

ABSTRACT
Stenotrophomonas maltophilia is an emerging, opportunistic nosocomial pathogen that can cause severe disease in immunocompromised individuals. We recently identified the StmPr1 and StmPr2 serine proteases to be the substrates of the Xps type II secretion system in S. maltophilia strain K279a. Here, we report that a third serine protease, StmPr3, is also secreted in an Xps-dependent manner. By constructing a panel of protease mutants in strain K279a, we were able to determine that StmPr3 contributes to the previously described Xps-mediated rounding and detachment of cells of the A549 human lung epithelial cell line as well as the Xps-mediated degradation of fibronectin, fibrinogen, and the cytokine interleukin-8 (IL-8). We also determined that StmPr1, StmPr2, and StmPr3 account for all Xps-mediated effects toward A549 cells and that StmPr1 contributes the most to Xps-mediated activities. Thus, we purified StmPr1 from the S. maltophilia strain K279a culture supernatant and evaluated the protease's activity toward A549 cells. Our analyses revealed that purified StmPr1 behaves more similarly to subtilisin than to trypsin. We also determined that purified StmPr1 likely induces cell rounding and detachment of A549 cells by targeting cell integrin-extracellular matrix connections (matrilysis) as well as adherence and tight junction proteins for degradation. In this study, we also identified anoikis as the mechanism by which StmPr1 induces the death of A549 cells and found that StmPr1 induces A549 IL-8 secretion via activation of protease-activated receptor 2. Altogether, these results suggest that the degradative and cytotoxic activities exhibited by StmPr1 may contribute to S. maltophilia pathogenesis in the lung by inducing tissue damage and inflammation.
KEYWORDS: A549 cells, PAR2, Stenotrophomonas maltophilia, StmPr1, StmPr3, Xps, anoikis, matrilysis, serine protease, type II secretion
INTRODUCTION
Stenotrophomonas maltophilia is a Gram-negative bacterium found in soil and aqueous sources that has emerged as an important, opportunistic human pathogen (1–3). Although considered relatively nonvirulent, S. maltophilia causes severe disease in immunocompromised individuals, such as those with severe burns, cystic fibrosis, and HIV infection, as well as patients receiving chemotherapy or immunosuppressive therapy (4). Pneumonia is the most common infection associated with S. maltophilia, and recently, surveillance studies found S. maltophilia to be the 6th most common cause of respiratory tract infections worldwide (5). Bacteremia is the second most common infection caused by S. maltophilia, but the pathogen has also been implicated in bone, soft tissue, eye, urinary tract, heart, and brain infections (1, 2, 5). S. maltophilia has long been considered a major problem in the hospital setting, especially in intensive care units, where S. maltophilia infection prevalence rates were most recently determined to be 1.4 to 3.0% (5). In the general population, S. maltophilia infection prevalence rates increased from 0.8% to 1.4% from 1997 to 2003 to 1.3 to 1.68% from 2007 to 2012 (5), indicating that S. maltophilia infections are on the rise in both hospitals and the community. Studies documenting community-acquired S. maltophilia infections (6–8), one of which recently described an infection in an immunocompetent individual (8), provide further evidence that S. maltophilia is also emerging as a community-associated pathogen. The intrinsic multidrug-resistant nature of S. maltophilia makes treatment of infections very difficult (5, 9). The antibiotic trimethoprim-sulfamethoxazole, which is currently the drug of choice for combating S. maltophilia infections, has been losing effectiveness (9–12), and the WHO recently listed S. maltophilia as one of the top drug-resistant pathogens found in hospitals worldwide (9). Much progress has been made with regard to understanding the antibiotic resistance mechanisms employed by S. maltophilia (5), but the virulence factors utilized by the pathogen to cause disease are still largely uncharacterized (13).
Our laboratory has recently described the effects of the Xps type II secretion (T2S) system from the S. maltophilia clinical isolate K279a on several cell lines, including the A549 lung epithelial cell line (14, 15). Specifically, we observed that the Xps T2S system causes A549 cell rounding, actin rearrangement, cell detachment, and cell death (15). We also ascribed a number of degradative activities to Xps, such as degradation of the extracellular matrix (ECM) components collagen type I, fibronectin, and fibrinogen, as well as degradation of the cytokine interleukin-8 (IL-8) (14). We went on to identify the serine proteases StmPr1 and StmPr2, which belong to the S8 peptidase family of subtilases, as secreted substrates of the Xps T2S system (14). StmPr1 and StmPr2 accounted for the Xps-mediated serine protease, gelatinase, and caseinolytic activities exhibited by strain K279a culture supernatants (14). StmPr1 accounted for all observed Xps-mediated collagen type I degradation and was also completely responsible for the Xps-mediated death of A549 cells (14). However, StmPr1 and StmPr2 did not explain all of the known Xps-mediated activities. A549 cell rounding and detachment were observed after 24 h of incubation with culture supernatants lacking StmPr1 and StmPr2 (14). Similarly, the Xps-mediated degradative activity toward fibronectin, fibrinogen, and IL-8 was not completely abolished in culture supernatants lacking StmPr1 and StmPr2 (14). These data indicate that an additional Xps substrate(s) has yet to be identified.
In this study, we show that another protease (StmPr3) contributes to Xps-mediated A549 cell rounding and detachment, as well as fibronectin, fibrinogen, and IL-8 degradation. Furthermore, with the exception of IL-8 degradation, StmPr1, StmPr2, and StmPr3 are completely responsible for these Xps activities, although we did confirm that SmPr1 contributes the most out of the three proteases. Herein, we also identify cell-ECM connections and tight and adherence junctions to be targets of StmPr1 degradation, determine that StmPr1 induces anoikis in A549 cells, and show StmPr1-mediated activation of protease-activated receptor 2 (PAR2).
RESULTS
S. maltophilia strain K279a produces a third Xps-secreted serine protease, StmPr3.
We first set out to determine what protease(s), in addition to StmPr1 and StmPr2, contributes to the previously described Xps-mediated cell rounding/detachment and degradative activities of S. maltophilia strain K279a (14). Two other S. maltophilia S8 peptidases, denoted StmPr3 and StmPr4, were previously identified, but they were studied only recombinantly in Escherichia coli with regard to their potential use as commercial detergents (16). BLAST analysis of the K279a genome indicated that the strain carries the stmPr3 (SMLT_RS20900) gene but not the stmPr4 gene. Alignment of StmPr3 with StmPr1 and StmPr2 from strain K279a revealed that StmPr3 shares 25.1% amino acid sequence identity and 48.2% amino acid sequence similarity to StmPr1 and shares 27.7% amino acid sequence identity and 50.7% amino acid sequence similarity to StmPr2 (Fig. S1). Like StmPr1 and StmPr2, StmPr3 is predicted to have a conserved catalytic triad (Asp-His-Ser) and an N-terminal signal sequence, making it a likely candidate for a type II secreted substrate (14, 16). To determine whether StmPr3 is secreted by a T2S system, we employed the same approach used previously to confirm that StmPr1 and StmPr2 are Xps substrates (14). StmPr3, which was engineered to have a C-terminal His tag, was expressed via a plasmid in wild-type (WT) K279a, as well as in the xpsF and gspF T2S mutants. SDS-PAGE and immunoblot analysis of early-stationary-phase culture supernatants revealed that StmPr3-His was secreted by the WT strain and the gspF mutant but not the xpsF mutant (Fig. 1A). These results indicate that StmPr3, like StmPr1 and StmPr2, is secreted in an Xps-dependent manner.
FIG 1.
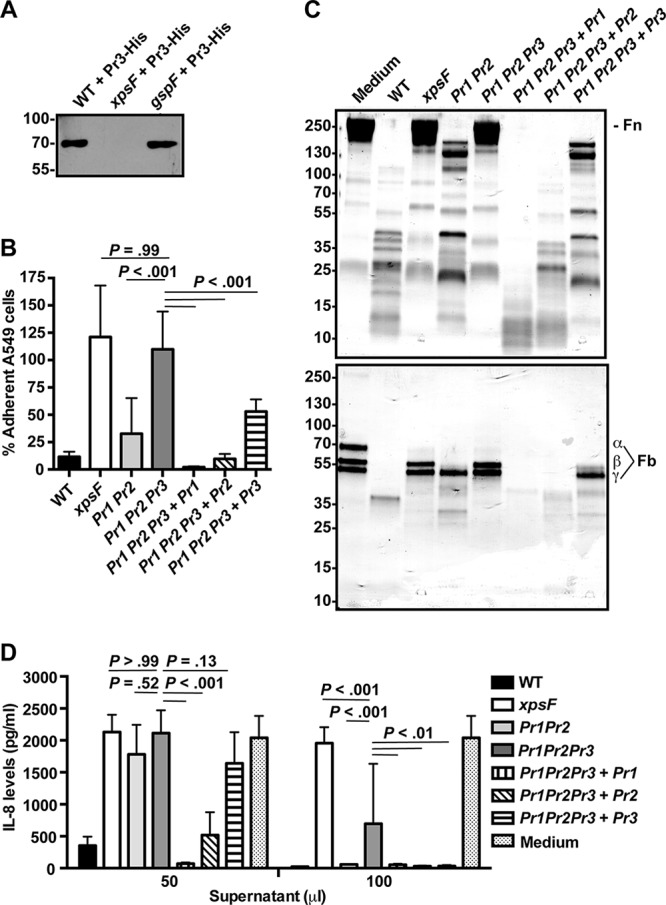
Contribution of StmPr3 to Xps-mediated A549 cell detachment and Xps-mediated degradative activities. (A) His-tagged StmPr3 (Pr3-His) was exogenously produced from a plasmid in strain K279a (WT), xpsF mutant NUS4 (xpsF), and gspF mutant NUS1 (gspF). Supernatants collected from the cultures of these strains were analyzed by SDS-PAGE, followed by immunoblotting for anti-His. The migration of molecular mass standards (in kilodaltons) is indicated to the left of the gel images. (B) A549 cells were incubated for 24 h with 25% (vol/vol) culture supernatant collected from the WT strain, xpsF mutant NUS4, stmPr1 stmPr2 mutant NUS7 (Pr1 Pr2), stmPr1 stmPr2 stmPr3 mutant NUS14 (Pr1 Pr2 Pr3), or the stmPr1 stmPr2 stmPr3 mutant complemented with stmPr1 (Pr1 Pr2 Pr3 + Pr1), stmPr2 (Pr1 Pr2 Pr3 + Pr2), or stmPr3 (Pr1 Pr2 Pr3 + Pr3). Cell detachment was determined by quantifying the remaining adherent cells. Data were normalized to those for cells treated with medium alone, for which adherence was set at 100%. (C) Ten micrograms of human fibronectin (Fn; top) or fibrinogen (Fb; bottom) was incubated at 37°C for 16 h with 25 μl of culture supernatant from the WT, the xpsF mutant, the indicated protease mutant and complemented strains, or a control treated with medium alone. ECM protein degradation was analyzed by SDS-PAGE and total protein staining with Coomassie. The fibrinogen α, β, and γ chains are indicated. The migration of molecular mass standards (in kilodaltons) is indicated to the left of the gel images. Data are representative of those from three independent experiments. (D) Recombinant IL-8 was incubated at 37°C for 16 h with 50 or 100 μl of culture supernatant collected from the WT strain, the xpsF mutant, the indicated protease mutant and complemented strains, or a control treated with medium alone. IL-8 levels were quantified by ELISA. For panels B and D, the data are represented as the mean and SD from three independent experiments.
StmPr3 contributes to Xps-mediated A549 cell detachment and Xps-mediated degradation of ECM components and IL-8.
We next constructed several stmPr3 mutants of strain K279a to evaluate the role of StmPr3 in a panel of assays where StmPr1 and StmPr2 did not account for all of the Xps-mediated activity. In addition to the previously constructed stmPr1, stmPr2, and stmPr1 stmPr2 mutants, for this study we made an stmPr3 mutant, stmPr1 stmPr3 and stmPr2 stmPr3 double mutants, and an stmPr1 stmPr2 stmPr3 triple mutant. All of the newly generated mutants grew similarly to the WT strain in broth culture (data not shown), indicating that the mutations do not result in a general growth defect. The secretion profiles of the stmPr3 mutants were analyzed by SDS-PAGE and total protein staining to confirm that these strains lacked StmPr3 (see Fig. S2 in the supplemental material). The parental strain and the stmPr1 stmPr2 mutant were also included in the analysis for comparison. An approximately 70-kDa band was observed in the WT and stmPr1 stmPr2 mutant but was absent from all mutants lacking stmPr3 (Fig. S2). The predicted size of StmPr3 is approximately 58 kDa, but the size of the StmPr3-His band observed by immunoblotting was also approximately 70 kDa (Fig. 1A). These data indicate that the 70-kDa band missing from the stmPr3 mutants corresponds to StmPr3 and that these strains indeed lack stmPr3.
In our previous study, we found that StmPr1 and StmPr2 did not account for all of the observed Xps-mediated A549 cell detachment at 24 h postincubation with strain K279a culture supernatants (14). Therefore, we were interested in seeing how the stmPr1 stmPr2 stmPr3 triple mutant compared to the stmPr1 stmPr2 mutant in the A549 cell detachment assay. As previously reported (14), incubation of A549 cells with culture supernatants from the stmPr1 stmPr2 mutant resulted in WT levels of cell detachment, whereas the xpsF mutant supernatant did not cause cell detachment (Fig. 1B). When the supernatant from the stmPr1 stmPr2 stmPr3 triple mutant was tested in this assay, it also did not promote cell detachment, and no significant difference between the stmPr1 stmPr2 stmPr3 mutant and the xpsF mutant was observed (Fig. 1B). The stmPr1 stmPr2 stmPr3 mutant's inability to cause A549 cell detachment could be restored by providing stmPr1, stmPr2, or stmPr3 in trans (Fig. 1B). These results indicate that StmPr1, StmPr2, and StmPr3 all contribute to A549 cell detachment and that these proteases account for all of the observed Xps-mediated A549 cell detachment caused by strain K279a.
Similar to the results of the A549 cell detachment assay, our previous study found that StmPr1 and StmPr2 did not account for all of the observed Xps-mediated degradation of the ECM components fibronectin and fibrinogen (14). To investigate whether StmPr3 contributes to the degradation of these ECM components, the stmPr1 stmPr2 stmPr3 mutant supernatant was compared to the WT, xpsF, and stmPr1 stmPr2 mutant supernatants upon incubation with purified human fibronectin and fibrinogen. When ECM component stability was monitored by SDS-PAGE and total protein staining, we observed that, unlike the stmPr1 stmPr2 mutant supernatant, the stmPr1 stmPr2 stmPr3 mutant supernatant behaved like the xpsF mutant supernatant and did not degrade fibronectin and fibrinogen (Fig. 1C). In this assay, the stmPr1 stmPr2 stmPr3 mutant could also be complemented with the genes for any of the three proteases that it lacked, although to various degrees. Complementation of the stmPr1 stmPr2 stmPr3 mutant with the stmPr1 gene restored the most degradative activity toward fibronectin and fibrinogen, followed by complementation with stmPr2 and complementation with stmPr3, in that order (Fig. 1C). We concluded that StmPr3 also contributes to fibronectin and fibrinogen degradation and that StmPr1, StmPr2, and StmPr3 are completely responsible for the Xps-mediated degradation of these ECM components.
Finally, we also tested whether StmPr3 contributes to Xps-mediated degradation of the cytokine IL-8, which we previously showed was predominantly, but not entirely, due to StmPr1 and StmPr2 (14). When purified recombinant IL-8 was incubated with 50 μl of supernatant and IL-8 stability was monitored by enzyme-linked immunosorbent assay (ELISA), we observed no difference between the xpsF, stmPr1 stmPr2, and stmPr1 stmPr2 stmPr3 culture supernatants, none of which caused degradation of IL-8 (Fig. 1D). However, at a high dose of supernatant (100 μl), the stmPr1 stmPr2 stmPr3 mutant supernatant caused significantly less IL-8 degradation than the stmPr1 stmPr2 mutant supernatant (Fig. 1D). The stmPr1 stmPr2 stmPr3 mutant supernatant also caused significantly more IL-8 degradation than the xpsF mutant supernatant at the 100-μl dose, indicating that an additional Xps-secreted substrate contributes to the degradation of IL-8 (Fig. 1D). At the 100-μl dose, the stmPr1 stmPr2 stmPr3 mutant was equally complemented with stmPr1, stmPr2, or stmPr3, but at the 50-μl dose, complementation of the stmPr1 stmPr2 stmPr3 mutant was achieved only with stmPr1 and stmPr2. From these data, we concluded that while StmPr3 does contribute to IL-8 degradation, StmPr1, StmPr2, and StmPr3 do not entirely account for the Xps-mediated degradation of IL-8.
StmPr1 is predominantly responsible for Xps-mediated activities.
While we were initially interested in comparing the stmPr1 stmPr2 stmPr3 mutant with the stmPr1 stmPr2 mutant to elucidate the role of StmPr3 (Fig. 1), performance of the cell detachment and degradation assays with the full panel of protease mutants revealed the relative importance of StmPr1, StmPr2, and StmPr3. Deletion of only stmPr1, stmPr2, or stmPr3 did not affect the ability of the strain K279a culture supernatant to cause A549 cell detachment upon 3 h (Fig. 2A) or 24 h (data not shown) of incubation. When the stmPr1 stmPr2, stmPr1 stmPr3, and stmPr2 stmPr3 double protease mutants were evaluated, all supernatants showed comparable cell detachment activity after 24 h of incubation with A549 cells (data not shown). However, after a 3-h incubation, the stmPr1 stmPr2 and stmPr1 stmPr3 mutant supernatants caused significantly less A549 cell detachment than the stmPr2 stmPr3 mutant supernatant, which behaved like the WT supernatant (Fig. 2A). Furthermore, there was no significant difference between the xpsF, stmPr1 stmPr3, stmPr1 stmPr2, and stmPr1 stmPr2 stmPr3 mutant supernatants at the 3-h time point (Fig. 2A). These data indicate that the loss of stmPr1 causes the greatest effect on the ability of the strain K279a culture supernatant to cause A549 cell detachment.
FIG 2.
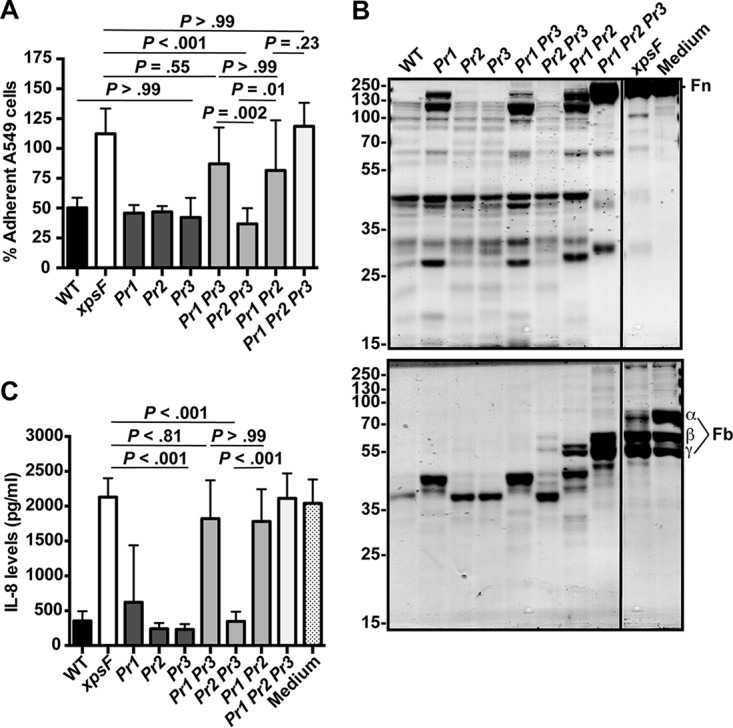
Predominant contribution of StmPr1 to Xps-mediated activities. (A) A549 cells were incubated for 3 h with 25% (vol/vol) culture supernatant collected from the WT, xpsF mutant NUS4, stmPr1 mutant NUS5 (Pr1), stmPr2 mutant NUS6 (Pr2), stmPr3 mutant NUS11 (Pr3), stmPr1 stmPr3 mutant NUS12 (Pr1 Pr3), stmPr2 stmPr3 mutant NUS13 (Pr2 Pr3), stmPr1 stmPr2 mutant NUS7 (Pr1 Pr2), or stmPr1 stmPr2 stmPr3 mutant NUS14 (Pr1 Pr2 Pr3). Cell detachment relative to that for control cells treated with medium alone was determined as described in the legend to Fig. 1. (B) Human fibronectin (Fn; top) and fibrinogen (Fb; bottom) were incubated at 37°C for 16 h with 25 μl of culture supernatant from the WT, the xpsF mutant, the indicated protease mutant strains, or a control treated with medium alone. ECM protein degradation was analyzed as described in the legend to Fig. 1. The fibrinogen α, β, and γ chains are indicated. The migration of molecular mass standards (in kilodaltons) is indicated to the left of the gel images. Although the samples were examined on the same gel, they were not in adjacent lanes, and therefore, we cropped out intervening lanes that were not pertinent to the analysis. Data are representative of those from three independent experiments. (C) Recombinant IL-8 was incubated at 37°C for 16 h with 50 μl of culture supernatant collected from the WT strain, the xpsF mutant, the indicated protease mutant strains, or a control treated with medium alone. IL-8 levels were quantified by ELISA. For panels B and D, data are represented as the mean and SD from three independent experiments.
The predominant contribution of StmPr1 to Xps-mediated activities was also evident when the degradation of fibronectin, fibrinogen, and IL-8 was evaluated using the full panel of protease mutants. The ECM degradation pattern caused by the stmPr2 and stmPr3 mutant supernatants was no different from that caused by the WT supernatant, and out of the single mutants, only the stmPr1 mutant supernatant was attenuated for degradation (Fig. 2B). As was observed in the A549 cell detachment assay, the double mutants lacking stmPr1 (the stmPr1 stmPr2 and stmPr1 stmPr3 double mutants) were more attenuated at causing ECM component degradation than the stmPr2 stmPr3 mutant (Fig. 2B). Moreover, these patterns seen among the single and double protease mutants in the ECM component degradation assays were also observed when the degradation of IL-8 was evaluated using the full panel of mutants (Fig. 2C). These data confirm that StmPr1 contributes the most to the known Xps-mediated activities. Therefore, we decided to focus our efforts on elucidating the mechanism by which StmPr1 acts.
Purified StmPr1 is sufficient to cause A549 cell rounding and detachment.
To better study the mechanism of action of the StmPr1 protease, we purified StmPr1 from the S. maltophilia strain K279a culture supernatant using benzamidine-Sepharose. This approach was employed to purify a serine protease from Vibrio cholerae on the basis of the property that benzamidine is a serine protease inhibitor that binds specifically to serine proteases (17). We chose to purify StmPr1 from the supernatant of the stmPr1 stmP2 stmPr3 mutant complemented with stmPr1 to avoid contamination by StmPr2 and StmPr3. We also performed a mock purification with supernatant from the stmPr1 stmP2 stmPr3 mutant with an empty vector. When purified StmPr1 was analyzed by SDS-PAGE and total protein staining with SYPRO Ruby, we observed a band at approximately 47 kDa that was not present in the mock purification (Fig. 3A and S3A) and that corresponds to the expected size of StmPr1 (14). We also noted the appearance of smaller bands in the purified material that were absent from the mock purification (Fig. 3A and S3A). These less abundant bands may be indicative of some StmPr1 degradation or a more processed version of StmPr1, as we have observed that StmPr1 is likely autoprocessed at both the N and C termini (14). Most serine proteases typically fall into one of two major clades, a trypsin-like clade or a subtilisin-like clade (18), but a StmPr1 variant purified from a S. maltophilia bronchoalveolar lavage (BAL) fluid isolate was previously classified as both trypsin-like and subtilisin-like with regard to cleavage specificity (19). Therefore, to determine if purified StmPr1 from strain K279a is more trypsin-like or subtilisin-like in its mechanism of action, we included subtilisin A purified from Bacillus licheniformis and trypsin purified from bovine pancreas in our analyses (Fig. 3A). To confirm the quality of our protein preparation, we tested the proteolytic activity of purified StmPr1 at various concentrations using N-succinyl–Ala–Ala–Pro-Phe–p-nitroanilide (pNA) as a substrate. Purified StmPr1 hydrolyzed the substrate in a dose-dependent manner, and the mock-purified sample exhibited no proteolytic activity toward the substrate (Fig. 3B and S3B). As expected, subtilisin A, but not trypsin, hydrolyzed the N-succinyl–Ala–Ala–Pro–Phe–pNA substrate, although not as efficiently as StmPr1 (Fig. 3B) (20, 21).
FIG 3.
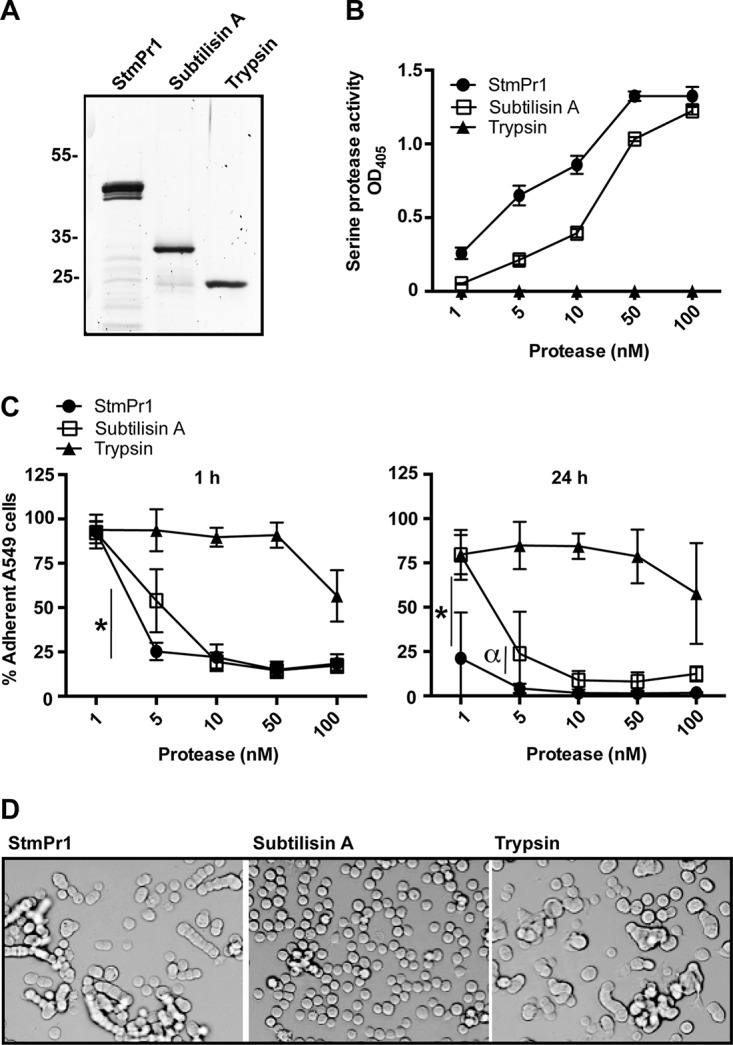
Characterization of the proteolytic and cell-detaching activities of purified StmPr1 compared to those of subtilisin A and trypsin. (A) StmPr1 was purified from the supernatant of the stmPr1 stmPr2 stmPr3 mutant complemented with stmPr1 using benzamidine-Sepharose. Purified StmPr1, subtilisin A, and trypsin (0.5 μg of each) were analyzed by SDS-PAGE, followed by SYPRO Ruby staining. (B) Purified StmPr1, subtilisin A, or trypsin was incubated with N-succinyl–Ala–Ala–Pro–Phe–pNA at the indicated equimolar concentrations for 60 min at 37°C to evaluate serine protease activity. The result for StmPr1 was statistically significantly different from the results for both subtilisin A and trypsin at all doses (P < 0.001). (C) A549 cells were incubated for 1 h or 24 h with the indicated equimolar concentrations of purified StmPr1, subtilisin A, or trypsin. Cell detachment was determined as described in the legend to Fig. 1. *, the difference between subtilisin A and StmPr1 was statistically significant (P < 0.001); α, the difference between subtilisin A and StmPr1 was statistically significant (P = 0.002). The results for both StmPr1 and subtilisin A were statistically significantly different from those for trypsin at the 5 to 100 nM doses (P < 0.001), and the results between StmPr1 and trypsin were also statistically significantly different at the 1 nM dose at 24 h (P < 0.001). (D) The morphology of A549 cells after 24 h of incubation with 100 nM StmPr1, subtilisin A, or trypsin was determined by phase-contrast light microscopy. Data are representative of those from three independent experiments. For panels B and C, data are represented as the mean and SD from three independent experiments.
We next tested whether purified StmPr1 was sufficient to cause A549 cell detachment. When incubated with purified StmPr1, approximately 75% of A549 cells rounded and detached from the tissue culture dish within 1 h of treatment with as little as 10 nM the protease, and similar results were observed with subtilisin A (Fig. 3C). StmPr1 at the 5 nM dose was also able to cause 75% of A459 cells to detach at 1 h, while subtilisin A at 5 nM caused significantly less A549 cell detachment (approximately 55%) at this time point (Fig. 3C). At 3 h postincubation, StmPr1 and subtilisin A behaved similarly with regard to A549 cell detachment at all doses tested (Fig. S4A). However, at 24 h postincubation with the proteases, StmPr1 again induced significantly more A549 cell detachment than subtilisin A at the 1 nM and 5 nM doses (Fig. 3C). Trypsin was able to induce A549 cell detachment only when 100 nM the protease was incubated with A549 cells for 1 h, and extending the incubation to 3 h (Fig. 3C) or 24 h (Fig. S4A) did not increase the amount of trypsin-induced detachment observed. When A549 cells were pretreated with the serine protease inhibitor phenylmethylsulfonyl fluoride (PMSF), the level of cell detachment induced by StmPr1 (and subtilisin A) was significantly reduced, indicating that the proteolytic activity of the proteases was responsible for the induced A549 cell detachment (Fig. S4B). Overall, these data indicate that StmPr1 is sufficient to cause A549 cell detachment and does so more efficiently than subtilisin A at low doses, while trypsin is the least efficient at inducing rounding and detachment of A549 cells. Interestingly, we did notice that the morphology of A549 cells after protease-induced rounding differed depending on which protease the cells were incubated with. StmPr1 (as well as trypsin) treatment resulted in chains of rounded and detached A549 cells, whereas subtilisin A treatment reduced the A549 cells to a single-cell suspension (Fig. 3D). We hypothesized that this result may indicate that these proteases degrade different cell targets or hydrolyze the same cell targets to various degrees. Therefore, we next wanted to determine what targets StmPr1 degrades to induce A549 cell rounding and detachment.
StmPr1 targets ECM connections as well as tight junction and adherence junctions to induce A549 cell rounding and detachment.
Based on the results of our earlier experiments (Fig. 1 and 2), we hypothesized that StmPr1 likely targets ECM connections to induce A549 cell rounding and detachment. Therefore, we next investigated whether StmPr1 degrades ECM proteins in the context of A549 cell culture and to compare the effects of StmPr1, subtilisin A, and trypsin on ECM protein degradation. A549 cells were incubated with equimolar concentrations of StmPr1, subtilisin A, and trypsin, and the stability of the major ECM proteins fibronectin and collagen type I was evaluated by immunoblot analysis with specific antibodies. Fibronectin was degraded by StmPr1 in a dose-dependent manner, as evidenced by the disappearance of full-length fibronectin (263 kDa) and the appearance of lower-molecular-mass fibronectin degradation products (Fig. 4A). Both subtilisin A and trypsin also degraded fibronectin, but StmPr1 and subtilisin A caused more extensive fibronectin degradation than trypsin. As little as 1 nM StmPr1 and subtilisin A degraded fibronectin when A549 cells were incubated with the proteases for 3 h, and the extent of fibronectin degradation observed with 1 nM StmPr1 and subtilisin A was comparable to that observed with 100 times the amount of trypsin (100 nM) (Fig. 4A). Moreover, degradation of fibronectin by StmPr1 was observed as early as 1 h postincubation, but at this time point subtilisin A caused more fibronectin degradation than StmPr1 (Fig. S5A). We were unable to detect collagen degradation when A549 cells were incubated with the proteases for 1 or 3 h (data not shown), but we did detect a dose-dependent degradation of collagen when collagen type I stability was assessed at 24 h postincubation (Fig. 4A). Similar to the findings for fibronectin, a loss of full-length collagen type I (400 kDa trimer) and the appearance of lower-molecular-mass degradation products were observed when A549 cells were incubated with StmPr1 (Fig. 4A). StmPr1 and subtilisin A degraded collagen to a greater extent than trypsin, with as little as 1 nM StmPr1 or subtilisin A inducing collagen degradation (Fig. 4A). Also, subtilisin A caused more collagen degradation than StmPr1 at the 10 nM dose of protease (Fig. 4A). From these data, we concluded that StmPr1 (and subtilisin A and trypsin) targets both fibronectin and collagen for degradation during A549 culture.
FIG 4.
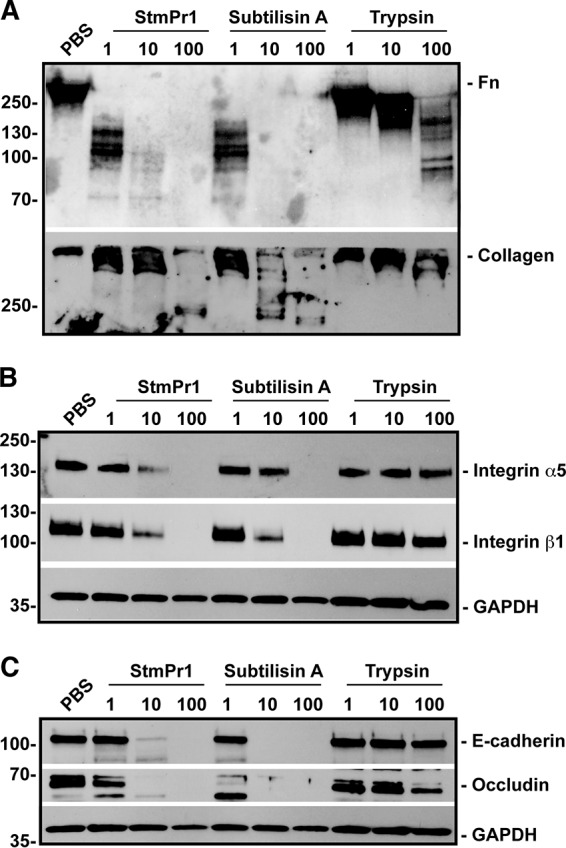
Effect of purified StmPr1 compared to the effects of subtilisin A and trypsin on ECM connections as well as adherence and tight junctions during A549 cell culture. A549 cells were incubated for 3 h (24 h for collagen analysis) with the indicated equimolar concentrations of purified StmPr1, subtilisin A, or trypsin, as well as a PBS-treated control. Cell culture supernatants (A) or cell lysates (B and C) were collected and analyzed by SDS-PAGE and immunoblot analysis with antifibronectin (Fn) and anti-collagen type I antibodies (A), anti-integrin α5 and anti-integrin β1 antibodies (B), and anti-E-cadherin and anti-occludin antibodies (C). The migration of molecular mass standards (in kilodaltons) is indicated to the left of the gel images. For cell lysates (B and C), immunoblot analysis with anti-GAPDH was also performed to confirm equal loading. Data are representative of those from three independent experiments.
Of the two ECM proteins evaluated, fibronectin was the most susceptible to degradation by StmPr1. Therefore, we decided to also examine the stability of the fibronectin receptor integrin α5β1, which is utilized by cells to anchor to fibronectin in the ECM and may also be targeted by StmPr1 to induce cell detachment. Similar to the results of the ECM degradation assays, we performed immunoblot analysis utilizing antibodies specific to the integrin α5 and integrin β1 subunits on A549 cell lysates after incubation of the cells with StmPr1, subtilisin A, or trypsin. StmPr1 degraded both integrin subunits, as evidenced by the loss or reduction of the band corresponding to full-length integrin α5 (115 kDa predicted, 145 kDa detected) and integrin β1 (88 kDa predicted, 115 kDa detected) following 1-h (Fig. S5B) and 3-h (Fig. 4B) incubations, although we did observe that the integrin was less susceptible than fibronectin to StmPr1-mediated degradation, as integrin degradation was observed only at a dose as low as 10 nM at 3 h (Fig. 4B). Subtilisin A degraded both integrin subunits to a similar degree as StmPr1, but trypsin did not degrade either of the integrin subunits at any of the doses or time points tested (Fig. 4B and S5B). These results demonstrate that StmPr1 (and subtilisin A) targets both fibronectin itself and the fibronectin receptor integrin α5β1 for degradation.
To investigate whether StmPr1 can also degrade tight and adherence junction proteins, we assessed the stability of E-cadherin and occludin in the A549 cell culture by immunoblot analysis with specific antibodies. The loss of a band corresponding to the full-length adherence junction protein E-cadherin (120 kDa) indicated that StmPr1 degrades E-cadherin as early as 1 h postincubation, although not as efficiently as subtilisin A (Fig. 4C and S5C). The tight junction protein occludin was also degraded by StmPr1, evidenced by the loss of full-length occludin (59 kDa) (Fig. 4C and S5C). However, subtilisin A degraded occludin to a greater extent than StmPr1, as subtilisin A, but not StmPr1, caused degradation of occludin at the 1 nM dose at 1 h and 3 h postincubation (Fig. 4C and S5C). Like the fibronectin receptor, both E-cadherin and occludin were resistant to degradation by trypsin at the doses and time points tested (Fig. 4C and S5C). These data indicate that StmPr1 and subtilisin A degrade the adherence and tight junction proteins E-cadherin and occludin.
StmPr1 induces anoikis in A549 cells.
In our previous study, we observed that A549 cells incubated with strain K279a culture supernatants for 24 h were less viable than cells treated with a medium control and that this drop in viability was dependent on StmPr1 (14). To determine if StmPr1 can directly cause a decrease in A549 viability, we incubated A549 cells with various concentrations of purified StmPr1, as well as subtilisin A and trypsin for comparison, and monitored the health of the cells over time using a metabolic dye. When cells were incubated for 3 h with purified StmPr1 or either of the other proteases, no loss of cell viability was observed (Fig. 5A). However, when A549 cells were incubated with StmPr1 for 24 h, we did observe a dose-dependent decrease in cell viability, with an approximately 30% decrease in viability occurring with the 50 and 100 nM doses of StmPr1 (P < 0.001) (Fig. 5A). These results were comparable to the StmPr1-mediated decrease in viability caused by the strain K279a culture supernatant, which was also approximately 30% (14). Subtilisin A also caused a loss of A549 cell viability, approximately 20% at the 100 nM dose (P < 0.001), though this was significantly less than the decrease in viability caused by StmPr1 (Fig. 5A). Trypsin treatment of the A549 cells for 24 h had the least effect, causing an approximately 10% decrease in cell viability at the 100 nM dose (P < 0.001) (Fig. 5A). From these data, we concluded that StmPr1 is sufficient to cause A549 cell death and that StmPr1 causes the most A549 cell death relative to that caused by subtilisin A and trypsin.
FIG 5.
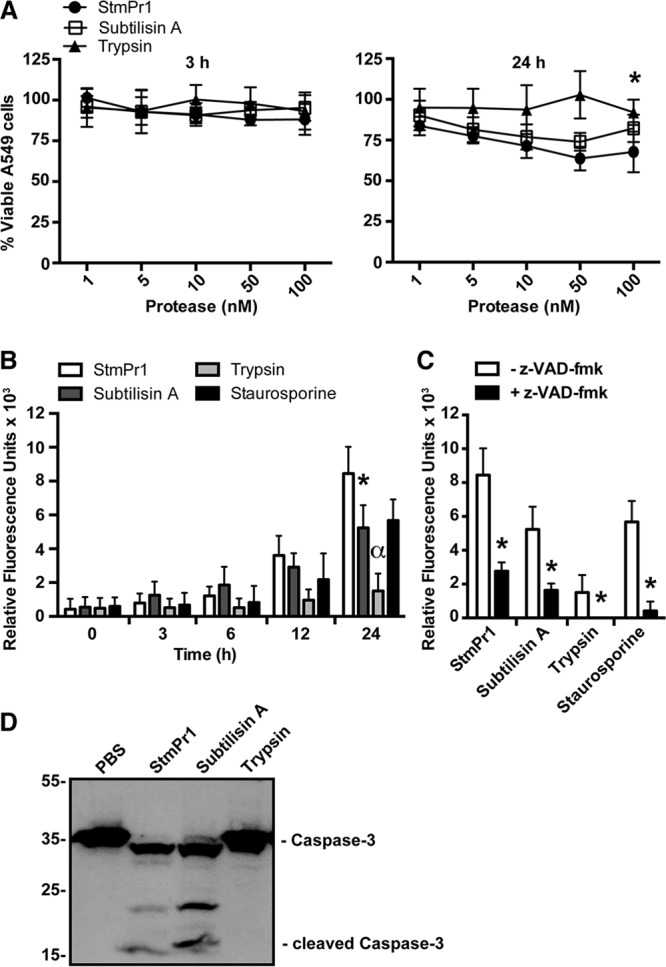
Effect of purified StmPr1 compared to the effects of subtilisin A and trypsin on A549 cell viability. (A) A549 cells were incubated for 3 h and 24 h with the indicated equimolar concentrations of purified StmPr1, subtilisin A, or trypsin. Cell viability was measured using the metabolic dye PrestoBlue. Data were normalized to those for cells treated with PBS alone, the viability of which was set at 100%. *, statistically significant difference from the result for StmPr1 for both subtilisin A and trypsin (P < 0.001). (B) A549 cells were incubated with 100 nM purified StmPr1, subtilisin A, or trypsin for 24 h. Caspase-3/7 activity was monitored at the indicated times over the 24-h time course using CellEvent caspase-3/7 green detection reagent, where the background level of caspase activity for a PBS-treated control was subtracted from the values for the experimental samples. Staurosporine was included as a positive control for caspase activation. *, statistically significant difference from the result for StmPr1 (P < 0.001); α, statistically significant difference from the results for StmPr1, subtilisin A, and staurosporine (P < 0.001). (C) Caspase-3/7 activity was monitored as described in the legend to panel B after 24 h of incubation with 100 nM purified StmPr1, subtilisin A, or trypsin in the presence or absence of the pan-caspase inhibitor Z-VAD-FMK. *, statistically significant difference from the result obtained in the absence of Z-VAD-FMK (P < 0.001). (D) A549 cells were incubated with 100 nM purified StmPr1, subtilisin A, or trypsin for 24 h. Cell lysates were collected and analyzed by SDS-PAGE and immunoblot analysis with an anti-caspase-3 antibody. The sizes of caspase-3 and cleaved caspase-3 are indicated. The migration of molecular mass standards (in kilodaltons) is indicated to the left of the gel images. Data are representative of those from three independent experiments. Data are represented as the mean and SD from three independent experiments.
Cells that lose connection with the ECM typically undergo a form of detachment-induced apoptosis known as anoikis (22, 23). Detachment is sensed by the cell through a loss of signaling through integrin-ECM connections, which then triggers apoptosis to occur through induction of the canonical caspase cascade (22, 23). The caspase cascade relies on initiator caspases to cleave and activate the executioner caspases, which include caspases 3, 6, and 7 (24). Because StmPr1 targets integrin-ECM connections for degradation (Fig. 4) and induces A549 cell detachment and death (Fig. 3 and 5A), we hypothesized that the A549 cells exposed to StmPr1 were likely dying through anoikis. To test this hypothesis, we kinetically monitored effector caspase activation in A549 cells incubated with StmPr1 using a DEVD peptide-linked fluorescent dye, which binds only DNA and fluoresces when the DEVD inhibitory peptide is cleaved by activated caspase-3 and caspase-7. Staurosporine is a well-known inducer of apoptosis and therefore was used as a positive control in this assay. Similar to the findings for the staurosporine-treated control, we observed that StmPr1 induced caspase activation in A549 cells over time, with maximum caspase activation occurring at 24 h postincubation (Fig. 5B). In line with the results from the cell viability assay, we observed that subtilisin A also induced caspase activation in A549 cells, although the level of caspase activation induced by subtilisin A was significantly less than the level of caspase activation induced by StmPr1 at the 24-h time point (Fig. 5B). Some caspase activation induced by trypsin was detected at the 24-h time point, but the level of caspase activation was significantly less than that induced by StmPr1, subtilisin A, and staurosporine (Fig. 5B). Importantly, when A549 cells were pretreated with the pan-caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp(O-methyl)-fluoromethylketone (Z-VAD-FMK) prior to incubation with StmPr1, subtilisin A, trypsin, or staurosporine, caspase-3/7 activation was significantly inhibited (Fig. 5C). We next examined whether we could detect cleaved caspase-3 in A549 cells incubated with StmPr1 by immunoblot analysis with a caspase-3-specific antibody. Consistent with the results of the kinetic caspase activation assay, when A549 cells were incubated with StmPr1 for 24 h, we detected a reduction in full-length caspase-3 (35 kDa) and the appearance of lower-molecular-mass bands corresponding to cleaved caspase-3 (17 to 19 kDa) (Fig. 5D). Subtilisin A also induced caspase-3 cleavage in A549 cells, but we did not detect cleaved caspase-3 when cells were incubated with trypsin (Fig. 5D). Together, these results indicate that A549 cells incubated with StmPr1 or subtilisin A undergo anoikis, characterized by caspase-3 cleavage and activation.
StmPr1 cleaves and activates PAR2 to induce IL-8 secretion by A549 cells.
We previously observed that strain K279a induces IL-8 secretion in A549 cells at early time points, but at later time points, the IL-8 secreted by A549 cells is degraded in an StmPr1-dependent manner (14). Similarly, here we observed that recombinant IL-8 is degraded by purified StmPr1 when a high dose of protease is used but not when a low dose of protease is tested, and this pattern was also observed for subtilisin A and trypsin (Fig. 6A). At low doses, trypsin has been shown to stimulate A549 cells to secrete IL-8 through activation of PAR2 (25, 26). A549 cells express all four PARs (PAR1, PAR2, PAR3, and PAR4) on their cell surface, although PAR2 is the most abundant (26). Typically, PARs are activated by a cleavage event involving host serine proteases like trypsin and thrombin (27). However, subtilisin A is also thought to activate PAR2, as subtilisin A-induced inflammation in the murine lung was dependent on PAR2 (28). Therefore, we hypothesized that StmPr1 may activate PAR2 to induce IL-8 secretion by A549 cells and first evaluated whether purified StmPr1 is sufficient to induce IL-8 secretion in A549 cells. A549 cells incubated with StmPr1 secreted significantly larger amounts of IL-8 than cells treated with the phosphate-buffered saline (PBS) control (Fig. 6B). Moreover, the IL-8 level detected in the culture medium of cells incubated with StmPr1 was comparable to that induced by subtilisin A and trypsin (Fig. 6B). As expected, treatment of A549 cells with the PAR2 agonist peptide SLIGKV-NH2 resulted in IL-8 secretion, similar to the results of treatment with StmPr1, subtilisin A, and trypsin (Fig. 6B). These data demonstrate that StmPr1 is sufficient to induce IL-8 secretion by A549 cells.
FIG 6.
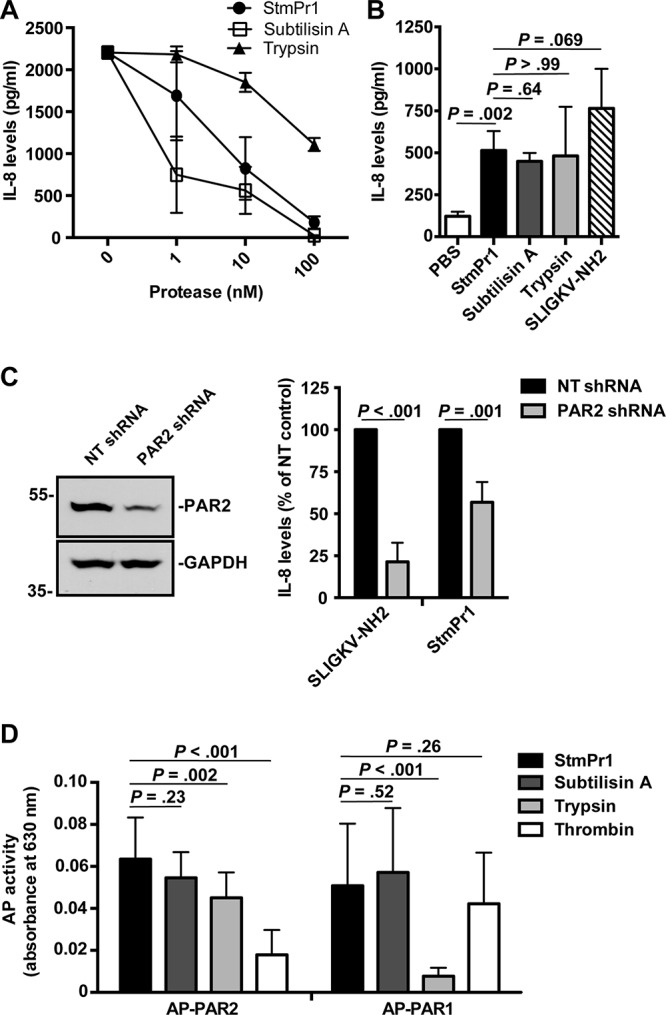
Activation of PAR2 by StmPr1 compared to that by subtilisin A and trypsin. (A) Recombinant IL-8 was incubated at 37°C for 16 h with the indicated concentrations of StmPr1, subtilisin A, or trypsin, and IL-8 levels were quantified by ELISA. (B) A549 cells were incubated with 3 nM purified StmPr1, subtilisin A, or trypsin or 100 μM the PAR2 agonist SLIGKV-NH2 and a PBS-treated control for 16 h. IL-8 levels in the cell culture supernatants were quantified by ELISA. (C) A549 cells that were transfected with PAR2-targeting shRNA and nontargeting (NT) shRNA were incubated with 100 μM SLIGKV-NH2 and 3 nM purified StmPr1 for 16 h. (Right) IL-8 levels in the cell culture supernatants were quantified by ELISA and normalized to the levels for control cells transfected with the NT shRNA for each cell line, which were set at 100%. (Left) Knockdown of PAR2 was confirmed via immunoblot analysis with an anti-PAR2 antibody and an anti-GAPDH loading control. The sizes of molecular mass standards (in kilodaltons) are indicated to the left of the gel images. (D) CHO cells that were transfected with alkaline phosphatase (AP)-labeled PAR1 and PAR2 reporter constructs were incubated with 10 nM purified StmPr1, subtilisin A, trypsin, or thrombin for 1 h. AP levels in the cell culture supernatants were quantified by measuring AP activity, where AP activity in the cell culture supernatant indicates that cleavage of the AP reporter from the PAR N terminus has occurred. The background level of AP activity measured for a PBS-treated control was subtracted from the values for the experimental samples. Data are represented as the mean and SD from three independent experiments.
To determine if the observed StmPr1-mediated induction of IL-8 secretion in A549 cells is due to PAR2 activation, we next performed RNA interference with A549 cells by stably transfecting the cells with a short hairpin RNA (shRNA) against PAR2. When PAR2 levels were compared in PAR2-knockdown cells and cells stably transfected with the nontargeting (NT) shRNA control, approximately 50% knockdown of PAR2 was observed by immunoblot analysis with an anti-PAR2 antibody (Fig. 6C). When the PAR2-knockdown cells and NT control cells were incubated with the PAR2 agonist for 16 h and secreted IL-8 levels were monitored by ELISA, we observed approximately 80% less IL-8 secretion by the PAR2-knockdown cells than the NT shRNA-transfected control cells (Fig. 6C and S6). Thus, the level of PAR2 knockdown achieved was sufficient to see a significant reduction in the level of A549 cell IL-8 secretion. Incubation of the PAR2-knockdown cells with StmPr1 also resulted in a significant reduction (approximately 45% less) in the level of IL-8 secretion by A549 cells compared to that by the NT shRNA-transfected control cells (Fig. 6C and S6). These results indicate that the StmPr1-induced IL-8 secretion by A549 cells is at least partially due to StmPr1-mediated PAR2 activation.
PARs are G-protein-coupled receptors that are activated by proteolytic cleavage at the N terminus, which creates an N-terminal tethered ligand that binds to and signals though the PAR (27). To investigate whether StmPr1 directly cleaves PAR2, we utilized a previously described alkaline phosphatase (AP)-PAR2 reporter construct transiently transfected into CHO-K1 cells (29, 30). Upon PAR2 cleavage, the AP reporter fused to the N terminus of PAR2 is released into the cell culture medium; thus, detection of AP activity in the cell culture medium is indicative of PAR2 cleavage (29, 30). The transfected CHO-K1 cells were incubated with StmPr1 for 1 h, and the AP activity in the culture medium was assayed at this time. Trypsin was included as a positive control for PAR2 cleavage, and thrombin, which cleaves PAR1 but not PAR2 (27), was included as a negative control. We found that the level of AP activity induced by StmPr1 cleavage of PAR2 was comparable to that induced by the trypsin control (Fig. 6D). Subtilisin A treatment of cells also resulted in a level of AP activity similar to that induced by StmPr1, indicating that subtilisin A also cleaves PAR2. Significantly less AP activity was observed when transfected CHO-K1 cells were incubated with thrombin (Fig. 6D). For comparison, an AP-PAR1 reporter construct was also tested. As expected, AP activity was detected in the cell culture medium of the CHO-K1 cells transfected with AP-PAR1 when they were treated with thrombin, but significantly less AP activity was detected when the cells were incubated with trypsin, which preferentially targets PAR2 for cleavage (27) (Fig. 6D). Interestingly, we found that incubation of AP-PAR1-expressing CHO-K1 cells with StmPr1 and subtilisin A induced AP activity in the cell culture medium at an amount comparable to that induced by thrombin (Fig. 6D), indicating that these two proteases also cleave PAR1. Altogether, these data demonstrate that StmPr1 cleaves PAR2 (and PAR1), with the cleavage of PAR2 explaining, at least in part, the StmPr1-induced IL-8 secretion.
DISCUSSION
Our understanding of the virulence mechanisms employed by S. maltophilia to cause disease is limited. A combination of S. maltophilia genome analyses and functional studies, including our own, have indicated that potential virulence factors may include flagella (31, 32), fimbriae (33, 34), a DNase (13), adhesins (13), a hemolysin (35, 36), exopolysaccharides and lipopolysaccharides (37), toxins (13, 38), a siderophore (36, 39), lipases (13), phospholipases (13, 40), hemagglutinin (13, 40), secretion systems (13–15, 40), and proteases (13, 14, 19). Building on our previous work describing the StmPr1 and StmPr2 proteases, here we identify the StmPr3 serine protease to be a third Xps T2S system substrate secreted by S. maltophilia strain K279a. BLASTP analysis revealed that StmPr3 from strain K279a is most closely related to an annotated serine protease from Shinella fusca (99% identity), followed by proteases from Xanthomonas retroflexus (95% identity) and Pseudomonas geniculata (86% identity). An updated BLASTP analysis of StmPr1 and StmPr2 also revealed that the amino acid sequences of these proteases also share the most homology with those of annotated serine proteases from S. fusca (70% identity and 97% identity, respectively). Among the 22 S. maltophilia strains previously analyzed for the presence of StmPr1 and StmPr2, StmPr3 is present in all of the genomes except for the draft genomes of RA8, Ab55555, EPMI, and the BAL fluid isolate described by Windhorst et al. (19) (Fig. S7). For comparison, BLASTP analysis revealed that StmPr4, which is not present in strain K279a, is found in only 5 of the 22 strains previously analyzed, and these include R551-3, RA8, RR-10, 53, and B418. StmPr3, like StmPr1 and StmPr2, is an S8 peptidase with a catalytic domain and catalytic triad, but it appears to have a short or nonexistent intermolecular chaperone (IMC) domain (14, 16). The IMC domain is essential for the proper folding of subtilisins and also serves as an inactivating peptide until autocleavage of the IMC domain by the catalytic domain converts the serine protease from the inactive proprotein form to the active mature form (41–43). Thus, it is unclear if StmPr3 is expressed as a proenzyme and/or undergoes autoprocessing in the same manner as StmPr1 and StmPr2. Unlike StmPr1 and StmPr2 (14), we did not detect both the proprotein and mature forms of secreted StmPr3 but detected only a single band of approximately 70 kDa, which most likely corresponds to the full-length StmPr3 without further processing.
Using a panel of mutants, we found that StmPr1, StmPr2, and StmPr3 are completely responsible for the observed Xps-mediated effects toward A549 cells and the Xps-mediated degradation of ECM components. However, we concluded that StmPr1 plays the biggest role, followed by StmPr2 and StmPr3, in that order. Out of the three proteases, StmPr1 is also the most abundant in the culture supernatant (Fig. S2), perhaps explaining its predominant role in Xps-mediated secretion activities. The StmPr1 purified from the strain K279a supernatant was sufficient to cause the rounding, detachment, and death of A549 cells, and when we compared the behavior of purified StmPr1 to that of subtilisin A and trypsin, we found that StmPr1 behaved more similarly to subtilisin A than to trypsin with regard to A549 cell detachment and death and degradative activities. Trypsin in combination with EDTA is typically used to passage adherent cell lines, and subtilisin (also known as Alcalase) is routinely used in the commercial detergent industry (44). With the exception of a few studies (28, 45), neither subtilisin A nor trypsin has been extensively studied for its effect on mammalian cells or degradative activities. Therefore, by including subtilisin A and trypsin in our analyses, we also learned about the effects of these proteases on A549 cells. In the absence of the calcium chelator EDTA, trypsin was not very efficient at inducing A549 cell detachment. EDTA greatly enhances the trypsin-induced detachment of cells by sequestering divalent cations, in turn destabilizing adhesion molecules like cadherins and integrins that rely on divalent cations to form cell-cell and cell-ECM interactions (46, 47). Interestingly, StmPr1 and subtilisin A successfully detached A549 cells in the absence of EDTA, suggesting that they degrade cadherins and integrins more efficiently than trypsin. In support of this hypothesis, we found that StmPr1 and subtilisin A both degraded E-cadherin and integrin α5β1 in A549 cell culture to a greater extent than trypsin. Moreover, subtilisin A degraded E-cadherin and the tight junction protein occludin to a greater extent than StmPr1, perhaps explaining why subtilisin A treatment reduces A549 cells to a single-cell suspension, while StmPr1 treatment (or trypsin treatment, in the absence of EDTA) results in chains of A549 cells.
The previously described StmPr1 variant from an S. maltophilia BAL fluid isolate was shown to round and detach human fibroblasts and degrade purified collagen type I and fibronectin in vitro, but the mechanism by which fibroblast rounding occurred was not identified (19). Here, we show that purified StmPr1 rapidly degrades fibronectin, integrin α5β1, E-cadherin, and occludin within 1 h in the context of A549 cell culture. These data suggest that the targeting of these proteins by StmPr1 leads to A549 cell rounding and detachment, as StmPr1-mediated cell rounding and detachment also occur within 1 h. In contrast, the degradation of collagen type I by StmPr1 was detected only at 24 h postincubation, and therefore, it is unlikely that the StmPr1-mediated degradation of collagen contributes to A549 cell rounding and detachment. A number of human bacterial pathogens exploit the host ECM for adhesion and colonization, and there are numerous examples of secreted bacterial proteases promoting pathogen invasion and host tissue destruction by targeting ECM components like collagen and fibronectin for degradation (48–50). Specifically, the Pseudomonas aeruginosa metalloprotease LasB, like StmPr1, degrades a number of ECM components and junction proteins in the context of mammalian cell culture to promote cell rounding and detachment (51, 52). However, LasB does not degrade integrins (52), while we observed that StmPr1 did target the fibronectin receptor integrin α5β1 for degradation.
Cell detachment induced by the targeting of cell-ECM connections, or matrilysis, causes cells to undergo a type of cell death referred to as anoikis. Anoikis, or detachment-induced apoptosis, is typically studied in the context of cancer, as cancer cells become resistant to the death signals triggered by the loss of ECM-integrin connections (22, 23). However, it is becoming clear that the proteases produced by human pathogens can also induce anoikis. Cell detachment induced by Entamoeba histolytica cysteine proteases was shown to cause anoikis in epithelial cells (53), and LasB-induced cell detachment led to anoikis in endothelial cells (51, 52). Here, we show that StmPr1 causes anoikis in A549 cells, as StmPr1-induced cell detachment caused cell death that was characterized by caspase activation, a hallmark of anoikis (22, 23). Interestingly, only about 30% of A549 cells succumbed to cell death after StmPr1-induced detachment, but due to the fact that immortalized cell lines are more resistant to anoikis (22, 23), this result is not unexpected and was also observed in epithelial cells treated with amoebal cysteine proteases (53). Our results also indicate that subtilisin A treatment can induce anoikis in A549 cells, but to a lesser extent than StmPr1 treatment. These data are the first to show that bacterial serine proteases induce anoikis, and it is likely that many other ECM-targeting bacterial proteases also induce anoikis.
In addition to targeting cell-cell and cell-ECM connections, we also observed that StmPr1 cleaves and activates PAR2. PAR2 has been linked to the lung inflammation caused by both pathogens and allergens (27) and is expressed by respiratory cells on their apical surface, where it is readily exposed to a variety of host and pathogen proteases (54). PAR2 activation is considered proinflammatory in vivo (27), and activation of PAR2 in epithelial cell culture results in the release of proinflammatory cytokines, such as IL-8 (55). Our data show that, like trypsin, StmPr1 and subtilisin A stimulate A549 cells to secrete IL-8 at low concentrations but degrade IL-8 at high concentrations. We previously found in the context of S. maltophilia-A549 cell coculture that IL-8 secretion was not dependent on Xps secretion, but IL-8 degradation over time was dependent on StmPr1 (14). However, it is possible that lipopolysaccharide-induced IL-8 secretion via Toll-like receptor signaling potentially masked any effect of StmPr1-induced PAR signaling in our previous study (56). Overall, the ability of proteases like StmPr1 to both induce IL-8 secretion by epithelial cells and degrade IL-8 sets up a complex dynamic in the lung where S. maltophilia may both cause and mitigate inflammation.
Due to the nature of PAR activation, cleavage of PAR2 by both host and pathogen serine proteases can be either activating or disabling (57). Trypsin is the prototypical activator of PAR2, but a number of other host proteases, such as neutrophil elastase, cleave PAR2 in a way that removes the N-terminal tethered ligand typically exposed by trypsin, thus disabling the receptor (57). LasB, also known as Pseudomonas elastase, and thermolysin from Bacillus thermoproteolyticus Rokko have also been shown to cleave and disable PAR2 (58, 59). Here we report that StmPr1 and subtilisin A, like trypsin, act as activators of PAR2. To our knowledge, the only other bacterial proteases shown to activate PARs are Porphyromonas gingivalis gingipains (60). Interestingly, we also found that StmPr1 and subtilisin A cleave PAR1, and like PAR2, PAR1 has also been associated with tissue damage in the murine lung (61, 62). It is yet to be determined if StmPr1- or subtilisin A-induced PAR1 cleavage is an activating event in A549 cells. However, the observation that PAR2 knockdown in A549 cells reduced PAR2 agonist-induced IL-8 secretion to a greater extent than StmPr1-induced IL-8 secretion suggests that PAR2 may not account for all of the StmPr1-induced IL-8 secretion by A549 cells.
S. maltophilia induces inflammation and neutrophil recruitment in the murine lung, and activation of both PAR1 and PAR2 has been shown to increase neutrophil recruitment to the lung in vivo, which results in increased tissue damage and impairs the host response (62, 63). Our data presented here suggest that StmPr1-induced PAR signaling may contribute to S. maltophilia-mediated inflammation in the lung. Moreover, we show that StmPr1 degrades lung epithelial cell collagen and fibronectin, and degradation of these ECM components has been shown to cause tissue damage and airway destruction in cystic fibrosis patients (64), who are commonly plagued by S. maltophilia infections. Taken together, the ability of StmPr1 to target cell-ECM and cell-cell connections for degradation, induce anoikis in lung epithelial cells, and stimulate lung epithelial cells to secrete IL-8 via PAR2 reinforces StmPr1's role as a potential virulence factor that likely contributes to S. maltophilia-mediated tissue destruction in the lung.
MATERIALS AND METHODS
Bacterial strains and media.
The multidrug-resistant isolate K279a (American Type Culture Collection [ATCC] strain BAA-2423) served as the wild-type (WT) S. maltophilia strain for these studies (Table 1). The mutants used in this study are also listed in Table 1. S. maltophilia strains were routinely cultured at 37°C on Luria-Bertani (LB) agar (Becton, Dickinson) or LB broth (14). In order to obtain supernatants for enzymatic assays and other analyses, S. maltophilia strains were cultured in buffered yeast extract (BYE) broth as previously described (14). Bacterial growth was monitored by measuring the optical density at 600 nm (OD600) of the cultures using a DU 720 spectrophotometer (Beckman Coulter). Escherichia coli strain DH5α (Life Technologies) served as the host strain for cloning and propagation of recombinant plasmids. E. coli cells were routinely grown on LB agar or in LB broth at 37°C, and when necessary, antibiotics were added as described before (14). All chemicals were from Sigma-Aldrich, unless otherwise noted.
TABLE 1.
S. maltophilia strains used in this study
| Strain | Description | Source or reference |
|---|---|---|
| K279a | Clinical isolate from blood | 15, 73 |
| NUS1(pBstmPr3-His) | gpsF mutant carrying pBstmPr3-His | This study |
| NUS4 | xpsF mutant of K279a | 15 |
| NUS4(pBstmPr3-His) | xpsF mutant carrying pBstmPr3-His | This study |
| NUS5 | stmPr1 mutant of K279a | 14 |
| NUS6 | stmPr2 mutant of K279a | 14 |
| NUS7 | stmPr1 stmPr2 mutant of K279a | 14 |
| NUS11 | stmPr3 mutant of K279a | This study |
| NUS12 | stmPr1 stmPr3 mutant of K279a | This study |
| NUS13 | stmPr2 stmPr3 mutant of K279a | This study |
| NUS14 | stmPr1 stmPr2 stmPr3 mutant of K279a | This study |
| NUS14(pBstmPr1) | stmPr1 stmPr2 stmPr3 mutant complemented with stmPr1 | This study |
| NUS14(pBstmPr2) | stmPr1 stmPr2 stmPr3 mutant complemented with stmPr2 | This study |
| NUS14(pBstmPr3) | stmPr1 stmPr2 stmPr3 mutant complemented with stmPr3 | This study |
DNA and protein sequence analysis.
S. maltophilia K279a genomic DNA was isolated as previously described (15). The primers used for sequencing and PCR were obtained from Integrated DNA Technologies (IDT). The primer names and sequences are listed in Table S1 in the supplemental material. DNA and protein sequences were analyzed using Lasergene software (DNAStar). BLASTN and BLASTP homology searches were done using the sequences in the GenBank database at the NCBI. Signal sequence prediction was performed using the SignalP (version 4.1) server.
Mutant construction and genetic complementation.
Mutants of strain K279a were constructed using the gene replacement vector pEX18Tc as described previously (14). All primers used to make the mutants and complemented strains in this study can be found in Table S1. A mutant (NUS11) from which the entire stmPr3 (SMLT_RS20900) coding region was deleted was obtained using a recombineering approach though E. coli and Flp excision as previously described (65). The stmPr3 gene with 800 bp of DNA flanking stmPr3 in both the 5′ and 3′ directions was PCR amplified from K279a DNA with Platinum Pfx DNA polymerase (Life Technologies) using the primer pair AD94 and AD95 and ligated into the pGEM-T Easy vector (Promega, Madison, WI), creating plasmid pGEMstmPr3. An Flp recombination target (FRT)-flanked chloramphenicol cassette with the 800-bp 5′ and 3′ stmPr3 flanking regions was PCR amplified from pKD3 (65) using primers AD86 and AD87 and High Fidelity Taq DNA polymerase (Life Technologies). Two micrograms of pGEMstmPr3 DNA and 700 ng of the FRT-flanked chloramphenicol cassette were transformed into electrocompetent cells of strain DY330, an E. coli strain with bacteriophage λ red recombinase genes (65). The transformed bacteria were recovered at 30°C, and the recombineered plasmid (pGEMΔstmPr3::frt-cat-frt) was selected on LB agar supplemented with chloramphenicol (30 μg/ml) at 30°C for 2 days. After PCR confirmation of the recombination event using M13 universal primers, the mutated stmPr3 gene was PCR amplified with High Fidelity Taq DNA polymerase using primers AD129 and AD95 and pGEMΔstmPr3::frt-cat-frt as the template DNA, followed by digestion with SacI and HindIII and ligation into pEX18Tc that had been digested with the same enzymes, yielding pEXΔstmPr3::frt-cat-frt. The newly made plasmid was moved into E. coli S17-1 (66) and then mobilized from there into S. maltophilia K279a via conjugation (15). Transconjugants were selected on LB agar supplemented with tetracycline (20 μg/ml), chloramphenicol (10 μg/ml), and norfloxacin (5 μg/ml). Resistant colonies were streaked onto LB agar supplemented with 10% (wt/vol) sucrose and chloramphenicol (10 μg/ml). To perform Flp-mediated excision of the chloramphenicol cassette in the stmPr3 mutant, pBSFlp (65) was electroporated into NUS11, and transformants were selected on LB agar supplemented with gentamicin (30 μg/ml) and IPTG (isopropyl-β-d-thiogalactopyranoside; 1 mM). Following an overnight incubation at 37°C, the plates were further incubated at room temperature for another 24 h. Small individual colonies that appeared after the 24-h incubation at room temperature were patched onto LB agar containing either chloramphenicol (10 μg/ml), gentamicin (30 μg/ml), or no selection. Colonies that were either chloramphenicol or gentamicin sensitive were streaked onto LB agar with 10% (wt/vol) sucrose. Deletion of stmPr3 was confirmed by PCR using the primer pair AD72 and AD73.
A mutant (NUS12) lacking both stmPr1 and stmPr3 was constructed by mobilizing the pEXΔstmPr3::frt-cat-frt plasmid carried by the E. coli S17-1 strain into mutant NUS5 containing the stmPr1 deletion. Transconjugants were selected as described above, and mutation of stmPr3 was confirmed by PCR using the primer pair AD72 and AD73. A mutant (NUS13) lacking both stmPr2 and stmPr3 was constructed by mobilizing the pEXstmPr2::Gmr plasmid carried by the E. coli S17-1 strain into mutant NUS11 containing the stmPr3 deletion. Transconjugants were selected as described above, and mutation of stmPr2 was confirmed by PCR using the primer pair AD01 and AD02. A mutant (NUS14) lacking stmPr1, stmPr2, and stmPr3 was constructed by mobilizing the pEXstmPr2::Gmr plasmid carried by the E. coli S17-1 strain into mutant NUS12 containing the stmPr1 and stmPr3 deletions. Transconjugants were selected as described above, and mutation of stmPr2 was confirmed by PCR using the primer pair AD01 and AD02.
For trans-complementation of the stmPr1 stmPr2 stmPr3 mutant (NUS14) with stmPr3, a 2.03-kb PCR fragment containing the stmPr3-coding region plus its 260-bp promoter region was PCR amplified from K279a DNA using primers AD108 and AD112. The stmPr3 fragment was digested with ApaI and SacI and cloned into pBBR1MCS (67) that had been digested with the same enzymes, yielding pBstmPr3. pBstmPr3, along with the previously made pBstmPr1 (15) and pBstmPr2 (14), was electroporated into the stmPr1 stmPr2 stmPr3 mutant (NUS14). Transformants were selected on LB agar supplemented with chloramphenicol (30 μg/ml). Cmr clones were confirmed to be carrying pBstmPr1, pBstmPr2, or pBstmPr3 by PCR using primer pairs SK251 and SK252, AD01 and AD02, or AD108 and AD112, respectively.
Detection of secreted bacterial proteins and degradative activities.
Culture supernatants were collected from bacteria grown to early stationary phase as previously described (14). To visualize secreted proteins, trichloroacetic acid precipitation of culture supernatants, followed by SDS-PAGE analysis and SYPRO Ruby total protein staining (Bio-Rad), was also performed as previously described (14). To specifically track the secretion of StmPr3 by S. maltophilia, a 6-histidine (6×His) tag was added to the C terminus with the primer pair AD108 and AD109, and pBstmPr3-His was electroporated into WT strain K279a, the xpsF mutant NUS4, and the gspF mutant NUS1, as previously described (14). To visualize the secretion of StmPr3, early-stationary-phase supernatants were collected, precipitated, and subjected to SDS-PAGE and immunoblot analysis with a mouse anti-6×His horseradish peroxidase (HRP)-conjugated antibody (Invitrogen) as previously described (14). Degradation of human fibronectin and fibrinogen by bacterial supernatants was analyzed by SDS-PAGE analysis and colloidal Coomassie staining as previously described (14), and the degradation of IL-8 by bacterial supernatants was determined by ELISA as previously described (14).
Purification of StmPr1.
StmPr1 was purified from the S. maltophilia culture supernatant with benzamidine-Sepharose as was done to purify VesB from V. cholerae (17). Early-stationary-phase culture supernatant was collected as described above from the stmPr1 stmPr2 stmPr3 mutant NUS14 with the empty complementation vector (mock purification) and NUS14 with pBstmPr1. The supernatant was filtered through a 0.2-μm-pore-size filter and then precipitated with 60% ammonium sulfate for 1 h at 4°C. Following precipitation, the supernatant was centrifuged at 10,000 × g at 4°C for 35 min. The precipitated pellet was then resuspended in 5 ml of 50 mM Tris-HCl, 150 mM NaCl, pH 8 (buffer A), and the sample was dialyzed in 3,500-molecular-weight-cutoff Slide-A-Lyzer dialysis cassettes (Thermo Fisher Scientific) against 2 liters of buffer A at 4°C for 16 h to remove the excess ammonium sulfate. StmPr1 was then purified by affinity chromatography using a 1 ml HiTrap benzamidine FF prepacked column (GE Healthcare) prepared according to the manufacturer's instructions. The flowthrough was collected and run through the column a second time, and then the column was washed 2 times with buffer A. StmPr1 was eluted from the column with 5 ml of 50 mM Tris-HCl, pH 8, buffer containing 200 mM benzamidine. To remove the benzamidine, the buffer was exchanged to 50 mM Tris-HCl, pH 8, using PD-10 columns (GE Healthcare) according to the manufacturer's instructions. The protein concentration was determined with a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific) using bovine serum albumin (BSA) as a reference.
Assays for A549 cell rounding, detachment, and viability.
Cells of the human A549 cell line (ATCC CCL-185) were routinely passaged in RPMI medium (Corning) containing 10% fetal bovine serum (FBS; Atlanta Biologicals) and maintained at 37°C in 5% CO2 (14). Detachment of A549 cells after 3 or 24 h of incubation with bacterial supernatants was determined as previously described (14). For analyzing the detachment of A549 cells following incubation with purified StmPr1, subtilisin A (catalog number P5380; Sigma-Aldrich), or trypsin (catalog number T1426; Sigma-Aldrich), cells were plated as described previously (14) and then incubated with the proteases for 1, 3, or 24 h at 37°C in 5% CO2. The wells were washed to remove nonadherent cells, and the remaining adherent cells were enumerated with PrestoBlue cell viability reagent (Life Technologies) as previously described (14). For cell rounding experiments, images were captured using a 40× objective on an Evos imaging system (AMG, Life Technologies) (14). To assess the effects of the protease inhibitors on cell detachment, prior to adding the proteases, A549 cells were pretreated with 0.25 mM the serine protease inhibitor phenylmethylsulfonyl fluoride (PMSF; MP Biomedicals) as described previously (14). To examine the effect of purified StmPr1 (and subtilisin A and trypsin) on host cell viability, A549 cells were seeded into a 96-well tissue culture plate and washed prior to addition of StmPr1, subtilisin A, or trypsin as described above. The proteases were then incubated with A549 cells for 3 or 24 h at 37°C with 5% CO2. Cellular metabolism was evaluated at this time point using PrestoBlue (Life Technologies), as previously described (14).
Collection of cell culture supernatant and cell lysates for immunoblot analysis.
A549 cell culture supernatants and cell lysates were collected as previously described (52). Briefly, A549 cells were seeded into a 24-well tissue culture plate at a density of 5 × 105 cells/well and allowed to adhere for 2 h in 1 ml of RPMI medium with 10% FBS. The monolayers were then washed with 0.5 ml of serum-free RPMI medium and serum-free RPMI medium was added to the cells. The cells were then incubated with StmPr1, subtilisin A, or trypsin for 1, 3, and 24 h at 37°C with 5% CO2. The cell culture medium was removed and centrifuged at 2,000 × g for 5 min at room temperature to pellet the detached cells and then centrifuged at 17,000 × g for 30 min at 4°C to pellet the debris. The remaining attached cells and the cell pellet containing the detached cells in the cell culture medium were lysed with radioimmunoprecipitation assay (RIPA; Thermo Fisher Scientific) buffer containing protease inhibitors (cOmplete protease inhibitor cocktail tablets; Roche) on ice for 20 min. The lysed cells were then centrifuged at 17,000 × g for 30 min at 4°C to pellet the debris. Both the cell culture supernatant and the cell lysates were stored at −80°C until immunoblot analysis.
For immunoblot analysis, the proteins in the cell culture supernatant or the cell lysate were mixed with 2× Laemmli sample buffer containing 5% β-mercaptoethanol, except for the samples for the collagen analysis, which were prepared and run under native conditions. Samples were heated for 5 min at 95°C before separation on a 4 to 20% SDS-polyacrylamide gel and then transferred to a polyvinylidene difluoride (PVDF) membrane. Proteins were visualized by immunoblotting with specific antibodies: antifibronectin (1:1,000; catalog number F3648; Sigma-Aldrich), anti-collagen I (1:1,000; catalog number ab34710; Abcam), anti-integrin α5 and anti-integrin β1 (1:1,000; catalog numbers 4705 and D2E5, respectively; Cell Signaling Technology), anti-E-cadherin (1:1,000; catalog number 610181; BD Biosciences), antioccludin (1:125; catalog number 71-1500; Invitrogen), and anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase; 1:2,000; Santa Cruz Biotechnology). For all immunoblots, the membranes were blocked for 1 h at room temperature in Tris-buffered saline solution with 0.1% Tween (TBST) and 5% milk, with the exception of the immunoblots probed with antifibronectin and anti-GAPDH, which were blocked overnight at 4°C. All primary antibody incubations were performed at 4°C overnight, where antibodies were diluted in TBST with 5% milk, with the exception of the antifibronectin and anti-GAPDH incubations, which were performed for 1 h at room temperature in TBST with 5% BSA and for 4 h at room temperature in TBST with 5% milk, respectively. All secondary antibody incubations were performed with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or rabbit anti-mouse immunoglobulin antibodies (Santa Cruz Biotechnology) at a 1:10,000 dilution in TBST with 5% milk for 1 h at room temperature. All immunoblots were visualized using an enhanced chemiluminescence (ECL) reagent (GE Healthcare) and exposure to film.
Caspase activation and cleavage assays.
A549 cells were seeded and washed as described above for the rounding, detachment, and viability assays but were plated in 96-well black clear-bottom plates (Corning) and in phenol red-free RPMI medium (Corning) to reduce the background. Cells were preloaded with 5 μM per well of CellEvent caspase-3/7 green detection reagent (Thermo Fisher Scientific) (68) and then incubated with 100 nM StmPr1, 100 nM subtilisin A, 100 nM trypsin, or 2 μM staurosporine (Santa Cruz) for 24 h at 37°C with 5% CO2. A subset of cells was also pretreated with the pan-caspase inhibitor Z-VAD-FMK (R&D Systems) for 30 min at 37°C with 5% CO2 and then loaded with CellEvent caspase-3/7 green detection reagent and treated with the proteases and staurosporine. At 3, 6, 12, and 24 h postincubation, caspase activation was quantified by measuring the fluorescence at 503 nm and 530 nm using a Synergy H1 plate reader (BioTek). To measure caspase-3 cleavage, A549 cells were seeded in a 24-well plate as described above and then incubated with 100 nM StmPr1, subtilisin A, or trypsin for 24 h at 37°C with 5% CO2. Cells were lysed with RIPA buffer and collected as described above. Proteins in the cell lysate were separated by SDS-PAGE on a 10% gel and transferred to a PVDF membrane as described above, followed by immunoblotting with anti-caspase-3 (1:2,000; catalog number AF-605-SP; R&D Systems). Blocking in TBST with 5% milk was performed at room temperature for 1 h, followed by primary incubation in TBST with 5% milk at 4°C overnight and secondary incubation with HRP-conjugated mouse anti-goat immunoglobulin (1:10,000; Santa Cruz) at room temperature for 1 h. Proteins were visualized using an enhanced ECL reagent and exposure to film.
Degradation of IL-8 and detection of IL-8 secretion by A549 cells.
As previously described (14), IL-8 degradation by StmPr1, subtilisin A, or trypsin was performed by incubating various concentrations of the proteases with carrier-free recombinant IL-8 for 16 h at 37°C and assessing IL-8 levels via ELISA. IL-8 secretion by A549 cells after protease stimulation was also evaluated as previously described (26). Briefly, cells were seeded into a 24-well tissue culture plate as described above. Monolayers were then washed with 0.5 ml of serum-free RPMI medium, 1 ml of serum-free RPMI medium was added back to the wells, and the cells were serum starved for 24 h. After serum starvation, the spent medium was removed and fresh serum-free RPMI medium was added to the cells. The cells were then incubated with 3 nM StmPr1, 3 nM subtilisin A, 3 nM trypsin, or 100 μM PAR2 agonist peptide SLIGKV-NH2 (Sigma-Aldrich) for 16 h at 37°C with 5% CO2. To measure the levels of secreted IL-8 by ELISA, the cell culture media were collected and centrifuged at 1,000 × g for 10 min to pellet any host cells, and then the supernatant was analyzed using a human IL-8 ELISA Ready-Set-Go! kit (eBioscience) as described previously (14).
Knockdown of PAR2 in A549 cells.
A549 cells were seeded into a 24-well tissue culture plate at a density of 2.5 × 105 cells/well and allowed to adhere for 16 h in RPMI medium with 10% FBS. To prepare the transfection reagent, 500 ng/well of plasmid DNA encoding shRNA against PAR2 (clone V2LHS_43134; GE Healthcare) or plasmid DNA harboring a nontargeting shRNA (clone RHS4346; GE Healthcare) and polyethylenimine (PEI) at a 4:1 ratio of PEI/DNA were mixed in Opti-MEM medium (Gibco) and incubated for 15 min at room temperature (69–71). The transfection mixture was added to the A549 cells for 16 h and then removed, and fresh RPMI medium with 10% FBS was added back to the cells. At 48 h posttransfection, the cells were selected with 1.5 μM puromycin (VWR) to kill any untransfected cells and create stable cell lines. Knockdown was confirmed by collecting cell lysates as described above, separating the proteins by SDS-PAGE on a 10% gel, transferring the proteins to a PVDF membrane as described above, and immunoblotting with anti-PAR2 (1:1,000; clone SAM11; Abcam). Blocking in TBST with 5% milk was performed at room temperature for 1 h, followed by primary incubation in TBST with 5% milk at 4°C overnight and secondary incubation with HRP-conjugated rabbit anti-mouse immunoglobulin (1:10,000; Santa Cruz) at room temperature for 1 h. Proteins were visualized using an enhanced ECL reagent and exposure to film.
PAR cleavage assay.
CHO-K1 cells (ATCC CCL-61), routinely passaged in Ham's F-12 medium (Corning) with 10% FBS, were seeded at 2 × 104 cells/well in a 96-well plate and transiently transfected with 50 ng/well alkaline phosphatase (AP)-labeled PAR1 or PAR2 reporter constructs (72) with the FuGENE6 transfection reagent (3:1 FuGENE/DNA ratio; Promega) in Opti-MEM medium. After 48 h, the transfected CHO-K1 cells were then incubated with 10 nM StmPr1, subtilisin A, trypsin, or thrombin (catalog number T6884; Sigma-Aldrich) for 1 h. After protease treatment, 25 μl of cell culture medium was collected from each well, the medium was centrifuged at 17,000 × g for 5 min at room temperature, and the AP activity in 20 μl of the supernatants was then assayed by using Quanti-Blue secreted alkaline phosphatase (InvivoGen) and reading of the absorbance at 630 nm using the plate reader.
Statistical analysis.
Data were analyzed using analysis of variance (ANOVA) and Tukey's multiple-comparison posttest (GraphPad Prism software, version 6.0; GraphPad Software).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the N. P. Cianciotto lab, past and present, for their helpful comments. We also thank Usha Pendurthi (University of Texas Health Center at Tyler) for generously providing the AP-PAR1 and AP-PAR2 reporter constructs. We also thank Richard Longnecker (Northwestern University) for providing the CHO-K1 cells.
This study was supported in part by NIAID grant F32AI114130, awarded to A.L.D. Overall, this work was supported by NIH grant AI117082, awarded to N.P.C.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00544-17.
REFERENCES
- 1.Looney WJ, Narita M, Muhlemann K. 2009. Stenotrophomonas maltophilia: an emerging opportunist human pathogen. Lancet Infect Dis 9:312–323. doi: 10.1016/S1473-3099(09)70083-0. [DOI] [PubMed] [Google Scholar]
- 2.Brooke JS. 2012. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev 25:2–41. doi: 10.1128/CMR.00019-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryan RP, Monchy S, Cardinale M, Taghavi S, Crossman L, Avison MB, Berg G, van der Lelie D, Dow JM. 2009. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat Rev Microbiol 7:514–525. doi: 10.1038/nrmicro2163. [DOI] [PubMed] [Google Scholar]
- 4.Paez JI, Tengan FM, Barone AA, Levin AS, Costa SF. 2008. Factors associated with mortality in patients with bloodstream infection and pneumonia due to Stenotrophomonas maltophilia. Eur J Clin Microbiol Infect Dis 27:901–906. doi: 10.1007/s10096-008-0518-2. [DOI] [PubMed] [Google Scholar]
- 5.Chang YT, Lin CY, Chen YH, Hsueh PR. 2015. Update on infections caused by Stenotrophomonas maltophilia with particular attention to resistance mechanisms and therapeutic options. Front Microbiol 6:893. doi: 10.3389/fmicb.2015.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falagas ME, Kastoris AC, Vouloumanou EK, Dimopoulos G. 2009. Community-acquired Stenotrophomonas maltophilia infections: a systematic review. Eur J Clin Microbiol Infect Dis 28:719–730. doi: 10.1007/s10096-009-0709-5. [DOI] [PubMed] [Google Scholar]
- 7.Chang YT, Lin CY, Lu PL, Lai CC, Chen TC, Chen CY, Wu DC, Wang TP, Lin CM, Lin WR, Chen YH. 2014. Stenotrophomonas maltophilia bloodstream infection: comparison between community-onset and hospital-acquired infections. J Microbiol Immunol Infect 47:28–35. doi: 10.1016/j.jmii.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 8.Trignano E, Manzo MJ, Fallico N, Maffei M, Marongiu F, Campus GV, Rubino C. 2014. First report of digital skin ulcer with Stenotrophomonas maltophilia infection in an immunocompetent patient. In Vivo 28:259–261. [PubMed] [Google Scholar]
- 9.Brooke JS. 2014. New strategies against Stenotrophomonas maltophilia: a serious worldwide intrinsically drug-resistant opportunistic pathogen. Expert Rev Anti Infect Ther 12:1–4. doi: 10.1586/14787210.2014.864553. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg E, Bishara J. 2012. Contemporary unconventional clinical use of co-trimoxazole. Eur J Clin Microbiol Infect Dis 18:8–17. doi: 10.1111/j.1469-0691.2011.03613.x. [DOI] [PubMed] [Google Scholar]
- 11.Milne KE, Gould IM. 2012. Combination antimicrobial susceptibility testing of multidrug-resistant Stenotrophomonas maltophilia from cystic fibrosis patients. Antimicrob Agents Chemother 56:4071–4077. doi: 10.1128/AAC.00072-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez MB, Martinez L. 2015. The efflux pump SmeDEF contributes to trimethoprim/sulfamethoxazole resistance in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 59:4347–4348. doi: 10.1128/AAC.00714-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adamek M, Linke B, Schwartz T. 2014. Virulence genes in clinical and environmental Stenotrophomonas maltophilia isolates: a genome sequencing and gene expression approach. Microb Pathog 67-68:20–30. doi: 10.1016/j.micpath.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 14.DuMont AL, Karaba SM, Cianciotto NP. 2015. Type II secretion-dependent degradative and cytotoxic activities mediated by Stenotrophomonas maltophilia serine proteases StmPr1 and StmPr2. Infect Immun 83:3825–3837. doi: 10.1128/IAI.00672-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karaba SM, White RC, Cianciotto NP. 2013. Stenotrophomonas maltophilia encodes a type II protein secretion system that promotes detrimental effects on lung epithelial cells. Infect Immun 81:3210–3219. doi: 10.1128/IAI.00546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribitsch D, Heumann S, Karl W, Gerlach J, Leber R, Birner-Gruenberger R, Gruber K, Eiteljoerg I, Remler P, Siegert P, Lange J, Maurer KH, Berg G, Guebitz GM, Schwab H. 2012. Extracellular serine proteases from Stenotrophomonas maltophilia: screening, isolation and heterologous expression in E. coli. J Biotechnol 157:140–147. doi: 10.1016/j.jbiotec.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 17.Gadwal S, Korotkov KV, Delarosa JR, Hol WG, Sandkvist M. 2014. Functional and structural characterization of Vibrio cholerae extracellular serine protease B, VesB. J Biol Chem 289:8288–8298. doi: 10.1074/jbc.M113.525261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siezen RJ, Leunissen JA. 1997. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci 6:501–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Windhorst S, Frank E, Georgieva DN, Genov N, Buck F, Borowski P, Weber W. 2002. The major extracellular protease of the nosocomial pathogen Stenotrophomonas maltophilia: characterization of the protein and molecular cloning of the gene. J Biol Chem 277:11042–11049. doi: 10.1074/jbc.M109525200. [DOI] [PubMed] [Google Scholar]
- 20.Huang HW, Chen WC, Wu CY, Yu HC, Lin WY, Chen ST, Wang KT. 1997. Kinetic studies of the inhibitory effects of propeptides subtilisin BPN′ and Carlsberg to bacterial serine proteases. Protein Eng 10:1227–1233. doi: 10.1093/protein/10.10.1227. [DOI] [PubMed] [Google Scholar]
- 21.DelMar EG, Largman C, Brodrick JW, Geokas MC. 1979. A sensitive new substrate for chymotrypsin. Anal Biochem 99:316–320. doi: 10.1016/S0003-2697(79)80013-5. [DOI] [PubMed] [Google Scholar]
- 22.Grossmann J. 2002. Molecular mechanisms of “detachment-induced apoptosis—anoikis”. Apoptosis 7:247–260. doi: 10.1023/A:1015312119693. [DOI] [PubMed] [Google Scholar]
- 23.Gilmore AP. 2005. Anoikis. Cell Death Differ 12(Suppl 2):1473–1477. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- 24.McIlwain DR, Berger T, Mak TW. 2013. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larsen AK, Seternes O-M, Larsen M, Aasmoe L, Bang B. 2008. Salmon trypsin stimulates the expression of interleukin-8 via protease-activated receptor-2. Toxicol Appl Pharmacol 230:276–282. doi: 10.1016/j.taap.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Zheng Y, He S. 2006. Induction of release and up-regulated gene expression of interleukin (IL)-8 in A549 cells by serine proteinases. BMC Cell Biol 7:22. doi: 10.1186/1471-2121-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ossovskaya VS, Bunnett NW. 2004. Protease-activated receptors: contribution to physiology and disease. Physiol Rev 84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 28.Florsheim E, Yu S, Bragatto I, Faustino L, Gomes E, Ramos RN, Barbuto JAM, Medzhitov R, Russo M. 2015. Integrated innate mechanisms involved in airway allergic inflammation to the serine protease subtilisin. J Immunol 194:4621–4630. doi: 10.4049/jimmunol.1402493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kothari H, Rao LVM, Pendurthi UR. 2011. Glycosylation of tissue factor is not essential for its transport or functions. J Thromb Haemost 9:1511–1520. doi: 10.1111/j.1538-7836.2011.04332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pendurthi UR, Rao LVM. 2010. Factor VIIa interaction with endothelial cells and endothelial cell protein C receptor. Thromb Res 125:S19–S22. doi: 10.1016/j.thromres.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Oliveira-Garcia D, Dall'Agnol M, Rosales M, Azzuz AC, Martinez MB, Giron JA. 2002. Characterization of flagella produced by clinical strains of Stenotrophomonas maltophilia. Emerg Infect Dis 8:918–923. doi: 10.3201/eid0809.010535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zgair AK, Chhibber S. 2011. Adhesion of Stenotrophomonas maltophilia to mouse tracheal mucus is mediated through flagella. J Med Microbiol 60:1032–1037. doi: 10.1099/jmm.0.026377-0. [DOI] [PubMed] [Google Scholar]
- 33.de Oliveira-Garcia D, Dall'Agnol M, Rosales M, Azzuz AC, Alcantara N, Martinez MB, Giron JA. 2003. Fimbriae and adherence of Stenotrophomonas maltophilia to epithelial cells and to abiotic surfaces. Cell Microbiol 5:625–636. doi: 10.1046/j.1462-5822.2003.00306.x. [DOI] [PubMed] [Google Scholar]
- 34.Zgair AK, Al-Adressi AM. 2013. Stenotrophomonas maltophilia fimbrin stimulates mouse bladder innate immune response. Eur J Clin Microbiol Infect Dis 32:139–146. doi: 10.1007/s10096-012-1729-0. [DOI] [PubMed] [Google Scholar]
- 35.Figueiredo PM, Furumura MT, Santos AM, Sousa AC, Kota DJ, Levy CE, Yano T. 2006. Cytotoxic activity of clinical Stenotrophomonas maltophilia. Lett Appl Microbiol 43:443–449. doi: 10.1111/j.1472-765X.2006.01965.x. [DOI] [PubMed] [Google Scholar]
- 36.Chhibber S, Gupta A, Sharan R, Gautam V, Ray P. 2008. Putative virulence characteristics of Stenotrophomonas maltophilia: a study on clinical isolates. World J Microbiol Biotechnol 24:2819–2825. doi: 10.1007/s11274-008-9812-5. [DOI] [Google Scholar]
- 37.Huang TP, Somers EB, Wong AC. 2006. Differential biofilm formation and motility associated with lipopolysaccharide/exopolysaccharide-coupled biosynthetic genes in Stenotrophomonas maltophilia. J Bacteriol 188:3116–3120. doi: 10.1128/JB.188.8.3116-3120.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hagemann M, Hasse D, Berg G. 2006. Detection of a phage genome carrying a zonula occludens like toxin gene (zot) in clinical isolates of Stenotrophomonas maltophilia. Arch Microbiol 185:449–458. doi: 10.1007/s00203-006-0115-7. [DOI] [PubMed] [Google Scholar]
- 39.Garcia CA, Passerini De Rossi B, Alcaraz E, Vay C, Franco M. 2012. Siderophores of Stenotrophomonas maltophilia: detection and determination of their chemical nature. Rev Argent Microbiol 44:150–154. [PubMed] [Google Scholar]
- 40.Crossman LC, Gould VC, Dow JM, Vernikos GS, Okazaki A, Sebaihia M, Saunders D, Arrowsmith C, Carver T, Peters N, Adlem E, Kerhornou A, Lord A, Murphy L, Seeger K, Squares R, Rutter S, Quail MA, Rajandream MA, Harris D, Churcher C, Bentley SD, Parkhill J, Thomson NR, Avison MB. 2008. The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol 9:R74. doi: 10.1186/gb-2008-9-4-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinde U, Inouye M. 1993. Intramolecular chaperones and protein folding. Trends Biochem Sci 18:442–446. doi: 10.1016/0968-0004(93)90146-E. [DOI] [PubMed] [Google Scholar]
- 42.Shinde U, Inouye M. 1995. Folding pathway mediated by an intramolecular chaperone: characterization of the structural changes in pro-subtilisin E coincident with autoprocessing. J Mol Biol 252:25–30. doi: 10.1006/jmbi.1995.0472. [DOI] [PubMed] [Google Scholar]
- 43.Shinde U, Inouye M. 1995. Folding mediated by an intramolecular chaperone: autoprocessing pathway of the precursor resolved via a substrate assisted catalysis mechanism. J Mol Biol 247:390–395. doi: 10.1006/jmbi.1994.0147. [DOI] [PubMed] [Google Scholar]
- 44.Vojcic L, Pitzler C, Korfer G, Jakob F, Ronny M, Maurer KH, Schwaneberg U. 2015. Advances in protease engineering for laundry detergents. N Biotechnol 32:629–634. doi: 10.1016/j.nbt.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 45.Huang HL, Hsing HW, Lai TC, Chen YW, Lee TR, Chan HT, Lyu PC, Wu CL, Lu YC, Lin ST, Lin CW, Lai CH, Chang HT, Chou HC, Chan HL. 2010. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J Biomed Sci 17:36. doi: 10.1186/1423-0127-17-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Powell K. 2005. The sticky business of discovering cadherins. J Cell Biol 170:514. doi: 10.1083/jcb1704fta1. [DOI] [Google Scholar]
- 47.Zhang K, Chen J. 2012. The regulation of integrin function by divalent cations. Cell Adh Migr 6:20–29. doi: 10.4161/cam.18702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh B, Fleury C, Jalalvand F, Riesbeck K. 2012. Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol Rev 36:1122–1180. doi: 10.1111/j.1574-6976.2012.00340.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhang YZ, Ran LY, Li CY, Chen XL. 2015. Diversity, structures, and collagen-degrading mechanisms of bacterial collagenolytic proteases. Appl Environ Microbiol 81:6098–6107. doi: 10.1128/AEM.00883-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henderson B, Nair S, Pallas J, Williams MA. 2011. Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol Rev 35:147–200. doi: 10.1111/j.1574-6976.2010.00243.x. [DOI] [PubMed] [Google Scholar]
- 51.Beaufort N, Corvazier E, Mlanaoindrou S, de Bentzmann S, Pidard D. 2013. Disruption of the endothelial barrier by proteases from the bacterial pathogen Pseudomonas aeruginosa: implication of matrilysis and receptor cleavage. PLoS One 8:e75708. doi: 10.1371/journal.pone.0075708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beaufort N, Corvazier E, Hervieu A, Choqueux C, Dussiot M, Louedec L, Cady A, de Bentzmann S, Michel JB, Pidard D. 2011. The thermolysin-like metalloproteinase and virulence factor LasB from pathogenic Pseudomonas aeruginosa induces anoikis of human vascular cells. Cell Microbiol 13:1149–1167. doi: 10.1111/j.1462-5822.2011.01606.x. [DOI] [PubMed] [Google Scholar]
- 53.Kumar S, Banerjee R, Nandi N, Sardar AH, Das P. 2012. Anoikis potential of Entamoeba histolytica secretory cysteine proteases: evidence of contact independent host cell death. Microb Pathog 52:69–76. doi: 10.1016/j.micpath.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 54.Ricciardolo FL, Steinhoff M, Amadesi S, Guerrini R, Tognetto M, Trevisani M, Creminon C, Bertrand C, Bunnett NW, Fabbri LM, Salvadori S, Geppetti P. 2000. Presence and bronchomotor activity of protease-activated receptor-2 in guinea pig airways. Am J Respir Crit Care Med 161:1672–1680. doi: 10.1164/ajrccm.161.5.9907133. [DOI] [PubMed] [Google Scholar]
- 55.Asokananthan N, Graham PT, Fink J, Knight DA, Bakker AJ, McWilliam AS, Thompson PJ, Stewart GA. 2002. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J Immunol 168:3577–3585. doi: 10.4049/jimmunol.168.7.3577. [DOI] [PubMed] [Google Scholar]
- 56.Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. 2004. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem 279:2712–2718. doi: 10.1074/jbc.M305790200. [DOI] [PubMed] [Google Scholar]
- 57.Chignard M, Pidard D. 2006. Neutrophil and pathogen proteinases versus proteinase-activated receptor-2 lung epithelial cells: more terminators than activators. Am J Respir Cell Mol Biol 34:394–398. doi: 10.1165/rcmb.2005-0250TR. [DOI] [PubMed] [Google Scholar]
- 58.Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, Hollenberg MD, Pidard D, Chignard M. 2005. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol 32:411–419. doi: 10.1165/rcmb.2004-0274OC. [DOI] [PubMed] [Google Scholar]
- 59.Ubl JJ, Grishina ZV, Sukhomlin TK, Welte T, Sedehizade F, Reiser G. 2002. Human bronchial epithelial cells express PAR-2 with different sensitivity to thermolysin. Am J Physiol Lung Cell Mol Physiol 282:L1339–L1348. doi: 10.1152/ajplung.00392.2001. [DOI] [PubMed] [Google Scholar]
- 60.Lourbakos A, Potempa J, Travis J, D'Andrea MR, Andrade-Gordon P, Santulli R, Mackie EJ, Pike RN. 2001. Arginine-specific protease from Porphyromonas gingivalis activates protease-activated receptors on human oral epithelial cells and induces interleukin-6 secretion. Infect Immun 69:5121–5130. doi: 10.1128/IAI.69.8.5121-5130.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jose RJ, Williams AE, Mercer PF, Sulikowski MG, Brown JS, Chambers RC. 2015. Regulation of neutrophilic inflammation by proteinase-activated receptor 1 during bacterial pulmonary infection. J Immunol 194:6024–6034. doi: 10.4049/jimmunol.1500124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schouten M, van't Veer C, Roelofs JJ, Levi M, van der Poll T. 2012. Protease-activated receptor-1 impairs host defense in murine pneumococcal pneumonia: a controlled laboratory study. Crit Care 16:R238. doi: 10.1186/cc11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lutfi R, Lewkowich IP, Zhou P, Ledford JR, Page K. 2012. The role of protease-activated receptor-2 on pulmonary neutrophils in the innate immune response to cockroach allergen. J Inflamm (Lond) 9:32. doi: 10.1186/1476-9255-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Twigg MS, Brockbank S, Lowry P, FitzGerald SP, Taggart C, Weldon S. 2015. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediators Inflamm 2015:293053. doi: 10.1155/2015/293053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bryan A, Harada K, Swanson MS. 2011. Efficient generation of unmarked deletions in Legionella pneumophila. Appl Environ Microbiol 77:2545–2548. doi: 10.1128/AEM.02904-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Howell HA, Logan LK, Hauser AR. 2013. Type III secretion of ExoU is critical during early Pseudomonas aeruginosa pneumonia. mBio 4:e00032-13. doi: 10.1128/mBio.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 68.Oroz-Parra I, Navarro M, Cervantes-Luevano KE, Alvarez-Delgado C, Salvesen G, Sanchez-Campos LN, Licea-Navarro AF. 2016. Apoptosis activation in human lung cancer cell lines by a novel synthetic peptide derived from Conus californicus venom. Toxins 8:38. doi: 10.3390/toxins8020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.White RC, Cianciotto NP. 2016. Type II secretion is necessary for optimal association of the Legionella-containing vacuole with macrophage Rab1B but enhances intracellular replication mainly by Rab1B-independent mechanisms. Infect Immun 84:3313–3327. doi: 10.1128/IAI.00750-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mallama CA, McCoy-Simandle K, Cianciotto NP. 2017. The type II secretion system of Legionella pneumophila dampens the MyD88 and Toll-like receptor 2 signaling pathway in infected human macrophages. Infect Immun 85:e00897-16. doi: 10.1128/IAI.00897-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A 92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LV. 2007. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem 282:11849–11857. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Avison MB, von Heldreich CJ, Higgins CS, Bennett PM, Walsh TR. 2000. A TEM-2 β-lactamase encoded on an active Tn1-like transposon in the genome of a clinical isolate of Stenotrophomonas maltophilia. J Antimicrob Chemother 46:879–884. doi: 10.1093/jac/46.6.879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.