

In this study, Miki et al. performed a genome-wide investigation of RNA polymerase II transcription termination in XRN2-deficient Caenorhabditis elegans and observed two distinct modes of termination. Their findings indicate that different termination mechanisms may work with different configurations of Pol II complexes dictated by promoters.
Keywords: transcription termination; RNA polymerase II; XRN2, Caenorhabditis elegans
Abstract
Transcription termination determines the ends of transcriptional units and thereby ensures the integrity of the transcriptome and faithful gene regulation. Studies in yeast and human cells have identified the exoribonuclease XRN2 as a key termination factor for protein-coding genes. Here we performed a genome-wide investigation of RNA polymerase II (Pol II) transcription termination in XRN2-deficient Caenorhabditis elegans and observed two distinct modes of termination. Although a subset of genes requires XRN2, termination of other genes appears both independent of, and refractory to, XRN2. XRN2 independence is not merely a consequence of failure to recruit XRN2, since XRN2 is present on—and promotes Pol II accumulation near the polyadenylation sites of—both gene classes. Unexpectedly, promoters instruct the choice of termination mode, but XRN2-independent termination additionally requires a compatible region downstream from the 3′ end cleavage site. Hence, different termination mechanisms may work with different configurations of Pol II complexes dictated by promoters.
Gene transcription involves three major steps: initiation, elongation, and termination. Transcription termination determines the ends of transcriptional units by disassembling the transcription elongation complex (TEC), thereby releasing RNA polymerases and nascent transcripts from DNA templates (Porrua et al. 2016; Proudfoot 2016). Failure in termination causes transcription readthrough, which yields wasteful and possibly harmful intergenic transcripts. It can also perturb expression of downstream genes when the unterminated TEC sweeps transcription initiation complexes off their promoters or collides with RNA polymerases that transcribe opposite strands (Shearwin et al. 2005). In eukaryotes, termination of protein-coding gene transcription by RNA polymerase II (Pol II) usually requires a functional polyadenylation (pA) signal, typically a variation of the AAUAAA hexamer. Nascent pre-mRNA is cleaved and the 5′ fragment is polyadenylated at the pA site shortly downstream from the hexamer by cleavage and pA factors (CPFs) (Shi and Manley 2015). Two mechanisms have been suggested for transcription termination of Pol II. In the allosteric model, the pA signal and/or other termination signals in the pA signal downstream region (PDR) induce reorganization of the Pol II complex; i.e., association or dissociation of components such as CPFs (Kim et al. 2004a; Zhang et al. 2005, 2015). This causes conformational changes in Pol II and thus TEC disassembly. In the kinetic model, also known as the “torpedo” model, cleavage at the pA site separates the pre-mRNA from the TEC, which continues synthesizing a downstream nascent transcript. This transcript is a substrate of XRN2/Rat1p, a processive 5′-to-3′ exoribonuclease that catches up with, and disassembles, the TEC by an unknown mechanism (Kim et al. 2004b; West et al. 2004). The two models are not mutually exclusive, and unified models have been proposed (Luo et al. 2006; Zhang et al. 2015).
Although transcription termination can occur in vitro without transcript cleavage at the pA site (Zhang et al. 2015), recent transcriptomics studies led to the conclusion that XRN2/Rat1p are broadly important for efficient transcription termination in human cells (Fong et al. 2015) and budding yeast (Baejen et al. 2017). At the same time, because XRN2 inactivation does not fully prevent but only delays termination in human cells and because Rat1p depletion in yeast caused globally less severe termination defects than depletion of CPFs, alternative termination mechanisms have been speculated to compensate partially for loss of XRN2 (Fong et al. 2015; Baejen et al. 2017). Here, we provide direct evidence for the operation of an XRN2-independent pathway of transcriptional termination in Caenorhabditis elegans. We demonstrate that the two pathways affect separate genes, with specific gene features (most notably promoters) instructing pathway choice. Hence, alternative termination pathways operate in C. elegans but appear distinct rather than redundant in function.
Results
Readthrough transcripts accumulate in the absence of XRN2
To examine the effect of XRN2 on the C. elegans transcriptome, we sequenced mRNA from animals depleted for XRN2 by RNAi. We noticed that a specific subset of genes showed elevated levels of downstream intergenic transcripts in XRN2-depleted samples as compared with mock RNAi control samples (Fig. 1A; Supplemental Fig. S1A,B), whereas other genes did not (Fig. 1B; Supplemental Fig. S1C). Since the sequencing libraries had been prepared from RNA purified by oligo-dT beads, we wondered whether these intergenic transcripts had been selectively captured due to pA or intrinsic adenine-rich sequences that specifically occurred in downstream regions of these genes. However, this was not the case, since specificity was retained when we sequenced total RNA depleted of ribosomal RNA (rRNA) (Supplemental Fig. S1D–F), suggesting the possibility of a selective readthrough defect upon XRN2 depletion.
Figure 1.

XRN2 inactivation yields readthrough transcripts on a specific subset of genes. Wild-type animals were treated with mock or xrn-2 RNAi from L1 to L4 stage and analyzed by RNA sequencing (RNA-seq). Snapshots of dpy-13 (A), col-180 (B), and a >50-kb region downstream from dpy-13 (C) genes are shown. RNA levels were normalized to total library size and are shown in green in a log2 scale (0–13). Genes are shown in blue. Only reads aligning to the same strand as the gene of interest are included, and genes on the same strand are marked with red arrows. (D) Schematic of the downstream and upstream regions used to identify XRN2-dependent termination. The upstream region consists of the 2 kb immediately upstream of the transcript start site (TSS), while the downstream region consists of the intergenic region downstream from the transcript end site (TES), extending either until the TSS of the next downstream gene on the same strand or up to 10 kb at the longest. Genes showing significantly up-regulated read number in downstream regions in xrn-2(RNAi) versus mock but no significantly up-regulated read number in upstream regions were classified as XRN2-dependent. Genes showing significant up-regulation in both upstream and downstream regions could not be classified accurately and were omitted from further analysis. (E) Volcano plot showing genome-wide analysis of read counts in regions downstream from all expressed genes. The X-axis represents the log2 fold change in reads between mock and xrn-2 RNAi conditions, while the Y-axis represents the adjusted −log10 P-value for each comparison. Cutoffs of P < 0.05 and log2 fold change ≥1 were used to define genes showing increased downstream readthrough in xrn-2 RNAi (red plus gray points). This set of genes was further filtered based on changes in upstream read counts and operon membership to result in the final set of high-confidence XRN2-dependent genes (red points; see the text).
Upon XRN2 knockdown, some regions showed intergenic transcription spanning distances of up to tens of kilobases (Fig. 1C). We found an instance in which the most upstream gene contributed to transcriptional readthrough over 50 kb, invading downstream gene loci (Fig. 1C; Supplemental Fig. S1G). In general, invasion is thought to reduce downstream gene expression by interfering with transcription initiation (Greger and Proudfoot 1998; Shearwin et al. 2005). In C. elegans, however, readthrough transcripts might give rise to functional mRNAs because trans-splicing can separate bicistronic or multicistronic transcripts and provide downstream transcripts with a 5′ cap required for translation (Krause and Hirsh 1987; Allen et al. 2011). Accordingly, we saw little evidence for accumulation of long transcripts, with downstream genes retaining extensive read coverage for exons but not for introns (Fig. 1C). Moreover, pre-mRNA 3′ end cleavage, which precedes degradation of nascent transcripts by XRN2 in the torpedo model, was not impaired by XRN2 depletion: Knockdown of a CPF PCF11 caused accumulation of both downstream transcripts (dpy-13 DS) and transcripts spanning the cleavage site (dpy-13 CS), whereas only the former accumulated upon XRN2 knockdown (Supplemental Fig. S1H,I). Finally, when we examined specifically transcripts of the ama-1 gene, located downstream from dpy-13, we found that levels of mature mRNA—i.e., 5′-capped (trans-spliced), cis-spliced, and polyadenylated transcripts—increased upon XRN2 knockdown (Supplemental Fig. S1J). Thus, transcription readthrough can cause ectopic expression and overexpression of downstream genes in C. elegans (see also Fig. 4, below).
Figure 4.

Promoters determine XRN2 dependence in transcription termination. (A) A transcription readthrough reporter. The indicated construct was integrated into an intergenic region with no active transcription. (B–G) Animals carrying the indicated constructs were treated with mock or xrn-2 RNAi from L1 to L4 stage and observed for GFP signal. The insets show differential interference contrast (DIC) images that confirm mid-L4 stage vulva morphology. Bar, 100 µm. (H) Summary of the reporter assay. All phenotypes, GFP-positive (+) or GFP-negative (−), were fully penetrant and scored for ≥10 animals per condition.
A large subset of genes fails to yield detectable accumulation of readthrough transcripts upon XRN2 inactivation
In order to determine a high-confidence set of genes that are dependent on XRN2 for their termination, we calculated the change in strand-specific read density downstream from as well as in the 2 kb of sequence immediately upstream of all genes upon XRN2 depletion (Fig. 1D). For genes with increased read density in both upstream and downstream regions, readthrough transcription might derive entirely or partially from a gene farther upstream, as evidenced above for the loci downstream from dpy-13. Hence, although this group might contain genuine targets of XRN2, we disregarded them for further analysis. We also discarded genes in operons, as the average intercistronic distance was too short to reliably quantify readthrough. Using a cutoff of at least twofold up-regulation, we identified 1693 XRN2-dependent genes (Fig. 1E; Supplemental Table S1).
Remarkably, out of 15,660 genes that were expressed in our experiment and for which we could clearly define a downstream region, 12,677 did not show increased read density in downstream regions upon XRN2 depletion by RNAi. As RNAi by feeding does not work efficiently in certain types of cells, such as most neurons (Tavernarakis et al. 2000), specific expression of genes in RNAi-resistant cells may falsely categorize them as XRN2-insensitive. To address this potential artifact, we performed RNA sequencing (RNA-seq) with a temperature-sensitive xrn-2 mutant strain, xrn-2(xe31) (see below), either on its own or additionally supplemented with a transgene encoding a catalytically dead xrn-2(D234/236A) mutant (xrn-2cd) (Miki et al. 2014a,b). The latter setup was chosen to achieve a particularly thorough inactivation of XRN2, mirroring an approach used previously for human cells (Fong et al. 2015). We found strongly correlated changes of downstream transcript levels among the three data sets, further supporting gene specificity in XRN2-dependent transcription termination (Fig. 2A–C). At the same time, each experiment also revealed a unique set of XRN2-dependent genes (Supplemental Fig. S2A), validating our approach to successfully reducing false identification of XRN2-insensitive genes.
Figure 2.

Assignment of XRN2 independence is robust to different XRN2 depletion strategies and not explained by gene expression levels or dynamics. (A–C) Scatter plots comparing log2 fold change of read density in downstream regions in three experiments inactivating XRN2 as indicated. In all experiments involving the xrn-2(xe31ts) allele, xrn-2 inactivation was achieved by a 10-h shift to restrictive temperature. XDT (XRN2-dependent for termination) genes as defined in the xrn-2(RNAi) experiment are colored red. (D,E) Scatter plots showing the log2 fold change of read density in downstream regions upon xrn-2(RNAi) versus the log2 mean mRNA expression levels in xrn-2(RNAi) conditions (D) or the log2 fold change of the corresponding mRNA expression levels upon xrn-2(RNAi) (E). XDT genes are shown in red.
It seemed possible that detection of readthrough was determined mostly by the expression levels of affected genes. However, we observed that although a weak positive relationship between gene expression and readthrough transcription exists (R2 = 0.058), XRN2-dependent genes are expressed at a wide range of levels, and, conversely, many highly expressed genes are not XRN2-dependent (Fig. 2D). Similarly, changes in gene expression upon XRN2 RNAi had a very weak ability to predict readthrough (R2 = 0.081) (Fig. 2E). We conclude that although high expression levels may aid detection of readthrough, our inability to detect readthrough for a large number of genes is not a technical consequence of their insufficient expression.
Nonetheless, in order to explore features of XRN2-dependent and XRN2-insensitive genes without any confounding influence of expression levels, we selected 1657 XRN2-insensitive genes from the pool of genes that showed no readthrough in any of our experiments with matching mRNA levels under mock conditions (Supplemental Fig. S2B). Unless noted otherwise, all following comparisons here use these matched groups of genes, which we call XDT (XRN2-dependent for termination) and non-XDT genes, respectively. The two groups did not show obvious differences in basic gene structure, such as gene body length (Supplemental Fig. S2C), number of introns (Supplemental Fig. S2D), the incidence of the canonical pA signal AAUAAA (61.6% and 66.8% of annotated 3′ untranslated regions [UTRs]) (Mangone et al. 2010), or the general nucleotide composition in the 50 base pairs (bp) upstream of and downstream from the cleavage site (Supplemental Fig. S2E), respectively.
Pol II termination selectively fails on XDT genes upon XRN2 knockdown
Since XRN2 degrades nascent transcripts from 3′ end cleavage sites, it was possible that our observations reflected selective stabilization of downstream transcripts from XDT genes rather than a genuine termination defect. To test this possibility, we examined Pol II binding to DNA by Pol II chromatin immunoprecipitation (ChIP) followed by sequencing (ChIP-seq) (Fig. 3A). As reported previously (Ahn et al. 2016), Pol II signal peaked around transcript start sites (TSSs) and shortly downstream from transcript end sites (TESs) (Fig. 3B,C), consistent with pausing at promoters and gene ends, respectively. For XDT genes, XRN2 knockdown extended Pol II binding toward more distal regions of PDRs (Fig. 3B, dark-gray shaded area), supporting a role of XRN2 in transcription termination. As no such effect occurred on the PDRs of non-XDT genes (Fig. 3C), we can conclude that the two groups genuinely differ in their reliance on XRN2 for termination.
Figure 3.

XRN2 is required for efficient transcription termination of a subset of genes (A) Wild-type animals were treated with mock or xrn-2 RNAi from L1 to L4 larval stages followed by Pol II ChIP-seq analysis. A snapshot of the ftn-2 gene is shown. ChIP signal is shown in blue in a linear scale (0–350), and RNA signal from both strands is shown in green in a log2 scale (0–12). Arrows indicate the 5′-to-3′ strand direction. (B,C) Metagene plots show Pol II ChIP signal on XDT (B) and non-XDT (C) genes with 1-kb upstream and 2-kb downstream regions. Gene bodies were scaled between the TSS and TES. ChIP signal represents the mean of two biological replicates from each condition, quantified in 50-bp bins across each gene and flanking regions. Proximal PDR is shaded in light gray, and distal PDR is shaded in dark gray. (D) XRN2 ChIP-seq was performed by immunoprecipitating transgenic XRN2-GFP from animals lacking endogenous XRN2. Metagene plots display XRN2 ChIP signals on XDT and non-XDT genes. Scaling is as in B and C. (E,F) Smoothed scatter plots showing mean XRN2 ChIP signal versus mean Pol II ChIP signal in 1-kb windows around the TSSs (E) or TESs (F) of protein-coding genes in wild-type animals. The blue density cloud represents all protein-coding genes, with darker-blue regions indicating a higher density of points, and lighter-blue regions indicating a lower density of points. The red points indicate XDT genes. The dashed line represents a linear regression of XRN2 ChIP signal against Pol II signal.
XRN2 binds to—and enhances Pol II accumulation at the TESs of—both XDT and non-XDT genes
We considered that the specificity of XRN2 for termination of only XDT genes was conferred by selective recruitment of XRN2 to only XDT but not non-XDT genes. To test this possibility, we performed XRN2 ChIP-seq on lysates from animals that carried a functional XRN2-GFP fusion protein as their only source of XRN2 (Miki et al. 2014b). Although XRN2 appeared more abundant on XDT genes, it was also readily detected on non-XDT genes, and its abundance correlated generally well with Pol II signal at both TSSs and TESs for both XDT and non-XDT genes (Fig. 3D–F). Because the XRN2 termination models imply that every TEC requires at least one XRN2 molecule for termination, selectivity in XRN2 recruitment thus does not appear to be the cause of differences in XRN2 sensitivity among genes. At the same time, binding of XRN2 to non-XDT genes appears to be functionally relevant: Although XRN2 was dispensable for termination of non-XDT gene transcription, its depletion decreased the peak of Pol II signal in the proximal PDR, near the TES, for both XDT and non-XDT genes (Fig. 3B,C).
A reporter system permits detection of termination defects in vivo
Based on the fact that readthrough transcripts can be 5′-capped, spliced, and polyadenylated (Supplemental Fig. S1J), we developed a reporter system to visualize transcription readthrough. We exploited the fact that intercistronic regions (ICRs; short sequences that separate C. elegans genes that are coexpressed in operons) promote trans-splicing of—and thus acquisition of a 5′ cap on—downstream transcripts (Huang et al. 2001). We generated a transgene where an ICR derived from an operon containing the gpd-2 and gpd-3 genes preceded a coding sequence for GFP fused to histone H2B and the tbb-2 3′ UTR. H2B was included to concentrate GFP in the nucleus, facilitating detection. We inserted the fragment into an intergenic region downstream from the endogenous dpy-13 gene (Supplemental Fig. S3A). Consistent with the lack of a promoter, no transgene expression occurred under mock RNAi conditions. However, upon XRN2 knockdown, GFP accumulated specifically in the hypodermis, the site of dpy-13 expression (Supplemental Fig. S3B), validating the reporter function.
Since the reporter system enabled detection of termination defects in living animals, we reasoned that it would facilitate not only examination of candidate protein factors or DNA/RNA sequence elements for involvement in the process (see below) but also unbiased screening. As a proof of principle, we performed a random mutagenesis screen using the dpy-13 readthrough reporter and recovered four temperature-sensitive alleles of xrn-2 (alleles xe31 through xe34) (Supplemental Fig. S3C,D; Supplemental Table S2). Moreover, although no CPF mutations were uncovered in our screen, possibly due to their detrimental effects on animal viability and fertility, targeted CPF knockdown caused a readthrough signal, as expected from the torpedo model (Supplemental Fig. S4).
XRN2 dependence is determined by gene elements, not genomic location
We exploited the reporter system to gain insight into the sequence or chromosomal features that determined the choice of termination mode. We made a construct composed of the dpy-13 promoter (Pdpy-13), its gene body (from TSS to pA signal), and a 0.5-kb-long PDR (Pdpy-13::dpy-13::dpy-13PDR) followed by ICR::GFP::H2B::tbb-23′ UTR (Fig. 4A) and inserted it into a transcriptionally inactive region on chromosome II. Like the endogenous dpy-13 locus on chromosome IV, the transgenic animals showed GFP signal in hypodermal cells specifically upon XRN2 knockdown (Fig. 4B). In contrast, animals carrying an analogous reporter based on the non-XDT gene col-180 (Fig. 1B), which is also expressed in hypodermal cells, did not show GFP signal upon XRN2 knockdown (Fig. 4C). Thus, XRN2 dependence for efficient transcription termination is determined by gene elements (namely, promoters, gene bodies, and/or PDRs) but not genomic locations.
Promoters determine XRN2 dependence in transcription termination
In order to address which gene element determines XRN2 dependence in transcription termination, we examined promoters, gene bodies, and PDRs of XDT (dpy-13) and non-XDT (col-180) genes using the readthrough reporter system (Fig. 4; Supplemental Fig. S5A–D). When we replaced dpy-13PDR by col-180PDR in the dpy-13 readthrough reporter, XRN2 knockdown, but not mock RNAi, yielded GFP signal in hypodermal cells (Fig. 4D). This was also true when both the gene body and the PDR were replaced by those of col-180 (Fig. 4E), suggesting that neither the gene body nor the PDR determines XRN2 dependence and implicating instead the promoter.
To test the role of the promoter, we generated four additional reporters with the gene body and the PDR of dpy-13 and the promoters of additional XDT genes (dpy-5 and sqt-1) or non-XDT genes (col-180 and dpy-18), respectively. Whereas the former retained XRN2 dependence (Supplemental Fig. S5A,B), the latter did not. Instead, when col-180 and dpy-18 promoters were tested, the reporter animals showed GFP signal even under mock RNAi conditions, and XRN2 knockdown did not enhance the signal (Fig. 4F; Supplemental Fig. S5C). In other words, the constructs that combined non-XDT promoters with the XDT gene body and PDR were constitutively termination-incompetent irrespective of XRN2 status. Their transcription is thus refractory to termination by XRN2 activity. We can exclude that this failure to terminate is caused by an incompatibility with the bicistronic reporter system because an additional reporter that contained the col-180 promoter, gene body, and PDR terminated both in the presence of XRN2 and upon its depletion (Fig. 4C). We conclude that the mode of termination, dependent on XRN2 or refractory to it, is determined at the gene promoter, which may endow TECs with specific properties.
To identify features that might distinguish between the different types of promoters, we searched for transcription factor motifs in promoters of XDT versus non-XDT genes using HOMER (Heinz et al. 2010). However, there were no significant enrichments of known transcription factor motifs. We did detect some enrichments of de novo motifs, but these were present in a small number of promoters of either class, making their biological relevance doubtful (Supplemental Table S3). Similarly, scanning each set of promoters for occurrences of core promoter motifs using OProf (Ambrosini et al. 2003) revealed a peak in occurrences of both the TATA-box and the Inr motif around the TSS, but these peaks did not differ between XDT and non-XDT promoters (Supplemental Fig. S5E,F). Hence, it appears that the different promoter functionalities are not due to differential usage of core promoter elements.
Non-XDT gene termination requires a compatible PDR
The fact that the Pcol-180::dpy-13::dpy-13PDR reporter showed transcription readthrough (Fig. 4F) while Pcol-180::col-180::col-180PDR did not (Fig. 4C) implicated either the gene body or the PDR of col-180 in non-XDT termination. We dissected this mechanism further by generating a Pcol-180::dpy-13::col-180PDR reporter and found that col-180PDR sufficed for termination of transcription initiated at the col-180 promoter (Fig. 4G). col-180PDR also terminated transcription initiated at the dpy-18 promoter (Supplemental Fig. S5D), suggesting that non-XDT genes share a downstream element (DSE)-dependent termination mechanism. However, this mechanism does not seem to function for Pol II complexes formed at promoters of XDT genes, since col-180PDR failed to terminate transcription initiated at the dpy-13 promoter in the absence of XRN2 (Fig. 4D). In other words, although col-180PDR supported XRN2-dependent termination from an XDT gene promoter, it did not suffice to confer non-XDT termination. Combined with XRN2 having failed to terminate transcription initiated at col-180 and dpy-18 promoters, this suggests that the two termination mechanisms are not interchangeable for transcription from two different types of promoters (Supplemental Fig. S6).
Reduced elongation rate is insufficient to permit termination on a non-XDT gene by XRN2
According to the kinetic termination model, high elongation rates may prevent or delay effective termination, suggesting that non-XDT genes might be refractory to XRN2-mediated termination because TECs that transcribe them are too fast for XRN2 to catch up with them (Fong et al. 2015). Since the relatively short length of C. elegans genes is an obstacle to direct measurements of transcription elongation rates (Jonkers and Lis 2015), we sought to test this model by artificially decreasing transcription elongation rates through the R743H mutation in the largest subunit of C. elegans Pol II (Bowman et al. 2011), which corresponds to the R749H mutation that reduces elongation rates of human Pol II (Fong et al. 2014, 2015). We examined its effect on the Pcol-180::dpy-13::dpy-13PDR reporter, where noncompatible dpy-13PDR prevents the non-XDT mechanism demanded by the Pcol-180 promoter, asking whether reduced elongation rates would promote termination and do so in an XRN2-dependent manner. This was not the case, as termination continued to fail even in the presence of XRN2 (Supplemental Fig. S7).
Discussion
Among few transcription termination factors known, XRN2 has been particularly well characterized. Thus, studies from both yeast and human cells support a major and conserved function of this ribonuclease in ending transcription (Kim et al. 2004b; West et al. 2004). Accordingly, we could readily observe termination defects when we depleted XRN2 in C. elegans in vivo. Surprisingly, however, termination continued to occur normally for a large fraction of genes even after extensive depletion of XRN2 through the combination of a temperature-sensitive endogenous and a dominant-negative transgene allele.
Even the stringent depletion protocol that we used might have failed to eliminate XRN2 entirely, and it might be argued that such residual amounts would suffice for termination of some genes. In this view, XRN2 would be highly potent rather than ineffective in terminating transcription of non-XDT genes. However, the fact that a reporter that combined a non-XDT promoter with an XDT PDR was termination-deficient even in an XRN2 wild-type background all but excludes this formal possibility. Specifically, it reveals that non-XDT genes are not merely insensitive to XRN2 depletion but in fact are refractory to XRN2-mediated termination. Hence, XDT and non-XDT genes are genuinely distinct in their termination modes.
Since recent studies revealed that loss of XRN2/Rat1p activities caused global termination defects in yeast and human cells (Fong et al. 2015; Baejen et al. 2017), it seems possible that termination in C. elegans might differ from that in previously studied systems. For instance, termination on genes with fast elongation rates requires distances of several kilobases past pA signal in human cells (Fong et al. 2015). A DSE-dependent termination mechanism might then be an adaptation to a compact genome, such as that of C. elegans, to effectively avoid invasive transcription into sensitive downstream genes. However, previous work (Fong et al. 2015; Baejen et al. 2017) focused on general principles derived by metagene analyses rather than individual genes and thus did not specifically address whether and to what extent XRN2-independent termination pathways might be operational. Indeed, individual gene responses to XRN2 depletion in yeast appear to differ greatly (Baejen et al. 2017). Hence, we consider it likely that non-XDT mechanisms function also in other organisms.
Our findings provide the first mechanistic insights into XRN2-independent termination and a framework for further investigation (Fig. 5). Specifically, since we could readily detect XRN2 on non-XDT genes, failure of XRN2 to terminate transcription on these genes is not explained by a lack of its recruitment. Rather, we propose that TECs that transcribe the two classes of genes differ in their susceptibility to XRN2 due to properties such as elongation rate or complex stability. The two promoter classes might then promote the recruitment of distinct factors to—and/or conformational changes of—TECs. This in turn would explain why promoter identity seems the key to the choice of termination mode.
Figure 5.
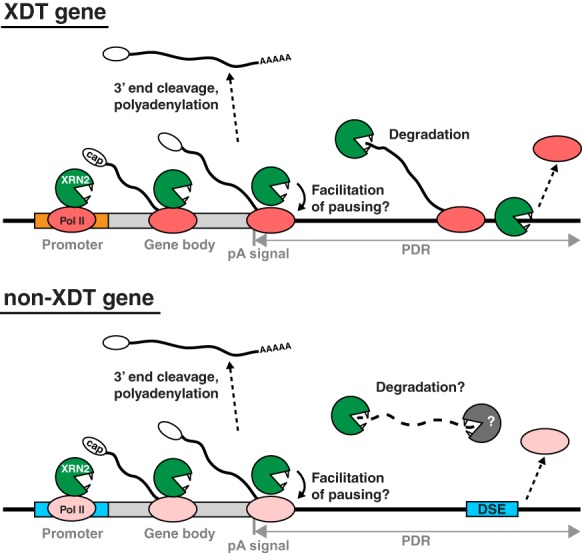
Models for XRN2-dependent and DSE-dependent transcription termination. XRN2 is recruited to Pol II on both XDT and non-XDT genes and facilitates Pol II accumulation at the gene ends, possibly by promoting pausing. On XDT genes, XRN2 degrades a nascent transcript following 3′ end cleavage and terminates Pol II. On non-XDT genes, Pol II transcribes beyond the pA site up to a DSE, which induces Pol II termination. Cleavage at the pA site separates the nascent mRNA from the downstream fragment; XRN2 might contribute, with other RNases, to the degradation of the latter. Different Pol II colors illustrate presumed differences in TEC properties and/or composition, determined by promoters, which result in differences in termination modes. See the text for details.
Differences in elongation rates are conceptually particularly appealing as a mechanism through which XRN2 sensitivity is determined. This is because previous work in mammalian cells showed that elongation rate can affect the location of transcription termination: Relative to the wild-type situation, termination occurred, on average, more distantly from the pA site in the presence of a “fast” Pol II and more proximally in the presence a “slow” Pol II (Fong et al. 2015). Hence, the currently unknown rate of RNA degradation by XRN2 and the rate of RNA synthesis by Pol II appear well balanced. A sufficiently large increase in synthesis rates could thus cause a complete failure of XRN2 to catch up with and terminate Pol II transcription. It is therefore intriguing that Pol II occupancy appears generally reduced on the expression-matched non-XDT relative to XDT genes, an observation consistent with, albeit not proof of, higher elongation rates on the former.
Despite conceptual appeal and circumstantial evidence, direct evidence in support of the elongation rate model is lacking. Specifically, we failed to convert a non-XDT gene into an XDT gene through use of a “slow” Pol II. It is possible that the mutation failed to reduce elongation rates sufficiently to permit XRN2-dependent termination. However, our reporter gene data further show that non-XDT promoters not only specify an inability to respond to XRN2-dependent termination but also positively determine a TEC's ability to respond to the alternative pathway: When we combined an XDT promoter with a non-XDT PDR, termination occurred normally in wild-type animals but was still fully dependent on XRN2 and failed in its absence. In other words, the non-XDT PDR, while required for non-XRN2-dependent termination, is not sufficient for it; it needs an appropriate promoter that specifies this termination mode. Hence, features additional to—or distinct from—elongation rate distinguish the XRN2-independent from the XRN2-dependent termination mechanism. Presumably, these features might include specific factors whose recruitment to Pol II at promoters promotes TEC disassembly at non-XDT PDRs.
Finally, we are intrigued by the observation that ChIP-seq analysis revealed the presence of XRN2 near TESs of non-XDT genes. Although further investigation will be needed to determine the functional relevance of this observation, we can imagine at least two, non-mutually exclusive, scenarios (Fig. 5). First, even in a non-XDT mechanism, Pol II transcribes beyond the pA site and thus generates a nonfunctional transcript requiring disposal. XRN2, although not essential for such “scavenger” activity, might contribute to it redundantly with other RNases. Second, and we think more interestingly, since lack of XRN2 also reduced Pol II occupancy near the TESs of both XDT and non-XDT genes, XRN2 might have a generic role in promoting Pol II pausing near pA sites. Speculatively, although not required for non-XRN2-dependent termination, XRN2-promoted stalling might then serve to make this termination pathway more robust. At any rate, we predict that further study of XRN2 function at gene ends will continue to provide novel insights into mechanisms of RNA metabolism.
Materials and methods
Strains
The Bristol N2 strain was used as wild type. The mutant strains used are listed in Supplemental Table S2.
Genetic screen
HW1604 strain animals carrying a dpy-13 transcription readthrough reporter were mutagenized with ethyl methanesulfonate (EMS), and GFP-positive F2 generation animals were identified. Mutations in xrn-2 were identified by direct sequencing or genome resequencing, respectively.
RNAi
RNAi was performed by the feeding method (Timmons and Fire 1998) at 20°C. Since xrn-2 RNAi causes slow growth, worms were treated with mock (empty vector) or xrn-2 RNAi from L1 stage for 40∼42 or 48∼52 h, respectively, to achieve the same mid-L4 developmental stage, confirmed by examination of vulval morphology. Depletion of XRN2 was confirmed by Western blot as described previously (Miki et al. 2014a).
Microscopy
Images were acquired using an Axio Observer Z1 microscope and AxioVision SE64 (release 4.8) software (Carl Zeiss). All phenotypes shown in Figure 4, GFP-positive (+) or GFP-negative (−), were fully penetrant and observed for ≥10 animals per condition.
RNA-seq
RNA-seq libraries were prepared using the TruSeq standard mRNA library preparation kit [for poly(A)-RNA-seq; Illumina] or the ovation universal RNA-seq system (for rRNA-depleted RNA-seq; NuGEN) and sequenced by the HiSeq 2500 sequencing system (Illumina). Note that except for the results shown in Supplemental Figure S1, D–F, all of our analyses were performed on poly(A)-selected libraries. This choice was made to minimize the number of falsely identified non-XDT genes: Poly(A)-selected libraries contained fewer sporadic intergenic reads than rRNA-depleted libraries in wild-type (XRN2-proficient) conditions. Since readthrough is defined via a comparison between xrn-2-depleted and control conditions, this lower background permitted more facile detection of readthrough (i.e., XDT genes) and thus helped to avoid false-positive identifications of non-XDT genes.
ChIP-seq
Pol II ChIP was performed as described previously with minor modifications using mouse anti-Pol II CTD antibody (clone 8WG16; Abcam) (Zhong et al. 2010). For XRN2 ChIP, a transgenic strain expressing XRN2-GFP fusion protein was used (see the Supplemental Material for details; Miki et al. 2014b). ChIP-seq libraries were prepared using the NEBNext Ultra DNA library preparation kit for Illumina (New England Biolabs) and sequenced by the HiSeq 2500 sequencing system.
Bioinformatics analysis
Sequencing reads were aligned to the October 2010 (ce10) C. elegans genome assembly from University of California at Santa Cruz (Rosenbloom et al. 2015). BigWig files of library size-normalized coverage were generated using the qExportWig function in QuasR (Gaidatzis et al. 2015). Gene-level expression for RNA-seq data was quantified using the exon union model of genes with annotated exons downloaded from WormBase (version WS220, corresponding to the ce10 assembly; ftp://ftp.wormbase.org/pub/wormbase/releases/WS220/species/c_elegans). Samples were normalized by the total number of reads mapping to exons, and very lowly expressed genes (mean normalized read count ≤16) were removed from further analysis. See the Supplemental Material for descriptions of downstream readthrough analysis and ChIP-seq analysis.
See the Supplemental Material for detailed methods information.
Data accession
All RNA-seq and ChIP-seq data sets generated for this study have been deposited at Gene Expression Omnibus (GSE79994: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79994 [Miki et al. 2016], and GSE97775: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97775 [this study]).
Supplementary Material
Acknowledgments
We thank Sebastien Smallwood, Kirsten Jacobeit, Stéphane Thiry, and Eliza Moreno Pandini Figueiredo of the Friedrich Miescher Institute Functional Genomics Facility for library preparation and sequencing; Donald Riddle and Stefan Taubert at the University of British Columbia for the strain with the dpy-13(m401) allele; Dimos Gaidatzis of the Friedrich Miescher Institute Computational Biology Platform and Mihaela Zavolan and Ralf Schmidt at the University of Basel for discussion and advice on data analysis; Wolfgang Maier at the University of Freiburg for introduction of the MimodD software; and Marc Bühler and Iskra Katic at Friedrich Miescher Institute and Domenico Libri at Institut Jacques Monod for critical comments on the manuscript. The DR786 strain with the ama-1(m322) allele was provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). This work was supported by the Swiss National Science Foundation (SNF 31003A_143313 to H.G.) and the Novartis Research Foundation through the Friedrich Miescher Institute for Biomedical Research Foundation.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.301093.117.
Freely available online through the Genes & Development Open Access option.
References
- Ahn JH, Rechsteiner A, Strome S, Kelly WG. 2016. A conserved nuclear cyclophilin is required for both RNA polymerase II elongation and co-transcriptional splicing in Caenorhabditis elegans. PLoS Genet 12: e1006227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Hillier LW, Waterston RH, Blumenthal T. 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Praz V, Jagannathan V, Bucher P. 2003. Signal search analysis server. Nucleic Acids Res 31: 3618–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baejen C, Andreani J, Torkler P, Battaglia S, Schwalb B, Lidschreiber M, Maier KC, Boltendahl A, Rus P, Esslinger S, et al. 2017. Genome-wide analysis of RNA polymerase II termination at protein-coding genes. Mol Cell 66: 38–49. [DOI] [PubMed] [Google Scholar]
- Bowman EA, Riddle DL, Kelly W. 2011. Amino acid substitutions in the Caenorhabditis elegans RNA polymerase II large subunit AMA-1/RPB-1 that result in α-amanitin resistance and/or reduced function. G3 1: 411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, Bentley DL. 2014. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev 28: 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Brannan K, Erickson B, Kim H, Cortazar MA, Sheridan RM, Nguyen T, Karp S, Bentley DL. 2015. Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol Cell 60: 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D, Lerch A, Hahne F, Stadler MB. 2015. QuasR: quantification and annotation of short reads in R. Bioinformatics 31: 1130–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Proudfoot NJ. 1998. Poly(A) signals control both transcriptional termination and initiation between the tandem GAL10 and GAL7 genes of Saccharomyces cerevisiae. EMBO J 17: 4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Kuersten S, Deshpande AM, Spieth J, MacMorris M, Blumenthal T. 2001. Intercistronic region required for polycistronic pre-mRNA processing in Caenorhabditis elegans. Mol Cell Biol 21: 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Lis JT. 2015. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16: 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Ahn SH, Krogan NJ, Greenblatt JF, Buratowski S. 2004a. Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J 23: 354–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. 2004b. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432: 517–522. [DOI] [PubMed] [Google Scholar]
- Krause M, Hirsh D. 1987. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell 49: 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Johnson AW, Bentley DL. 2006. The role of Rat1 in coupling mRNA 3′-end processing to transcription termination: implications for a unified allosteric-torpedo model. Genes Dev 20: 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangone M, Manoharan AP, Thierry-Mieg D, Thierry-Mieg J, Han T, Mackowiak SD, Mis E, Zegar C, Gutwein MR, Khivansara V, et al. 2010. The landscape of C. elegans 3′UTRs. Science 329: 432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki TS, Richter H, Rüegger S, Großhans H. 2014a. PAXT-1 promotes XRN2 activity by stabilizing it through a conserved domain. Mol Cell 53: 351–360. [DOI] [PubMed] [Google Scholar]
- Miki TS, Rüegger S, Gaidatzis D, Stadler MB, Großhans H. 2014b. Engineering of a conditional allele reveals multiple roles of XRN2 in Caenorhabditis elegans development and substrate specificity in microRNA turnover. Nucleic Acids Res 42: 4056–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki TS, Carl SH, Stadler MB, Großhans H. 2016. XRN2 autoregulation and control of polycistronic gene expresssion in Caenorhabditis elegans. PLoS Genet 12: e1006313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Boudvillain M, Libri D. 2016. Transcription termination: variations on common themes. Trends Genet 32: 508–522. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ. 2016. Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science 352: aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. 2015. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 43: D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearwin KE, Callen BP, Egan JB. 2005. Transcriptional interference—a crash course. Trends Genet 21: 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Manley JL. 2015. The end of the message: multiple protein–RNA interactions define the mRNA polyadenylation site. Genes Dev 29: 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernarakis N, Wang SL, Dorovkov M, Ryazanov A, Driscoll M. 2000. Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat Genet 24: 180–183. [DOI] [PubMed] [Google Scholar]
- Timmons L, Fire A. 1998. Specific interference by ingested dsRNA. Nature 395: 854. [DOI] [PubMed] [Google Scholar]
- West S, Gromak N, Proudfoot NJ. 2004. Human 5′ → 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432: 522–525. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Fu J, Gilmour DS. 2005. CTD-dependent dismantling of the RNA polymerase II elongation complex by the pre-mRNA 3′-end processing factor, Pcf11. Genes Dev 19: 1572–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Rigo F, Martinson HG. 2015. Poly(A) signal-dependent transcription termination occurs through a conformational change mechanism that does not require cleavage at the poly(A) site. Mol Cell 59: 437–448. [DOI] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HY, Preston E, et al. 2010. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.