of chromatin looping impairs 3’RR function. Our studies indicate that the long range looping interactions anchored by 3’Eα:hs3a are dependent on 53BP1 and AID. Our findings are in accord with an earlier study showing that the 3’Eα is a target for AID induced DSBs (42) and indicate that 53BP1 in conjunction with AID induced DNA damage play a functional role in this looping interaction.
Based on our observation that Eμ:3’Eα looping is 53BP1 dependent we assessed the chromatin distribution pattern of 53BP1 in lymphocytes. We hypothesized that AID induced DNA damage was the dominant factor driving 53BP1 chromatin binding. Unexpectedly, we found that 53BP1 chromatin binding is elevated in resting B cells, reduced in activated B cells and absent in T cells. This was unanticipated since AID induced DNA damage increases significantly in activated B cells. These observations prompted us to consider the possibility that several variables modulate the pattern of 53BP1 chromatin occupancy including AID expression, histone modifications, and chromatin spreading from sites of initial DNA DSBs. Expression of transgenic AID in T cells that are ordinarily devoid of AID leads to a substantial increase of 53BP1 chromatin occupancy indicating that AID induced DNA lesions could stimulate 53BP1 binding at multiple sites. Small amounts of active phosphorylated AID (pS38) have been reported in resting B cells and not in normal T lymphocytes (46, 67) suggesting that DNA damage might accumulate and promote 53BP1 chromatin association. Indeed, low levels of AID cause DNA damage since γH2AX repair foci have been visualized in ~5% of resting B cells (15). However, comparison of 53BP1 binding in AID sufficient and deficient resting B cells indicates that some 53BP1 chromatin binding is AID independent and implies that additional pathways are involved. We conclude that elevated 53BP1 chromatin binding is B cell specific.
53BP1 methyl-histone binding Tudor domains constitutively associate with chromatin via H4K20me2 in cells free of DNA damage (5-7). Strikingly, we found that 53BP1 chromatin association is correlated with elevated levels of H4K20me2 in resting B cells and that this histone mark significantly diminishes in activated B cells. Furthermore, we note that the chromatin landscape of the Igh locus is profoundly remodeled upon B cell activation. 53BP1 shifts from direct chromatin binding via H4K20me2 in resting B cells to indirect association through γH2AX and MDC1 during the AID mediated DNA damage response in activated B cells. Because indirect chromatin association is technically more difficult to detect it is possible that ChIP assays under-report actual 53BP1 chromatin occupancy in activated B cells. Finally, 53BP1 has the propensity to spread long distances from the original site of DNA damage. The spreading phenomenon obscures focal enrichment of 53BP1 at S regions in comparison to NBS1, a DNA repair factor that does not spread. Accordingly, 53BP1 was detected spanning the Iμ-Sμ-Cμ region in CH12 cells activated to undergo CSR (68).
In summary, 53BP1 is a regulator of Eμ:3’Eα looping interactions in activated B cells. These findings provide a plausible mechanistic explanation for the requirement of 53BP1 in enforcing deletional- as opposed to inversional- CSR. 53BP1 chromatin association changes as a function of B cell activation status. However, the unique distribution properties of 53BP1 in activated B cell chromatin make it difficult to detect focal accumulation and to directly demonstrate that 53BP1 chromatin binding mediates Eμ:3’Eα looping.
Supplementary Material
Acknowledgments
We thank Drs. T. Honjo for the AID-/- mice, F. Alt for 53bp1-/- and 53bp1-/-×AID-/- mice, S. Franco for 53bp1+/- and 53bp1-/- mice, Oltz for the G9a∆/∆ mice, T. Jenuwein for antisera to histone H4K20Me1 and H4K20Me2.
This work was supported by the National Institutes of Health (RO1AI052400 to A. L. K., 5R21AI076747-01 to P.B.C.) and the Welch Foundation (AU-1569 to P.B.C.).
Abbreviations
- 3’RR
3’ regulatory region
- AID
activation induced cytidine deaminase
- 3C
chromosome conformation capture
- CSR
class switch recombination
- CH
constant region
- ChIP
chromatin immunoprecipitation
- DSBs
DNA double strand breaks
- hs
DNase hypersensitive sites
- GLT
germ line transcription
- H4K20me2
histone 4 di-methyl lysine 20
- I exon
intervening
- MRN
Mre11/Rad50/Nbs1
- NHEJ
nonhomologous end joining
- γH2AX
phosphorylated H2AX
- Pr
promoter
- q
quantitative
- SHM
somatic hypermutation
- S
switch
- 3D
three dimensional
- Tg
transgenic
- TF
transcription factors
Footnotes
The authors declare that they have no competing financial interests.
LITERATURE CITED
- 1.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, Manis J, Alt FW. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 3.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, Alt FW. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 4.Jankovic M, Nussenzweig A, Nussenzweig MC. Antigen receptor diversification and chromosome translocations. Nat Immunol. 2007;8:801–808. doi: 10.1038/ni1498. [DOI] [PubMed] [Google Scholar]
- 5.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J Cell Biol. 2005;170:201–211. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 8.DiTullio RA, Jr, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 9.Mochan TA, Venere M, DiTullio RA, Jr, Halazonetis TD. 53BP1, an activator of ATM in response to DNA damage. DNA Repair (Amst) 2004;3:945–952. doi: 10.1016/j.dnarep.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 10.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 12.Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem. 2003;278:19579–19582. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- 13.Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reina-San-Martin B, Chen J, Nussenzweig A, Nussenzweig MC. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1-/- B cells. Eur J Immunol. 2007;37:235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- 15.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 18.Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med. 2004;200:1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, Espejo A, Bedford MT, Nussenzweig A, Busslinger M, Jenuwein T. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22:2048–2061. doi: 10.1101/gad.476008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 22.Stavnezer J. Molecular processes that regulate class switching. Curr Top Microbiol Immunol. 2000;245:127–168. doi: 10.1007/978-3-642-59641-4_6. [DOI] [PubMed] [Google Scholar]
- 23.Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, Alt FW, Cogne M, Pinaud E, Kenter AL. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ju Z, Volpi SA, Hassan R, Martinez N, Giannini SL, Gold T, Birshtein BK. Evidence for physical interaction between the immunoglobulin heavy chain variable region and the 3’ regulatory region. J Biol Chem. 2007;282:35169–35178. doi: 10.1074/jbc.M705719200. [DOI] [PubMed] [Google Scholar]
- 25.Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med. 2009;206:1073–1087. doi: 10.1084/jem.20082311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feldman S, Achour I, Wuerffel R, Kumar S, Gerasimova T, Sen R, Kenter AL. Constraints contributed by chromatin looping limit recombination targeting during Ig class switch recombination. J Immunol. 2015;194:2380–2389. doi: 10.4049/jimmunol.1401170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig F, Livak MC, Nussenzweig A, Chen J. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong J, Panchakshari RA, Zhang T, Zhang Y, Hu J, Volpi SA, Meyers RM, Ho YJ, Du Z, Robbiani DF, Meng F, Gostissa M, Nussenzweig MC, Manis JP, Alt FW. Orientation-specific joining of AID-initiated DNA breaks promotes antibody class switching. Nature. 2015;525:134–139. doi: 10.1038/nature14970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, Sleckman BP, Nussenzweig A. 53BP1 facilitates long-range DNA end-joining during VDJ recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams MM, Carpenter PB. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div. 2006;1:19. doi: 10.1186/1747-1028-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saintamand A, Rouaud P, Saad F, Rios G, Cogne M, Denizot Y. Elucidation of IgH 3’ region regulatory role during class switch recombination via germline deletion. Nat Commun. 2015;6:7084. doi: 10.1038/ncomms8084. [DOI] [PubMed] [Google Scholar]
- 34.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. J Exp Med. 2006;203:215–226. doi: 10.1084/jem.20051774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, Kellems RE, Hittelman WN, Elledge SJ, Carpenter PB. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J Biol Chem. 2003;278:14971–14977. doi: 10.1074/jbc.M212484200. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009;206:1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuang FL, Luo Z, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc Natl Acad Sci U S A. 2009;106:5288–5293. doi: 10.1073/pnas.0901368106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garrett FE, Emelyanov AV, Sepulveda MA, Flanagan P, Volpi S, Li F, Loukinov D, Eckhardt LA, Lobanenkov VV, Birshtein BK. Chromatin architecture near a potential 3’ end of the igh locus involves modular regulation of histone modifications during B-Cell development and in vivo occupancy at CTCF sites. Mol Cell Biol. 2005;25:1511–1525. doi: 10.1128/MCB.25.4.1511-1525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J Exp Med. 2005;202:561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wuerffel RA, Du J, Thompson RJ, Kenter AL. Ig Sgamma3 DNA-specifc double strand breaks are induced in mitogen-activated B cells and are implicated in switch recombination. J Immunol. 1997;159:4139–4144. [PubMed] [Google Scholar]
- 42.Peron S, Laffleur B, Denis-Lagache N, Cook-Moreau J, Tinguely A, Delpy L, Denizot Y, Pinaud E, Cogne M. AID-driven deletion causes immunoglobulin heavy chain locus suicide recombination in B cells. Science. 2012;336:931–934. doi: 10.1126/science.1218692. [DOI] [PubMed] [Google Scholar]
- 43.Kumari G, Sen R. Chromatin Interactions in the Control of Immunoglobulin Heavy Chain Gene Assembly. Adv Immunol. 2015;128:41–92. doi: 10.1016/bs.ai.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, Lou Z, Bassing CH, Manis JP, Chen J, Carpenter PB, Alt FW. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Meier A, Fiegler H, Munoz P, Ellis P, Rigler D, Langford C, Blasco MA, Carter N, Jackson SP. Spreading of mammalian DNA-damage response factors studied by ChIP-chip at damaged telomeres. EMBO J. 2007;26:2707–2718. doi: 10.1038/sj.emboj.7601719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen HM, Bozek G, Pinkert CA, McBride K, Wang L, Kenter A, Storb U. Expression of AID transgene is regulated in activated B cells but not in resting B cells and kidney. Mol Immunol. 2008;45:1883–1892. doi: 10.1016/j.molimm.2007.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197:1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2-/- ung-/- mice. J Exp Med. 2006;203:2085–2094. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 50.Sarg B, Helliger W, Talasz H, Koutzamani E, Lindner HH. Histone H4 hyperacetylation precludes histone H4 lysine 20 trimethylation. J Biol Chem. 2004;279:53458–53464. doi: 10.1074/jbc.M409099200. [DOI] [PubMed] [Google Scholar]
- 51.Wang L, Wuerffel R, Kenter AL. NF-kappa B binds to the immunoglobulin S gamma 3 region in vivo during class switch recombination. Eur J Immunol. 2006;36:3315–3323. doi: 10.1002/eji.200636294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y, Shimizu A. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 53.Li Z, Luo Z, Scharff MD. Differential regulation of histone acetylation and generation of mutations in switch regions is associated with Ig class switching. Proc Natl Acad Sci U S A. 2004;101:15428–15433. doi: 10.1073/pnas.0406827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, Alt FW, Losson R, Reina-San-Martin B. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. The Journal of experimental medicine. 2011;208:1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chowdhury M, Forouhi O, Dayal S, McCloskey N, Gould HJ, Felsenfeld G, Fear DJ. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc Natl Acad Sci U S A. 2008;105:15872–15877. doi: 10.1073/pnas.0808462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 58.Thomas LR, Miyashita H, Cobb RM, Pierce S, Tachibana M, Hobeika E, Reth M, Shinkai Y, Oltz EM. Functional analysis of histone methyltransferase g9a in B and T lymphocytes. Journal of immunology. 2008;181:485–493. doi: 10.4049/jimmunol.181.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.FitzGerald JE, Grenon M, Lowndes NF. 53BP1: function and mechanisms of focal recruitment. Biochem Soc Trans. 2009;37:897–904. doi: 10.1042/BST0370897. [DOI] [PubMed] [Google Scholar]
- 60.Santos MA, Huen MS, Jankovic M, Chen HT, Lopez-Contreras AJ, Klein IA, Wong N, Barbancho JL, Fernandez-Capetillo O, Nussenzweig MC, Chen J, Nussenzweig A. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J Exp Med. 2010;207:973–981. doi: 10.1084/jem.20092308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, Zaheen A, Durocher D, Scharff MD, Martin A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc Natl Acad Sci U S A. 2010;107:809–814. doi: 10.1073/pnas.0913790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol. 2007;179:6064–6071. doi: 10.4049/jimmunol.179.9.6064. [DOI] [PubMed] [Google Scholar]
- 64.Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, Nussenzweig A, Nussenzweig MC. Regulation of DNA End Joining, Resection, and Immunoglobulin Class Switch Recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morvan CL, Pinaud E, Decourt C, Cuvillier A, Cogne M. The immunoglobulin heavy-chain locus hs3b and hs4 3’ enhancers are dispensable for VDJ assembly and somatic hypermutation. Blood. 2003;102:1421–1427. doi: 10.1182/blood-2002-12-3827. [DOI] [PubMed] [Google Scholar]
- 66.Pinaud E, Khamlichi AA, Le Morvan C, Drouet M, Nalesso V, Le Bert M, Cogne M. Localization of the 3’ IgH locus elements that effect long-distance regulation of class switch recombination. Immunity. 2001;15:187–199. doi: 10.1016/s1074-7613(01)00181-9. [DOI] [PubMed] [Google Scholar]
- 67.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 68.Stanlie A, Yousif AS, Akiyama H, Honjo T, Begum NA. Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining. Mol Cell. 2014;55:97–110. doi: 10.1016/j.molcel.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 69.Khamlichi AA, Pinaud E, Decourt C, Chauveau C, Cogne M. The 3’ IgH regulatory region: a complex structure in a search for a function. Adv Immunol. 2000;75:317–345. doi: 10.1016/s0065-2776(00)75008-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.