

ABSTRACT
Cryptococcus neoformans, the causative agent of cryptococcosis, is an opportunistic fungal pathogen that kills over 200,000 individuals annually. This yeast may grow freely in body fluids, but it also flourishes within host cells. Despite extensive research on cryptococcal pathogenesis, host genes involved in the initial engulfment of fungi and subsequent stages of infection are woefully understudied. To address this issue, we combined short interfering RNA silencing and a high-throughput imaging assay to identify host regulators that specifically influence cryptococcal uptake. Of 868 phosphatase and kinase genes assayed, we discovered 79 whose silencing significantly affected cryptococcal engulfment. For 25 of these, the effects were fungus specific, as opposed to general alterations in phagocytosis. Four members of this group significantly and specifically altered cryptococcal uptake; one of them encoded CaMK4, a calcium/calmodulin-dependent protein kinase. Pharmacological inhibition of CaMK4 recapitulated the observed defects in phagocytosis. Furthermore, mice deficient in CaMK4 showed increased survival compared to wild-type mice upon infection with C. neoformans. This increase in survival correlated with decreased expression of pattern recognition receptors on host phagocytes known to recognize C. neoformans. Altogether, we have identified a kinase that is involved in C. neoformans internalization by host cells and in host resistance to this deadly infection.
KEYWORDS: Cryptococcus neoformans, fungal pathogenesis, image-based screen
INTRODUCTION
Cryptococcus neoformans is a ubiquitous encapsulated fungal pathogen that causes pneumonia and meningitis in immunocompromised individuals. Cryptococcal infection is responsible for significant morbidity and mortality in AIDS patients and is the third most common invasive fungal infection in organ transplant recipients (1, 2), causing an estimated one million cases of meningitis and over 200,000 deaths each year (3–5). Current antifungal therapy is hampered by toxicity, the emergence of drug-resistant organisms, and the inability of the host's immune system to aid in resolution of the disease; treatment is further limited by drug cost and availability in the resource-limited settings where this disease is rampant (6, 7).
Cryptococcosis is primarily an opportunistic infection, highlighting the vital role of host immune responses in control of this infection. Once C. neoformans enters the host, the first line of defense it encounters is the innate immune system. Phagocytic cells, including macrophages and dendritic cells (DCs), engulf C. neoformans and present antigen to initiate the adaptive immune response.
Fungal internalization by host phagocytes may benefit either the pathogen or the host, depending on microbial virulence factors, host species, host immune status, and the stage of infection. Cryptococci can survive and even proliferate in macrophage phagosomes (8, 9), potentially using these host cells to disseminate. This idea is supported by the reduced C. neoformans burden observed in multiple tissues when clodronate liposomes were used to deplete phagocytes in mice before infection (10). Engulfment by host cells is not always a benefit, however, as internalized fungi may also be destroyed by classically activated phagocytes (11–13).
C. neoformans enters host phagocytes via multiple receptor-mediated interactions. Cryptococcal cells may be opsonized by complement deposited on the capsule surface, leading to recognition by host complement receptors. They may also be opsonized by antibody binding, with subsequent recognition by host Fc receptors (14). Finally, the polysaccharide capsule itself may directly interact with macrophage cell surface proteins, including multiple pattern recognition receptors (PRRs) (15). We speculated that additional host factors are important for specific fungal internalization and designed a screen to identify the host genes encoding such proteins.
We used an automated imaging-based assay that we previously developed (16) to screen an RNA interference (RNAi) library that targets genes encoding host phosphatases and kinases. We identified four genes whose products are involved in C. neoformans internalization by human phagocytes; one of these encodes calcium/calmodulin-dependent protein kinase 4 (CaMK4). Mice lacking CaMK4 survived longer than wild-type (WT) mice after infection with C. neoformans, a result that was partly recapitulated by pharmacological inhibition of CaMK4. Immune profiling revealed that the increase in survival time was not due to changes in host cytokine responses or infiltrating immune cells. However, CaMK4 knockout mice infected with C. neoformans showed significantly reduced numbers of TLR2, Dectin-1, and CD206-positive phagocytes compared to infected wild-type animals. All three of these pattern recognition receptors have previously been shown to recognize C. neoformans (see below). Our studies therefore support a role for this kinase in C. neoformans internalization by host cells and in host resistance to this infection.
RESULTS
RNAi screening for host factors that influence cryptococcal engulfment.
We hypothesized that host phosphatases and kinases would be among the factors that regulate engulfment of C. neoformans by host phagocytes. We took an unbiased approach to identify such proteins and screened human short interfering RNA (siRNA) libraries that target the corresponding sequences. Using a human monocytic cell line, THP-1, as the host cell population, we achieved an average silencing efficiency of over 65% (see Fig. S1 in the supplemental material). Following silencing, the treated THP-1 cells were cultured with stained and serum-opsonized fungi for 1 h and then washed, fixed, and stained for imaging (16). Images were automatically collected and analyzed (see Materials and Methods), and the phagocytic index (fungi internalized per 100 host cells) was determined for each sample.
For our primary screen, we targeted each host gene with a pool of two siRNAs, performing two biological replicates with assays in triplicate. We then analyzed the results using a quartile-based threshold method to account for nonnormal data distribution and control for false positives (Fig. 1A). We identified 79 host genes that, when silenced, consistently and significantly affected fungal adherence, uptake, or both (Table S1). The goal of our screen was to identify host factors with specific roles in interactions with C. neoformans rather than in general phagocytosis. To rule out the latter, we compared the uptake of cryptococcal cells by siRNA-treated THP-1 cells to that of inert particles (1-μm fluorescent latex beads; Fig. 1B). Of the 79 candidates assayed, 25 showed differences in uptake or adherence with the yeast but not with latex beads (Table S2). We next tested these 25 targets with three additional organisms: the pathogenic yeast Candida albicans, the model yeast Saccharomyces cerevisiae, and the bacterium Escherichia coli. Four host genes showed specific alterations in uptake or adherence of C. neoformans compared to the other particles (highlighted in Table S2). Results for one of these genes (CaMK4) is shown in Fig. 1C.
FIG 1.

RNAi screening for regulators of fungal uptake. (A) Fungal uptake by THP-1 cells treated with siRNA pools targeting CamK4 (orange) or 867 other kinases and phosphatases (dark blue symbols), no siRNA (light blue symbols), or scrambled siRNA (white symbols); see Materials and Methods for additional controls. The red lines indicate the upper and lower boundaries of the plate averages using the quartile-based threshold method. Candidate genes outside those bounds (79) advanced to the next round of screening. (B) A second round of screening identified gene depletions that altered uptake of cryptococcal cells but not of an inert particle control (latex beads). Results were normalized to no-siRNA control assays with the appropriate particle and assessed by Student's t test. Controls (as described for panel A) fitted to the diagonal (red line) had an R2 value of 0.76. (C) THP-1 cells were treated with siRNA targeting CaMK4, and uptake of C. neoformans (Cn), C. albicans (Ca), S. cerevisiae (Sc), E. coli (Ec), or latex beads (LB) was assayed. Phagocytosis of C. neoformans was significantly decreased compared to that of the other particles (P < 0.1 for C. albicans and P < 0.05 for all other particles). Red line, normalized uptake value of 1.
CaMK4 influences cryptococcal infection.
We chose CaMK4 for follow-up, because silencing the corresponding gene yielded the most marked and reproducible reduction in cryptococcal uptake and it had not previously been implicated in cryptococcal interactions with host macrophages. To further investigate its role in C. neoformans infection, we treated THP-1 cells with KN93, a competitive inhibitor of CaMK4 and a related kinase, CaMK2 (17). Treatment with 10 nM KN93 did not compromise host cell growth or viability (not shown), but it did specifically reduce C. neoformans uptake in standard assays, similar to the effects of silencing. It had no effect on the uptake of C. albicans (Fig. 2A) or latex beads (not shown). We also tested whether treatment with this compound would alter the survival of mice after intranasal infection with C. neoformans. We found that treatment of C57BL/6 mice with KN93 modestly but significantly increased survival compared to control treatment with phosphate-buffered saline (PBS) alone (Fig. 2B and Fig. S2).
FIG 2.

Inhibition or knockout of CamK4 influences fungal uptake and pathogenesis. (A) THP-1 cells were assayed for uptake of the organisms indicated after treatment with PBS alone (blue) or 10 nM KN93 in PBS (orange). The averages and standard errors of the means (SEM) from five independent studies are shown (*, P < 0.05 by Student's t test). (B) Survival of C57BL/6 mice treated with either PBS alone (blue) or with 250 μg/g body weight KN93 in PBS (orange). Mice were treated 1 day prior to infection with 5 × 104 cryptococci and three times a week postinfection. Results are representative of two independent experiments with 6 mice per group; P < 0.05 by log-rank test. (C) Peritoneal macrophages from WT (blue) and CaMK4−/− (orange) mice were assayed for uptake of C. neoformans. The averages and SEM from four independent studies are shown (***, P < 0.0001 by Student's t test). (D) Wild-type (WT; n = 16) and CaMK4 knockout (n = 9) mice were infected with C. neoformans and monitored as detailed in Materials and Methods. Results are representative of four independent experiments; P < 0.05 by log-rank test.
KN93 inhibits both CaMK4 and CaMK42. To more definitively address the role of CamK4 in C. neoformans infection, we isolated peritoneal macrophages from WT and CaMK4−/− mice and compared them in uptake assays. Consistent with the results of siRNA silencing and KN93 treatment experiments, primary macrophages deficient in CaMK4 showed significantly reduced uptake of C. neoformans compared to that of WT microphages (Fig. 2C). Based on this defect, we next examined infection kinetics in CaMK4−/− mice. Notably, these mice survived significantly longer than WT mice (Fig. 2D and Fig. S2). Interestingly, despite the survival difference, total fungal burden in lung, spleen, and brain did not differ between the two strains at any time after infection (Fig. S3).
The extended survival of CaMK4−/− mice is not due to differences in cytokine levels or immune cell infiltrate.
Our infection studies indicated that decreased fungal burden was not responsible for the prolonged survival of CaMK4−/− mice. We wondered whether the survival difference was instead due to adverse effects of the host immune response, which is stimulated by wild-type infection. We first tested this by cytokine profiling of WT and CaMK4−/− mice infected with KN99α. Analysis of pulmonary homogenates from naive mice and from infected mice on days 7 and 14 postinoculation revealed no significant differences in any of the 23 cytokines or chemokines assayed (Table 1).
TABLE 1.
Pulmonary cytokine and chemokine levels
| Cytokine or chemokine | Level on daya: |
|||||
|---|---|---|---|---|---|---|
| 0 (naïve) |
7 |
14 |
||||
| WT | CaMK4−/− | WT | CaMK4−/− | WT | CaMK4−/− | |
| Th-1 type | ||||||
| IL-2 | 12.3 ± 2.0 | 37.4 ± 10.5 | 57.5 ± 9.9 | 64.3 ± 12.7 | 43.9 ± 7.5 | 27.0 ± 3.5 |
| IL-12p70 | 29.2 ± 2.2 | 42.6 ± 3.4 | 301.1 ± 36.0 | 287.8 ± 41.6 | 182.7 ± 21.1 | 149.7 ± 5.8 |
| IFN-γ | 8.8 ± 0.6 | 11.8 ± 2.0 | 44.1 ± 6.9 | 45.3 ± 7.9 | 23.2 ± 4.0 | 18.1 ± 2.5 |
| Th-2 type | ||||||
| IL-4 | ND | ND | 168.5 ± 23.4 | 268.0 ± 24.4 | 644.5 ± 60.6 | 765.3 ± 56.5 |
| IL-5 | ND | ND | 123.5 ± 12.6 | 433.5 ± 71.8 | 169.4 ± 24.1 | 330.0 ± 40.9 |
| IL-13 | 32.2 ± 7.4 | 48.3 ± 6.3 | 637.4 ± 100.6 | 1,013.0 ± 153.8 | 657.4 ± 78.5 | 977.6 ± 213.7 |
| Proinflammatory | ||||||
| IL-1α | 16.1 ± 2.9 | 35.9 ± 8.3 | 161.5 ± 17.1 | 104.7 ± 11.6 | 121.6 ± 19.7 | 106.2 ± 11.6 |
| IL-1β | 67.3 ± 8.3 | 107.2 ± 24.6 | 1,975.0 ± 261.7 | 1,916.0 ± 296.7 | 1,510.0 ± 74.5 | 1,436.0 ± 122.1 |
| IL-17a | 5.4 ± 0.3 | 5.4 ± 0.3 | 45.4 ± 7.7 | 46.9 ± 5.2 | 22.8 ± 2.2 | 20.5 ± 1.5 |
| G-CSF | 2.1 ± 0.3 | 2.4 ± 0.7 | 161.8 ± 27.4 | 173.2 ± 29.0 | 68.3 ± 56.1 | 56.1 ± 8.2 |
| TNF-α | 32.8 ± 1.9 | 42.5 ± 5.9 | 343.6 ± 65.7 | 388.0 ± 68.9 | 225.3 ± 54.0 | 131.6 ± 15.8 |
| Chemokines | ||||||
| MIP-1α | 20.3 ± 1.9 | 19.8 ± 4.6 | 1,108.0 ± 119.3 | 1,002.0 ± 116.4 | 1,991.0 ± 143.2 | 1,974.0 ± 144.0 |
| MIP-1β | 3.2 ± 0.6 | 6.2 ± 2.6 | 203.3 ± 32.9 | 166.2 ± 25.9 | 74.7 ± 8.9 | 37.4 ± 6.1 |
| MCP-1 | 124.4 ± 14.9 | 137.5 ± 17.2 | 2,078.0 ± 285.4 | 2,046.0 ± 258.8 | 1,503.0 ± 124.4 | 1,862.0 ± 209.0 |
| KC | 8.2 ± 0.9 | 9.2 ± 1.4 | 362.7 ± 61.4 | 344.8 ± 44.4 | 181.8 ± 16.5 | 133.7 ± 17.8 |
| RANTES | 167.4 ± 24.5 | 157.3 ± 42.9 | 719.9 ± 96.4 | 516.5 ± 40.2 | 183.6 ± 13.7 | 159.2 ± 19.6 |
Results are given as means ± standard errors of the means. ND, not detected.
We next assayed wild-type and CaMK4−/− mice for differences in infiltrating immune cells after infection. To do this, we isolated pulmonary leukocytes on days 7 and 14 postinoculation and used flow cytometry to characterize the immune cells recruited to the lungs. We observed no significant differences in percent (Fig. 3) or absolute number (not shown) of any of the cell types assayed. The extended survival of CaMK4−/− mice thus is not likely due to changes in cytokine or immune cell profiles.
FIG 3.

CaMK4 knockout mice exhibit no difference in leukocyte infiltration in response to C. neoformans infection. Shown are leukocyte profiles of WT (blue circles) and CaMK4 knockout mice (orange squares) infected with 104 C. neoformans KN99α for the times shown. Each symbol represents data from an individual mouse. Results from 2 independent experiments with 4 to 5 mice per group per time point are shown; means ± SEM are also plotted. Leukocytes were labeled with antibodies to identify B cells (CD45+ and CD19+), CD4+ T cells (CD45+, CD4+, and CD8−), CD8+ T cells (CD45+, CD8+, and CD4−), DCs (CD45+, CD11b+, CD11c+, and F4/80−), eosinophils (CD45+, CD11b+, and SiglecF+), exudate or recruited macrophages (CD45+, CD11b+, CD11c+, and F4/80+), macrophages (CD45+, CD11b+, CD11c−, and F4/80+), and polymorphonuclear leukocytes (PMNs; CD45+, CD11b+, and Ly6Ghi) and then analyzed by flow cytometry (see Table S3 in the supplemental material for antibodies and Fig. S4 for gating strategy).
CaMK4 ablation leads to reduced pattern recognition receptor expression.
Our studies using RNAi, a kinase inhibitor, and knockout mice all showed that lowering CaMK4 activity leads to reduced cryptococcal engulfment by host phagocytes. Because lack of CaMK4 did not alter fungal burden, cytokine profile, or leukocyte distribution during infection, we hypothesized that the connection between this regulatory protein and fungal uptake occurs at the level of immune cell surface receptor expression. To test this, we used surface staining and flow cytometry to analyze pulmonary leukocytes from wild-type and CaMK4−/− mice on day 7 postinoculation for expression of 10 receptors that have been implicated in interactions with cryptococcal cells. In CaMK4−/− mice we observed a significant reduction in the percentage of exudate/recruited macrophages (F4/80+, CD11b+, and CD11c+) positive for CD282/TLR2, Dectin-1, and CD206/mannose receptor and the percentage of dendritic cells (F4/80−, CD11b+, and CD11c+) positive for CD206 (Fig. 4), although for those cells that were positive, the fluorescence intensity was comparable (Fig. S5). In contrast, we found no significant differences in receptor expression between uninfected WT and CaMK4−/− mice (Fig. S6). Thus, the reduced fungal uptake we observe in vitro likely is caused by surface receptor expression that is altered in the absence of CaMK4.
FIG 4.
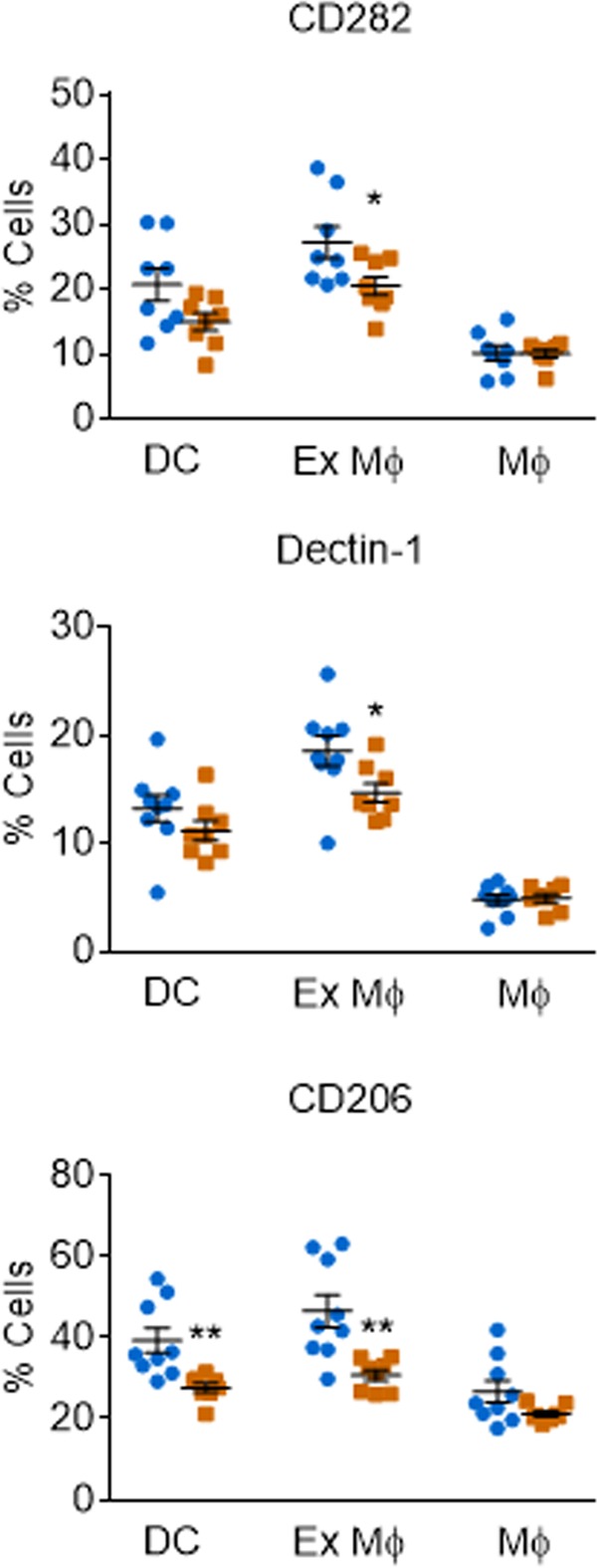
Leukocytes from CaMK4−/− mice show reduced expression of PRRs implicated in cryptococcal recognition. PRR profiling of leukocytes from WT (blue circles) and CaMK4 knockout (orange squares) mice 7 days after infection with 104 C. neoformans KN99α. Antibody labeling and flow cytometry was used to classify pulmonary leukocytes as DCs (CD45+, CD11b+, CD11c+, and F4/80−), exudate or recruited macrophages (Ex Mϕ) (CD45+, CD11b+, CD11c+, and F4/80+), or macrophages/monocytes (CD45+, CD11b+, CD11c−, and F4/80+) and to assess PRR expression. Symbols indicate data from individual mice in 2 independent experiments with 4 to 5 mice per group; means ± SEM are also shown (*, P < 0.05; **, P < 0.01 by Student's t test).
DISCUSSION
Understanding host-pathogen interactions is vital to the development of new drugs and/or immune therapies. C. neoformans, a facultative intracellular pathogen, interacts with multiple facets of the immune system. Because the patients most susceptible to this infection are severely immunocompromised, identifying key host factors potentially can lead to ways to tip the balance in favor of the host. One way to identify such host factors is by screening host genes for their impact on pathogen interactions. We probed these complex interactions using siRNA gene silencing in a human macrophage-like cell line.
Qin et al. previously performed a manual siRNA screen for host factors involved in cryptococcal uptake (18), although multiple key features distinguish our study from that work. Most central to the project design, the prior study targeted genes that were preselected for known involvement in membrane traffic, membrane remodeling, phagosome establishment, or phagosome maturation and thus were expected to affect fungal engulfment or survival. The screen also used insect cells, rather than mammalian cells, to represent the host. Finally, fungi in the earlier study were not opsonized, although some form of opsonization likely occurs in all disease contexts (14), and were not exposed to host-like growth conditions (e.g., 37°C and 5% CO2) that are known to dramatically alter virulence factor expression (19) and engulfment by host cells (16). A subset of the genes targeted by Qin et al. did, as hypothesized, alter fungal uptake; several of them, initially selected for their role in autophagosome biogenesis, were also shown to influence uptake and/or replication of unopsonized fungi in a mammalian system (murine RAW264.7 macrophages).
We performed an automated, unbiased screen of RNAi-treated human phagocytes, which were exposed to fungi opsonized with human serum under host-like conditions. We targeted 868 genes encoding two classes of host proteins that frequently act in regulatory processes, kinases and phosphatases, because we hypothesized that some of these would regulate host-fungus interactions. From 79 first-round hits of this screen, we refined our results based on specificity for C. neoformans interactions.
Of the four cryptococcus-specific genes that we identified, one prominent hit was the gene encoding calcium/calmodulin-dependent protein kinase 4 (CaMK4). CaMKs are serine/threonine kinases sensitive to changes in intracellular calcium. They are involved in cellular processes ranging from gene regulation, apoptosis, and cytokine release to differentiation and survival of T-lymphocytes and monocyte-derived cells (20, 21). Interestingly, a recent study based on phosphoproteomic analysis identified another member of this family, CaMK2, as a protein involved in C. neoformans phagocytosis (22). This work did not yield CaMK2, which may reflect the very distinct approaches of the two studies.
Multiple isoforms of CaMK display tissue-specific expression, with CaMK4 predominantly expressed in cells of the nervous and immune systems (20, 21). Both of these are significant lineages for cryptococcal infection, as C. neoformans shows tropism for the central nervous system. CaMK4 occupies a key position between upstream Ca2+/calmodulin-dependent protein kinase kinase (CaMKK) and downstream transcription factors in a Ca2+/calmodulin-regulated pathway or CaM kinase cascade (20). Once activated by phosphorylation, CaMK4 translocates to the nucleus and can phosphorylate multiple proteins, including CREB (21).
In phagocytic cells CaMKs are involved in several PRR signaling pathways, including those initiated by TLR2 and TLR4, where they act to modulate the transduced signal by phosphorylating CREB (20, 23). CREB activation is also involved in expression and signaling of the C-type lectin receptors Dectin-1 and CD206/mannose receptor (24, 25). We observed a significant reduction in cells positive for TLR2, Dectin-1, and CD206 across multiple phagocytic cell types during infection in CaMK4−/− mice compared to WT mice (Fig. 4). Notably, all three of these PRRs can recognize cryptococcal cells. TLR2 recognizes components of the cryptococcal capsule (26–28). Peripheral blood mononuclear cells (PBMCs) isolated from patients with cryptococcal meningitis also exhibit significantly less TLR2 than healthy control PBMCs, which has been interpreted as demonstrating a role for TLR2 in cryptococcal infection (29). TLR2 in combination with Dectin-1 can recognize fungal β-glucan and is important in protection against Aspergillus, Candida, and Pneumocystis infections (30–32). While Dectin-1 alone can recognize cryptococcal cells, there is no significant difference in disease progression in Dectin-1 KO mice compared to that of WT mice, suggesting that Dectin-1 is not required for host defense against C. neoformans (33, 34). CD206 recognizes heavily mannosylated cryptococcal mannoproteins (35), and the corresponding knockout mice succumb to C. neoformans infection significantly faster than WT mice (36). Increased surface expression of CD206, which is upregulated on M2 macrophages, results in increased phagocytosis, consistent with our results, but is also accompanied by decreased intracellular killing and tumor necrosis factor alpha (TNF-α) production (36). Although the roles of these three PRRs are complex, the changes observed in their expression could explain the reduced uptake of cryptococci by cells lacking CaMK4.
As discussed above and recently reviewed (15), host phagocytes play disparate roles in C. neoformans infection. These range from killing the fungi to providing them with a replicative niche or acting as Trojan horses to convey them to the brain, but all depend on interactions between the yeast and host cells. Alanio et al. tested the relationship between cryptococcus-macrophage interactions and disease outcome by measuring the engulfment and intracellular proliferation of clinical isolates upon exposure to murine phagocytes in vitro (37). They found that low phagocytic and intracellular proliferation indices of C. neoformans strains correlated with longer patient survival (37). The reduced uptake of fungi by innate immune cells with lower CaMK4 (Fig. 1A) therefore might account for the increased survival of CaMK4−/− mice compared to that of WT mice. Given this difference in survival, we were surprised that we observed no significant change in fungal burden between the WT and the CaMK4−/− mice (see Fig. S3 in the supplemental material). Importantly, assessment of organ burden by CFU (as is standard in the field and done here) does not distinguish between free and internalized fungi. It is possible that this is a key parameter in infection outcome.
CaMK4 has been implicated in neurological disorders (38, 39) and the level of Foxp3(+) regulatory T cells (Treg) (40). We considered the possibility that changes in the neuroimmune response or in the number of pulmonary Treg in CaMK4−/− mice could explain the difference in mouse survival, but we found no significant differences in these parameters (data not shown). Finally, it is also possible that the global CaMK4 deletion influences mouse survival by altering processes other than those of the hematopoietic compartment; these remain to be investigated.
In this study, we combined siRNA screening and an automated imaging-based assay to identify host phosphatases and kinases that are involved in C. neoformans internalization by human phagocytes. We identified four genes that, when silenced, demonstrated C. neoformans-specific alterations in uptake and verified the role of one of them in vivo. Unbiased strategies thus may be a powerful tool for identifying host factors involved in cryptococcal pathogenesis and potentially inform efforts to develop improved therapies that are desperately needed.
MATERIALS AND METHODS
Ethics statement.
All animal protocols were reviewed and approved by the Animal Studies Committee of the Washington University School of Medicine and conducted according to National Institutes of Health guidelines for housing and care of laboratory animals. Human serum was obtained from healthy donors with informed consent under a protocol approved by the Washington University in St. Louis Institutional Review Board (IRB).
Reagents.
Unless otherwise stated, chemicals were purchased from Sigma-Aldrich (St. Louis, MO), tissue culture medium from Invitrogen (Grand Island, NY), and plasticware from Fisher Scientific (Pittsburgh, PA).
Strains and media.
Cryptococcus neoformans strains H99α (from Joe Heitman) and KN99α (from Kirsten Nielsen), Saccharomyces cerevisiae strain W303 (from Randy Schekman), and a Candida albicans efg1Δ cphΔ strain (a nonhyphenating mutant; from Aaron Mitchell) were recovered from 15% glycerol stocks stored at −80°C and maintained on YPD plates (1% yeast extract, 2% peptone, 2% dextrose, and 2% Bacto agar). For all studies yeast cells were grown for 15 to 17 h at 30°C with shaking in YPD broth, collected by centrifugation, washed three times with sterile phosphate-buffered saline (PBS), and counted on a Cellometer Auto M10 (Nexcelom Bioscience, Lawrence, MA).
Cell culture.
Mammalian cells were grown at 37°C and 5% CO2. THP-1, a male human monocytic cell line (ATCC TIB-202), was cultured in RPMI 1640 with 10% heat-inactivated fetal bovine serum (FBS), 100 μg/ml penicillin, 100 U/ml streptomycin, 1 mM sodium pyruvate, and 48 μM β-mercaptoethanol and passaged every 3 days to maintain cell density of 1.5 × 105 to 9 × 105/ml. Prior to uptake assays, cells were seeded in flat-bottomed polystyrene 96-well microtiter plates (Costar) at 1.5 × 104 cells/well in THP-1 differentiation medium (the growth medium described above with 0.2 μg/ml phorbol 12-myristate 13-acetate) and grown for 48 h.
RNAi library testing.
For treatment with Qiagen siRNA libraries targeting human phosphatases and kinases (v 4.0), THP-1 cells were seeded in 96-well plates using a BioMek FX laboratory automation workstation. Pools of two siRNAs per target (10 nM final concentration) were added to each well with 100 μl of Polyplus INTERFERin transfection reagent (Polyplus Transfection, New York, NY), and the cells were incubated for 2 h before the addition of THP-1 differentiation medium and continued growth for 48 h. Host cell viability was monitored using alamarBlue (Life Technologies). Twenty-four hours prior to uptake assays the transfected cells were washed and the medium replaced with RPMI 1640 alone to induce starvation. For some studies, THP-1 cells were treated with 10 nM CamK4 inhibitor KN93 (dimethyl sulfoxide [DMSO]-soluble form; Sigma) for 24 h prior to uptake assays (during the starvation phase). Cells were then washed and fresh RPMI 1640 was added before initiation of the assays.
Uptake assays.
Four particles were used for uptake assays: C. neoformans cells stained for 30 min at room temperature (RT) with Lucifer yellow (Sigma) in McIlvaine's solution (pH 6.0; final concentration, 100 μg/ml); S. cerevisiae and C. albicans stained for 30 min at RT with FUN-1 (2.0 μl of 10 mM stock; Invitrogen) in 100 μl 1× PBS; and fluorescent yellow-green 1-μm latex beads (Sigma). All particles were opsonized for 30 min at 37°C with 40% human serum before addition at a multiplicity of infection (MOI) of 16 to the plates of transfected and starved THP-1 cells, and the plates then were incubated for 1 h at 37°C and 5% CO2. Human serum was obtained from individual healthy volunteers by following a protocol approved by the Washington University School of Medicine IRB.
Postinfection, plates were processed as described in Srikanta et al. (16). Briefly, they were washed with a BioTek ELx405 microplate washer (BioTek, Winooski, VT) to remove nonadherent cells, fixed in 4% formaldehyde, and permeabilized with 0.1% saponin. Host cells then were stained with 2 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) to label host nuclei and 250 ng/ml CellMask plasma membrane deep red to stain host cell bodies (both from Sigma). Plates were imaged using an IN Cell analyzer (GE Healthcare) with scanning on 3 channels (wavelengths of 360/460, 475/535, and 620/460 nm to detect DAPI, Lucifer yellow, and CellMask, respectively). Fifteen to 20 images were captured for each channel/well and analyzed using the IN Cell Developer Toolbox (GE Healthcare). Each sample was assayed in one well in the same position on 3 separate 96-well plates, and all experiments were repeated at least twice. All studies included controls for uptake (fungi with or without serum opsonization) and for RNAi treatment (no siRNA, scrambled siRNA, and siRNA targeting proteins required for general phagocytosis).
Quantitative real-time PCR.
Total RNA was isolated using the RNeasy Plus minikit (Qiagen), and first-strand cDNA was synthesized using SuperScript II (Invitrogen) per the manufacturer's instructions. mRNA levels were quantified with SYBR green-based detection using a Bio-Rad CFX96 instrument and 40 cycles of PCR (94°C for 15 s, 60°C for 30 s, and 72°C for 30 s). Gene expression was normalized to ACT1 levels and expressed as a percentage of WT control expression.
Pulmonary infections.
Four- to 6-week-old female C57BL/6J mice or CaMK4−/− mice (The Jackson Laboratory, Bar Harbor, ME) were anesthetized by intraperitoneal (i.p.) injection (of 150 μl of 2 mg/ml xylazine [VEDCO] and 10 mg/ml of ketaset [Fort Dodge Animal Health]) and intranasally inoculated with the indicated number of CFU (verified by quantitative culture on YPD agar) of C. neoformans strain KN99α in 50 μl of sterile PBS. The mice were fed ad libitum and monitored daily for symptoms. For organ burden, cytokine analysis, and flow cytometry studies, mice were euthanized at specific time points postinoculation by CO2 inhalation, and the lungs, brain, and spleen were harvested. For survival studies, mice were sacrificed when body weight fell below 80% of peak weight. For some survival studies, mice were injected i.p. with water-soluble KN93 (250 μg/g body weight; EMD Millipore) in 100 μl of PBS or PBS alone three times a week, beginning 1 day prior to infection.
Pulmonary leukocyte isolation.
For leukocyte isolation, lungs were enzymatically digested at 37°C for 30 min in 10 ml of digestion buffer (RPMI 1640 containing 1 mg/ml of collagenase type IV). The digested tissues then were successively passed through sterile 70- and 40-μm-pore nylon strainers (BD Biosciences, San Jose, CA). Erythrocytes in the strained suspension were lysed by incubation in NH4Cl buffer (0.859% NH4Cl, 0.1% KHCO3, 0.0372% Na2EDTA; pH 7.4; Sigma-Aldrich) for 3 min on ice, followed by the addition of a 2-fold excess of PBS. The unlysed leukocytes then were collected by centrifugation, resuspended in sterile PBS, and counted with a Cellometer Auto M10 (Nexcelom Bioscience).
Isolation and assay of peritoneal macrophages.
Four- to 6-week-old female mice were injected i.p. with 1 ml 5 mM sodium periodate (Sigma) in sterile PBS to induce cell recruitment into the peritoneal cavity. Mice were euthanized 72 h postinjection, and peritoneal lavage was performed with ice-cold sterile PBS. Macrophages were isolated from the lavage fluid by positive selection using biotinylated α-F4/80 antibody (eBioscience) and α-biotin-conjugated magnetic beads (Miltenyi Biotec). Uptake assays were performed as described above, except that C57BL/6 mouse serum was used for opsonization.
Flow cytometry.
All flow cytometry steps were performed at 4°C. Pulmonary leukocytes in PBS were stained using the LIVE/DEAD fixable blue dead cell stain kit (Invitrogen, Carlsbad, CA) for 30 min. Following incubation, samples were washed and resuspended at 107/ml in 100 μl PBS plus 2% fetal bovine serum (fluorescence-activated cell sorting buffer), and 100 μl was dispensed into wells of a 96-well U-bottom plate. Samples where Fc receptors were not to be directly analyzed then were incubated with CD16/CD32 (Fc block; BD Biosciences, San Jose, CA) for 5 min. For flow cytometry, cells next were incubated with the optimal concentrations of fluorochrome-conjugated antibodies (see Table S3 in the supplemental material for antigen, clone, and source) in various combinations for 30 min, washed, and fixed in 2% ultrapure formaldehyde. For data acquisition, >50,000 events were collected on a BD LSRFortessa X-20 flow cytometer (BD Biosciences, San Jose, CA), and the data were analyzed with FlowJo V10 (TreeStar, Ashland, OR).
Cytokine analysis.
Cytokine levels in lung tissues were analyzed using the Bio-Plex protein array system (Bio-Rad Laboratories, Hercules, CA) as described previously (41). Briefly, lung tissue was excised and homogenized in ice-cold PBS (1 ml) and mixed with an equal volume of PBS containing 2× Pierce protease inhibitors (Thermo Scientific, Rockford, IL) and 0.05% Triton X-100, and the samples were clarified by centrifugation. Supernatant fractions from the pulmonary homogenates then were assayed using the Bio-Plex Pro mouse cytokine 23-plex (Bio-Rad Laboratories) for the presence of interleukin-1α (IL-1α), IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-17A, granulocyte colony-stimulating factor (G-CSF), granulocyte monocyte colony-stimulating factor (GM-CSF), interferon gamma (IFN-γ), CXCL1/keratinocyte-derived chemokine (KC), CCL2/monocyte chemotactic protein-1 (MCP-1), CCL3/macrophage inflammatory protein-1α (MIP-1α), CCL4/MIP-1β, CCL5/RANTES, and TNF-α.
Statistics.
All in vitro experiments were performed independently in triplicate. Candidates from the RNAi screen were assessed using the quartile-based threshold method described previously (42). To compare results between experiments, the data from each plate were normalized to the plate controls. Significance was assessed using the Student t test for individual paired comparison and by one-way analysis of variance and Tukey's test when multiple groups were compared. Mouse survival was analyzed using Kaplan-Meier analysis and log-rank statistics. All statistical analyses were confirmed using GraphPad Prism software, version 6.0 (GraphPad Software). P values of <0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Center for Human Immunology and Immunotherapy Programs and the High-Throughput Screening Center at Washington University Medical School, where flow cytometry and screening were performed, respectively. We also thank Jayne Marasa and members of the Doering laboratory for helpful discussions, Mary Dinauer and Rachel Idol for assistance with peritoneal macrophages, and Joe Heitman, Aaron Mitchell, Kirsten Nielsen, and Randy Schekman for yeast strains.
These studies were supported by NIH grants AI102882 and AI078795 to T.L.D. D.S. was partially supported by F32 AI100481 and T32 AI007172, and C.R.H. was partially supported by T32 AI007172. S.A.K. was supported by R01 HL105427.
We have no conflicts of interest to declare.
D.S. and T.L.D. conceived of the project; D.S., C.R.H., and M.W. performed experiments; D.S., C.R.H., S.A.K., and T.L.D. analyzed data; and D.S., C.R.H., and T.L.D. wrote the text and all authors reviewed it.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00195-17.
REFERENCES
- 1.Andama AO, den Boon S, Meya D, Cattamanchi A, Worodria W, Davis JL, Walter ND, Yoo SD, Kalema N, Haller B, Huang L, International HIV-Associated Opportunistic Pneumonias Study . 2013. Prevalence and outcomes of cryptococcal antigenemia in HIV-seropositive patients hospitalized for suspected tuberculosis in Uganda. J Acquir Immune Defic Syndr 63:189–194. doi: 10.1097/QAI.0b013e3182926f95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heitman J, Kozel TR, Kwon-Chung KJ, Perfect JR, Casadevall A. 2010. Cryptococcus: from human pathogen to model yeast. ASM Press, Washington, DC. [Google Scholar]
- 3.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 4.Denning DW. 2016. Minimizing fungal disease deaths will allow the UNAIDS target of reducing annual AIDS deaths below 500,000 by 2020 to be realized. Philos Trans R Soc Lond B Biol Sci 371:20150468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, Denning DW, Loyse A, Boulware DR. 2017. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis 17:873–881. doi: 10.1016/S1473-3099(17)30243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarvis JN, Bicanic T, Loyse A, Namarika D, Jackson A, Nussbaum JC, Longley N, Muzoora C, Phulusa J, Taseera K, Kanyembe C, Wilson D, Hosseinipour MC, Brouwer AE, Limmathurotsakul D, White N, van der Horst C, Wood R, Meintjes G, Bradley J, Jaffar S, Harrison T. 2014. Determinants of mortality in a combined cohort of 501 patients with HIV-associated Cryptococcal meningitis: implications for improving outcomes. Clin Infect Dis 58:736–745. doi: 10.1093/cid/cit794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bicanic T, Meintjes G, Wood R, Hayes M, Rebe K, Bekker LG, Harrison T. 2007. Fungal burden, early fungicidal activity, and outcome in cryptococcal meningitis in antiretroviral-naive or antiretroviral-experienced patients treated with amphotericin B or fluconazole. Clin Infect Dis 45:76–80. doi: 10.1086/518607. [DOI] [PubMed] [Google Scholar]
- 8.Levitz SM, Nong SH, Seetoo KF, Harrison TS, Speizer RA, Simons ER. 1999. Cryptococcus neoformans resides in an acidic phagolysosome of human macrophages. Infect Immun 67:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker SC, Casadevall A. 2002. Replication of Cryptococcus neoformans in macrophages is accompanied by phagosomal permeabilization and accumulation of vesicles containing polysaccharide in the cytoplasm. Proc Natl Acad Sci U S A 99:3165–3170. doi: 10.1073/pnas.052702799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kechichian TB, Shea J, Del Poeta M. 2007. Depletion of alveolar macrophages decreases the dissemination of a glucosylceramide-deficient mutant of Cryptococcus neoformans in immunodeficient mice. Infect Immun 75:4792–4798. doi: 10.1128/IAI.00587-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith LM, Dixon EF, May RC. 2015. The fungal pathogen Cryptococcus neoformans manipulates macrophage phagosome maturation. Cell Microbiol 17:702–713. doi: 10.1111/cmi.12394. [DOI] [PubMed] [Google Scholar]
- 12.Perfect JR. 2014. Cryptococcosis: a model for the understanding of infectious diseases. J Clin Investig 124:1893–1895. doi: 10.1172/JCI75241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabiiti W, Robertson E, Beale MA, Johnston SA, Brouwer AE, Loyse A, Jarvis JN, Gilbert AS, Fisher MC, Harrison TS, May RC, Bicanic T. 2014. Efficient phagocytosis and laccase activity affect the outcome of HIV-associated cryptococcosis. J Clin Investig 124:2000–2008. doi: 10.1172/JCI72950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly RM, Chen JM, Yauch LE, Levitz SM. 2005. Opsonic requirements for dendritic cell-mediated responses to Cryptococcus neoformans. Infect Immun 73:592–598. doi: 10.1128/IAI.73.1.592-598.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leopold Wager CM, Hole CR, Wozniak KL, Wormley FL Jr. 2016. Cryptococcus and phagocytes: complex interactions that influence disease outcome. Front Microbiol 7:105. doi: 10.3389/fmicb.2016.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srikanta D, Yang M, Williams M, Doering TL. 2011. A sensitive high-throughput assay for evaluating host-pathogen interactions in Cryptococcus neoformans infection. PLoS One 6:e22773. doi: 10.1371/journal.pone.0022773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vest RS, Davies KD, O'Leary H, Port JD, Bayer KU. 2007. Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell 18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin QM, Luo J, Lin X, Pei J, Li L, Ficht TA, de Figueiredo P. 2011. Functional analysis of host factors that mediate the intracellular lifestyle of Cryptococcus neoformans. PLoS Pathog 7:e1002078. doi: 10.1371/journal.ppat.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaragoza O, Fries BC, Casadevall A. 2003. Induction of capsule growth in Cryptococcus neoformans by mammalian serum and CO(2). Infect Immun 71:6155–6164. doi: 10.1128/IAI.71.11.6155-6164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Illario M, Giardino-Torchia ML, Sankar U, Ribar TJ, Galgani M, Vitiello L, Masci AM, Bertani FR, Ciaglia E, Astone D, Maulucci G, Cavallo A, Vitale M, Cimini V, Pastore L, Means AR, Rossi G, Racioppi L. 2008. Calmodulin-dependent kinase IV links Toll-like receptor 4 signaling with survival pathway of activated dendritic cells. Blood 111:723–731. doi: 10.1182/blood-2007-05-091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, Means AR, Billiar TR, Angus DC, Rosengart MR. 2008. Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol 181:5015–5023. doi: 10.4049/jimmunol.181.7.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandey A, Ding SL, Qin QM, Gupta R, Gomez G, Lin F, Feng X, Fachini da Costa L, Chaki SP, Katepalli M, Case ED, van Schaik EJ, Sidiq T, Khalaf O, Arenas A, Kobayashi KS, Samuel JE, Rivera GM, Alaniz RC, Sze SH, Qian X, Brown WJ, Rice-Ficht A, Russell WK, Ficht TA, de Figueiredo P. 2017. Global reprogramming of host kinase signaling in response to fungal infection. Cell Host Microbe 21:637–649. doi: 10.1016/j.chom.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang W, Ghisletti S, Perissi V, Rosenfeld MG, Glass CK. 2009. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol Cell 35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elcombe SE, Naqvi S, Van Den Bosch MW, MacKenzie KF, Cianfanelli F, Brown GD, Arthur JS. 2013. Dectin-1 regulates IL-10 production via a MSK1/2 and CREB dependent pathway and promotes the induction of regulatory macrophage markers. PLoS One 8:e60086. doi: 10.1371/journal.pone.0060086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barminko JA, Nativ NI, Schloss R, Yarmush ML. 2014. Fractional factorial design to investigate stromal cell regulation of macrophage plasticity. Biotechnol Bioeng 111:2239–2251. doi: 10.1002/bit.25282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shoham S, Huang C, Chen JM, Golenbock DT, Levitz SM. 2001. Toll-like receptor 4 mediates intracellular signaling without TNF-alpha release in response to Cryptococcus neoformans polysaccharide capsule. J Immunol 166:4620–4626. doi: 10.4049/jimmunol.166.7.4620. [DOI] [PubMed] [Google Scholar]
- 27.Yauch LE, Mansour MK, Levitz SM. 2005. Receptor-mediated clearance of Cryptococcus neoformans capsular polysaccharide in vivo. Infect Immun 73:8429–8432. doi: 10.1128/IAI.73.12.8429-8432.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yauch LE, Mansour MK, Shoham S, Rottman JB, Levitz SM. 2004. Involvement of CD14, toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect Immun 72:5373–5382. doi: 10.1128/IAI.72.9.5373-5382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Liu T, Kong W, Zhang W, Gu M, Chen Y, Deng A, Chen S. 2015. Decreased TLR2 signal expression in peripheral blood mononuclear cell from patients with cryptococcal meningitis. Microbiol Immunol 59:357–364. doi: 10.1111/1348-0421.12264. [DOI] [PubMed] [Google Scholar]
- 30.Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD. 2005. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog 1:e42. doi: 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, Iwakura Y. 2007. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol 8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- 32.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura K, Kinjo T, Saijo S, Miyazato A, Adachi Y, Ohno N, Fujita J, Kaku M, Iwakura Y, Kawakami K. 2007. Dectin-1 is not required for the host defense to Cryptococcus neoformans. Microbiol Immunol 51:1115–1119. doi: 10.1111/j.1348-0421.2007.tb04007.x. [DOI] [PubMed] [Google Scholar]
- 34.Giles SS, Dagenais TR, Botts MR, Keller NP, Hull CM. 2009. Elucidating the pathogenesis of spores from the human fungal pathogen Cryptococcus neoformans. Infect Immun 77:3491–3500. doi: 10.1128/IAI.00334-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansour MK, Latz E, Levitz SM. 2006. Cryptococcus neoformans glycoantigens are captured by multiple lectin receptors and presented by dendritic cells. J Immunol 176:3053–3061. doi: 10.4049/jimmunol.176.5.3053. [DOI] [PubMed] [Google Scholar]
- 36.Dan JM, Kelly RM, Lee CK, Levitz SM. 2008. Role of the mannose receptor in a murine model of Cryptococcus neoformans infection. Infect Immun 76:2362–2367. doi: 10.1128/IAI.00095-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alanio A, Desnos-Ollivier M, Dromer F. 2011. Dynamics of Cryptococcus neoformans-macrophage interactions reveal that fungal background influences outcome during cryptococcal meningoencephalitis in humans. mBio 2:e00158-11. doi: 10.1128/mBio.00158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen SD, Lin TK, Lin JW, Yang DI, Lee SY, Shaw FZ, Liou CW, Chuang YC. 2010. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res 88:3144–3154. doi: 10.1002/jnr.22469. [DOI] [PubMed] [Google Scholar]
- 39.Wei YP, Ye JW, Wang X, Zhu LP, Hu QH, Wang Q, Ke D, Tian Q, Wang JZ. 23 June 2017. Tau-induced Ca2+/calmodulin-dependent protein kinase-IV activation aggravates nuclear Tau hyperphosphorylation. Neurosci Bull doi: 10.1007/s12264-017-0148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koga T, Mizui M, Yoshida N, Otomo K, Lieberman LA, Crispin JC, Tsokos GC. 2014. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3(+) regulatory T cells in MRL/lpr mice. Autoimmunity 47:445–450. doi: 10.3109/08916934.2014.915954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wozniak KL, Ravi S, Macias S, Young ML, Olszewski MA, Steele C, Wormley FL. 2009. Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS One 4:e6854. doi: 10.1371/journal.pone.0006854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, Smith Q, Beijersbergen RL, Ghazal P, Shamu CE. 2009. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 6:569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.