

ABSTRACT
Locus of enterocyte effacement (LEE)-negative Shiga toxin (Stx)-producing Escherichia coli (STEC) strains are human pathogens that lack the LEE locus, a pathogenicity island (PAI) involved in the intimate adhesion of LEE-positive strains to the host gut epithelium. The mechanism used by LEE-negative STEC strains to colonize the host intestinal mucosa is still not clear. The cell invasion determinant tia, previously described in enterotoxigenic E. coli strains, has been identified in LEE-negative STEC strains that possess the subtilase-encoding pathogenicity island (SE-PAI). We evaluated the role of the gene tia, present in these LEE-negative STEC strains, in the invasion of monolayers of cultured cells. We observed that these strains were able to invade Caco-2 and HEp-2 cell monolayers and compared their invasion ability with that of a mutant strain in which the gene tia had been inactivated. Mutation of the gene tia resulted in a strong reduction of the invasive phenotype, and complementation of the tia mutation with a functional copy of the gene restored the invasion activity. Moreover, we show that the gene tia is overexpressed in bacteria actively invading cell monolayers, demonstrating that tia is involved in the ability to invade cultured monolayers of epithelial cells shown by SE-PAI-positive E. coli, including STEC, strains. However, the expression of the tia gene in the E. coli K-12 strain JM109 was not sufficient, in its own right, to confer to this strain the ability to invade cell monolayers, suggesting that at least another factor must be involved in the invasion ability displayed by the SE-PAI-positive strains.
KEYWORDS: Escherichia coli, LEE negative, STEC, cell invasion, tia
INTRODUCTION
Shiga toxin-producing Escherichia coli (STEC) strains are human pathogens that cause a wide range of symptoms, from self-limited watery diarrhea to hemorrhagic colitis and the life-threatening hemolytic-uremic syndrome (HUS) (1). Ruminants are the main natural reservoir of STEC, and its transmission to humans can occur through the consumption of contaminated food as well as by interhuman cycles and contacts with infected animals (1). The Shiga toxin (Stx) produced by STEC strains is the main virulence factor of these bacteria, but other bacterial features, such as an efficient mechanism of colonization and persistence in the gut, are required to establish and maintain the infection (1). The most virulent STEC strains isolated from human infections, such as those belonging to the O157:H7 serotype, can colonize the intestinal mucosa through the attaching and effacing (A/E) lesion, in which colonization occurs through the intimate adhesion of bacteria to the gut epithelium, mediated by the outer membrane protein intimin and its receptor, Tir, translocated into the host cell by a type III secretion system (1). The A/E lesion is characterized by the localized destruction of brush border microvilli, intimate bacterial adhesion, and the formation of actin pedestals beneath the attached bacteria. The genes responsible for the A/E lesion are carried by the pathogenicity island (PAI) termed locus of enterocyte effacement (LEE) (1). Although most STEC strains isolated from patients with severe infections are able to induce the A/E lesion, LEE-negative STEC strains belonging to numerous serotypes, such as O113:H21, O91:H21, O8:H19, and O104:H4, have been associated with both sporadic cases and outbreaks of severe disease (2). With the exception of the infamous STEC O104:H4 serotype that caused the German outbreak in 2011 and that was an enteroaggregative E. coli strain (3), the means of colonization and persistence in the human gut by LEE-negative STEC strains are still not clear. Previous studies have shown that certain STEC strains present an invasive phenotype on epithelial cell lines derived from the intestinal mucosa (Caco-2 and T-84 cells) (2). Moreover, several LEE-negative STEC strains isolated from humans harbor the gene tia (4), an invasion determinant previously described in enterotoxigenic E. coli (ETEC), in which it appears to facilitate the invasion of cells derived from intestinal cell lines (HCT-8, HCT-116, and T-84 cells) (5). In this group of LEE-negative STEC strains, the gene tia is carried by the subtilase-encoding pathogenicity island (SE-PAI), which carries the genes coding for the subtilase SubAB2, an AB5 toxin that induces a toxic effect on Vero cells comparable to that exerted by Stx (4, 6). The SE-PAI was first described and characterized in the Stx-negative E. coli strain ED32 and further identified to be a common PAI of LEE-negative STEC strains isolated from human cases of diarrhea (6). In this work, we investigated the role of the gene tia conveyed by the SE-PAI in the invasion of monolayers of epithelial (Caco-2 and HEp-2) cells using the prototype SE-PAI-positive E. coli strain ED32 and the LEE-negative SE-PAI-positive STEC strain ED97 as models.
RESULTS
The gene tia confers to SE-PAI-positive E. coli strains ED32 and ED97 the ability to invade Caco-2 and HEp-2 cells.
To investigate the hypothesis that the SE-PAI-positive strains may invade cell monolayers through a tia-mediated mechanism, we inactivated the gene tia in strain ED32 by site-specific mutagenesis and compared the invasion ability of the ED32 wild-type strain with that of its mutant, ED32 Δtia, on Caco-2 and HEp-2 cell monolayers. We observed that the ED32 wild-type strain was able to invade both Caco-2 and HEp-2 cell monolayers, whereas E. coli K-12 strain JM109, used as a negative control, was not (Fig. 1). The ability to invade cell monolayers was abolished in the ED32 Δtia strain on Caco-2 cell monolayers (Fig. 1A) and strongly reduced (about 90%) on HEp-2 cell monolayers (Fig. 1B). With both cell types, the invasion ability of the wild-type ED32 strain was restored by transforming an intact copy of the gene tia into mutant strain ED32 Δtia (Fig. 1). Similar to what was observed for the ED32 wild-type strain, the SE-PAI-positive STEC strain ED97 was also able to invade both the Caco-2 cell monolayers and the HEp-2 cell monolayers (Fig. 1). In all the experiments, enteroinvasive E. coli (EIEC) strain 6.81 was used as a positive control for the invasion assay, and the ability of ED32 and ED97 to penetrate the cell monolayer was about 10% of the invasion ability of EIEC 6.81 (data not shown). To assess whether the product of the gene tia was sufficient in its own right to govern the invasion of Caco-2 and HEp-2 cell monolayers, we compared the invasion ability of E. coli K-12 strain JM109/pGEM_tia, which expressed a cloned copy of the tia gene amplified from ED32, with the invasion ability of the same K-12 strain carrying the empty vector, pGEM-T Easy (JM109/pGEM-T Easy).
FIG 1.
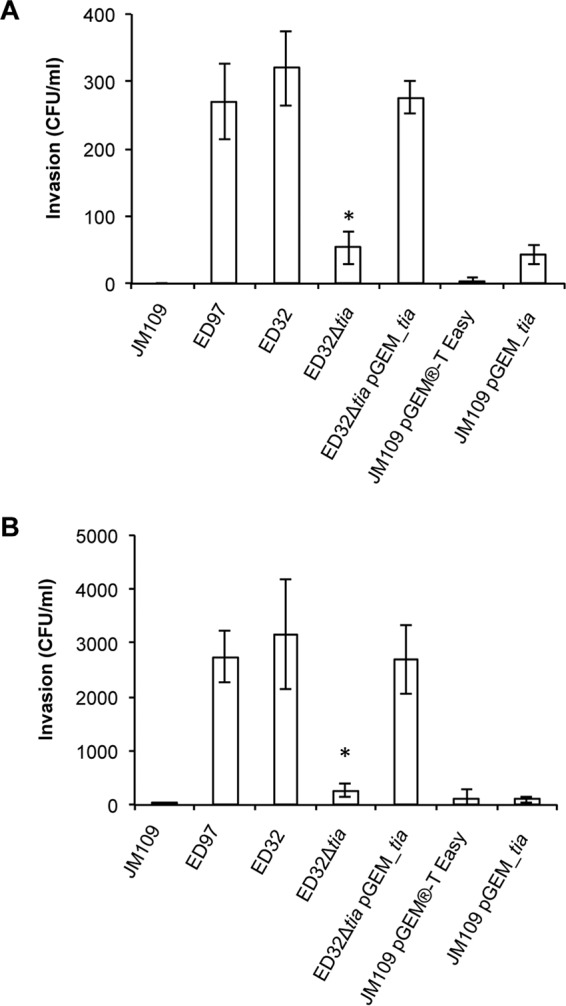
Invasion of Caco-2 and HEp-2 cell monolayers. The level of invasion of both the Caco-2 and HEp-2 cell monolayers by the strains is expressed as the number of CFU per milliliter. ED97 and ED32 displayed the ability to invade the monolayers of both Caco-2 (A) and HEp-2 (B) cells, and the inactivation of the gene tia in strain ED32 Δtia strongly reduced the invasion ability. Complementation of the tia mutation restored the invasive phenotype of ED32. The difference between strain ED32 Δtia and both the ED32 and ED32 Δtia/pGEM_tia strains was statistically significant with a P value of <0.01, indicated by the asterisk. The expression of tia in the laboratory strain JM109 conferred only a moderate ability to invade the Caco-2 cell monolayers (A) but did not confer any ability to invade the HEp-2 cell monolayers (B).
Even though the ability of strain JM109/pGEM_tia to invade Caco-2 cells was slightly greater than that of the negative-control strain, JM109/pGEM-T Easy, the number of CFU of this strain recovered from the cell lysate was comparable to that of strain ED32 Δtia (Fig. 1A). The ability of strain JM109/pGEM_tia to invade HEp-2 cells was the same as that of its control strain, JM109/pGEM-T Easy (Fig. 1B). Our result indicates that the gene tia carried by the SE-PAI was not able to confer to a K-12 strain the ability to invade Caco-2 and HEp-2 cell monolayers, at least under our experimental conditions. Since we could not exclude the possibility that in strain JM109/pGEM_tia the gene tia was not sufficiently expressed to confer the invasive phenotype, we retrotranscribed the total RNA from strains JM109/pGEM_tia and ED32 and used the same amount of cDNA as the amount of template to amplify the gene tia with the primers tiaUP and tiaLO (4) by PCR. The gene tia was apparently expressed in strain JM109/pGEM_tia at a rate higher than that at which it was expressed in wild-type strain ED32 (Fig. 2). Such a result could be explained by considering that in strain JM109/pGEM_tia the tia gene is harbored on the high-copy-number plasmid pGEM-T Easy. Our results indicate that the absence of the invasive phenotype in the JM109/pGEM_tia strain was not due to the insufficient expression of the gene tia.
FIG 2.
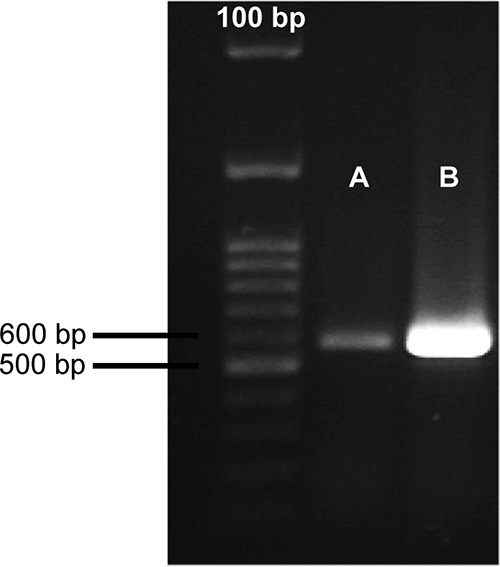
Amplification of the cDNA of the gene tia. The expression of the gene tia by strain ED32 (A) and by the K-12 strain JM109/pGEM_tia (B) was evaluated by reverse transcriptase PCR amplification of a 571-bp fragment within the coding sequence with the primers tiaUP and tiaLO (4) using the same amount of cDNA from the two strains as the template.
The gene tia is overexpressed in strains ED32 and ED97 within HEp-2 cells.
Previous results showed that strains ED32 and ED97 are able to invade monolayers of epithelial cells. To further investigate the role of the gene tia in conferring the invasive phenotype to these two strains, we performed a quantitative real-time PCR on the total RNA extracted from the bacteria recovered from inside the HEp-2 cells and from the bacteria that were incubated with the HEp-2 cell monolayer but that did not invade the bacteria. Our results showed that the gene tia was overexpressed in both the ED32 and ED97 strains recovered from inside the HEp-2 cell monolayers compared with its level of expression in the bacteria recovered from the medium outside the monolayer with fold changes of +48.5 and +47.0, respectively (P < 0.01). The overexpression of tia in the invasive bacteria suggests that tia plays an active role during the process of invasion of these strains.
DISCUSSION
We showed that the SE-PAI-positive prototype E. coli strain ED32 (4) is able to invade Caco-2 and HEp-2 cell monolayers upon functional tia gene expression, as shown by the inability to invade the cell monolayers displayed upon inactivation of the gene tia. So far, the implication that tia has a role in cell invasion processes has been demonstrated only for ETEC strains (5), and to the best of our knowledge, our results represent the first evidence that this gene is involved in the invasion of epithelial cell lines in other pathogenic E. coli strains, such as LEE-negative, SE-PAI-positive STEC strain ED97. Moreover, this work also presents the first evidence that the tia function can be restored in a tia-deficient mutant. The evidence provided suggests that the gene tia harbored by the SE-PAI-positive strains, including STEC, is involved in the mechanism leading to intracellular localization. This hypothesis is strongly supported by the finding that tia is overexpressed in bacteria internalized in cell monolayers by about 48% compared with the level of expression by bacteria that were incubated with the monolayers but that did not engage in the process of invasion. However, the role of this internalization in infection processes still needs to be elucidated. In fact, different from what was observed for typical invasive pathogens, like EIEC, Salmonella species, and Shigella species, STEC strains seem to not replicate inside the host cells and their invasion does not culminate with the destruction of the epithelial cells (2). As a matter of fact, in our invasion experiments, the number of CFU of EIEC strain 6.81, used as a positive control, recovered from inside the cell cultures was much higher (by about 90% [data not shown]) than the number of CFU of both the ED32 and ED97 strains recovered. Altogether, these data suggest that these strains use tia as part of their mechanism of invasion, which differs from that used by typical invasive E. coli strains.
It has already been suggested that the intracellular localization of STEC might serve as a mechanism that protects the bacteria from the host defense mechanisms that operate on mucosal surfaces rather than as a virulence mechanism involving a massive invasion of the mucosa of the host, as is the case for typical invasive pathogens (2). A hypothesis for the existence of such a mild invasive phenotype could be that the survival of STEC within slightly damaged enterocytes might allow a more persistent colonization of the intestinal mucosa of animals, which in turn could favor the shedding of STEC and the consequent contamination of the environment and of other animals (2). As a matter of fact, the tia-mediated internalization of SE-PAI-positive strains could represent a mechanism to escape the host defenses that operate on the intestinal mucosal surface. LEE-negative SE-PAI-positive STEC strains, in the absence of other mechanisms of colonization, might use a tia-mediated invasion mechanism to resort to the intracellular localization in order to limit the effect of the defense mechanisms of the host intestinal mucosal surface. This mechanism, if verified, could lead to an increased fitness of the microbes not only in the gut of the reservoir animals, as has been previously suggested (2), but also in humans during the course of infection, favoring persistence in the human gut.
In conclusion, our work provides evidence linking the observation that some STEC strains show an invasive phenotype (2) with the presence of the ETEC invasion determinant tia, carried on the SE-PAI, in a group of LEE-negative STEC strains (4, 5). We demonstrate that in the ED32 strain the gene tia is required to invade cell monolayers and suggest that in SE-PAI-positive LEE-negative STEC strains, such as ED97, the invasive phenotype relies, at least partly, on this factor. Our data also provide evidence that the product of the gene tia from the SE-PAI is not sufficient in its own right to govern the invasion process on cell monolayers and that at least another factor seems to cooperate in this process. Identification of the latter factor and comprehension of the full mechanism of invasion operated by SE-PAI-positive E. coli strains are under investigation.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and plasmids.
The E. coli strains used in this study (Table 1) were grown in tryptone soya broth (TSB) or on tryptone soya agar (TSA) plates at 37°C. For the invasion experiments, in order to avoid the loss of the invasive plasmid pINV, enteroinvasive E. coli strain 6.81 (7) was grown at 30°C in all steps before incubation with the cell monolayers (G. Prosseda, personal communication). SOB and SOC media were prepared as previously described (8). Kanamycin (Km) at 50 μg/ml, ampicillin (Ap) at 100 μg/ml, and gentamicin (Gm) at 100 μg/ml were added to the media when required. Plasmid pGEM-T Easy (Promega, Madison, WI, USA) and its derivative expressing the gene tia (pGEM_tia) were used for the expression of tia in JM109 competent cells as described below.
TABLE 1.
Strains used in this study
| Strain | Description | Reference or source |
|---|---|---|
| JM109 | K-12 laboratory strain | 13 |
| EIEC 6.81 | Enteroinvasive strain E. coli 6.81 | 7 |
| ED32 | ED32 wild-type strain: stx negative, LEE negative, and SE-PAI positive | 6 |
| ED32 Δtia | ED32 strain (6) in which the gene tia has been inactivated by the partial replacement of the gene with a Km resistance cassette | This study |
| ED32 Δtia/pGEM_tia | ED32 Δtia strain carrying the plasmid pGEM_tia to complement the tia mutation | This study |
| ED97 | ED97 wild-type strain: stx positive, LEE negative, and SE-PAI positive | 6 |
Expression of tia in JM109 competent cells.
A DNA region encompassing the gene tia and its promoter region was amplified from the genome of strain ED32 with primers tia_pGEM_FW and tia_pGEM_RV (Table 2) using the GoTaq Green master mix (Promega, Madison, WI, USA). The amplified fragment was purified with a Wizard SV gel and PCR cleanup system (Promega, Madison, WI, USA) and cloned into the plasmid pGEM-T Easy (Promega, Madison, WI, USA), resulting in plasmid pGEM_tia. This plasmid was transformed into electrocompetent JM109 cells (9), which were then grown on plates supplemented with 100 μg/ml ampicillin to select transformed clones. The positive clones were screened by PCR using the primers pGEM_check_FW and pGEM_check_RV (Table 2), which anneal on the plasmid pGEM-T Easy, generating in the empty vector a fragment of 319 bp. The insertion of the expected 973-bp fragment in the plasmid pGEM-T Easy resulted in an amplification product of 1,292 bp. The expression of tia in this strain and in the ED32 wild-type strain was evaluated by the extraction of total RNA, followed by PCR amplification of the gene tia using cDNA as a template. Briefly, total RNA was extracted from the ED32 wild-type strain and strain JM109/pGEM_tia using a total RNA extraction kit (Norgen Biotek Corp., Thorold, ON, Canada) according to the manufacturer's instructions. An additional DNA removal step was performed with an RNase-free DNase set (Qiagen, Chatsworth, CA, USA) as suggested by the kit instructions. A DNA wipeout kit (Qiagen, Chatsworth, CA, USA) was used to remove genomic DNA from a total of 1 μg of total RNA, which was retrotranscribed into cDNA with a QuantiTect reverse transcription kit (Qiagen, Chatsworth, CA, USA) according to the manufacturer's instructions. One hundred nanograms of cDNA, quantified with a Qubit (version 3.0) fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), was used as the template for a PCR with primers tiaUP and tiaLO (4). To verify that the amount of cDNA was the same in the two strains, 10 ng of the same cDNA was used to amplify a 215-bp fragment of the constitutive gene gapA, coding for glyceraldehyde-3-phosphate dehydrogenase, using the primers gapAF and gapAR (10). To verify that the amplification of tia and of gapA did not occur because of the presence of residual genomic DNA, the total RNA after the wipeout step was used as the template in a PCR with primers tiaUP and tiaLO, resulting in no amplification.
TABLE 2.
Primers used in this studya
| Name | Sequence (5′–3′) | Source of reference sequence | Position of primer in the sequence (bp) |
|---|---|---|---|
| tia_pGEM_FW | GGAGTAGTTTCGTCAGAAGT | GenBank accession no. JQ994271.1 | 4714 |
| tia_pGEM_RV | TCAATGCCTGTGTCAGTACT | GenBank accession no. JQ994271.1 | 5686 |
| FW_Kmb | GCGGACACCTGCCCCAAAACTCCATGCAAAATTGTTATCATACATGGCGATAGCTAGACT | Thermo Fisher Scientificc | 1135 |
| GenBank accession no. JQ994271.1 | 4885 | ||
| RV_Kmb | ATGAAAAAGATTATTGCGGTTTCAGCGCTTGCTGTGGTGTTCAATTCAGAAGAACTCGTC | Thermo Fisher Scientificc | 2119 |
| GenBank accession no. JQ994271.1 | 5472 | ||
| Km_check_RV | TAAAGCACGAGGAAGCGGTC | Thermo Fisher Scientificc | 2049 |
| pGEM_check_FW | CGAAAGGGGGATGTGCTGC | Promegad | 2910 |
| pGEM_check_RV | AGCGGATAACAATTTCACACAGGA | Promegad | 211 |
| tia_RT_F | TGTTCTGTGTGTCTTCCCGA | GenBank accession no. JQ994271.1 | 5109 |
| tia_RT_R | GGCGGCGATAAGAACGATAC | GenBank accession no. JQ994271.1 | 5295 |
| tia_RT_probe | GTGCGCACAGGAACATTGTA | GenBank accession no. JQ994271.1 | 5210 |
The primers designed for this study are listed. Primers used in previous studies are indicated with the corresponding reference in the text.
The nucleotides underlined in the sequence of primers FW_Km and RV_Km anneal on the plasmid pCR2.1 TOPO to amplify the Km resistance cassette, while the rest of the sequence (not underlined) anneals to the gene tia in order to amplify two regions of the gene and allow recombination (GenBank accession no. JQ994271.1).
The sequence of the plasmid pCR2.1 TOPO is available on the Thermo Fisher Scientific website, at the following address: http://tools.thermofisher.com/content/sfs/vectors/pcr2_1topo_seq.txt.
The sequence of the plasmid pGEM-T Easy is available on the Promega website, at the following address: www.promega.com/vectors/.
Inactivation of the gene tia in strain ED32.
The mutant strain ED32 Δtia was generated by deletion of 509 bp out of 747 bp of the gene tia in strain ED32 and replacement with a Km resistance cassette, as previously described with some modifications (8). Briefly, the Km resistance cassette was amplified with its promoter region from plasmid pCR2.1 TOPO (Thermo Fisher Scientific, Waltham, MA, USA) using the primers FW_Km and RV_Km (Table 2). PCR was performed by using GC-platinum Taq polymerase (Thermo Fisher Scientific, Waltham, MA, USA), and the amplification product was purified with a QIAquick gel extraction kit (Qiagen, Chatsworth, CA, USA) after electrophoresis on a 1% agarose gel. Four micrograms of the linear fragments, obtained in a final volume of 7 μl, was transformed into electrocompetent E. coli ED32 cells that had previously been transformed with plasmid pKD46, which facilitates the recombination of linear fragments (8). Recombinant strains carrying the gene tia interrupted by the Km resistance cassette were selected on TSA plates with 50 μg/ml Km. The partial replacement of the gene tia with the Km resistance cassette was verified by PCR amplification, considering that, when successful, a fragment of 1,047 bp instead of the 571-bp amplicon expected from the wild-type strain is produced with primers tiaUP and tiaLO (4). Moreover, another PCR was performed using the primers tiaLO (4) and Km_check_RV (Table 2), which anneal on a nondeleted region of the gene tia and on the Km resistance cassette, respectively. This PCR resulted in a fragment of 935 bp and confirmed that the Km resistance cassette had been inserted in the correct position.
Invasion assay.
The invasion assay was performed on Caco-2 cell monolayers as previously described (11) with some modification. Briefly, Caco-2 cells were grown in flasks in Dulbecco modified Eagle medium supplemented with 4.5 g/liter d-glucose, 20% fetal bovine serum, 1% glutamine, 1% minimal essential medium (MEM; with nonessential amino acids), and 1% penicillin-streptomycin (EuroClone, Milan, Italy). The medium was changed every day until a healthy monolayer was observed. The cells from the monolayer were detached, seeded in a 24-well plate with 500 μl/well of the medium described above but without the penicillin and streptomycin, and incubated at 37°C with 5% CO2 to form the monolayers used in the invasion assays. Single colonies of the E. coli strains were grown overnight in 2 ml of TSB at 37°C, except the positive-control strain, enteroinvasive E. coli (EIEC) strain 6.81, was grown at 30°C. Overnight cultures were then diluted 1:20 in fresh TSB and grown at 37°C (EIEC 6.81 was grown at 30°C) with shaking until they reached an optical density at 600 nm of 0.5, which corresponds to approximately 108 CFU/ml. Each strain was diluted 1:100 to reach a final concentration of 106 CFU/ml. The Caco-2 cell monolayers were washed with TSB and incubated for 3 h at 37°C in 5% CO2 with 106 CFU of each E. coli strain. After 3 h of incubation, the supernatant was removed and the monolayers were incubated with 1 ml of TSB with 100 μg/ml of gentamicin for 2 h at 37°C in 5% CO2. The supernatant was then removed and the monolayers were lysed with 1 ml phosphate-buffered saline (PBS)–1% Triton X-100 to release the internalized bacteria. Different dilutions of the cell lysate were plated on TSA plates to determine the number of CFU.
The invasion assay on HEp-2 cell monolayers was performed as described above for Caco-2 cells but with the following modification. HEp-2 cells were grown in a 24-well plate with 500 μl/well of RPMI 1640 supplemented with 10% fetal bovine serum, 1% glutamine, 1% sodium pyruvate, and 1% MEM (with nonessential amino acids) but not penicillin and streptomycin. Cells were incubated at 37°C with 5% CO2 for 1 or 2 days, until they formed a monolayer.
The quantitative data reported for the invasion assays are the means from three independent experiments, each of which was performed in triplicate. Statistical analysis was performed using the two-tailed Student's t test.
Quantitative analysis of tia expression.
To compare the expression of the gene tia in the bacteria inside or outside the cell monolayers, we extracted the total RNA from the bacteria from these two locations. Bacteria outside the cells were recovered after incubation with HEp-2 cell monolayers during the invasion experiments, and the total RNA was extracted with a total RNA extraction kit (Norgen Biotek Corp., Thorold, ON, Canada) according to the manufacturer's instructions, including the optional on-column DNase digestion step, performed using an RNase-free DNase set (Qiagen, Chatsworth, CA, USA) to eliminate genomic DNA. The RNA obtained in this step was further purified of genomic DNA contamination by using the RNA cleanup protocol of the RNeasy minikit (Qiagen, Chatsworth, CA, USA). The bacteria inside the HEp-2 cell monolayers were recovered after treatment with Gm, and the total RNA was extracted with the total RNA extraction kit with some modification. Briefly, after treatment with Gm, the HEp-2 cell monolayer was washed two times with PBS and then lysed with 350 μl of lysis solution according to the manufacturer's instructions. The cell lysates from both the HEp-2 cells and the internalized bacteria were centrifuged for 10 min at 12,000 rpm, and the pellet was resuspended in 100 μl of TE (Tris-EDTA) buffer with 1 mg/ml lysozyme. From this step, the protocol provided by the RNA extraction kit in the section on lysate preparation from bacteria, including the optional on-column DNA removal protocol, performed with an RNase-free DNase set (Qiagen, Chatsworth, CA, USA), was followed. The synthesis of cDNA from the total purified RNA was performed by using random hexamer primers and a QuantiTect reverse transcription kit (Qiagen, Chatsworth, CA, USA). Quantitative real-time reactions were performed using a SensiMix II probe kit (Bioline, London, UK) and the tia_RT primers and probes listed in Table 2, which were designed using Primer3web software (http://primer3.ut.ee/). The reaction involved incubation at 95°C for 10 min and 40 cycles of amplification at 95°C for 15 s and 55°C for 60 s. The housekeeping gene gapA (10) was used as the internal control to calculate the relative fold change in gene expression by the 2−ΔΔCT threshold cycle (CT) method (12). The analysis was performed in duplicate, and statistical significance was calculated by the chi-square test.
ACKNOWLEDGMENTS
R.B. generated the strains, set up the experimental settings, and drafted the manuscript, P.C. performed the invasion assays, V.M. contributed to the scientific discussion and to the setup of some experiments and also helped with the revision of the draft manuscript, F.M. isolated and characterized the wild-type strains, A.C. contributed to the revision of the draft manuscript, and S.M. conceived the study and strongly contributed to the scientific discussion and to the revision of the manuscript.
REFERENCES
- 1.Caprioli A, Morabito S, Brugère H, Oswald E. 2005. Enterohaemorrhagic Escherichia coli: emerging issues on virulence and modes of transmission. Vet Res 36:289–311. doi: 10.1051/vetres:2005002. [DOI] [PubMed] [Google Scholar]
- 2.Cordeiro F, da Silva RI, Vargas-Stampe TL, Cerqueira AM, Andrade JR. 2013. Cell invasion and survival of Shiga toxin-producing Escherichia coli within cultured human intestinal epithelial cells. Microbiology 159:1683–1694. doi: 10.1099/mic.0.064204-0. [DOI] [PubMed] [Google Scholar]
- 3.Bielaszewska M, Mellmann A, Zhang W, Köck R, Fruth A, Bauwens A, Peters G, Karch H. 2011. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect Dis 11:671–676. doi: 10.1016/S1473-3099(11)70165-7. [DOI] [PubMed] [Google Scholar]
- 4.Michelacci V, Tozzoli R, Caprioli A, Martínez R, Scheutz F, Grande L, Sánchez S, Morabito S. 2013. A new pathogenicity island carrying an allelic variant of the subtilase cytotoxin is common among Shiga toxin producing Escherichia coli of human and ovine origin. Clin Microbiol Infect 19:E149–E156. doi: 10.1111/1469-0691.12122. [DOI] [PubMed] [Google Scholar]
- 5.Fleckenstein JM, Kopecko DJ, Warren RL, Elsinghorst EA. 1996. Molecular characterization of the tia invasion locus from enterotoxigenic Escherichia coli. Infect Immun 64:2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tozzoli R, Caprioli A, Cappannella S, Michelacci V, Marziano ML, Morabito S. 2010. Production of the subtilase AB5 cytotoxin by Shiga toxin-negative Escherichia coli. J Clin Microbiol 48:178–183. doi: 10.1128/JCM.01648-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sansonetti PJ, d'Hauteville H, Formal SB, Toucas M. 1982. Plasmid-mediated invasiveness of “Shigella-like” Escherichia coli. Ann Microbiol (Paris) 133:351–355. [PubMed] [Google Scholar]
- 8.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 10.Fitzmaurice J, Glennon M, Duffy G, Sheridan JJ, Carroll C, Maher M. 2004. Application of real-time PCR and RT-PCR assays for the detection and quantitation of VT 1 and VT 2 toxin genes in E. coli O157:H7. Mol Cell Probes 18:123–132. doi: 10.1016/j.mcp.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Elsinghorst EA. 1994. Measurement of invasion by gentamicin resistance. Methods Enzymol 236:405–420. doi: 10.1016/0076-6879(94)36030-8. [DOI] [PubMed] [Google Scholar]
- 12.Schmittgen TD. 2001. Real-time quantitative PCR. Methods 25:383–385. doi: 10.1006/meth.2001.1260. [DOI] [PubMed] [Google Scholar]
- 13.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]