

ABSTRACT
Hypervirulent group A streptococcus (GAS) can inhibit neutrophil recruitment and cause systemic infection in a mouse model of skin infection. The purpose of this study was to determine whether platelet-activating factor acetylhydrolase Sse and streptolysin S (SLS) have synergistic contributions to inhibition of neutrophil recruitment and systemic infection in subcutaneous infection of mice by MGAS315, a hypervirulent genotype emm3 GAS strain. Deletion of sse and sagA in MGAS315 synergistically reduced the skin lesion size and GAS burden in the liver and spleen. However, the mutants were persistent at skin sites and had similar growth factors in nonimmune blood. Thus, the low numbers of Δsse ΔsagA mutants in the liver and spleen were likely due to their reduction in the systemic dissemination. Few intact and necrotic neutrophils were detected at MGAS315 infection sites. In contrast, many neutrophils and necrotic cells were present at the edge of Δsse mutant infection sites on day 1 and at the edge of and inside Δsse mutant infection sites on day 2. ΔsagA mutant infection sites had massive numbers of and few intact neutrophils at the edge and center of the infection sites, respectively, on day 1 and were full of intact neutrophils or necrotic cells on day 2. Δsse ΔsagA mutant infection sites had massive numbers of intact neutrophils throughout the whole infection site. These sse and sagA deletion-caused changes in the histological pattern at skin infection sites could be complemented. Thus, the sse and sagA deletions synergistically enhance neutrophil recruitment. These findings indicate that both Sse and SLS are required but that neither is sufficient for inhibition of neutrophil recruitment and systemic infection by hypervirulent GAS.
KEYWORDS: esterase Sse, PAF acetylhydrolase, streptolysin S, group A streptococcus, inhibition of neutrophil recruitment, innate immune evasion, neutrophil, skin invasion, virulence
INTRODUCTION
Group A streptococcus (GAS) is a major human pathogen that commonly causes relatively mild pharyngitis and superficial skin infections (1). GAS can also cause potentially lethal severe invasive infections, with about 12,000 cases occurring annually in the United States, and these severe infections include bacteremic skin and soft tissue infections, pneumonia, necrotizing fasciitis, and bacteremia without focus (2). From 2005 to 2012 in the United States, these severe invasive infections were most frequently associated with GAS of M protein gene-based genotypes emm1, emm12, emm3, emm28, and emm89 (2). Invasive emm3 GAS strains cause higher rates of mortality than invasive strains of other serotypes (3).
Necrotizing fasciitis (NF) is a rapidly progressive infection of the skin, subcutaneous and deep soft tissue, and muscle and leads to systemic dissemination (4). Some NF patients have numerous bacteria but few or no neutrophilic responses at infection sites (5, 6), which is classified as stage III NF (6), and other histopathologic types include a moderate-to-severe neutrophilic response and a positive Gram staining (stage II) and an intense neutrophilic response with the absence of bacteria (stage I) in infected tissues (6). Patients with stage III NF have a higher mortality rate than patients with stage I and II NF (5, 6).
Animals in a murine model of NF caused by hypervirulent, invasive M1T1 GAS displays stage III histopathologic features of few or no neutrophils at sites of bacterial infection (7, 8). Hypervirulent M1T1 GAS isolates are usually natural CovRS mutants (8–10). CovRS (also known as CsrRS) is the two-component regulatory system that negatively regulates multiple virulence factors (11–14), including those involved in innate immune evasion, such as the capsule synthase HasA (11), interleukin-8 (IL-8)/CXC chemokine peptidase SpyCEP (15), platelet-activating factor (PAF) acetylhydrolase Sse (7, 16), opsonophagocytosis-inhibiting Mac (17), NAD+ glycohydrolase (18), and streptolysin O (19). Natural CovRS mutations in a M1T1 clone of serotype M1 GAS enhance the expression of CovRS-controlled virulence factors and downregulate the expression of the protease SpeB, resulting in a high capacity to cause skin invasion, innate immune evasion, systemic dissemination, and hypervirulence (8, 14, 20, 21). The selection of CovRS M1T1 mutations during infection has been readily demonstrated in invasive M1T1 GAS during experimental infections of mice (10, 20–26). Neutrophils are required for in vivo selection of covRS mutants, and M1T1 CovRS mutants exhibit greater resistance to clearance by neutrophils in vivo than wild-type (wt) strains (27).
The severe inhibition of neutrophil responses by M1T1 GAS requires CovRS mutations, and the PAF acetylhydrolase Sse, but not the IL-8 peptidase SpyCEP or C5a peptidase ScpA, plays a critical role in inhibition of neutrophil recruitment by hypervirulent M1T1 GAS CovS mutants (7, 8). Streptolysin S (SLS) has been shown to inhibit neutrophil recruitment in a zebrafish model of GAS infection and delays the exodus of neutrophils from the vessel lumen into tissue at the early stage of GAS skin infection in mice (28). The purpose of this study was to determine whether Sse and SLS have a synergistic effect on the capacity of MGAS315, a hypervirulent emm3 strain (29, 30), to inhibit neutrophil recruitment and to cause systemic infection in subcutaneous infection of mice. We generated MGAS315 mutants with a single deletion of sagA, which encodes the peptide component of SLS, or sse or a double deletion of both sagA and sse. We found that the sse and sagA deletions synergistically reduce the level of skin invasion and the GAS load in the liver and spleen and that both sse and sagA are required for inhibition of neutrophil recruitment by MGAS315.
RESULTS
Synergistic effects of Sse and SLS on skin invasion, systemic infection, and neutrophil recruitment in a murine model of subcutaneous MGAS315 infection.
MGAS315 caused large lesions in female CD-1 mice at 48 h after subcutaneous inoculation (mean lesion area ± standard deviation, 934 ± 165 mm2), whereas the MGAS315 Δsse, ΔsagA, and Δsse ΔsagA mutants caused lesion sizes of 277 ± 64, 380 ± 126, and 188 ± 58 mm2, respectively (Fig. 1A and B). There were (4.0 ± 2.6) × 104 neutrophils at the MGAS315 skin infection site, as estimated by the myeloperoxidase (MPO) assay, and the Δsse, ΔsagA, and Δsse ΔsagA mutants recruited (2.8 ± 1.5) × 105, (2.5 ± 0.8) × 105, and (5.1 ± 1.3) × 105 neutrophils at the infection sites, respectively (Fig. 1C). There were (2.5 ± 3.6) × 108 and (2.0 ± 1.1) × 107 GAS bacteria per gram of tissue in the spleen and liver, respectively, in MGAS315-infected mice, and deletion of sse or sagA in MGAS315 reduced the GAS loads in the spleen by ≥4 orders of magnitude and in the liver by 2 orders of magnitude (Fig. 1D and E). More importantly, there were no GAS in the liver or spleen of the majority of mice infected with the Δsse ΔsagA mutant on day 2 after inoculation (Fig. 1D and E). These data indicate that Sse and SLS significantly contribute to skin invasion, inhibition of neutrophil recruitment, and systemic infection in subcutaneous infection of mice and that Sse and SLS have a synergistic contribution to skin invasion, inhibition of neutrophil recruitment, and systemic infection in skin infections of CD-1 mice.
FIG 1.

Effects of sse and sagA deletions on skin invasion, GAS loads in liver and spleen, and neutrophil recruitment in subcutaneous MGAS315 infections of CD-1 mice. Six-week-old female CD-1 mice were subcutaneously inoculated with 1.7 × 108 CFU of MGAS315, 2.0 ×108 CFU of the Δsse mutant, 2.1 × 108 CFU of the ΔsagA mutant, or 1.7 × 108 CFU of the Δsse ΔsagA mutant and euthanized at 48 h after inoculation for analyses. Shown are representative inside-out images of the skin infection sites (A), lesion size (B), neutrophil recruitment (C), GAS loads in spleen (D), and GAS loads in liver (E). P values were determined by the Mann-Whitney t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. PMN, polymorphonuclear leukocytes.
The effects of these strains in subcutaneously infected C57BL/6J mice were also compared at day 1 after inoculation. The effects of the sse and/or sagA deletion in MGAS315 on skin invasion, neutrophil recruitment, and the bacterial loads in the liver and spleen of C57BL/6J mice were similar to the effects in CD-1 mice (Fig. 2). In particular, the sse and sagA deletions also showed a synergistic contribution to enhancement of neutrophil recruitment and reduction of the GAS loads in the liver and spleen. We also compared the effects of infection of female and male C57BL/6J mice with the Δsse mutant and observed no difference in lesion size, neutrophil levels, or GAS loads in the liver and spleen by gender (Fig. 2).
FIG 2.

Effects of sse and sagA deletions on skin invasion, GAS loads in liver and spleen, and neutrophil recruitment in subcutaneous MGAS315 infection of C57BL/6J mice. Six-week old C57BL/6J mice were subcutaneously inoculated with 1.5 × 108 CFU of MGAS315, 1.9 ×108 CFU of the Δsse mutant, 1.8 × 108 CFU of the ΔsagA mutant, or 1.7 × 108 CFU of the Δsse ΔsagA mutant and euthanized at 24 h after inoculation for analyses. Shown are the lesion size (A), neutrophil recruitment (B), GAS loads in liver (C), and GAS loads in spleen (D). P values were determined by the Mann-Whitney t test. *, P < 0.05; **, P < 0.01; ns, not significant.
Resistance of MGAS315 and its sse and sagA deletion mutants to clearance from skin infection sites and nonimmune human and mouse blood.
The bacterial loads of the Δsse ΔsagA mutant in the liver and spleen on days 1 and 2 after inoculation were >1,000-fold lower than those of MGAS31 and the Δsse and ΔsagA mutants. This difference could be due to the effective clearance of the Δsse ΔsagA mutant from the skin infection sites because of the more robust neutrophil recruitment to Δsse ΔsagA mutant infection sites. Alternatively, the growth and survival in blood of the double mutant might be compromised. These possibilities were examined. The numbers of viable GAS bacteria at the skin infection sites at 24 h after inoculation were 286%, 156%, 55%, and 85% of those at 1 h after inoculation in MGAS315 and Δsse, ΔsagA, and Δsse ΔsagA mutant infections, respectively (Fig. 3A). The numbers of bacteria of the wt strain and the Δsse mutant at skin infection sites slightly increased with time, whereas the numbers of the ΔsagA and Δsse ΔsagA mutants slightly decreased with time. The data indicate that the mutants with enhanced neutrophil responses, especially Δsse ΔsagA mutants, were not efficiently cleared at 24 h after inoculation. To determine whether the mutants were compromised in growth and survival in blood, the apparent growth factors for MGAS315, its Δsse, ΔsagA, and Δsse ΔsagA mutants, and the complemented mutant strains in heparinized blood and serum from two persons and in pooled heparinized mouse blood were measured. The two persons were healthy and lacked anti-SLO and anti-NADase antibodies in their serum (data not shown). The numbers of MGAS315, mutant, and complemented mutant strain bacteria after a 3-h incubation in blood and serum were more than 40-fold higher than the numbers in the inoculum (Fig. 3B). The apparent growth factors for the test strains in each test were not significantly different, as multiple-comparison analyses of the data by 1-way analysis of variance (ANOVA) using GraphPad Prism software (version 7.03) resulted in P values of ≥0.1184. Thus, the sse and/or sagA deletion did not alter the growth or survival of MGAS315 in human and mouse blood. These data suggest that the lower loads of the sse and sagA deletion mutants in the liver and spleen appear to be at least partially due to reduced systemic dissemination.
FIG 3.
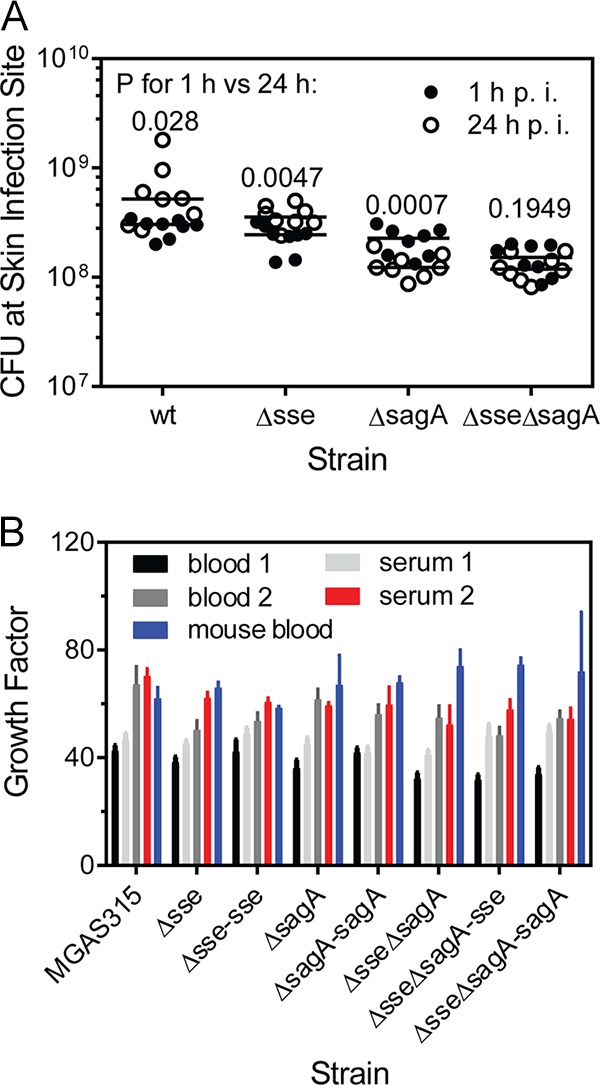
Lack of a detrimental effect of sse and sagA deletion on the clearance of MGAS315 from skin infection sites and from human blood. (A) Persistence of MGAS315 and the Δsse, ΔsagA, and Δsse ΔsagA mutants at skin infection sites. About 108 CFU of bacteria of each strain was subcutaneously inoculated into groups of 16 6-week-old female C57BL/6J mice. Eight mice from each group were euthanized at 1 h and 24 h after inoculation. Presented are the numbers and median values of the numbers of viable GAS at the skin infection sites. The P values are for 1 h versus 24 h by comparison of each strain by the Mann-Whitney t test. p.i., postinoculation. (B) Growth factors for MGAS315 and its derivative strains in nonimmune blood and serum. The strains were inoculated (∼105 CFU) into 0.5 ml of nonimmune blood or serum in triplicate and incubated for 3 h at 37°C with end-to-end rotation. The growth factor was defined as the ratio of the number of viable CFU of GAS in each sample over the number of CFU in the inoculum. P values were determined by 1-way ANOVA for multiple-comparison analyses (P ≥ 0.1184).
Inhibition of neutrophil recruitment inside skin infection sites by MGAS315.
The MPO assay data in Fig. 1 and 2 indicate that there was no robust neutrophil response at MGAS315 infection sites in the skin. The Gram and hematoxylin and eosin (H&E) staining patterns were consistent with this finding. Representative images of Gram- and H&E-stained sections covering the center of the whole subcutaneous infection site at days 1 and 2 after GAS inoculation are shown in Fig. S1 and S2 in the supplemental material, respectively. Bacterial infection sites are referred as the GAS zone, and a thin layer of neutrophils, referred to as the neutrophil zone, was present at the edge of the GAS zone. Higher-magnification images show that the bacterial infection site had very few neutrophils or necrotic material, which is indicated by the boxes with a dashed red outline in Fig. 4 and 5, on both days 1 and 2 after inoculation of MGAS315. There was a thin layer of neutrophils on day 1 and more neutrophils on day 2, which are indicated by the boxes with a dashed yellow outline in Fig. 4 and 5. The neutrophil zone indicated by the boxes with a dashed yellow outline in Fig. 4 and 5 was at the edge of the GAS site, and the boxes with dashed red and yellow outlines did not overlap each other. These results indicate that MGAS315 can inhibit neutrophil recruitment within GAS infection sites.
FIG 4.

Histological analyses of skin infection sites infected with MGAS315 and the Δsse, ΔsagA, and Δsse ΔsagA mutants in mice at day 1 after inoculation. Groups of 10 6-week-old female C57BL/6J mice were subcutaneously inoculated with 1.5 × 108 CFU of MGAS315, 1.8 × 108 CFU of the Δsse mutant, 1.9 × 108 CFU of the ΔsagA mutant, or 2.0 × 108 CFU of the Δsse ΔsagA mutant in 0.2 ml DPBS. Five mice from each group were sacrificed on days 1 and 2 after inoculation to collect the skin infection sites. The skin infection sites were fixed and analyzed with Gram and H&E stains, as described in Materials and Methods. Shown are representative Gram- and H&E-stained images of the regions in the skin infection sites indicated by the boxes in Fig. S1, S3, S5, and S7 in the supplemental material. Bars, 20 μm.
FIG 5.

Histological analyses of skin infection sites infected with MGAS315 and the Δsse, ΔsagA, and Δsse ΔsagA mutants in mice at day 2 after inoculation. The infection conditions are described in the legend to Fig. 4. Shown are representative Gram- and H&E-stained images of the regions in the skin infection site indicated by the boxes in Fig. S2, S4, S6, and S8 in the supplemental material. Bars, 20 μm.
Synergistic effect of sse and sagA deletions on neutrophil recruitment inside GAS infection sites.
The skin infection sites in C57BL/6J mice infected with the Δsse, ΔsagA, and Δsse ΔsagA mutants were analyzed at 24 h (5 mice) and 48 h (5 mice) after inoculation by Gram and H&E staining for subcutaneous infections. A series of sections through each skin infection site was analyzed by Gram and H&E staining, and representative stained images of whole-skin infection sites obtained on days 1 and 2 are presented in Fig. S3 to S8. The staining pattern of the Δsse mutant infection site at day 1 (Fig. S3) was largely similar to that of the MGAS315 infection site (Fig. S1), with neutrophils mainly appearing at the edge of the bacterial infection sites. However, the higher-magnification images show a couple of differences between the MGAS315 and Δsse mutant infection sites. More neutrophils were present at the edge of the Δsse mutant infection site and were engaged with bacteria (Fig. 4, boxes with pink dashed outlines), and a zone of bacteria and necrotic materials with intense staining (Fig. 4, boxes with green dashed outlines) was seen between the GAS zone with many neutrophils (Fig. 4, boxes with pink dashed outlines) and sites with free bacteria and few neutrophils (Fig. 4, boxes with red dashed outlines). While the staining patterns of the MGAS315 infection site on day 2 were very similar to those of the MGAS315 infection site on day 1, the staining patterns for the Δsse mutant infection site on day 2 were totally different from those of the Δsse mutant infection site on day 1 and the MGAS315 infection site on day 2, showing robust neutrophil recruitment throughout the whole infection site (Fig. S4). The higher-magnification images of the Δsse mutant infection site on day 2 show intense staining of bacteria and necrotic neutrophils (Fig. 5, boxes with green dashed outlines) and staining of intact neutrophils and bacteria at the edges of the infection site (Fig. 5, boxes with yellow dashed outlines). Thus, deletion of sse abolishes MGAS315 inhibition of neutrophil recruitment inside the GAS infection site.
The deletion of sagA had a more profound effect on neutrophil recruitment on day 1 than deletion of sse did (Fig. S5). The ΔsagA mutant infection site on day 1 showed intense neutrophil staining at the edge (Fig. 4, boxes with pink dashed outlines), and free bacteria and fewer neutrophils were observed at the center of the ΔsagA mutant infection site, which contained a region with bacteria and necrotic material (Fig. 4, boxes with green dashed outlines) and a region with mainly free bacteria (Fig. 4, boxes with red dashed outlines). On day 2, the ΔsagA mutant infection site showed more intense staining of intact neutrophils (Fig. 5, boxes with pink dashed outlines, and S6) than the Δsse mutant infection site and had necrotic materials only at the center of the infection site (Fig. 5, boxes with green dashed outlines, and S6). Thus, deletion of sagA also abolishes MGAS315 inhibition of neutrophil recruitment inside the GAS infection site.
In contrast to the Δsse and ΔsagA mutant infection sites, in which the center of the sites had sparsely scattered neutrophils at day 1 after inoculation, the Δsse ΔsagA mutant infection site was almost full of neutrophils at day 1 after inoculation (Fig. 4 and S7). The staining patterns of the Δsse ΔsagA mutants on day 2 were similar to those on day 1 (Fig. 5 and S8). These results indicate that the sse and sagA deletions synergistically promote neutrophil recruitment inside GAS infection sites in the skin. Thus, neither Sse nor SLS is sufficient to cause the inhibition of neutrophil infiltration by MGAS315, and both Sse and SLS are required for and synergistically contribute to inhibition of neutrophil recruitment by MGAS315. In addition, on both days the Δsse ΔsagA mutant infection sites did not have many necrotic neutrophils, suggesting that both Sse and SLS can contribute to apoptosis or necrosis of recruited neutrophils.
Complementation of Δsse, ΔsagA, and Δsse ΔsagA mutants.
To confirm that the phenotype of the Δsse, ΔsagA, and Δsse ΔsagA mutants was due to the sse and sagA deletions and not a spurious mutation, the sse gene was put back into the Δsse and Δsse ΔsagA mutants and the sagA gene was put back into the ΔsagA and Δsse ΔsagA mutants, yielding the Δsse-sse, ΔsagA-sagA, Δsse ΔsagA-sse, and Δsse ΔsagA-sagA mutants. Complementation of the sse and sagA deletion mutants restored Sse and SLS production, respectively, according to the PAF acetylhydrolase activity in the culture supernatant of the Δsse-sse and Δsse ΔsagA-sse mutants and the beta-hemolytic activity of the ΔsagA-sagA and Δsse ΔsagA-sagA mutants on blood agar plates (data not shown). The histological patterns of these complemented strains in subcutaneously infected C57BL/6J mice were determined on days 1 and 2 after inoculation. The Gram and H&E staining patterns of the Δsse-sse strain (Fig. S9) and the ΔsagA-sagA strain (Fig. S10) were similar to those of MGAS315 (Fig. S1 and S2), and the staining patterns of the Δsse ΔsagA-sse strain (Fig. S11) and the Δsse ΔsagA-sagA strain (Fig. S12) were similar to those of the ΔsagA mutant (Fig. S5 and S6) and the Δsse mutant (Fig. S3 and S4), respectively. In addition, the levels of the emm3, hasA, spyCEP, and scpA transcripts of the Δsse, ΔsagA, and Δsse ΔsagA mutants at the exponential growth phase in Todd-Hewitt broth supplemented with 0.2% yeast extract (THY) were similar to those of MGAS315 (data not shown). Thus, the phenotype of the Δsse, ΔsagA, and Δsse ΔsagA mutants is caused by the sse and/or sagA deletion.
For a better understanding of the histopathological features of the skin infections in the figures showing the histological findings, images of Gram- and H&E-stained skin of C57BL/6J mice without GAS infection are presented in Fig. S13 for comparison.
DISCUSSION
This study was designed to examine the role of Sse and SLS in the inhibition of neutrophil recruitment by hypervirulent emm3 GAS. The findings are as follows: (i) deletion of sse or sagA enhanced neutrophil recruitment and reduced skin invasion and GAS loads in the liver and spleen of mice subcutaneously infected with MGAS315; (ii) MGAS315 inhibited neutrophil recruitment inside GAS infection sites, and both sse and sagA were required for the inhibition of neutrophil recruitment; and (iii) sse and sagA deletions had synergistic effects on skin invasion, neutrophil recruitment, and systemic infection. These findings indicate that Sse and SLS are required but that either of them is not sufficient for the inhibition of neutrophil recruitment by hypervirulent emm3 GAS and synergistically contribute to GAS skin invasion and systemic GAS infections.
Bakleh et al. have classified necrotizing fasciitis caused by GAS into 3 stages according to its histopathologic features (6). Stage I NF has an intense neutrophilic response and an absence of bacteria according to the findings of Gram staining, stage II NF has a moderate-to-severe neutrophilic response and positive Gram staining results, and stage III NF is characterized by the presence of few or no neutrophils and Gram staining results positive for bacteria. Patients with stage III NF have a higher mortality rate than patients with stage I and II NF. The subcutaneous MGAS315 infection site in mice displayed histopathologic features of stage III NF. Sites of skin MGAS5005 infection in mice where the Gram staining result is positive for GAS also have few neutrophils (7). MGAS5005, an M1T1 isolate, and MGAS315 are both hypervirulent CovS mutants, and the correction of their covS mutation enhances neutrophil recruitment by more than 10-fold (8, 30). Thus, the findings in our current and previous (7) studies indicate that hypervirulent M1T1 and M3 GAS CovRS mutants can cause the histopathologic pattern of stage III NF in skin infections by inhibiting neutrophil recruitment.
The enhancement of neutrophil recruitment by deletion of sse in MGAS315 confirms our previous findings on the function of Sse in inhibition of neutrophil responses. The deletion of sse in M1T1 strain MGAS5005 enhances neutrophil recruitment (7). Sse is a potent PAF acetylhydrolase, and the function of Sse is mediated by the targeting of PAF (7, 31). Passive immunization with an enzymatic activity-neutralizing Sse monoclonal antibody enhances neutrophil recruitment (32). Sse, but not the CXC chemokine peptidase SpyCEP or C5a peptidase ScpA, is critical for MGAS5005 inhibition of neutrophil recruitment in skin infections in mice (8). Our results indicate that Sse also plays a critical role in the inhibition of neutrophil recruitment by hypervirulent emm3 GAS.
SLS is another factor critical for inhibition of the neutrophil responses induced by MGAS315 in skin infections in mice. SLS is a cytolytic toxin, and its peptide component is encoded by sagA (33). GAS sagA deletion mutants are attenuated in virulence and skin invasion (34). SLS-negative mutants are associated with the robust recruitment of neutrophils and a significantly reduced incidence of lethal myositis in zebrafish, and the extravasation of neutrophils in the early stage of subcutaneous infection is quicker in mice infected with SLS-negative mutant GAS than in mice infected with wild-type GAS (28). In this study, we showed that SLS is required for the inhibition of neutrophil recruitment by hypervirulent M3 GAS. Thus, both sse and sagA are essential but not sufficient for the inhibition of neutrophil recruitment by hypervirulent M3 GAS CovS mutants in subcutaneous infections in mice.
The essential but insufficient role of each of Sse and SLS in MGAS315 inhibition of neutrophil recruitment implies that Sse and SLS evade neutrophil responses through different mechanisms. The synergistic effects of sse and sagA deletions on neutrophil recruitment, in comparison with that of single sse or sagA deletion, support this implication. Sse has potent PAF acetylhydrolase activity, and neutrophil recruitment is lower in PAF receptor knockout mice infected with MGAS5005 Δsse than in wild-type control mice infected with the same strain (7, 31). PAF is an important lipid mediator in inflammation and a chemoattractant for neutrophils (35, 36). Deletion of sse leads to a reduction of the capacity of MGAS315 to inhibit neutrophil recruitment inside GAS infection sites, suggesting that PAF may play a critical role in neutrophil recruitment in the presence of SLS. Whether PAF mainly functions as a chemoattractant for neutrophils in GAS infections is currently being investigated in our laboratory. The difference in the transepithelial migration of neutrophils between HaCaT human keratinocyte cells treated with wild-type GAS and HaCaT cells treated with an SLS-negative GAS mutant suggests that SLS inhibits host cell production of signals chemotactic for neutrophils (28). SLS can promote the programmed cell death of epithelial keratinocytes during GAS infection (37). In MGAS315 Δsse infections, necrotic cells were present at the edge of the bacterial infection sites on day 1 and inside the GAS infection sites on day 2 after inoculation, suggesting that SLS may kill neutrophils when they encounter GAS bacteria, affect neutrophil recruitment at the early stage of infection, and result in a histological pattern of Δsse mutant infection at day 1 after inoculation.
The deletion of both sse and sagA has a synergistic effect on GAS loads in the liver and spleen in subcutaneously infected mice. The sse and sagA deletion mutants were not efficiently cleared from the skin infection sites, and the growth factors for the mutants in nonimmune human blood and serum were similar to those for the parent strain. These observations suggest that Sse and SLS synergistically contribute to GAS dissemination and systemic infections by hypervirulent GAS.
MATERIALS AND METHODS
Declaration of ethical approval.
All animal procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (38). The protocols for the experiments were approved by the Institutional Animal Care and Use Committee at Montana State University (permit number 2014-45). Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at MSU (protocol no. BL120513). Written informed consent was provided by the study participants.
Bacterial strains and growth.
Genotype emm3 strain MGAS315 has been described previously (29), and this hypervirulent clinical isolate has the function-losing CovS G457V mutation (30). MGAS315 and its derivative strains were grown in Todd-Hewitt broth supplemented with 0.2% yeast extract (THY).
Generation of MGAS315 Δsse, ΔsagA, and Δsse ΔsagA mutants.
MGAS315 Δsse was generated using the suicide plasmid pGRV-Δsse (16), as described previously (16). The mutant lacked a 621-bp fragment that encodes amino acids 55 to 261 of Sse and had no foreign DNA, including no antibiotic selection marker.
In the deletion of the sagA gene, a DNA fragment containing the whole sagA gene and its 23-bp upstream and 164-bp downstream regions was deleted, whereas the downstream sagB gene was intact. The 5′and 3′ ∼1,000-bp flanking fragments of the sagA fragment to be deleted were amplified from MGAS315 chromosomal DNA using high-fidelity Phusion DNA polymerase and primer pairs 5′-AGATCTGCAAACGTGTTCAATTGGTTG-3′/5′-CTCGAGTAACTGATAAGAACACGAG-3′ and 5′-TTCTCGAGCTAGATAGTACCTGCTAATTAC-3′/5′-TTGGATCCGAAATGTCACGACACGAAC-3′. The PCR products of the upstream and downstream flanking fragments were sequentially cloned into pGRV (39) at the BglII/XhoI and XhoI/BamHI sites, respectively, yielding the suicide plasmid pGRV-ΔsagA. The plasmid was introduced into MGAS315 using electroporation and inserted into the sagA locus of the MGAS315 chromosome through a homologous recombination event at one flanking fragment. Bacteria with this first crossover were selected on THY agar plates with 10 mg/liter chloramphenicol. One strain with the first crossover was grown on a THY agar plate. At every 12 h after plating, bacteria were recovered from a whole plate, resuspended in 10 ml THY, and vortexed for 2 min, and 10 μl of the vortexed GAS suspension was streaked onto a THY agar plate. This procedure was regarded as one passage. Deletion mutants were identified after >12 passages. Bacteria were plated on THY agar plates after the last passage, and the resultant colonies were spotted in parallel on THY agar with and without chloramphenicol to identify potential double-crossover strains that were chloramphenicol sensitive. Chloramphenicol-sensitive strains were analyzed to identify deletion mutants by PCR using primers 5′-GTGATAAGAACTAGATAGTTG-3′ and 5′-GGCATTAAATGTTGAGCAAC-3′. The sagA gene in MGAS315 Δsse was similarly deleted to yield Δsse ΔsagA mutants.
The sse and sagA deletions in the mutants were confirmed by DNA sequencing of the sse and sagA loci and by the loss of Sse activity in the culture supernatant of Δsse mutants using the PAF acetylhydrolase activity assay (31) and by the loss of beta-hemolytic activity of ΔsagA mutants on blood agar plates.
Construction of complemented strains.
The sse gene was put back into the Δsse and Δsse ΔsagA mutants using pGRV-sse as previously described (16), yielding Δsse-sse and Δsse ΔsagA-sse mutants complemented with sse. For complementation of the ΔsagA and Δsse ΔsagA mutants with sagA, a fragment containing the full-length sagA gene and its flanking regions was amplified by PCR using a Phusion High Fidelity PCR kit from New England BioLabs, MGAS315 genomic DNA, and primers 5′-TTGGATCCGCAAACGTGTTCAATTGGTTG-3′ and 5′-TTGGATCCGAAATGTCACGACACGAAC-3′. The PCR product was cloned into pGRV at the BamHI site, yielding suicide plasmid pGRV-sagA. This plasmid was introduced into the ΔsagA and Δsse ΔsagA mutants by electroporation. The procedures described above to generate the ΔsagA mutant were followed to generate complemented ΔsagA-sagA and Δsse ΔsagA-sagA strains. These complemented strains were confirmed by sequencing of the sse or sagA locus. The production of Sse was restored in the Δsse-sse and Δsse ΔsagA-sse strains, which were assayed for Sse enzymatic activity in their culture supernatants using the Sse PAF acetylhydrolase activity assay (31), and SLS production was restored in the ΔsagA-sagA and Δsse ΔsagA-sagA strains, as the strains displayed beta-hemolytic activity on blood agar plates.
Subcutaneous mouse infections.
Six-week-old female CD-1 mice and female and male C57BL/6J mice were subcutaneously infected with MGAS315, the Δsse, ΔsagA, and Δsse ΔsagA mutants, and the complemented strains of the mutants. CD-1 mice were purchased from Charles River Laboratories, and C57BL/6J mice were bred at the Animal Resources Center at Montana State University using breeding mice from The Jackson Laboratory. GAS bacteria grown in THY were harvested at the exponential growth phase, washed with pyrogen-free Dulbecco's phosphate-buffered saline (DPBS) three times, and resuspended in DPBS. Groups of mice were subcutaneously inoculated with 0.2 ml of GAS in DPBS at an optical density at 600 nm of 1.0, and the actual inoculum size was determined by plating and usually contained about 108 CFU. Mice were euthanized at the times after inoculation indicated above and below to collect skin samples for measurement of the lesion size, the numbers of viable bacteria, and neutrophil recruitment and histological analyses, and the liver and spleen were also harvested to measure the numbers of viable GAS. The mice were euthanized by a gradual fill method at a rate of CO2 displacement of 30% of the chamber volume per minute, as recommended in the 2013 American Veterinary Medical Association guidelines.
Measurement of skin lesion size, neutrophil recruitment, and numbers of viable GAS in tissues.
The skin around the infection site was peeled off, and the skin lesion was recognized by the boundary of the inflammation area. The size of the skin lesions was measured by analyzing the lesion pictures using the area measurement tool of the Adobe Acrobat 9 software program. The skin containing the infection area was excised for neutrophil measurement. The numbers of recruited neutrophils in the infected skin samples were determined by a myeloperoxidase assay, as described previously (7). The numbers of viable GAS in the skin infection sites, liver, and spleen were determined by homogenizing the tissues in DPBS using Kontes pestles and then plating at appropriate dilutions.
Histological analyses.
GAS-infected skin samples collected at 24 h and 48 h after inoculation were fixed in 10% neutral buffered formalin for 24 h. The samples were dehydrated with ethanol, cleared with xylene, and infiltrated with paraffin using a tissue-embedding console system (Sakura Finetek, Inc.). The paraffin blocks were processed to obtain 4-μm sections, which were stained with hematoxylin and eosin (H&E) or with Gram stain from a kit from Becton, Dickinson and Company. Stained slides were examined using a Nikon Eclipse 80i microscope.
GAS growth in nonimmune human blood and pooled mouse blood.
GAS growth in nonimmune blood and serum from two persons and pooled blood from female C57BL/6J mice was determined as previously described (7). Bacteria of MGAS315, the Δsse, ΔsagA, and Δsse ΔsagA mutants, and the complemented mutant strains were harvested at the exponential growth phase in THY, washed three times with DPBS, and inoculated in triplicate at ∼105 CFU/ml into 0.5 ml of heparinized nonimmune human blood or serum and into 0.2 ml of pooled heparinized mouse blood. The samples were rotated end to end for 3 h at 37°C, and the numbers of viable GAS in the samples and inocula were determined by plating. The growth factor was defined as the ratio of the number of viable CFU in each sample after a 3-h incubation over the number of CFU of the corresponding inoculum.
Other analyses.
DNA sequencing of the amplified PCR products was performed by using a BigDye Terminator (v3.1) cycle sequencing kit and an Applied Biosystems 3130 genetic analyzer. Sequence data were analyzed by using Sequencer (v5.1) software (Gene Codes Corporation). The PAF acetylhydrolase activity in the culture supernatant of MGAS315 and its isogenic mutants was assayed using the 2-thio PAF-based colorimetric assay as described previously (31).
Statistical analyses.
Statistical analyses were done using GraphPad Prism software (version 7.03). The data on the lesion size, neutrophil recruitment, and GAS loads in infected mice were analyzed by the Mann-Whitney t test, and the growth factors in human blood and serum and mouse blood were analyzed by multiple-comparison analyses by 1-way ANOVA.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grants AI095704, AI097703, and GM110732 from the National Institutes of Health, Montana University System Research Initiative 51040-MUSRI2015-03, and the Montana State Agricultural Experimental Station.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00530-17.
REFERENCES
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Nelson GE, Pondo T, Toews KA, Farley MM, Lindegren ML, Lynfield R, Aragon D, Zansky SM, Watt JP, Cieslak PR, Angeles K, Harrison LH, Petit S, Beall B, Van Beneden CA. 2016. Epidemiology of invasive group A streptococcal infections in the United States, 2005-2012. Clin Infect Dis 63:478–486. doi: 10.1093/cid/ciw248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharkawy A, Low DE, Saginur R, Gregson D, Schwartz B, Jessamine P, Green K, McGeer A, Ontario Group A Streptococcal Study Group . 2002. Severe group A streptococcal soft-tissue infections in Ontario: 1992-1996. Clin Infect Dis 34:454–460. doi: 10.1086/338466. [DOI] [PubMed] [Google Scholar]
- 4.Olsen RJ, Musser JM. 2010. Molecular pathogenesis of necrotizing fasciitis. Annu Rev Pathol 5:1–31. doi: 10.1146/annurev-pathol-121808-102135. [DOI] [PubMed] [Google Scholar]
- 5.Dahl PR, Perniciaro C, Holmkvist KA, O'Connor MI, Gibson LE. 2002. Fulminant group A streptococcal necrotizing fasciitis: clinical and pathologic findings in 7 patients. J Am Acad Dermatol 47:489–492. doi: 10.1067/mjd.2002.120536. [DOI] [PubMed] [Google Scholar]
- 6.Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. 2005. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis 40:410–414. doi: 10.1086/427286. [DOI] [PubMed] [Google Scholar]
- 7.Liu M, Zhu H, Li J, Garcia CC, Feng W, Kirpotina LN, Hilmer J, Tavares LP, Layton AW, Quinn MT, Bothner B, Teixeira MM, Lei B. 2012. Group A Streptococcus secreted esterase hydrolyzes platelet-activating factor to impede neutrophil recruitment and facilitate innate immune evasion. PLoS Pathog 8:e1002624. doi: 10.1371/journal.ppat.1002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Zhu H, Feng W, Liu M, Song Y, Zhang X, Zhou Y, Bei W, Lei B. 2013. Regulation of inhibition of neutrophil infiltration by the two-component regulatory system CovRS in subcutaneous murine infection with group A Streptococcus. Infect Immun 81:974–983. doi: 10.1128/IAI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyoshi-Akiyama T, Ikebe T, Watanabe H, Uchiyama T, Kirikae T, Kawamura Y. 2006. Use of DNA arrays to identify a mutation in the negative regulator, csrR, responsible for the high virulence of a naturally occurring type M3 group A streptococcus clinical isolate. J Infect Dis 193:1677–1684. doi: 10.1086/504263. [DOI] [PubMed] [Google Scholar]
- 10.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30:209–219. doi: 10.1046/j.1365-2958.1998.01057.x. [DOI] [PubMed] [Google Scholar]
- 12.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun 67:5298–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Federle MJ, McIver KS, Scott JR. 1999. A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J Bacteriol 181:3649–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA III, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun 77:3141–3149. doi: 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards RJ, Taylor GW, Ferguson M, Murray S, Rendell N, Wrigley A, Bai Z, Boyle J, Finney SJ, Jones A, Russell HH, Turner C, Cohen J, Faulkner L, Sriskandan S. 2005. Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J Infect Dis 192:783–790. doi: 10.1086/432485. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H, Liu M, Sumby P, Lei B. 2009. The secreted esterase of group A streptococcus is important for invasive skin infection and dissemination in mice. Infect Immun 77:5225–5232. doi: 10.1128/IAI.00636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lei B, DeLeo FR, Hoe NP, Graham MR, Mackie SM, Cole RL, Liu M, Hill HR, Low DE, Federle MJ, Scott JR, Musser JM. 2001. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nat Med 7:1298–1305. doi: 10.1038/nm1201-1298. [DOI] [PubMed] [Google Scholar]
- 18.Bricker AL, Carey VJ, Wessels MR. 2005. Role of NADase in virulence in experimental invasive group A streptococcal infection. Infect Immun 73:6562–6566. doi: 10.1128/IAI.73.10.6562-6566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Seaghdha M, Wessels MR. 2013. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from xenophagic killing. PLoS Pathog 9:e1003394. doi: 10.1371/journal.ppat.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 21.Kansal RG, Datta V, Aziz RK, Abdeltawab NF, Rowe S, Kotb M. 2010. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis 201:855–865. doi: 10.1086/651019. [DOI] [PubMed] [Google Scholar]
- 22.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. 2001. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis 183:1043–1054. doi: 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 23.Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1:e00191-10. doi: 10.1128/mBio.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G, Feng W, Li D, Liu M, Nelson DC, Lei B. 2015. The Mga regulon but not deoxyribonuclease Sda1 of invasive M1T1 group A Streptococcus contributes to in vivo selection of CovRS mutations and resistance to innate immune killing mechanisms. Infect Immun 83:4293–4303. doi: 10.1128/IAI.00857-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng W, Liu M, Chen DG, Yiu R, Fang FC, Lei B. 2016. Contemporary pharyngeal and invasive emm1 and invasive emm12 group A Streptococcus isolates exhibit similar in vivo selection for CovRS mutants in mice. PLoS One 11:e0162742. doi: 10.1371/journal.pone.0162742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng W, Minor D, Liu M, Li J, Ishaq SL, Yeoman C, Lei B. 2017. Null mutations of group A streptococcus orphan kinase RocA: selection in mouse infection and comparison with CovS mutations in alteration of in vitro and in vivo protease SpeB expression and virulence. Infect Immun 85:e00790-16. doi: 10.1128/IAI.00790-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Liu G, Feng W, Zhou Y, Liu M, Wiley JA, Lei B. 2014. Neutrophils select hypervirulent CovRS mutants of M1T1 group A Streptococcus during subcutaneous infection of mice. Infect Immun 82:1579–1590. doi: 10.1128/IAI.01458-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin A, Loughman JA, Zinselmeyer BH, Miller MJ, Caparon MG. 2009. Streptolysin S inhibits neutrophil recruitment during the early stages of Streptococcus pyogenes infection. Infect Immun 77:5190–5201. doi: 10.1128/IAI.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. 2002. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci U S A 99:10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stetzner ZW, Li D, Feng W, Liu M, Liu G, Wiley J, Lei B. 2015. Serotype M3 and M28 group A streptococci have distinct capacities to evade neutrophil and TNF-α responses and to invade soft tissues. PLoS One 10:e0129417. doi: 10.1371/journal.pone.0129417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu G, Liu M, Xie G, Lei B. 2013. Characterization of streptococcal platelet-activating factor acetylhydrolase variants that are involved in innate immune evasion. Infect Immun 81:3128–3138. doi: 10.1128/IAI.00398-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Feng W, Zhu H, Lei B. 2015. A neutralizing monoclonal IgG1 antibody of platelet-activating factor acetylhydrolase SsE protects mice against lethal subcutaneous group A Streptococcus infection. Infect Immun 83:2796–2805. doi: 10.1128/IAI.00073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nizet V, Beall B, Bast DJ, Datta V, Kilburn L, Low DE, De Azavedo JC. 2000. Genetic locus for streptolysin S production by group A streptococcus. Infect Immun 68:4245–4254. doi: 10.1128/IAI.68.7.4245-4254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, Kotb M, Nizet V. 2005. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol Microbiol 56:681–695. doi: 10.1111/j.1365-2958.2005.04583.x. [DOI] [PubMed] [Google Scholar]
- 35.Venable ME, Zimmerman GA, McIntyre TM, Prescott SM. 1993. Platelet-activating factor: a phospholipid autacoid with diverse actions. J Lipid Res 34:691–702. [PubMed] [Google Scholar]
- 36.Shaw JO, Pinckard RN, Ferrigni KS, McManus LM, Hanahan DJ. 1981. Activation of human neutrophils with 1-O-hexadecyl/octadecyl-2-acetyl-sn-glycerol-3-phosphorylcholine (platelet activating factor). J Immunol 127:1250–1255. [PubMed] [Google Scholar]
- 37.Flaherty RA, Puricelli JM, Higashi DL, Park CJ, Lee SW. 2015. Streptolysin S promotes programmed cell death and enhances inflammatory signaling in epithelial keratinocytes during group A Streptococcus infection. Infect Immun 83:4118–4133. doi: 10.1128/IAI.00611-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 39.Liu M, Hanks TS, Zhang J, McClure MJ, Siemsen DW, Elser JL, Quinn MT, Lei B. 2006. Defects in ex vivo and in vivo growth and sensitivity to osmotic stress of group A Streptococcus caused by interruption of response regulator gene vicR. Microbiology 152:967–978. doi: 10.1099/mic.0.28706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.