

ABSTRACT
Small-colony variants (SCVs) of Staphylococcus aureus typically lack a functional electron transport chain and cannot produce virulence factors such as leukocidins, hemolysins, or the antioxidant staphyloxanthin. Despite this, SCVs are associated with persistent infections of the bloodstream, bones, and prosthetic devices. The survival of SCVs in the host has been ascribed to intracellular residency, biofilm formation, and resistance to antibiotics. However, the ability of SCVs to resist host defenses is largely uncharacterized. To address this, we measured the survival of wild-type and SCV S. aureus in whole human blood, which contains high numbers of neutrophils, the key defense against staphylococcal infection. Despite the loss of leukocidin production and staphyloxanthin biosynthesis, SCVs defective for heme or menaquinone biosynthesis were significantly more resistant to the oxidative burst than wild-type bacteria in human blood or the presence of purified neutrophils. Supplementation of the culture medium of the heme-auxotrophic SCV with heme, but not iron, restored growth, hemolysin and staphyloxanthin production, and sensitivity to the oxidative burst. Since Enterococcus faecalis is a natural heme auxotroph and cause of bloodstream infection, we explored whether restoration of the electron transport chain in this organism also affected survival in blood. Incubation of E. faecalis with heme increased growth and restored catalase activity but resulted in decreased survival in human blood via increased sensitivity to the oxidative burst. Therefore, the lack of functional electron transport chains in SCV S. aureus and wild-type E. faecalis results in reduced growth rate but provides resistance to a key immune defense mechanism.
KEYWORDS: small-colony variant, Staphylococcus aureus, Enterococcus faecalis, neutrophil, oxidative burst, bacteremia, enterococcus
INTRODUCTION
Staphylococcus aureus is responsible for a multitude of different infections of humans and animals (1–3). The key host defense against infection is the neutrophil, which phagocytoses S. aureus and exposes it to a cocktail of reactive oxygen species (ROS) during a process known as the oxidative (or respiratory) burst (4–6). While this is often sufficient to clear infection, invasive staphylococcal diseases frequently lead to persistent or recurrent infections of the bones, joints, heart, or implanted devices (1, 7–9). The development of these hard-to-treat infections is often associated with the presence of small-colony variants (SCVs) (10–17). As the name suggests, SCVs form small colonies on agar plates, typically due to metabolic defects caused by mutations that abrogate the electron transport chain or biosynthetic pathways (16–21). For example, several clinical studies have isolated SCVs with mutations in genes required for heme or menaquinone biosynthesis, including from the bloodstream (17–20). The slow growth of SCVs provides a strong selection pressure for reversion to the wild type, either by repair of the causative mutation or acquisition of a suppressor mutation (18, 19, 22). This presents challenges to their study, and so targeted deletion of genes within the hem or men operons, which confer a phenotype that is identical to that of clinical SCVs, has been used to enable their study without the problem of reversion to the wild type (23–26). SCVs can also arise in the absence of mutation, resulting in a very unstable phenotype, although the molecular basis for this is unknown (27). The emergence of SCVs is a rare but consistent consequence of S. aureus replication, which generates a small subpopulation of the variants (22). However, SCV emergence is significantly increased in response to diverse environmental stresses, including antibiotics, reactive oxygen species, low pH within host cell vacuoles, and exoproducts from Pseudomonas, which frequently causes coinfections with S. aureus (26–33).
Despite their diverse molecular bases, most SCVs have similar phenotypic characteristics. For example, activity of the Agr quorum-sensing system is weak or absent, and therefore cytolytic toxin production is negligible while surface proteins are strongly expressed (25, 34–36). These properties enable SCVs to persist in nonimmune host cells and form robust biofilms, which has been hypothesized to contribute to their ability to persist in host tissues (27, 37–39). Furthermore, SCVs are typically resistant to antibiotics, including the aminoglycosides, sulfonamides, and fusidic acid, and are often less susceptible to other antibiotics than wild-type bacteria (40–44).
While these phenotypic properties very likely contribute to staphylococcal persistence in the host, the ability of SCVs to resist phagocytic cells, the key host defense against S. aureus, is poorly understood. Respiration-defective SCVs are resistant to the ROS H2O2, and suppression of respiration by the Pseudomonas exoproduct HQNO confers ROS resistance upon wild-type bacteria (26). However, SCV S. aureus lacks several defenses used by wild-type bacteria to protect against immune cells (26). For example, staphyloxanthin pigment, which promotes wild-type survival of both the oxidative burst and antimicrobial peptides (AMPs), is absent in SCVs (15, 18, 45–47). Furthermore, wild-type bacteria secrete numerous cytolytic toxins that kill neutrophils and enable bacterial survival, but this is absent in SCVs (15, 18, 25, 34). SCVs also exhibit reduced coagulase activity and some isolates lack catalase, both of which have been linked to survival of wild-type bacteria in the host (15, 18, 26, 48–50). Therefore, the effect of a defective electron transport chain on the susceptibility of SCV S. aureus to the oxidative burst of neutrophils is unclear.
Enterococcus faecalis, another major cause of bloodstream infections, shares some of the phenotypic properties of S. aureus SCVs, since it is naturally defective for heme production and therefore lacks a functional electron transport chain (51–53). However, E. faecalis encodes type a and b cytochromes, and the presence of exogenous heme promotes E. faecalis growth in air, confirming the presence of an otherwise intact respiratory chain (51–53). Exogenous heme also restores catalase activity, which has been shown to promote H2O2 resistance (54, 55). As such, it is unclear whether E. faecalis gains an advantage from being defective for heme biosynthesis, particularly with respect to host defenses that generate reactive oxygen species such as neutrophils.
Therefore, the aim of this work was to determine how the absence of the electron transport chain affects the survival of S. aureus and E. faecalis exposed to the oxidative burst of neutrophils.
RESULTS
The loss of the electron transport chain promotes survival of S. aureus in human blood.
To study the susceptibility of electron transport chain-deficient SCVs to the oxidative burst, we employed the well-established ex vivo whole human blood model of infection. This model is appropriate because S. aureus is a major cause of bacteremia and blood contains a high density of neutrophils, as well as the required opsonins and other relevant immune factors such as platelets (4, 56, 57). In this model system, S. aureus is rapidly phagocytosed by neutrophils and exposed to the oxidative burst (4, 56, 57).
Freshly drawn human blood containing anticoagulant (EDTA) was incubated with wild-type S. aureus USA300, or mutants with deletions of hemB or menD, and survival was determined over time by CFU counts. Preliminary experiments determined that individual donors had slightly different antistaphylococcal activities, and so at least three different donors were used for each experiment (Fig. 1A). However, for each of the five donors, we observed a consistent decrease in CFU counts of wild-type bacteria over time, with just 1 to 5% of the inoculum surviving after 6 h (Fig. 1A). In contrast, SCVs defective for heme or menaquinone biosynthesis survived at much higher levels than the wild type over the entire duration of the assay, with 70% of the ΔhemB mutant inoculum and 69% of the ΔmenD mutant inoculum viable after 6 h of incubation in blood (Fig. 1B and C). To ensure that the presence of EDTA did not affect bacterial viability, each of the strains described above was incubated in phosphate-buffered saline (PBS) containing an identical concentration of the cation chelator for 6 h, and viability was determined. In each case, bacterial viability was unchanged by the presence of EDTA (data not shown).
FIG 1.

Survival of SCV S. aureus in blood is greater than that of wild-type (WT) bacteria. (A) Survival of wild-type S. aureus USA300 in blood from individual donors. Data are the mean survival from three independent experiments from each donor. (B and C) Survival of wild-type S. aureus USA300 and ΔhemB (B) and ΔmenD (C) mutants, and complemented strains, in human blood. Data are the mean of results of four independent experiments using blood from at least three different donors. (D) Images of pelleted stationary-phase S. aureus strains highlighting differences in pigmentation. Images are representative of three independent assays. (E and F) Growth profiles of S. aureus wild-type and ΔhemB (E) and ΔmenD (F) mutants and complemented strains. Data are the mean of results of three independent experiments. Where shown, error bars represent the standard deviations of the mean. Data in panels B and C were analyzed by a two-way repeated-measures analysis of variance (ANOVA) and Sidak's post hoc test. *, P < 0.01, compared with the wild type. Since data points overlap in panels E and F, error bars were omitted for clarity, but standard deviations were within 5% of the mean.
Complementation of the hemB or menD mutations conferring the SCV phenotype restored the wild-type phenotype for growth and staphyloxanthin production and resulted in significantly decreased survival in blood (Fig. 1B to F). This confirmed that enhanced SCV survival in blood was due to the loss of heme or menaquinone biosynthesis, rather than the acquisition of adventitious mutations during genetic manipulation. Therefore, despite the lack of staphyloxanthin pigment and cytolysin production, loss of the electron transport chain confers a survival advantage to S. aureus in blood.
Wild-type S. aureus is more sensitive to the oxidative burst than SCVs.
Having demonstrated that survival of SCVs in blood is greater than that of the wild type, we sought to understand why. First, to confirm that the survival of SCVs in whole blood was due to resistance to killing by neutrophils, each of the staphylococcal strains was incubated with polymorphonuclear leukocytes (PMNs) purified from blood. As for whole human blood, the survival of wild-type S. aureus (9%) was lower than that of the ΔhemB mutant (36%) and the ΔmenD mutant (46%) after 3 h of incubation (Fig. 2A). Assays could not be extended beyond this point due to extensive formation of neutrophil extracellular traps that made accurate CFU determination difficult.
FIG 2.

SCVs survive the oxidative burst better than wild-type S. aureus. (A) Survival of wild-type S. aureus USA300 (WT) and ΔhemB and ΔmenD mutants in the presence of PMNs purified from human blood. (B) Percentages of S. aureus USA300 wild-type, ΔhemB, and ΔmenD bacteria internalized into phagocytic cells 2 h after inoculation into whole human blood. (C) Percentages of phagocytic cells that contained S. aureus strains, and had impaired membrane integrity, as determined using the Zombie Violet reagent after 6 h in whole human blood. (D) Survival of S. aureus USA300 wild-type, ΔhemB, and ΔmenD bacteria after 6 h in blood pretreated with the NADPH oxidase inhibitor diphenyleneiodonium (DPI) or an identical volume of DMSO solvent alone (DMSO). In all cases, data are the mean of results of four independent experiments using blood from at least three different donors. Data in panel A were analyzed by a two-way repeated-measures ANOVA and Sidak's post hoc test. *, P < 0.01, compared with the wild type. For panels B to D, data were analyzed via a one-way ANOVA with Tukey's post hoc test. This revealed no significant differences between values in panels B and C. In panel D, an asterisk indicates a P of <0.01 and NS (nonsignificant) indicates a P of >0.05 when the indicated comparisons were made.
Although S. aureus encodes several immune evasins, several previous studies have shown rapid phagocytic uptake of the bacterium by PMNs (4, 56, 57). We confirmed those findings and found no differences in the phagocytosis of the wild type and ΔhemB or ΔmenD mutants in whole blood (Fig. 2B). We also demonstrated that the viability of neutrophils that phagocytosed S. aureus did not differ between the wild type and SCVs (Fig. 2C). Therefore, both immune evasion and killing of immune cells by SCVs were ruled out as an explanation for their ability to survive in human blood.
The principle mechanism by which neutrophils kill S. aureus is the oxidative burst (4–6). To confirm that this was the case in our model system, we measured bacterial viability in human blood treated with diphenyleneiodonium (DPI), which blocks the oxidative burst, or the dimethyl sulfoxide (DMSO) solvent alone. Suppression of NADPH with DPI, but not DMSO alone, resulted in significantly elevated survival of wild-type S. aureus, confirming that the oxidative burst is the key defense against S. aureus in human blood (Fig. 2D) (4–6). The addition of DPI to blood did not significantly alter SCV CFU counts, since survival was already very high (Fig. 2C). Therefore, SCV S. aureus bacteria appear to be significantly less susceptible to the oxidative burst than wild-type bacteria. This is in agreement with our previously reported finding that both the ΔhemB and ΔmenD SCVs were more resistant to H2O2 than wild-type bacteria and provides an explanation for the increased survival of SCVs in blood (26).
Agr activity promotes the survival of wild-type but not SCV S. aureus in blood.
Although Agr-regulated toxins have been shown to kill neutrophils, several clinical studies have shown an association of Agr dysfunction with persistent bacteremia (58). Therefore, we considered the possibility that the weak Agr activity of SCVs contributed to their survival in blood.
To test this, we compared the survival of wild-type and Agr-defective strains in whole human blood. Previous work has shown that these USA300 ΔagrA and ΔagrC mutants are completely defective for hemolysin production, while the ΔRNAIII mutant retains a low level of hemolytic activity due to the production of phenol-soluble modulins (PSMs) (59, 60). Incubation of agr mutants in blood revealed a significantly greater loss of viability of Agr-defective strains than of the wild type (Fig. 3A). In particular, mutants lacking quorum-sensing components of Agr (ΔagrA or ΔagrC) were approximately 4-fold more susceptible to immune cells in blood than the wild type, while the RNAIII mutant was 2-fold more susceptible than the wild type (Fig. 3A). This finding is in keeping with previous work that showed that AgrA-regulated PSMs contribute to the survival of S. aureus within the phagocytic vacuole of neutrophils, in addition to RNAIII-regulated toxins (59).
FIG 3.

Survival of wild-type but not SCV S. aureus is enhanced by Agr. (A) Survival of S. aureus USA300 wild-type (WT), ΔagrA, ΔagrC, and ΔRNAIII strains in whole human blood over 6 h. (B) Survival of S. aureus USA300 ΔagrC transformed with pCL55 (CTL), pCL55 containing the wild-type agrC gene (WT), and three mutated variants of agrC that result in Q305H, M234L, or R238H substitutions conferring a constitutively active phenotype. (C) Survival of S. aureus USA300 wild-type (WT), ΔhemB, ΔhemB ΔagrA, ΔhemB ΔagrC, and ΔhemB ΔRNAIII strains in whole human blood over 6 h. For all panels, data are the mean of results of four independent experiments using blood from at least three different donors. Data were analyzed by a two-way repeated-measures ANOVA with Dunnett's post hoc test to compare strains to the WT (A), CTL (B), or the ΔhemB mutant (C). *, P < 0.01. In panel A, all mutants were significantly more susceptible to immune defenses than the wild type at 4 and 6 h. In panel B, all strains expressing agrC (wild type or mutated) survived better than the ΔagrC mutant at the 4- and 6-h time points. In panel C, all ΔhemB mutants (with or without agr) survived equally well and significantly better than the wild type.
Complementation of the ΔagrC mutant with a wild-type copy of the gene increased survival in blood (Fig. 3B). However, complementation of ΔagrC with mutant copies of agrC which confer constitutive Agr activity, even in the presence of serum (61), did not promote bacterial survival above that of the wild-type gene (Fig. 3B).
Although Agr activity is extremely weak in SCVs, we explored whether this contributed to their survival by generating ΔhemB mutants defective for agrA, agrC, or RNAIII and measuring their survival in blood (Fig. 3C). This revealed that survival of each of the ΔhemB Δagr mutants was as high as that for the ΔhemB mutant with an intact agr operon. Therefore, while loss of Agr activity in the wild type reduces survival in human blood, the lack of Agr activity in SCVs is not detrimental for their survival. This indicates that toxin production is an important mechanism by which wild-type S. aureus survives phagocytosis. In contrast, since SCVs can survive the oxidative burst, they do not need toxins to survive phagocytosis.
Restoration of the electron transport chain with heme results in decreased survival of SCVs in blood.
During infection, S. aureus acquires iron from the host, predominantly via the acquisition of heme liberated from erythrocytes via hemolytic toxins (62). In addition to acting as an iron source, heme can also be utilized by heme-auxotrophic SCVs to restore the electron transport chain (18, 26). To determine how heme influenced the phenotype of heme- and menquinone-defective SCVs, and their susceptibility to the oxidative burst, we grew wild-type or SCV S. aureus in medium deficient for heme and containing minimal free iron (1 μM FeCl3) or abundant iron (10 μM FeCl3) or in medium in the presence of heme (10 μM).
The growth rate of wild-type S. aureus was not significantly affected by the presence of the higher concentration of FeCl3 or heme, although the latter led to a slight increase in the length of the lag phase (Fig. 4A). Similarly, abundant iron did not affect the growth of the ΔmenD SCV, but heme caused slight growth retardation (Fig. 4A). In contrast, abundant iron slightly promoted the growth rate of the ΔhemB SCV, while heme enhanced the growth to almost wild-type levels (Fig. 4A). In addition to the growth rate, heme supplementation restored hemolytic activity and pigmentation to the ΔhemB mutant (Fig. 4B). However, heme supplementation of the ΔhemB mutant also resulted in significantly increased susceptibility to the oxidative burst of neutrophils in blood (Fig. 4C), which is in keeping with our previous finding that heme supplementation renders heme-auxotrophic SCVs sensitive to H2O2 (26). In contrast, supplementation of the medium with iron had no effect on susceptibility of the ΔhemB mutant to the oxidative burst or H2O2 (Fig. 4C). This is in agreement with previous work showing that iron loading of S. aureus does not alter susceptibility to the oxidative burst of neutrophils (63, 64).
FIG 4.

Heme promotes growth and virulence factor production of the ΔhemB mutant but decreases survival in blood. (A) Growth profiles (as determined by OD600 readings) of the WT and ΔhemB and ΔmenD mutants in metal-adjusted TSB containing iron in the form of 1 or 10 μM FeCl3 or 10 μM heme. Note that the open triangles are largely obscured by the filled triangles. (B) Graph showing hemolytic activities of the WT and ΔhemB and ΔmenD mutants grown in the presence of 1 or 10 μM FeCl3 or 10 μM heme. The panel above the graph illustrates the pigmentation of the ΔhemB mutant grown in the absence or presence of 10 μM heme. The WT is shown for comparison. There was no effect of heme on the pigmentation of the WT or ΔmenD strain. (C) Survival of the WT and ΔhemB and ΔmenD mutants, grown in the presence of 1 or 10 μM FeCl3 or 10 μM heme, after 6 h of incubation in whole human blood. Data in panels B and C were analyzed via a one-way ANOVA with Tukey's post hoc test. For each strain, comparisons were made between 1 μM FeCl3 and 10 μM FeCl3 or 10 μM heme. *, P < 0.01.
To ensure that experiments in whole human blood were not confounded by the presence of free heme from lysed erythrocytes, we examined serum recovered from blood incubated with bacteria, as described for survival assays. We were unable to detect hemolysis in blood incubated with either the wild type or SCVs. Although the wild type is hemolytic, the suppression of Agr activity by serum likely explains why we failed to detect hemolysis in whole human blood assays (61, 65).
In contrast to the ΔhemB mutant, the susceptibility of both the wild type and ΔmenD mutant to the oxidative burst was unchanged by growth in the presence of heme. Therefore, at the concentration used (10 μM), heme does not directly sensitize S. aureus to the oxidative burst. Rather, it appeared that the restoration of the electron transport chain in the ΔhemB mutant conferred sensitivity to the oxidative burst. To confirm this, we restored the electron transport chain in the ΔmenD mutant by supplementing the growth medium with menadione (1 μg ml−1), which resulted in a drop in survival of the SCV from 86% ± 10% to just 4% ± 3%.
The absence of an electron transport chain enables survival of Enterococcus faecalis in human blood.
The elevated survival of the S. aureus ΔhemB mutant, relative to the wild type, led us to consider whether a similar phenomenon occurred with Enterococcus faecalis, which despite producing cytochromes lacks a functional electron transport chain due to an inability to synthesize heme (51–53). However, E. faecalis employs heme uptake systems to scavenge heme from the environment, and therefore supplementation of the culture medium with heme results in increased growth under aerobic conditions. We confirmed this in two different E. faecalis strains (Fig. 5A and B), which grew to a higher optical density in the presence of heme. In addition, E. faecalis grown in the presence of heme produces a functional catalase, which we observed in both of the strains examined (Fig. 5C and D). However, as observed for the ΔhemB SCV, growth of E. faecalis in the presence of heme led to significantly diminished survival in human blood by increasing sensitivity to the oxidative burst (Fig. 5E and F). Therefore, as for SCV S. aureus, the absence of the electron transport chain in E. faecalis promotes survival in the bloodstream by reducing sensitivity to oxidative stress generated by host immune cells.
FIG 5.
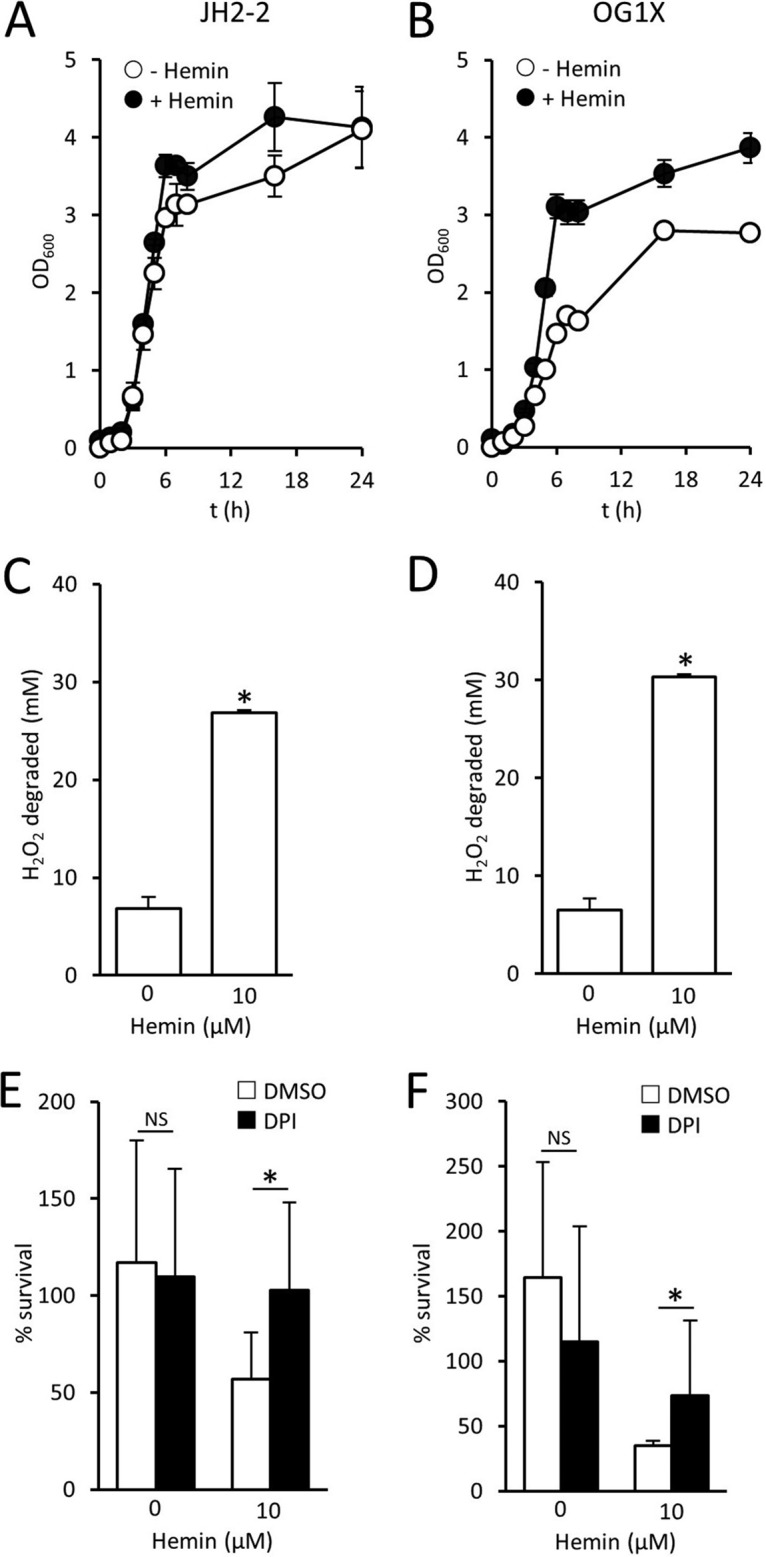
Heme promotes susceptibility of E. faecalis to host defenses. (A and B) Growth profiles (as determined by OD600 readings) of E. faecalis JH2-2 (A) and OG1X (B) grown in the absence or presence of 10 μM heme. (C and D) Catalase activity (expressed as mM H2O2 degraded in 1 h by 107 CFU) of E. faecalis JH2-2 (C) and OG1X (D) grown in the absence or presence of 10 μM heme. (E and F) Survival of E. faecalis JH2-2 (E) and OG1X (F) after 6 h in blood pretreated with DPI or an identical volume of DMSO solvent alone. In each case, data are the mean of results of four experiments in duplicate. For panels E and F, three different blood donors were used. Error bars represent the standard deviations of the mean. Data were analyzed by a one-way ANOVA with Tukey's post hoc test, which revealed significant differences (P < 0.01) in panels C and D between bacteria grown in the absence or presence of heme. In panels E and F, an asterisk indicates a P of <0.01 and NS (nonsignificant) indicates a P of >0.05 when the indicated comparisons were made.
DISCUSSION
During infection, S. aureus faces two major threats, host defenses and antibiotic therapy. Previous work has shown that SCVs of S. aureus are less susceptible to antibiotics than wild-type bacteria. Our data demonstrate that SCV S. aureus is also less susceptible to host immune defenses. These data fit with a previous study that revealed that SCVs are less sensitive than the wild type to host-derived AMPs (66). However, the resistance of SCVs to both the oxidative burst and AMPs is surprising given the lack of staphyloxanthin pigment, which contributes to the resistance of wild-type S. aureus to both ROS and AMPs (4, 47).
We do not currently understand the molecular basis of ROS resistance in SCVs. However, the damaging effects of ROS are proposed to occur via the Fenton reaction, which involves the reaction of H2O2 with free iron, leading to the generation of highly reactive hydroxyl radicals (67, 68). The lack of an electron transport chain, together with the associated decreased tricarboxylic acid activity (which utilizes iron-containing enzymes such as aconitase), in SCVs is therefore hypothesized to result in decreased iron content relative to that of wild-type bacteria. Furthermore, there is evidence that the electron transport chain generates superoxide radicals that liberate iron from iron-sulfur clusters, making it available for the Fenton reaction (69). However, the role of iron in susceptibility to the oxidative burst is far from clear, since previous work revealed that iron loading of S. aureus resulted in increased susceptibility to H2O2 but not killing by neutrophils (63, 64).
What is clear is that the ability of S. aureus SCVs to survive the oxidative burst comes at a cost. The electron transport chain enables aerobic respiration, rapid bacterial growth, and toxin production. These toxins include hemolysins that enable S. aureus to access heme, the bacterium's primary source of iron during infection (62). Therefore, the absence of hemolysin production by the ΔhemB mutant enables maintenance of the SCV phenotype in the presence of red blood cells. The menaquinone-defective SCV cannot restore the wild-type phenotype using host-derived materials and therefore maintains its phenotype regardless of hemolysin production.
E. faecalis lacks the necessary biosynthetic machinery to synthesize heme, making it a heme auxotroph (51–53). However, some strains secrete a cytolysin with hemolytic activity that provides a mechanism of heme acquisition (70, 71). The liberation of heme from erythrocytes would be expected to promote growth and restore catalase activity but would also increase susceptibility to host defenses. The maintenance of cytochromes and catalase that are restored by exogenous heme suggests that heme acquisition is a consistent and beneficial event during colonization and/or infection. What is not clear, however, is when and where heme acquisition occurs. For example, isolates recovered from patients with infective endocarditis, an infection of the heart valves that persists despite a robust immune response, are typically defective for hemolysin production (70, 71). This may indicate that hemolysin production, and thus heme acquisition, is undesirable at this site. In contrast, 30 to 40% of E. faecalis isolates carried in the gut or isolated from urinary tract infections are hemolytic (71). However, further work is needed to understand the basis for this observation and whether heme-mediated susceptibility to the oxidative burst plays a role.
Previous work reported that heme supplementation enabled E. faecalis to survive H2O2 challenge by restoring catalase activity (54, 55). However, while we also observed restoration of catalase activity in E. faecalis supplied with heme, this did not correlate with increased resistance to the oxidative burst.
In summary, SCV S. aureus sacrifices fast growth and toxin production for enhanced resistance to host defenses and antibiotics. This dramatic change in phenotype may enable the transition from a highly damaging, acute infection to a less pathogenic but persistent infection type. Our data indicate that the lack of heme production in E. faecalis also promotes survival in human blood, suggesting a common survival mechanism for these two pathogens.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are detailed in Table 1. Staphylococci were grown in tryptic soy broth (TSB) at 37°C with shaking (180 rpm) for 18 h to late stationary phase. Enterococci were grown in Todd-Hewitt broth supplemented with 0.5% yeast extract (THY) at 37°C with shaking (180 rpm) for 18 h to late stationary phase. For assays involving human blood, bacteria were plated onto Columbia blood agar (CBA) or THY supplemented with 5% sterile defibrinated sheep's blood to neutralize any remaining oxidants from the assay. For some experiments, iron (and other cations) was removed from TSB (100 ml) by incubation with Chelex resin (6 g) for 16 h at 4°C with stirring. The following individual metals were then replaced: ZnCl2 (25 μM), CaCl2 (1 mM), MgCl2 (1 mM), and MnCl2 (25 μM). Iron was added in the form of FeCl3 (1 or 10 μM) or heme (10 μM, >97% purity; Sigma).
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| S. aureus strains | ||
| USA300 LAC | LAC strain of the USA300 CA-MRSA lineage | 25 |
| USA300 ΔhemB | USA300 in which hemB has been deleted; heme auxotroph, SCV phenotype | 26 |
| USA300 ΔhemB geh::pCL55 | USA300 hemB mutant with pCL55 integrated into the geh locus; heme auxotroph, SCV phenotype | 26 |
| USA300 ΔhemB geh::phemB | USA300 hemB mutant with phemB integrated into the geh locus, restoring wild-type phenotype | 26 |
| USA300 ΔhemB ΔagrA | USA300 in which hemB and agrA have been deleted; Agr defective, SCV phenotype | This study |
| USA300 ΔhemB ΔagrC | USA300 in which hemB and agrC have been deleted; Agr defective, SCV phenotype | This study |
| USA300 ΔhemB ΔRNAIII | USA300 in which hemB and RNAIII have been deleted; SCV phenotype and defective for most secreted cytolysins | 25 |
| USA300 ΔmenD | USA300 in which menD has been deleted; menadione auxotroph, SCV phenotype | 26 |
| USA300 ΔmenD geh::pCL55 | USA300 menD mutant with pCL55 integrated into the geh locus; menadione auxotroph, SCV phenotype | 26 |
| USA300 ΔmenD geh::pmenD | USA300 menD mutant with pmenD integrated into the geh locus, restoring wild-type phenotype | 26 |
| USA300 ΔagrA | USA300 in which agrA has been deleted; Agr-defective phenotype | 60 |
| USA300 ΔagrC | USA300 in which agrC has been deleted; Agr-defective phenotype | 60 |
| USA300 ΔagrC pCN34 | USA300 in which agrC has been deleted, transformed with pCN34 | 60 |
| USA300 ΔagrC pagrCWT | USA300 ΔagrC transformed with pagrCWT | 60 |
| USA300 ΔagrC pagrCM234L | USA300 ΔagrC transformed with pagrCM234L | This study |
| USA300 ΔagrC pagrCR238H | USA300 ΔagrC transformed with pagrCR238H | This study |
| USA300 ΔagrC pagrCQ305H | USA300 ΔagrC transformed with pagrCQ305H | This study |
| E. faecalis strains | ||
| JH2-2 | Gelatinase deficient | 75 |
| OG1X | Gelatinase deficient | 76 |
| Plasmids | ||
| pEmpty | pCL55 E. coli-S. aureus shuttle vector that inserts as a single copy at the staphylococcal geh locus; Ampr Chlr | 77 |
| phemB | pCL55 containing the hemB gene under the control of the hem operon promoter | 26 |
| pmenD | pCL55 containing the menD gene under the control of the men operon promoter | 26 |
| pCN34 | E. coli-S. aureus shuttle vector; Ampr Kanr | |
| pagrCWT | pCN34 containing a wild-type copy of agrC under the control of the P3 promoter, restoring wild-type Agr phenotype | 61 |
| pagrCM234L | pCN34 containing a mutated copy of agrC resulting in M234L substitution, under the control of the P3 promoter, conferring constitutive Agr phenotype | 61 |
| pagrCR238H | pCN34 containing a mutated copy of agrC resulting in R238H substitution, under the control of the P3 promoter, conferring constitutive Agr phenotype | 61 |
| pagrCQ305H | pCN34 containing a mutated copy of agrC resulting in Q305H substitution, under the control of the P3 promoter, conferring constitutive Agr phenotype | 61 |
Genetic manipulation of S. aureus.
The construction of ΔmenD, ΔhemB ΔagrA, ΔagrC, and ΔRNAIII mutants was achieved using pIMAY as described previously (25, 26, 72). To construct the ΔhemB Δagr double mutants, the three agr mutants (ΔagrA, ΔagrC, and ΔRNAIII) were made electrocompetent and the hemB gene was deleted using pIMAY as described previously (25).
Mutants lacking hemB or menD were complemented with pCL55 containing the relevant gene under the control of the hem or men operon promoters, respectively (26). To control for pleiotropic effects of plasmid insertion into geh, pCL55 alone was transformed into hemB and menD mutant strains. The ΔagrC mutant was complemented with pCN34 containing a copy of the agrC gene under the control of the agr P3 promoter, and pCN34 alone (pEmpty) was used to control for pleiotropic effects of the plasmid. In addition to wild-type agrC, plasmids containing mutated forms of agrC which confer a constitutively active phenotype were also transformed into the ΔagrC mutant strain (61).
Whole human blood survival assay.
The survival of bacteria in whole human blood was assayed as described previously (73). Ethical approval for drawing and using human blood was obtained from the Regional Ethics Committee and Imperial NHS Trust Tissue Bank (REC Wales approval no. 12/WA/0196, ICHTB HTA license no. 12275). Blood was drawn from healthy human donors into tubes containing EDTA and used immediately in assays based on a previously described protocol (4). Suspensions of bacteria (105 CFU in 10 μl PBS) were mixed with blood (90 μl) and incubated for up to 6 h at 37°C with mixing. At the time points indicated in the figures, aliquots were taken, diluted serially in PBS, and plated onto CBA plates to enumerate CFU. In some assays, blood was pretreated (10 min) with diphenyleneiodonium (DPI) or an identical volume of DMSO alone to control for solvent effects (4).
Neutrophil survival assay.
Blood (20 ml) freshly collected in EDTA-treated tubes was layered over a 20-ml room temperature Polymorph preparation (Alere Limited). Cells were separated by centrifugation (500 × g, 45 min, brake off) until a clear separation of blood, peripheral blood mononuclear cells (PBMCs), and polymorphonuclear leukocytes (PMNs) was seen. The PBMCs were discarded, and the PMNs were transferred to a fresh 50-ml polypropylene tube. Then, 50 ml of Hanks' balanced salt solution (HBSS) without calcium or magnesium was added to the PMNs and the cells were pelleted (400 × g, 10 min, brake off). The supernatant was removed, and the PMNs were resuspended in 5 ml of red blood cell lysing buffer (eBioscience) before incubation at 37°C for 5 min. Next, 50 ml of HBSS (without calcium or magnesium) was added to the PMNs and cells were pelleted again (400 × g, 10 min, brake off). The PMNs were adjusted to 1 × 107 cells ml−1in HBSS supplemented with calcium and magnesium (1 mM) and 10% human serum. Stationary-phase bacteria were washed twice in PBS, and 106 CFU was added to 1 ml of the neutrophil suspension (multiplicity of infection [MOI], 1:10). The bacterial and neutrophil suspension was then incubated at 37°C with tumbling. At each time point, 50 μl of the suspension was transferred to a 96-well plate and serially diluted in PBS to enable enumeration of CFU on CBA plates after 48 h of incubation at 37°C. Survival was calculated as a percentage of the number of bacteria in the inoculum.
Measurement of bacterial growth.
Stationary-phase bacteria were diluted 1:50 into a final volume of 200 μl TSB in microtiter plates (Corning) before incubation at 37°C with shaking (500 rpm) in a POLARstar Omega multiwell plate reader. Bacterial growth was measured using measurements of optical density at 600 nm (OD600) every 30 min for a total of 17 h (61).
Hemolysin production.
The hemolytic activity of bacterial culture supernatants was determined as described previously (25). Briefly, culture supernatants were recovered by centrifugation (13,000 × g, 10 min) of stationary-phase cultures. The supernatant was then diluted in 2-fold steps using fresh TSB. Aliquots from each dilution (100 μl) were mixed with an equal volume of 2% sheep blood suspension in PBS and incubated at 37°C for 1 h in a static incubator. Subsequently, unlysed blood cells were removed by centrifugation and the supernatant containing lysed erythrocytes was transferred to a new microtiter plate. The degree of erythrocyte lysis was quantified by measuring the absorbance of the supernatant at 450 nm and reference to controls. Erythrocytes incubated with TSB alone or TSB containing 1% Triton X-100 (TX-100) served as negative and positive controls, respectively.
Whole blood hemolysis assay.
To determine whether the presence of bacteria in whole blood resulted in hemolysis, human blood was incubated with S. aureus strains for 6 h at 37°C as described above for survival assays. The serum was then recovered by centrifugation of blood at 1,000 × g for 5 min, and the presence of heme was detected by measuring the A450 as described above for hemolysin production assays. Blood lysed with 1% TX-100 acted as a positive control, while blood incubated without bacteria served as a negative control.
Measurement of phagocytosis and immune cell viability.
Phagocytosis of bacteria in whole human blood was determined using a protocol based on that described previously (74). Stationary-phase bacteria (1 ml) were pelleted (17,000 × g, 3 min) and washed twice with PBS. The pellet was then resuspended in 200 μl of 1.5 mM fluorescein isothiocyanate (FITC) dissolved in freshly prepared carbonate buffer (0.05 M NaCO3 and 0.1 M NaCl). Bacteria were then incubated for 60 min (room temperature with tumbling) in the dark. FITC-labeled bacteria were then washed three times in carbonate buffer and adjusted to 1 × 106 CFU ml−1 in PBS. FITC-labeled bacteria (10 μl, 1 × 104 CFU) were added to 96-well plates prior to the addition of 90 μl of freshly isolated blood, as described for the whole blood killing assay. At each time point (0, 2, 4, and 6 h), the blood/bacterium mixture (100 μl) was added to 900 μl of red blood cell lysis solution (eBioscience) and incubated at room temperature in the dark for 10 min. Samples were then centrifuged (500 × g, 10 min), the resulting pellet was washed once in PBS (1 ml) before a final centrifugation step (500 × g, 10 min), and then the pellet containing immune cells and bacteria was resuspended in 100 μl PBS or 1% paraformaldehyde (PFA; Affymetrix) if no further staining was required. Where samples were to be analyzed for host cell death, samples were incubated in PBS containing the Zombie Violet live-dead dye (Biolegend) at a 1:500 dilution in the dark. Free primary amine groups were quenched using 1.4 ml 1% bovine serum albumin (BSA), and samples were centrifuged (500 × g, 10 min) before resuspension in 100 μl 1% PFA. Positive controls were generated by heat-killing host cells (100°C, 10 min) prior to Zombie staining. Samples were then fixed overnight (12 to 16 h) in 1% paraformaldehyde at 4°C. Immune cell/bacterium samples were analyzed on a Fortessa flow cytometer (BD), and at least 10,000 events were captured. Green (FITC-bacteria) and violet (Zombie-labeled host cells) fluorescences were detected at 488/530 nm (30) and 404/450 nm, respectively. Based on preliminary analyses and using the methodology of Surewaard et al. (59), free bacteria (i.e., bacteria not phagocytosed) were identified as events with a side scatter of <50,000. In contrast, host cells were identified as events with a side scatter of >50,000. Samples were analyzed alongside controls, which consisted of bacteria without FITC labeling, host cells with or without Zombie stain, uninfected host cells, and heat-killed host cells as appropriate. Data were analyzed using FlowJo software (version 10). Compensation was not necessary, as the spectra of the fluorescent signals did not overlap.
Catalase assay.
Catalase activity of bacterial cells was determined as described previously (26). Overnight bacterial cultures (1 ml) were washed three times in PBS, and 107 CFU was added to 100 μM H2O2 in PBS (1 ml). Bacteria were incubated in the H2O2 in the dark at 37°C. At the start of the assay and every 15 min, 200 μl of sample was pelleted (17,000 × g) and 20 μl was added to a 96-well microtiter plate. The concentration of the remaining H2O2 was determined using a Pierce quantitative peroxide assay (aqueous compatible) kit.
ACKNOWLEDGMENTS
The following are gratefully acknowledged for providing bacterial strains, phage, or reagents: Ruth Massey (University of Bath), Malcolm Horsburgh (University of Liverpool), Tim Foster (Trinity College Dublin), Angela Nobbs (University of Bristol), and the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) Program under NIAID/NIH contract no. HHSN272200700055C.
A.M.E. acknowledges funding from the Royal Society, Department of Medicine (Imperial College), and from the Imperial NIHR Biomedical Research Centre, Imperial College London. K.L.P. was supported by a Ph.D. studentship from the Faculty of Medicine, Imperial College London. K.P.H. is supported by an MRC-funded Ph.D. studentship awarded to the Centre for Molecular Bacteriology and Infection, Imperial College London.
REFERENCES
- 1.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Gordon RJ, Lowy FD. 2008. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis 5:S350–359. doi: 10.1086/533591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aires-de-Sousa M. 2017. Methicillin-resistant Staphylococcus aureus among animals: current overview. Clin Microbiol Infect 23:373–380. doi: 10.1016/j.cmi.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. 2005. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buvelot H, Posfay-Barbe KM, Linder P, Schrenzel J, Krause KH. 2017. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol Rev 41:139–157. [DOI] [PubMed] [Google Scholar]
- 6.Ellson CD, Davidson K, Ferguson GJ, O'Connor R, Stephens LR, Hawkins PT. 2006. Neutrophils from p40phox−/− mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med 203:1927–1937. doi: 10.1084/jem.20052069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen CJ, Su LH, Lin TY, Huang YC. 2010. Molecular analysis of repeated methicillin-resistant Staphylococcus aureus infections in children. PLoS One 5:e14431. doi: 10.1371/journal.pone.0014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sreeramoju P, Porbandarwalla NS, Arango J, Latham K, Dent DL, Stewart RM, Patterson JE. 2011. Recurrent skin and soft tissue infections due to methicillin-resistant Staphylococcus aureus requiring operative debridement. Am J Surg 201:216–220. doi: 10.1016/j.amjsurg.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 9.Peyrani P, Allen M, Seligson D, Roberts C, Chen A, Haque N, Zervos M, Wiemken T, Harting J, Christensen D, Ramirez R. 2012. Clinical outcomes of osteomyelitis patients infected with methicillin-resistant Staphylococcus aureus USA-300 strains. Am J Orthop 41:117–122. [PubMed] [Google Scholar]
- 10.Abele-Horn M, Schupfner B, Emmerling P, Waldner H, Göring H. 2000. Persistent wound infection after herniotomy associated with small-colony variants of Staphylococcus aureus. Infection 2:53–54. doi: 10.1007/s150100050014. [DOI] [PubMed] [Google Scholar]
- 11.Acar JF, Goldstein FW, Lagrange P. 1978. Human infections caused by thiamine- or menadione-requiring Staphylococcus aureus. J Clin Microbiol 8:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agarwal H, Verrall R, Singh SP, Tang YW, Wilson G. 2007. Small colony variant Staphylococcus aureus multiorgan infection. Pediatr Infect Dis J 26:269–271. doi: 10.1097/01.inf.0000256749.29244.67. [DOI] [PubMed] [Google Scholar]
- 13.Besier S, Ludwig A, Ohlsen K, Brade V, Wichelhaus TA. 2007. Molecular analysis of the thymidine-auxotrophic small colony variant phenotype of Staphylococcus aureus. Int J Med Microbiol 297:217–225. doi: 10.1016/j.ijmm.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Kahl B, Herrmann M, Everding AS, Koch HG, Becker K, Harms E, Proctor RA, Peters G. 1998. Persistent infection with small colony variant strains of Staphylococcus aureus in patients with cystic fibrosis. J Infect Dis 177:1023–1029. doi: 10.1086/515238. [DOI] [PubMed] [Google Scholar]
- 15.Kahl BC. 2014. Small colony variants (SCVs) of Staphylococcus aureus—a bacterial survival strategy. Infect Genet Evol 21:515–522. doi: 10.1016/j.meegid.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Proctor RA, van Langevelde P, Kristjansson M, Maslow JN, Arbeit RD. 1995. Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin Infect Dis 20:95–102. doi: 10.1093/clinids/20.1.95. [DOI] [PubMed] [Google Scholar]
- 17.Kim NH, Kang YM, Han WD, Park KU, Park KH, Yoo JI, Lee DG, Park C, Song KH, Kim ES, Park SW, Kim NJ, Oh MD, Kim HB. 2016. Small-colony variants in persistent and recurrent Staphylococcus aureus bacteremia. Microb Drug Resist 22:538–544. doi: 10.1089/mdr.2015.0262. [DOI] [PubMed] [Google Scholar]
- 18.Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol 4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 19.Dean MA, Olsen RJ, Long SW, Rosato AE, Musser JM. 2014. Identification of point mutations in clinical Staphylococcus aureus strains that produce small-colony variants auxotrophic for menadione. Infect Immun 82:1600–1605. doi: 10.1128/IAI.01487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lannergård J, von Eiff C, Sander G, Cordes T, Seggewiss J, Peters G, Proctor RA, Becker K, Hughes D. 2008. Identification of the genetic basis for clinical menadione-auxotrophic small-colony variant isolates of Staphylococcus aureus. Antimicrob Agents Chemother 52:4017–4022. doi: 10.1128/AAC.00668-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriegeskorte A, Block D, Drescher M, Windmüller N, Mellmann A, Baum C, Neumann C, Lorè NI, Bragonzi A, Liebau E, Hertel P, Seggewiss J, Becker K, Proctor RA, Peters G, Kahl BC. 2014. Inactivation of thyA in Staphylococcus aureus attenuates virulence and has a strong impact on metabolism and virulence gene expression. mBio 5:e01447-. doi: 10.1128/mBio.01447-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards AM. 2012. Phenotype switching is a natural consequence of Staphylococcus aureus replication. J Bacteriol 194:5404–5412. doi: 10.1128/JB.00948-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Eiff C, Heilmann C, Proctor RA, Woltz C, Peters G, Götz F. 1997. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J Bacteriol 179:4706–4012. doi: 10.1128/jb.179.15.4706-4712.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bates DM, von Eiff C, McNamara PJ, Peters G, Yeaman MR, Bayer AS, Proctor RA. 2003. Staphylococcus aureus menD and hemB mutants are as infective as the parent strains, but the menadione biosynthetic mutant persists within the kidney. J Infect Dis 187:1654–1661. doi: 10.1086/374642. [DOI] [PubMed] [Google Scholar]
- 25.Pader V, James EH, Painter KL, Wigneshweraraj S, Edwards AM. 2014. The agr quorum-sensing system regulates fibronectin binding but not hemolysis in the absence of a functional electron transport chain. Infect Immun 82:4337–4347. doi: 10.1128/IAI.02254-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Painter KL, Strange E, Parkhill J, Bamford KB, Armstrong-James D, Edwards AM. 2015. Staphylococcus aureus adapts to oxidative stress by producing H2O2-resistant small-colony variants via the SOS response. Infect Immun 83:1830–1844. doi: 10.1128/IAI.03016-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuchscherr L, Medina E, Hussain M, Völker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, Peters G, Löffler B. 2011. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med 3:129–141. doi: 10.1002/emmm.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vesga O, Groeschel MC, Otten MF, Brar DW, Vann JM, Proctor RA. 1996. Staphylococcus aureus small colony variants are induced by the endothelial cell intracellular milieu. J Infect Dis 173:739–742. doi: 10.1093/infdis/173.3.739. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman LR, Déziel E, D'Argenio DA, Lépine F, Emerson J, McNamara S, Gibson RL, Ramsey BW, Miller SI. 2006. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 103:19890–19895. doi: 10.1073/pnas.0606756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Massey RC, Buckling A, Peacock SJ. 2001. Phenotypic switching of antibiotic resistance circumvents permanent costs in Staphylococcus aureus. Curr Biol 11:1810–1814. doi: 10.1016/S0960-9822(01)00507-3. [DOI] [PubMed] [Google Scholar]
- 31.Schaaff F, Bierbaum G, Baumert N, Bartmann P, Sahl HG. 2003. Mutations are involved in emergence of aminoglycoside-induced small colony variants of Staphylococcus aureus. Int J Med Microbiol 293:427–435. doi: 10.1078/1438-4221-00282. [DOI] [PubMed] [Google Scholar]
- 32.Vestergaard M, Paulander W, Ingmer H. 2015. Activation of the SOS response increases the frequency of small colony variants. BMC Res Notes 8:749. doi: 10.1186/s13104-015-1735-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leimer N, Rachmühl C, Palheiros Marques M, Bahlmann AS, Furrer A, Eichenseher F, Seidl K, Matt U, Loessner MJ, Schuepbach RA, Zinkernagel AS. 2016. Nonstable Staphylococcus aureus small-colony variants are induced by low pH and sensitized to antimicrobial therapy by phagolysosomal alkalinization. J Infect Dis 213:305–313. doi: 10.1093/infdis/jiv388. [DOI] [PubMed] [Google Scholar]
- 34.Moisan H, Brouillette E, Jacob CL, Langlois-Bégin P, Michaud S, Malouin F. 2006. Transcription of virulence factors in Staphylococcus aureus small-colony variants isolated from cystic fibrosis patients is influenced by SigB. J Bacteriol 188:64–76. doi: 10.1128/JB.188.1.64-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell G, Fugère A, Pépin Gaudreau K, Brouillette E, Frost EH, Cantin AM, Malouin F. 2013. SigB is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS One 8:e65018. doi: 10.1371/journal.pone.0065018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaudaux P, Francois P, Bisognano C, Kelley WL, Lew DP, Schrenzel J, Proctor RA, McNamara PJ, Peters G, Von Eiff C. 2002. Increased expression of clumping factor and fibronectin-binding proteins by hemB mutants of Staphylococcus aureus expressing small colony variant phenotypes. Infect Immun 70:5428–5437. doi: 10.1128/IAI.70.10.5428-5437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Eiff C, Becker K, Metze D, Lubritz G, Hockmann J, Schwarz T, Peters G. 2001. Intracellular persistence of Staphylococcus aureus small-colony variants within keratinocytes: a cause for antibiotic treatment failure in a patient with Darier's disease. Clin Infect Dis 32:1643–1647. doi: 10.1086/320519. [DOI] [PubMed] [Google Scholar]
- 38.Singh R, Ray P, Das A, Sharma M. 2010. Enhanced production of exopolysaccharide matrix and biofilm by a menadione-auxotrophic Staphylococcus aureus small-colony variant. J Med Microbiol 59:521–527. doi: 10.1099/jmm.0.017046-0. [DOI] [PubMed] [Google Scholar]
- 39.Tuchscherr L, Heitmann V, Hussain M, Viemann D, Roth J, von Eiff C, Peters G, Becker K, Löffler B. 2010. Staphylococcus aureus small-colony variants are adapted phenotypes for intracellular persistence. J Infect Dis 202:1031–1040. doi: 10.1086/656047. [DOI] [PubMed] [Google Scholar]
- 40.Baumert N, von Eiff C, Schaaff F, Peters G, Proctor RA, Sahl HG. 2002. Physiology and antibiotic susceptibility of Staphylococcus aureus small colony variants. Microb Drug Resist 8:253–260. doi: 10.1089/10766290260469507. [DOI] [PubMed] [Google Scholar]
- 41.Brouillette E, Grondin G, Lefebvre C, Talbot BG, Malouin F. 2004. Mouse mastitis model of infection for antimicrobial compound efficacy studies against intracellular and extracellular forms of Staphylococcus aureus. Vet Microbiol 101:253–262. doi: 10.1016/j.vetmic.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Norstrom T, Lannergard J, Hughes D. 2007. Genetic and phenotypic identification of fusidic acid-resistant mutants with the small-colony-variant phenotype in Staphylococcus aureus. Antimicrob Agents Chemother 51:4438–4446. doi: 10.1128/AAC.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuji BT, von Eiff C, Kelchlin PA, Forrest A, Smith PF. 2008. Attenuated vancomycin bactericidal activity against Staphylococcus aureus hemB mutants expressing the small-colony-variant phenotype. Antimicrob Agents Chemother 52:1533–1537. doi: 10.1128/AAC.01254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia LG, Lemaire S, Kahl BC, Becker K, Proctor RA, Denis O, Tulkens PM, Van Bambeke F. 2013. Antibiotic activity against small-colony variants of Staphylococcus aureus: review of in vitro, animal and clinical data. J Antimicrob Chemother 68:1455–1464. doi: 10.1093/jac/dkt072. [DOI] [PubMed] [Google Scholar]
- 45.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. 2008. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clauditz A, Resch A, Wieland KP, Peschel A, Götz F. 2006. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect Immun 74:4950–4953. doi: 10.1128/IAI.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra NN, Liu GY, Yeaman MR, Nast CC, Proctor RA, McKinnell J, Bayer AS. 2011. Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob Agents Chemother 55:526–531. doi: 10.1128/AAC.00680-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cosgrove K, Coutts G, Jonsson IM, Tarkowski A, Kokai-Kun JF, Mond JJ, Foster SJ. 2007. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol 189:1025–1035. doi: 10.1128/JB.01524-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaupp R, Ledala N, Somerville GA. 2012. Staphylococcal response to oxidative stress. Front Cell Infect Microbiol 2:33. doi: 10.3389/fcimb.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McAdow M, Kim HK, Dedent AC, Hendrickx AP, Schneewind O, Missiakas DM. 2011. Preventing Staphylococcus aureus sepsis through the inhibition of its agglutination in blood. PLoS Pathog 7:e1002307. doi: 10.1371/journal.ppat.1002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bryan-Jones DG, Whittenbury R. 1969. Hematin-dependent oxidative phosphorylation in Streptococcus faecalis. J Gen Microbiol 58:247–260. doi: 10.1099/00221287-58-2-247. [DOI] [PubMed] [Google Scholar]
- 52.Ritchey TW, Seeley HW. 1974. Cytochromes in Streptococcus faecalis var. zymogenes grown in a hematin-containing medium. J Gen Microbiol 85:220–228. doi: 10.1099/00221287-85-2-220. [DOI] [PubMed] [Google Scholar]
- 53.Winstedt L, Frankenberg L, Hederstedt L, von Wachenfeldt C. 2000. Enterococcus faecalis V583 contains a cytochrome bd-type respiratory oxidase. J Bacteriol 182:3863–3866. doi: 10.1128/JB.182.13.3863-3866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frankenberg L, Brugna M, Hederstedt L. 2002. Enterococcus faecalis heme-dependent catalase. J Bacteriol 184:6351–6356. doi: 10.1128/JB.184.22.6351-6356.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baureder M, Reimann R, Hederstedt L. 2012. Contribution of catalase to hydrogen peroxide resistance in Enterococcus faecalis. FEMS Microbiol Lett 331:160–164. doi: 10.1111/j.1574-6968.2012.02567.x. [DOI] [PubMed] [Google Scholar]
- 56.Smith EJ, Visai L, Kerrigan SW, Speziale P, Foster TJ. 2011. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect Immun 79:3801–3809. doi: 10.1128/IAI.05075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malachowa N, Whitney AR, Kobayashi SD, Sturdevant DE, Kennedy AD, Braughton KR, Shabb DW, Diep BA, Chambers HF, Otto M, DeLeo FR. 2011. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One 6:e18617. doi: 10.1371/journal.pone.0018617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Painter KL, Krishna A, Wigneshweraraj S, Edwards AM. 2014. What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia? Trends Microbiol 22:676–685. doi: 10.1016/j.tim.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 59.Surewaard BG, de Haas CJ, Vervoort F, Rigby KM, DeLeo FR, Otto M, van Strijp JA, Nijland R. 2013. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol 15:1427–1437. doi: 10.1111/cmi.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pader V, Hakim S, Painter KL, Wigneshweraraj S, Clarke TB, Edwards AM. 2016. Staphylococcus aureus inactivates daptomycin by releasing membrane phospholipids. Nat Microbiol 2:16194. doi: 10.1038/nmicrobiol.2016.194. [DOI] [PubMed] [Google Scholar]
- 61.James EH, Edwards AM, Wigneshweraraj S. 2013. Transcriptional downregulation of agr expression in Staphylococcus aureus during growth in human serum can be overcome by constitutively active mutant forms of the sensor kinase AgrC. FEMS Microbiol Lett 349:153–162. doi: 10.1111/1574-6968.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O. 2004. Iron-source preference of Staphylococcus aureus infections. Science 305:1626–1628. doi: 10.1126/science.1099930. [DOI] [PubMed] [Google Scholar]
- 63.Repine JE, Fox RB, Berger EM, Harada RN. 1981. Effect of staphylococcal iron content on the killing of Staphylococcus aureus by polymorphonuclear leukocytes. Infect Immun 32:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoepelman IM, Bezemer WA, Vandenbroucke-Grauls CM, Marx JJ, Verhoef J. 1990. Bacterial iron enhances oxygen radical-mediated killing of Staphylococcus aureus by phagocytes. Infect Immun 58:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peterson MM, Mack JL, Hall PR, Alsup AA, Alexander SM, Sully EK, Sawires YS, Cheung AL, Otto M, Gresham HD. 2008. Apolipoprotein B is an innate barrier against invasive Staphylococcus aureus infection. Cell Host Microbe 4:555–566. doi: 10.1016/j.chom.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gläser R, Becker K, von Eiff C, Meyer-Hoffert U, Harder J. 2014. Decreased susceptibility of Staphylococcus aureus small-colony variants toward human antimicrobial peptides. J Investig Dermatol 134:2347–2350. doi: 10.1038/jid.2014.176. [DOI] [PubMed] [Google Scholar]
- 67.Fang FC. 2011. Antimicrobial actions of reactive oxygen species. mBio 2:e00141-. doi: 10.1128/mBio.00141-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Keyer K, Imlay JA. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci U S A 93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coque TM, Patterson JE, Steckelberg JM, Murray BE. 1995. Incidence of hemolysin, gelatinase, and aggregation substance among enterococci isolated from patients with endocarditis and other infections and from feces of hospitalized and community-based persons. J Infect Dis 171:1223–1229. doi: 10.1093/infdis/171.5.1223. [DOI] [PubMed] [Google Scholar]
- 71.Creti R, Imperi M, Bertuccini L, Fabretti F, Orefici G, Di Rosa R, Baldassarri L. 2004. Survey for virulence determinants among Enterococcus faecalis isolated from different sources. J Med Microbiol 53:13–20. doi: 10.1099/jmm.0.05353-0. [DOI] [PubMed] [Google Scholar]
- 72.Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edwards AM, Potts JR, Josefsson E, Massey RC. 2010. Staphylococcus aureus host cell invasion and virulence in sepsis is facilitated by the multiple repeats within FnBPA. PLoS Pathog 6:e1000964. doi: 10.1371/journal.ppat.1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ko YP, Kuipers A, Freitag CM, Jongerius I, Medina E, van Rooijen WJ, Spaan AN, van Kessel KP, Höök M, Rooijakkers SH. 2013. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog 9:e1003816. doi: 10.1371/journal.ppat.1003816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacob AE, Hobbs SJ. 1974. Conjugal transfer of plasmid-borne multiple antibiotic resistance in Streptococcus faecalis var. zymogenes. J Bacteriol 117:360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ike Y, Craig RA, White BA, Yagi Y, Clewell DB. 1983. Modification of Streptococcus faecalis sex pheromones after acquisition of plasmid DNA. Proc Natl Acad Sci U S A 80:5369–5373. doi: 10.1073/pnas.80.17.5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee CY, Buranen SL, Ye ZH. 1991. Construction of single-copy integration vectors for Staphylococcus aureus. Gene 103:101–105. doi: 10.1016/0378-1119(91)90399-V. [DOI] [PubMed] [Google Scholar]