

ABSTRACT
Development of approaches to genetically manipulate Chlamydia is fostering important advances in understanding pathogenesis. Fluorescence-reported allelic exchange mutagenesis (FRAEM) now enables the complete deletion of specific genes in C. trachomatis L2. We have leveraged this technology to delete the coding sequences for a known type III effector. The evidence provided here indicates that CT694/CTL0063 is a virulence protein involved in chlamydial invasion. Based on our findings, we designate the gene product corresponding to ct694-ctl0063 translocated membrane-associated effector A (TmeA). Deletion of tmeA did not impact development of intracellular chlamydiae. However, the absence of TmeA manifested as a decrease in infectivity in both tissue culture and murine infection models. The in vitro defect was reflected by impaired invasion of host cells. TmeA binds human AHNAK, and we demonstrate here that AHNAK is transiently recruited by invading chlamydiae. TmeA, however, is not required for endogenous AHNAK recruitment. TmeA also impairs AHNAK-dependent actin bundling activity. This TmeA-mediated effect likely does not explain impaired invasion displayed by the tmeA strain of Chlamydia, since AHNAK-deficient cells revealed no invasion phenotype. Overall, our data indicate the efficacy of FRAEM and reveal a role of TmeA during chlamydial invasion that manifests independently of effects on AHNAK.
KEYWORDS: cytoskeleton, invasion, type III secretion, AHNAK
INTRODUCTION
Chlamydia trachomatis remains the leading agent of bacterial sexually transmitted disease in the United States (serovars D to K) (1) and the leading cause of preventable blindness globally (serovars A to C) (2). Chlamydiae are Gram-negative bacteria that preferentially infect mucosal columnar epithelial cells and replicate within a parasitophorous vacuole termed an inclusion. A biphasic developmental cycle (3) is manifested during infections in which a nonmetabolic elementary body (EB) invades a host cell and subsequently transitions to the replicative, yet noninfectious, reticulate body (RB). The cycle is completed by asynchronous differentiation of RBs back into EBs and subsequent extrusion of the inclusion or lysis of the host cell (4). Historically challenged by a lack of genetic tools, the molecular mechanisms governing these processes are now coming into better focus due to development of approaches to transform Chlamydia (5, 6), ectopically express chlamydial genes (7–10), and create random (11) or targeted (12, 13) mutations. These advances have culminated in the ability to completely delete targeted chlamydial genes via fluorescence-reported allelic exchange mutagenesis (FRAEM) (14).
As an obligate intracellular pathogen, invasion of new host cells represents the first essential step in initiating and perpetuating chlamydial infection. Given the importance of this process, chlamydial entry into nonphagocytic epithelial cells involves multiple, likely redundant pathways (15). Indeed, serovar- and species-specific differences in attachment and entry requirements have been described (16), and interference with any one pathway does not lead to complete inhibition of chlamydial invasion. Despite complexities, key themes have emerged regarding chlamydial entry into epithelial cells. First, chlamydial EBs are completely competent for invasion, since neither de novo transcription nor translation are required for entry (17). This is consistent with data (18) indicating the EBs are preloaded with proteins known to be necessary for orchestrating the invasion process. Attachment in cell culture occurs through a two-step process involving an initial, reversible electrostatic interaction between glycan moieties on the EB and host cell heparan sulfate-containing glycosaminoglycans (19–23). Initial association is followed by irreversible binding to the host cell surface through specific receptors (24, 25). Following attachment, Chlamydia and host proteins converge to create an infection synapse where assembled signaling platforms remodel host cytoskeleton-associated factors to promote invasion (26). C. trachomatis-induced remodeling of the cytoskeleton is localized to points of chlamydial attachment and is required for entry (27). Manipulation of host cell actin is a conserved requirement for efficient chlamydial invasion (16, 26), as treatment of epithelial cells with cytochalasin D (28) or jasplakinolide (26, 29) or expression of dominant-negative Rac1 (30, 31) interferes with entry of C. trachomatis. Subsequent to entry, nascent inclusions remain segregated from the endosomal-lysosomal compartment, a process that requires manipulation of host Rab GTPases and vesicle fusion machinery (32). Inclusions traffic to the microtubule-organizing center, where they remain anchored and intercept lipids and other resources required for further development (33).
Similar to other Gram-negative pathogens (34), C. trachomatis employs a type III secretion system (T3SS) to accomplish secretion and translocation of anti-host effector proteins into an associated host cell (35). Prior to invasion, EBs possess secretion-capable T3SSs (36, 37) and contain pools of previously synthesized effectors (18) capable of being secreted during the invasion process. The most thoroughly characterized effector is the translocated actin recruiting protein (TarP), which directly nucleates and polymerizes actin (29, 38–43). The central repeat units of TarP also serve as scaffolding for host guanine nucleotide exchange factors which regulate signaling pertaining to actin dynamics and recruitment (43). Microinjection of host cells with TarP-specific antibodies reduces the efficiency of chlamydial entry (41). Efficient translocation of TarP requires SycE-like chaperone-1(Slc1) (44), and Slc1 binds additional effector proteins originally designated CT694, CT695, and CT875 (45, 46). CT875, now designated translocated early phosphoprotein (TepP), is secreted during invasion and recruits host CrkL and phosphatidylinositol 3-kinase (47) to entry sites, yet chlamydial null mutants lacking TepP were not defective for entry (46). Previously referred to using nomenclature first established in C. trachomatis serovar D (48), CT694, CT695, and CT696 correspond to C. trachomatis L2 proteins designated CTL0063, CTL0064, and CTL0065, respectively. The proteins are highly conserved among trachomatis serotypes, exhibiting 99% identity. Both C. trachomatis CT694/CTL0063 (49) and CT695/CTL0064 (9) are secreted during chlamydial entry and are capable of peripherally associating with eukaryotic membranes (9, 50). Based on their described properties, we propose the designation translocated membrane-associated effectors A (TmeA) and B (TmeB) for CT694/CTL0063 and CT695/CTL0064, respectively.
TmeA binds human AHNAK (49), a ubiquitously expressed 700-kDa phosphoprotein capable of localizing to the cytoplasm, nucleus, and plasma membrane of cells in response to a variety of stimuli (51–56). Importantly, AHNAK is localized to the apical membranes of polarized epithelial cells and contributes to maintenance of apical cytoarchitecture (57). AHNAK has a tripartite structure consisting of a short 251-residue amino-terminal (N-terminal) domain and a 1,002-residue carboxyl-terminal (C-terminal) domain connected by a 4,300-residue central region comprised of 39 conserved central repeat units (CRU) (58). C. trachomatis TmeA was found to be capable of binding either the CRUs or the C-terminal domain of AHNAK in a yeast two-hybrid screen (49). AHNAK CRUs are predicted to form β-propeller-like structures that confer scaffolding function for interactions of AHNAK with other host proteins (59). CRUs contribute to membrane-associated signaling events through direct interactions with protein kinase C-alpha (PKCα) and phospholipase C-gamma (PLCγ) (59–61). The C-terminal domain of AHNAK exerts roles in actin dynamics by serving as a scaffold for assembly of cytoskeletal elements and membrane-associated signaling (58, 60, 62–65). AHNAK functions directly in actin dynamics via C-terminal-dependent bundling of filamentous actin (64) and indirectly through interaction with the tetrameric annexin A2-S100A10 (A2t) complex (66, 67). The AHNAK-A2t complex has been implicated in maintaining the cortical actin cytoskeleton (57) and serving as scaffolding for membrane repair (68).
Based on previous data, we hypothesized that TmeA (and possibly TmeB) is required for chlamydial invasion. Given the newly acquired tractability in chlamydial genetics, this hypothesis is now directly testable. Indeed, we recently reported the successful deletion of both tmeA and tmeB in C. trachomatis L2 using FRAEM (14). Although no developmental phenotype was detected for tmeB chlamydiae, the tmeA deletion resulted in decreased infectivity in a murine model and manifested as a reproducible defect at the level of host cell invasion. Interestingly, AHNAK colocalizes with invading chlamydiae, and the TmeA interaction with AHNAK disrupts AHNAK-dependent actin bundling activity. However, our data are consistent with an AHNAK-independent role of TmeA in C. trachomatis invasion, reinforcing a working model where TmeA represents a multifunctional effector.
RESULTS
Phenotypic characterization of C. trachomatis tmeA strain.
Recent advances in the genetic manipulation of chlamydiae have enabled more definitive functional characterization of chlamydial gene products. We recently reported the FRAEM-mediated construction of C. trachomatis L2 strains lacking coding sequences for CTL0063 (TmeA), CTL0064 (TmeB), and the downstream, but independently expressed, CTL0065. Both TmeA and TmeB are type III secreted effectors (TS3Es), whereas secretion of CTL0065 has not been demonstrated. We began our phenotypic assessment by testing whether developmental defects were manifested in null strains (Fig. 1). The ctl0065 strain was included for comparison. HeLa cells were infected with equal inclusion-forming units (IFUs) of C. trachomatis wild-type (WT) or mutant strains, and inclusions were visualized at 24 h postinfection (hpi) by staining for chlamydiae and the inclusion membrane protein IncG. Consistent levels of infection were confirmed by enumeration of inclusions (data not shown). We did not detect any overt qualitative differences in inclusion morphology (Fig. 1A), and measurement of inclusion area did not reveal differences in the ability of strains to establish mature-sized inclusions (Fig. 1B). We next tested the ability of strains to produce progeny EBs by equally infecting HeLa cells, harvesting cultures at times corresponding to early-cycle (1 and 6 h), midcycle (12 h), and late-cycle (24 and 48 h) development, and enumerating subsequent second-passage IFUs (Fig. 1C). Under these conditions, progeny counts were similar at all tested time points when comparing C. trachomatis WT and tmeA and tmeB strains. In contrast, production of progeny chlamydiae was attenuated for ctl0065 chlamydiae, with the 48-h burst size being reduced ca. 10-fold. Finally, we chose to enumerate chlamydial genomes during serial passage of cultures as a mechanism to detect more subtle differences in infectivity. HeLa cultures were initially infected with equal IFUs, and material was subsequently passaged onto new monolayers every 24 h. Samples were removed and processed for quantitative PCR (qPCR)-based quantitation of chlamydial genomes at every passage (Fig. 1D). Growth of the tmeB strain did not differ significantly from that of the WT, whereas levels of genomes from the tmeA strain steadily decreased and were 34.3% (±11.1%) of that of the WT by the fourth sampling. As expected, infectivity of the ctl0065 strain was also reduced, yielding 3.4% (±1.7%) as many progeny as the WT by the final time point. Linear regression analysis revealed a statistically significant, progressive decrease in chlamydial genomes for tmeA and ctl0065 but not tmeB chlamydiae. These data indicate that disruption of either tmeA or ctl0065 negatively impacts the ability of chlamydiae to sustain infection. The distinctive phenotype exhibited by the ctl0065 strain confirms our earlier observations that the mutations in tmeA and tmeB did not affect CTL0065 expression (14). Since there is currently no evidence that CTL0065 is a T3SE, we proceeded with characterization of only the tmeA and tmeB strains.
FIG 1.

Deletion of tmeA impacts infectivity independent of growth rate. HeLa cells were infected with equal IFUs of WT, tmeA, tmeB, or ctl0065 strains of C. trachomatis L2. Cultures were methanol fixed at 24 hpi and stained to visualize chlamydial inclusions via indirect immunofluorescence. (A) Inclusions were visualized by staining with Hsp60 (red) and IncG (green). Bar, 2 μm. (B) Areas of 50 representative inclusions were measured and plotted individually and with respective means and standard deviations shown. (C) Cultures were harvested over time (1, 6, 24, and 48 h) for enumeration of progeny chlamydiae. Two-way ANOVA was performed to establish statistical significance (*, P < 0.0001). (D) Alternatively, cultures were serially passaged onto fresh HeLa monolayers every 24 h and genome content was quantitated via qPCR at each passage. Direct and progeny inclusion counts (B and C) were derived from triplicate cultures, and data are reported as means ± SD. In panel D, the 16S copy number for each 24-h passage number is presented relative to the wild type as means ± SD (n = 3). Linear regression analysis indicates significant differences in slope over time (*, P = 0.0059; **, P = 0.0002).
We normalized infections for numbers of bacteria to more directly test for defects in development. Genome content was deduced for WT, tmeA, and tmeB strains of C. trachomatis via qPCR, and duplicate sets of HeLa monolayers were infected by rocking cultures for 1 h at 37°C with inocula containing equal numbers of genomes. IFU content was determined in primary cultures (Fig. 2A) and in parallel cultures harvested for progeny counts (Fig. 2B). Under these infection conditions, the tmeA, but not tmeB, strain of C. trachomatis yielded significantly fewer inclusions than the WT. The C. trachomatis tmeA strain direct IFU counts in primary cultures were reduced ca. 52% while progeny counts were ca. 43% of WT levels. Similar results were obtained when infections were normalized by particle counts (data not shown). Since no phenotype was detected with the tmeB strain, we conclude from these data that only TmeA is required for efficient infection of HeLa cells.
FIG 2.
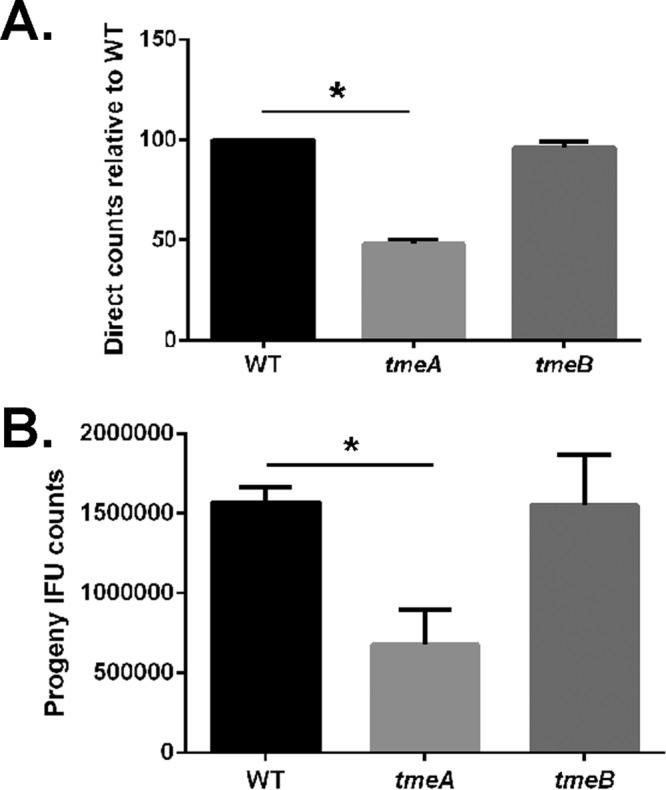
Infections using equivalent numbers of bacteria reveal an infectivity defect for C. trachomatis lacking TmeA. Duplicate sets of HeLa cultures were infected in triplicate with equal numbers (as determined by genome copy number) of C. trachomatis WT, tmeA, or tmeB strain to achieve an approximate MOI of 0.1. At 24 h postinfection, one set of cultures was methanol fixed and stained for chlamydiae (A), while the second was processed for enumeration of progeny EBs by passage onto fresh HeLa monolayers (B). All inclusions were enumerated by fluorescence staining of chlamydiae in fixed samples, and data for direct and progeny counts are represented as means ± standard deviations from triplicate samples. Student's t test with Welch's correction was used to address significance (*, P < 0.008; **, P < 0.009).
Lack of TmeA results in decreased infectivity and invasion.
We have previously demonstrated that the bla-gfp marker inserted in place of tmeA exerts a polar effect on TmeB expression (14). Although our tmeB-deficient strain showed no decrease in infectivity, we could not rule out the possibility that infectivity defects manifested by our tmeA strain were due to lack of both effectors. Complementation studies therefore were performed to address this question. We cloned tmeA and 292 nucleotides (nt) of upstream DNA into our expression vector, pCompAII, and mobilized this plasmid into the tmeA strain. TmeA expression was first confirmed by immunoblotting of EBs (Fig. 3A) or whole-culture material harvested 12 or 24 hpi (Fig. 3B). TmeA was detected in levels comparable to those of the WT in both EBs and 24-h whole-culture material. As expected, TmeB was not detected in either the tmeA or tmeA+ptemA strains. Having established comparable expression of ectopic tmeA, HeLa monolayers were infected with equal particles of WT, tmeA, and tmeA+ptmeA C. trachomatis strains, and inclusions were enumerated 24 hpi (Fig. 3C). Inclusion abundance was reduced in tmeA, but not tmeA+ptmeA, cultures compared to that of the WT. Therefore, we conclude that the infection defect is due solely to loss of tmeA. We next extended this analysis to the murine infection model to gauge the requirement of TmeA during in vivo infection (Fig. 3D). Mice were infected with equal IFUs of WT, tmeA, and tmeA+ptmeA C. trachomatis strains, and shed IFUs were enumerated over time. Although infectious loads were similar at days 3 and 7, the tmeA strain was attenuated compared to the WT, since tmeA strain-infected mice resolved infections by day 11. In contrast, mice remained infected with the complemented strain, and IFU counts were similar to those of the WT until day 19. Hence, TmeA is required for in vivo virulence and the tmeA mutation can be partially complemented with tmeA alone.
FIG 3.

Complementation with TmeA restores infectivity to WT levels. Material from equivalent particles of purified WT, tmeA, or complemented tmeA (tmeA+ptmeA) EBs (A) or cultures infected at an MOI of 1 were probed in immunoblots for TmeA and TmeB (B). Chlamydial Hsp60 and HeLa cell actin were examined as loading controls. All proteins were visualized by chemiluminescent development. (C) HeLa cells were infected with equal numbers of WT, tmeA, or tmeA+ptmeA EBs and processed for enumeration of IFUs at 24 hpi. Data are represented as means ± standard deviations from triplicate samples. Student's t test with Welch's correction was used to address significance (*, P < 0.008). (D) Groups of 5 female C3H/HeJ mice were infected intravaginally with equal input IFUs, and shed IFUs were enumerated at days 3, 7, 11, 15, 19, and 23 postinfection. Two-way ANOVA was performed to establish statistical significance (*, P < 0.0001).
We next sought to establish the step at which chlamydial development was impaired. Intracellular tmeA strain organisms develop similarly to WT organisms (Fig. 1C), and we did not detect a difference in tmeA strain attachment efficiency (data not shown). We therefore considered the possibility that invasion efficiency was altered in our tmeA strain. HeLa monolayers were infected with the WT or tmeA strain at 4°C to allow attachment, shifted to 37°C to promote entry, and processed to differentiate intracellular and extracellular bacteria, as described previously (27), via indirect immunofluorescence (Fig. 4). We also included cultures supplemented with cytochalasin D as positive controls for inhibition of invasion. As expected, cytochalasin D treatment reduced invasion of both strains. In addition, the level of intracellular tmeA bacteria was reduced ca. 37% relative to the WT. These data implicate invasion as the developmental step that is impaired in the absence of TmeA. We could not assess invasion of the complementation strain, since the assay requires differential staining and the tmeA+ptmeA strain is already green and red.
FIG 4.
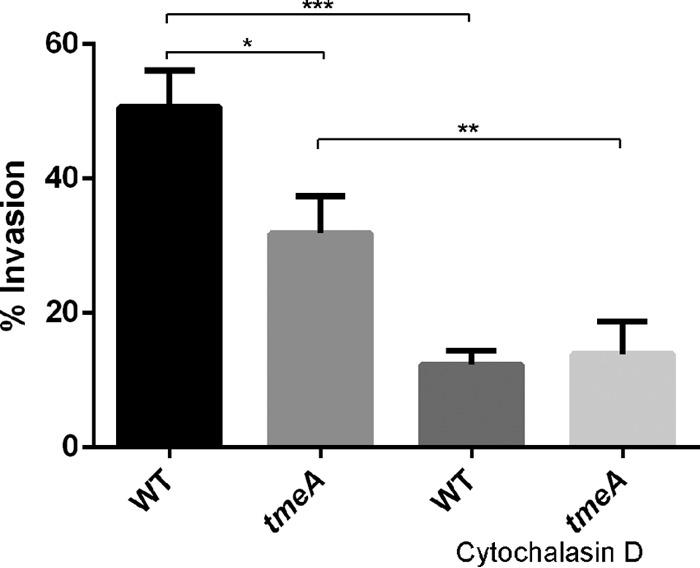
TmeA contributes to infection during the invasion process. HeLa monolayers were infected for 1 h at 4°C with the WT or tmeA strain of C. trachomatis L2 at an MOI of 10. Infections were carried out in the presence of vehicle control or with cytochalasin D. Cultures were shifted to 37°C for 30 min and then paraformaldehyde fixed. External EBs were specifically labeled with MOMP-specific antibodies, and internalized bacteria were labeled in subsequently permeabilized cultures using Chlamydia-specific antibodies. Data are represented as mean values for percentage of internalized chlamydiae and are shown with standard deviations. Statistical significance was computed using Student's t test with Welch's correction (*, P = 0.0134; **, P = 0.0131; ***, P = 0.0028).
Endogenous AHNAK is recruited transiently to nascent inclusions independently of TmeA.
We have previously identified an interaction of TmeA with human AHNAK (49) and wondered if disruption of this interaction could explain the invasion defect manifested by the C. trachomatis tmeA strain. We began our investigation of AHNAK by infecting HeLa cells with WT C. trachomatis and visualizing endogenous AHNAK in cultures fixed and stained 1 or 8 hpi (Fig. 5). Chlamydiae were stained with major outer membrane protein (MOMP)-specific antibodies, and we detected clear colocalization of Chlamydia and AHNAK at 1 hpi when focusing on the apical surface of HeLa cells (Fig. 5A). No colocalizing AHNAK signal was detected in cultures fixed 8 hpi (Fig. 5B). To narrow the duration of AHNAK colocalization with WT chlamydiae, we enumerated AHNAK-positive EBs in cultures fixed at 0.5, 1, 2, and 4 hpi (Fig. 5C). Colocalization was maximal at 30 min, with punctate AHNAK signal adjacent to 48.3% (±3.5%) of EBs. AHNAK-EB colocalization decreased steadily, and 89.7% (±1.0%) of EBs were not associated with detectible AHNAK signal at 4 hpi. Finally, we tested whether EB-AHNAK colocalization was required TmeA by similarly examining tmeA strain-infected cultures at 0.5 hpi (Fig. 5D). As with WT C. trachomatis, AHNAK staining was readily detectible in association with tmeA EBs. In aggregate, these data indicate that endogenous AHNAK is transiently recruited to the site of invading EBs, yet this process does not require TmeA.
FIG 5.

Endogenous AHNAK is transiently recruited to the site of chlamydial entry independently of TmeA. HeLa cells were infected at an MOI of 20 with the C. trachomatis WT or tmeA strain at 4°C. Cultures were shifted to 37°C for the indicated times and subsequently fixed with paraformaldehyde and processed for indirect immunofluorescence analyses. Merged-color epifluorescence micrographs are shown where chlamydiae were detected with anti-MOMP and AHNAK was detected with specific antibodies. Individual channels are depicted for insets and demonstrate positions of EBs and AHNAK. Scale bars, 5 μm. (A) AHNAK (green) position relative to WT C. trachomatis (red) after 30 min at 37°C. (B) AHNAK (green) position relative to WT C. trachomatis (red) after 8 h at 37°C. (C) AHNAK colocalization with EBs was directly quantitated in cultures fixed 30, 60, 120, and 240 min after temperature shift. Representative data are presented as the mean values (with standard deviations) for percentages of EBs associated with AHNAK-specific signal. (D) AHNAK (red) position relative to tmeA strain of C. trachomatis (green) after 30 min at 37°C.
TmeA interacts with AHNAK during invasion.
TmeA interacts with the C-terminal domain of AHNAK, and this 1,000-residue portion of AHNAK has been successfully expressed for functional studies (64, 65). We further examined AHNAK recruitment by invading EBs by expressing the C-terminal domain of AHNAK fused to mCherry (Fig. 6A). HeLa cells expressing mCherry or mCherry AHNAK were infected with the WT or tmeA strain and fixed 30 min after infection. Immunofluorescence analysis indicated a clear colocalization of EBs with mCherry-AHNAK but not mCherry. Interestingly, no colocalization was detected when cells were infected with the tmeA strain, raising the possibility that TmeA and AHANK do indeed interact during entry. Proximity labeling (69) has recently been applied to study the interaction of chlamydial effectors with host factors (70). We were unable to create TmeA- and BirA-containing chimeras for expression in Chlamydia, since N-terminal fusions would block T3S and a C-terminal fusion was inactive (data not shown). Therefore, we performed the reciprocal experiment by infecting HeLa cells expressing a chimeric protein containing the C-terminal domain of AHNAK fused to BirA. Perforin-2 associates with the nascent chlamydial inclusion (71) and was similarly fused to BirA as a negative control. Cultures were preinduced with biotin for 15 h prior to infection, and immunoblot analysis of whole-culture material from parallel cultures confirmed that fusion proteins contained active BirA (Fig. 6B). HeLa cells were infected in the presence of biotin for 5 h with WT Chlamydia and then harvested, and biotinylated proteins were purified via immunoprecipitation (Fig. 6C). Samples of the cleared lysate and immunoprecipitates were probed with TmeA-specific antibodies to test for protein proximity. Blots were probed for nonsecreted (Hsp60) or secreted (TarP) chlamydial proteins as negative controls. AHNAK interacts with annexin A2 (57, 66), and this protein served as a positive control. Signals in lysate material indicated the abundant presence of all proteins. As expected, TmeA was biotinylated by AHNAK-BirA but not P2-BirA, whereas no evidence of TarP or Hsp60 biotinylation was observed. Annexin A2 was detected in immunoprecipitation material from both P2- and AHNAK-BirA. Collectively, these data indicate an interaction of TmeA and AHNAK during early events in chlamydial infection.
FIG 6.

Association of AHNAK and TmeA during chlamydial invasion. (A) HeLa cells were nucleofected with pmCherry or pmCherry-Ahnak. At 24 h cells were infected at 4°C with C. trachomatis L2 WT or tmeA strain at an MOI of 20. Cultures were shifted to 37°C for 30 min and subsequently were fixed with paraformaldehyde and processed for direct and indirect immunofluorescence. Chlamydia organisms were detected using anti-MOMP (green). mCherry or mCherry-Ahnak (red) is shown in the mCherry channel. Scale bar, 5 μm. (B) HeLa cells or cells expressing P2-BirA or AHNAK-BirA were cultivated in the presence of biotin for 15 h, and whole-culture material was resolved via SDS-PAGE. Material was probed with NeutrAvidin-HRP to visualize biotin- or actin-specific antibodies as a loading control. Positions of molecular size markers are indicated. (C) Biotinylated proteins were immunoprecipitated (IP) from cleared lysates of cultures infected for 5 h and expressing P2-BirA or AHNAK-BirA. Material from cleared lysates (lysate) or immunoprecipitates was probed with Hsp60-, TarP-, TmeA-, or annexin A2-specific antibodies in immunoblots and visualized via chemiluminescent detection.
C. trachomatis TmeA interferes with AHNAK-dependent actin bundling.
The carboxyl-terminal domain of AHNAK binds both G- and F-actin (65) and mediates bundling of F-actin (64). We asked whether TmeA is capable of altering AHNAK-actin interactions and began by testing whether TmeA affected AHNAK-dependent actin bundling activity in an in vitro sedimentation assay. Filamentous (F)-α-actin from rabbit skeletal muscle was subjected to differential centrifugation in the presence of full-length, glutathione S-transferase (GST)-tagged TmeA and/or GST-tagged AHNAK C-terminal domain corresponding to the terminal 1,000 residues. The resulting pellet and supernatant fractions were resolved by SDS-PAGE, and actin was visualized by Coomassie blue staining while GST-TmeA and GST-AHNAK were detected via immunoblotting (Fig. 7A). As expected, actin was detected predominately in the supernatant fraction when GST-AHNAK was not present (21.3% in pellet). Consistent with its role in actin bundling, inclusion of GST-AHNAK resulted in a shift of actin into the pellet (60.6%) fraction. GST-TmeA alone did not affect actin partitioning. However, inclusion of GST-TmeA with GST-AHNAK resulted in actin partitioning similar to that of the actin-alone treatment (24.8% in the pellet). Neither bovine serum albumin (BSA) nor GST affected actin partitioning (data not shown). The actin bundling protein α-actinin was used as a positive control (Fig. 7B). As expected, actin distribution was most abundant in pellet fractions in the presence of α-actinin. Moreover, addition of GST-TmeA did not affect the α-actinin-mediated shift in actin partitioning. In aggregate, these data indicate that TmeA is able to disrupt AHNAK-dependent actin bundling. Furthermore, GST-AHNAK and α-actinin were detected in pellet fractions. This is consistent with observations that proteins which bind F-actin will copellet with the bundled actin. Although GST-TmeA was detectable in pellet fractions, this did not require the presence of actin, suggesting that the localization is not indicative of an interaction between actin and GST-TmeA.
FIG 7.

TmeA inhibits AHNAK-dependent F-actin bundling activity in vitro. Rabbit skeletal muscle F-actin was subjected to low-speed (12,000 × g) differential centrifugation in the presence or absence of GST-tagged or control proteins. All recombinant proteins were mixed at 1:1 molar ratios. Samples subsequently were separated into supernatant (S) and pellet (P) fractions. Equal volumes of material were resolved by SDS-PAGE, and actin was visualized by Coomassie blue staining. (A) GST-AHNAK and GST-TmeA were detected by immunoblotting with AHNAK-specific or TmeA-specific antibodies and visualized via chemiluminescence after probing with secondary antibodies coupled to HRP. Numerical values represent the relative percentage of actin in respective fractions. (B) Actin, α-actinin, and GST-TmeA were visualized via Coomassie blue staining. (C) F-actin alone or in combination with GST-694 and/or GST-AHNAK in a 1:1 or 1:1:1 molar ratio was processed for visualization by transmission electron microscopy. Samples were visualized at 10,500× and 64,000× magnification, and representative images are shown. Scale bar, 1 μm.
Analysis of material via transmission electron microscopy was used to confirm the impact of GST-tagged proteins on actin structure (Fig. 7C). Filamentous α-actin was incubated with GST-TmeA, GST-AHNAK, or both proteins at a 1:1 molar ratio and negatively stained. Compared with F-actin only and consistent with previous reports (64), web-like bundles of actin were detected in the sample of F-actin incubated with GST-AHNAK. While GST-TmeA alone did not seem to have an impact on gross actin structure, inclusion of GST-TmeA with GST-AHNAK resulted in actin fibers, similar to the negative controls. These data are consistent with our conclusion that C. trachomatis TmeA is capable of interfering with the actin bundling activity of AHNAK.
Evidence for AHNAK-independent functions for TmeA.
Knockdown of AHNAK via short interfering RNA (siRNA) treatment of epithelial cells has been reported (57), yet depletion of HeLa cell AHNAK is neither efficient nor complete (data not shown). Lee et al. (60) generated and characterized an ahnak knockout (KO) murine line that has proven efficacious in studying the roles of AHNAK in eukaryotic cell biology. We therefore leveraged these mice by isolating murine embryonic fibroblasts (MEFs) from embryos derived from heterozygous (HET) matings. PCR and immunoblot analyses were employed to confirm MEF genotype, and WT, HET, and KO cells appeared to grow equivalently in tissue culture (see Fig. S1 in the supplemental material). Passage-matched MEFs derived from littermates then were used to compare growth and development of C. trachomatis in the presence or absence of AHNAK (Fig. 8). Parallel WT and KO MEF monolayers were grown to equal confluence and infected at 37°C with C. trachomatis L2 at a multiplicity of infection (MOI) of 1. When cultures were fixed 24 hpi and processed to quantitate direct IFU counts, we did not detect any difference in the ability of Chlamydia to establish infection in KO cells (Fig. 8A). With no difference distinguishable in infectivity, we progressed to test whether invasion defects where apparent in the absence of AHNAK (Fig. 8B). Parallel cultures of MEF monolayers were infected with C. trachomatis serovar L2 at an MOI of 10 at 4°C and processed to examine invasion. Addition of cytochalasin D was used as a positive control for reduction of chlamydial invasion. A proportion of 67.9% ± 9.1% of C. trachomatis organisms invaded WT MEFs, and this was significantly reduced by the presence of cytochalasin D (5.6% ± 2.3%). Similarly, chlamydial invasion of untreated KO MEFs was 60.8% ± 8.8%, and this was reduced to 11.2% ± 5.1% in the presence of cytochalasin D. Collectively, these data indicate that AHNAK-dependent functions are not required to support chlamydial invasion. However, the data would be consistent with a situation where TmeA negatively impacts AHNAK to promote invasion, perhaps via disruption of the AHNAK-dependent actin bundling activity. Therefore, we infected KO MEFs with WT and tmeA strains to test whether the tmeA strain-specific infectivity defect was manifested in these cells (Fig. 8C). Similar to HeLa cell infections, we observed a ca. 45% decrease in tmeA strain IFUs compared to those of the WT. These data indicate that the infectivity defect manifested by the lack of TmeA occurs independently of AHNAK and is consistent with alternative TmeA-mediated activities.
FIG 8.

Analysis of C. trachomatis infection in AHNAK-deficient MEFs. (A) WT or ahnak homozygous knockout (KO) MEFs were infected at an MOI of 1 with WT C. trachomatis. Cultures were fixed at 24 h postinfection for direct IFU counts. (B) WT KO MEFs were infected for 1 h at 4°C with C. trachomatis L2 at an MOI of 10. Infections were carried out in the presence of vehicle control or cytochalasin D (CytoD). Cultures were shifted to 37°C for 30 min and then paraformaldehyde fixed. External EBs were specifically labeled with MOMP-specific antibodies, and internalized bacteria were labeled in subsequently permeabilized cultures using Chlamydia-specific antibodies (**, P < 0.0001). (C) KO MEFs were infected with equal particles of WT or tmeA mutant C. trachomatis at an MOI of 0.5. Cultures were processed for direct enumeration of inclusions at 24 hpi (*, P < 0.003). All data are represented as means with standard deviations calculated from triplicate cultures. Student's t test with Welch's correction was employed to assess statistical significance.
DISCUSSION
Increased efficacy of genetic manipulation in C. trachomatis is steadily progressing. Random, chemical-induced mutagenesis coupled with tilling (12) was originally employed to inactivate chlamydial genes. Whole-genome sequencing has now provided a useful platform for forward genetic approaches (11). Reproducible transformation of Chlamydia with a stably maintained plasmid represents perhaps the most significant breakthrough to date (5). This ability enabled targeted gene disruption via insertion mutagenesis using a group II intron (13). We developed FRAEM for complete gene deletion, since the most definitive functional studies require complete deletion of target genes. This approach was used to successfully delete coding sequences for CTL0063, CTL0064, and CTL0065 (14). In the present study, we applied a reverse genetic approach to examine the impact of these mutations on chlamydial infectivity. Our data indicate that TmeA (CTL0063) is essential for efficient infection in both animal and tissue culture models.
Proteins secreted via the T3S mechanism directly impact virulence in numerous Gram-negative pathogens (72). TmeA (CTL0063) and TmeB (CTL0064) represent established effectors of the chlamydial T3SS (9, 46, 49, 73). Both effectors appear to be Chlamydia-specific, yet tmeA is unique to C. trachomatis genomes and belongs to a family of cytoskeleton-interactive effectors due to the presence of an N-terminally positioned membrane localization domain (50). Both chlamydial effectors have been implicated in invasion due to deployment during chlamydial entry. Although below detection in the host cytosol after 4 h, TmeA-specific signal is detectable via immunoblotting throughout development, even at times prior to de novo synthesis (49). In contrast, TmeB is detectible via indirect immunofluorescence and clearly colocalizes with inclusion membranes during late-cycle development (9). It was therefore initially possible that tmeA- or tmeB-specific phenotypes could manifest at any point in chlamydial development. Indeed, we detected a reproducible negative impact on chlamydial infectivity in the absence of TmeA that correlated with a defect in invasion. This was in contrast to the delayed development phenotype apparent in the ctl0065 strain. We did not detect any overt developmental defect for tmeB chlamydiae. This does not rule out a role in the invasion process due to several possibilities. For example, functional redundancy among effectors could provide an explanation (see below). Since columnar epithelial cells represent the in vivo replication niche for Chlamydia, it is also possible that TmeB-mediated functions are not manifested in the HeLa cell infection model. Finally, TmeB may function independently of processes required for physical entry of EBs. For example, TepP represents another effector secreted during invasion. TepP recruits Crk-II during entry, yet a tepP-deficient strain was not impaired for entry (46).
Application of FRAEM results in replacement of targeted chromosomal coding sequences with an antibiotic resistance gene for selection and GFP for detection. Our immunoblot analysis confirmed that this insertion in tmeA has a polar effect on tmeB expression. This was not surprising given that tmeA and tmeB are cotranscribed (9). There is no evidence that the tmeA or tmeB mutation impacted expression of ctl0065. Although encoded downstream, ctl0065 transcription occurs independently (9) and wild-type levels of CTL0065 are detectable by immunoblotting in material from tmeA and tmeB chlamydiae (14). Furthermore, the delayed development observed here for the ctl0065 mutant was distinct from tmeA and tmeB strain phenotypes. The polar effect of the tmeA mutation on TmeB, however, necessitated complementation studies. We mobilized tmeA and 292 nt of upstream DNA, containing the endogenous promoter and transcription start site (74), into pComAII for complementation studies. Trans-expressed TmeA was detected in both EBs and during the late cycle, indicating an expression profile consistent with endogenous tmeA. TmeB was not detected in the complemented strain, ruling out the possibility that TmeA expression is required for TmeB stability. Complementation with tmeA restored WT levels of inclusion counts, verifying that the developmental phenotype manifested in the mutant was due solely to loss of tmeA. We were surprised that trans-expression of tmeA also complemented the infectivity defect observed in mice. Shed EBs for the complemented strain were roughly decreased compared to the level for the WT after day 15, indicating that complementation was not entirely complete. These data suggest that loss of tmeB manifests as a more modest defect for C. trachomatis L2 intravaginal infection of mice.
The decreased ability to produce inclusions by tmeA chlamydiae could result from deficiencies in attachment, entry, or postentry survival. We did not detect reductions in tmeA C. trachomatis attachment (data not shown), and the WT levels of development observed during IFU-normalized infections make it unlikely that postentry survival is reduced in the absence of TmeA. Importantly, we did observe a decrease in tmeA invasion efficiency. This decrease was reproducible but modest in magnitude. One possible explanation for this modest impact is that, similarly to Legionella pneumophila (75), deletion of a single gene may not reveal overt phenotypes due to potential redundancy in effector function. As noted above, Chlamydia deploy multiple known effectors during the invasion process, and others are likely waiting to be identified. We believe, however, that the most likely explanation is that Chlamydia employ multiple invasion mechanisms to gain access to the interior of host cells. C. trachomatis invasion remains incompletely characterized but involves initial electrostatic association with target cells (16). This is followed by irreversible engagement of host receptors, such as platelet-derived growth factor receptor (76), protein disulfide isomerase (77), integrin β1 (78), epidermal growth factor receptor (79), and EphA2 (80), which have been implicated in C. trachomatis attachment and/or invasion. Numerous potential chlamydial adhesins have been implicated. For example, polymorphic outer membrane protein D (81) and Ctad1 (78) are important for attachment of C. trachomatis. Although specific host factor requirements can vary by chlamydial species, entry of attached EBs is largely driven by recruitment and rearrangement of the actin-based cytoskeleton (16). As recently outlined by Stallmann and Hegemann (78), the range of bacterial and host factors that have been identified implicate multiple, redundant entry mechanisms. Interestingly, interference with any one of these pathways results in only modest decreases in invasion, similar in magnitude to that of the tmeA phenotype. Hence, we conclude that TmeA plays an important role in T3S-mediated invasion but is not absolutely essential for chlamydial entry due to redundant mechanisms. In contrast, TmeA is absolutely required during intravaginal infections, since tmeA strain infections of mice rapidly resolved. These data would be consistent with a more profound invasion impact in the comparatively more complex in vivo scenario or additional functions of TmeA.
TmeA is secreted during invasion, and we previously provided in vitro evidence for an interaction of TmeA with host AHNAK (49). An involvement of AHNAK in bacterial infection has precedent. During Salmonella enterica serovar Typhimurium infection, AHNAK is phosphorylated (82) and recruited to membrane ruffles (83) via a mechanism dependent on the S. Typhimurium T3SS-1 effectors SopB and SopE/E2. Infection of WT and KO MEFs revealed that AHNAK was essential for invasion of these cells by Salmonella (83). Our data using the same ahnak KO MEFs indicate that AHNAK is not required for chlamydial invasion or intracellular development. Furthermore, the invasion defect detected during infection of HeLa cells with the tmeA strain was still manifested in KO MEFs. These data argue against an active requirement of the TmeA-AHNAK interaction for invasion.
However, our data do provide evidence for an interaction of TmeA and AHNAK during early events. Although colocalization of endogenous AHNAK with invading EBs did not require TmeA, recruitment of the C-terminal domain was not apparent for the tmeA strain. Proximity labeling experiments were also consistent with a TmeA-AHNAK interaction. AHNAK is a large, multidomain protein (58), and our data indicate that other chlamydial or host factors could mediate AHNAK colocalization. It is unclear what the role is for TmeA-independent AHNAK recruitment. Numerous functions have been ascribed to AHNAK, including regulation of calcium channels, cellular architecture, and membrane repair (reviewed in reference 84).
AHNAK is also involved in cytoskeleton dynamics, and this function appears to be impacted by the TmeA-AHNAK interaction. The C terminus of AHNAK actively engages the actin cytoskeleton and is capable of bundling actin fibers (64), and we employed a standard in vitro actin bundling assay to demonstrate that TmeA interferes with this bundling activity. Actin bundling proteins are defined by their ability to directly bind and organize filamentous actin. While roles are diverse, actin bundling is important in assembly of filopodia that are exploited by multiple bacterial pathogens to gain entry into epithelial cells (85). Although chlamydiae do not induce membrane ruffles, EB attachment to host cells is accompanied by formation of a pedestal-like structure (86) and surrounding microvillar hypertrophy (27) on the cellular surface. We did not detect a deficiency in actin recruitment by the tmeA strain (data not shown), and it should be noted that C. trachomatis TarP has actin bundling activity (87). Since AHNAK was not essential for invasion, these observations raise the possibility that TmeA-mediated disruption of AHNAK-containing actin bundles is instrumental in the overall cytoskeletal reorganization orchestrated subsequent to entry. This function might not directly impact chlamydial fitness. As noted above, it is formally possible that redundant entry pathways could also mask any functional impact of this activity in the tissue culture infection model.
In aggregate, we have leveraged a novel genetic approach that enables generation of bona fide null mutations to reveal an active role of TmeA in chlamydial infection and invasion. This activity is independent of the interaction with AHNAK and is consistent with TmeA representing a multifunctional effector. Indeed, a multifunctional character would be consistent with evidence indicating differing functional domains within TmeA (50). Ongoing studies are aimed at elucidating the molecular mechanisms governing the role of TmeA in invasion and understanding how TmeA-dependent manipulation of AHNAK function contributes to chlamydial infection.
MATERIALS AND METHODS
Cell cultures and organisms.
C. trachomatis was routinely cultivated in HeLa 229 epithelial cell monolayers (ATCC CCL-1.2), and chlamydial transformations were accomplished using McCoy cell monolayers (CRL-1696; ATCC) as described previously (88). All cultures were maintained at 37°C in an atmosphere of 5% CO2 and 95% humidified air in RPMI 1640 containing 2 mM l-glutamine (Life Technologies) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (HIFBS; Sigma). All infection studies employed HeLa cultures below passage 20 from the original stock. Murine embryonic fibroblasts (MEFs) were derived from ahnak-deficient mice according to University of Kentucky IACUC-approved protocols essentially as described previously (60). Fibroblasts were isolated at day 13.5 from heterozygous ahnak knockout (KO) females that were bred to heterozygous ahnak KO males. Individual MEF isolates derived from littermates were used for experiments. Each isolated culture was evaluated for AHNAK production after a single passage by PCR and immunoblot analysis of isolated genomic DNA or whole-cell protein, respectively. PCR-based genotyping was accomplished using an antisense primer (5′-CCTCAGGAGCAGATGTCTTC-3′) paired with a separate sense primer (5′-CTGGATGGCCAGATTCCAGAG-3′ or 5′-CAGCTCATTCCTCCCACTCAT-3′) to discriminate between homozygous full-length (WT), heterozygous ahnak KO (HET), or homozygous KO alleles. Whole-culture proteins isolated from respective MEF isolates were probed with AHNAK-specific (49) or β-actin-specific (Sigma) antibodies to evaluate AHNAK levels via immunoblotting. MEFs were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/liter (25 mM) d-glucose, 4 mM l-glutamine, and 110 mg/liter (1 mM) sodium pyruvate (Life Technologies) and supplemented with 10% (vol/vol) HIFBS (Sigma) and 1% (vol/vol) 100× MEM nonessential amino acids solution (Life Technologies). All infection experiments were performed using MEFs from passages 2 to 6.
These studies employed C. trachomatis serovar L2 (LGV 434) and previously described strains harboring complete deletion of ctl0063, ctl0064, or ctl0065 (14). A complementation strain was generated by first mobilizing tmeA with its endogenous promoter into pComA II (14) using engineered primers (694pro@NmPgfp F, 5′-CGGTTCCTGGCCTTTTGCTGGGTACGGAAATACTATCTCCAGCTCAAAGC-3′, and 694@NmPgfp R, 5′-GCCCCGCCCTGCCACTCATCGGGACCGAACACCGTATACCTTCT-3′). The plasmid ptmeA was transformed into Chlamydia, and clonal strains were isolated exactly as described previously (88). All EBs were purified from HeLa cells by centrifugation through MD-76R (Mallinckrodt Pharmaceuticals) density gradients (DG-purified) as previously described (89) and were used as the infection source for all experiments. Escherichia coli NEB-10β (New England BioLabs) was utilized for routine cloning procedures, while E. coli BL21-Al (Invitrogen) was used for production of GST and His-tagged proteins. E. coli was routinely grown at 37°C in Luria-Bertani (Amresco) broth or LB agar plates supplemented with 50 μg/ml carbenicillin (Teknova) or 100 μg/ml spectinomycin (Alfa Aesar) as appropriate.
Chlamydia infectivity and immunolocalization.
Infection of cell monolayers with C. trachomatis was carried out for the indicated time points at 37°C as previously described (89, 90) or as otherwise indicated. Inocula for respective strains were normalized by either direct inclusion-forming units (IFUs) or genome content. Chlamydial genomes were quantitated via qPCR of the 16S rRNA region as described previously (14). Direct IFUs were assessed by processing cultures for indirect immunofluorescence and visualization of chlamydiae with Hsp60-specific antibodies (Santa Cruz) after 24 h of infection. Enumeration of progeny IFUs was accomplished via harvesting infected monolayers at the indicated times postinfection in cold Hanks' balanced salt solution (HBSS) (Life Technologies), passage of serial dilutions onto fresh HeLa monolayers, and detection of inclusions via indirect immunofluorescence with Hsp60-specific antibodies as described previously (91). Where indicated, relative inclusion areas were computed from images acquired utilizing using a Zeiss epifluorescence microscope equipped with a 100× PlanApo objective and AxioCam MRM camera. Images were imported into ImageJ, and areas were computed for 50 inclusions using the area measurement function. All IFU counts were assessed from triplicate cultures plated in triplicate.
For immunolocalization studies, HeLa or MEF cells were cultivated on 12-mm glass coverslips. For studies involving mCherry-tagged AHNAK, HeLa cells were transfected with pmCherry-AHNAK or vector only using AMAXA (Lonza, Corp) nucleofection. Cells were infected with C. trachomatis at the indicated MOIs. Samples were prepared for indirect immunofluorescence by fixation at the indicated time points with 4% (wt/vol) paraformaldehyde (PFA; EM Science). Permeabilization was accomplished using 0.5% Triton X-100 (Sigma-Aldrich) in phosphate-buffered saline (PBS) containing 3% (wt/vol) BSA (Sigma) for 20 min at room temperature. Fixed samples were blocked at room temperature for 1 h in 5% BSA in PBS supplemented with 0.05% (vol/vol) Tween 20. Endogenous AHNAK was detected with anti-AHNAK monoclonal antibody (Santa Cruz) and chlamydiae with anti-MOMP polyclonal (49) or anti-HSP60 (Santa Cruz). Where indicated, IncG-specific antibodies (92) were used to define the inclusion periphery. Proteins were subsequently visualized with secondary antibodies conjugated to Alexa Fluor 488 or 594 (Invitrogen). For epifluorescence microscopy, images were acquired using a 100× oil-immersion objective on a Zeiss AX10 upright light microscope (Carl Zeiss Foundation, Oberkochen, Germany) with Zeiss AxioVision software (Carl Zeiss Foundation). All images were processed equivalently using Adobe Photoshop 6.0 (Adobe Systems).
Murine infection studies.
Groups of 5 female C3H/HeJ mice (Jackson Laboratory), 6 to 8 weeks old, were intravaginally infected essentially as described previously (93). Mice were pretreated with 2.5 mg medroxyprogesterone 5 days prior to infection. Mice were infected with 5 × 105 IFU of each chlamydial strain, and shedding was monitored beginning on day 3 and then every 4 days until 2 consecutive time points failed to yield EBs for the mutant strain. Recovered IFUs were enumerated on fresh HeLa cells as described above. All manipulations were reviewed and approved by the University of Kentucky Institutional Animal Care and Use Committee.
Invasion studies.
The ability of chlamydiae to invade HeLa or MEF cells was conducted as described previously (27), with modifications. Infections were carried out when cells reached ca. 70% confluence. Where appropriate, cells were pretreated for 30 min with 2.5 μg/ml cytochalasin D (Sigma). Plates were cooled to 4°C for 10 min prior to inoculation and then infected with respective C. trachomatis strains at an MOI of 10 in cold HBSS (containing dimethyl sulfoxide [DMSO] or cytochalasin D where appropriate) for 1 h at 4°C to allow EBs to attach but not enter cells. Inoculate was replaced with 37°C medium (containing drugs as appropriate) and maintained for 30 min. After thorough washes, cells were fixed on ice with 4% (wt/vol) PFA and blocked for 20 min with 3% BSA. For probing, primary antibodies were diluted in 3% BSA (Sigma-Aldrich) in PBS. External EBs were probed with α-MOMP (49) and visualized with secondary antibody conjugated to Alexa Fluor 488 (Invitrogen). Samples then were permeabilized with 0.5% Triton X-100, stained with α-Chlamydia lipopolysaccharide (LPS) (Novus Biologicals), and visualized with secondary antibodies conjugated to Alexa Fluor 594 (Invitrogen). Percentages of invaded chlamydiae where computed by enumeration of internal and external chlamydiae for a minimum of 100 bacteria in 10 fields of view. Percent EB internalization was calculated via the formula (total EBs − external EBs)/total red EBs × 100 = percent invasion.
DNA methods.
The final 1,000 residues (4889 to 5890) representing the carboxyl-terminal domain of the eukaryotic AHNAK gene were cloned into pmCherry (Clontech Laboratories, Inc., Mountain View, CA) and pcDNA3.1 mycBioID (69) via amplification of the insert from HeLa cell cDNA isolated from the ProQuest premade HeLa cDNA library (Invitrogen). DNA was amplified with Q5 high-fidelity (New England BioLabs, Inc., Ipswich, MA) DNA polymerase according to the manufacturer's protocols using specified buffers and associated reagents. For pmCherry, a pair of custom oligonucleotide primers, 5′-CCGGAATTCCACGTCTGTTCGAAGGCC-3′ and 5′-CCCGAATTCCTACTCTTTCTTTGTGGAAACTGACAGCTCC-3′ (Integrated DNA Technologies, Coralville, IA), were designed with specified engineered EcoRI restriction sites. The primers 5′-AAAGCTAGCCTGGTATATGGATTTCGAAGGCCC-3′ and 5′-AAACCGAATTCCTCTTTCTTTGTGGAAACTGACAGCTCC-3′, containing an NheI or EcoRI restriction site, respectively, were used for mobilization into pcDNA3.1 mycBioID. Cloning protocols were performed essentially as previously described (94). The full-length gene for Perforin-2 was also mobilized into pcDNA3.1 mycBioID via amplification from RFP-P2 (95) using engineered primers (5′-AAAGCTAGCCTGGTATATGGCCAAAACC-3′ and 5′-AAACCGAATTCAGCTGGACTTGGATCCTC-3′) containing NheI and EcoRI restriction sites. Upon sequencing verification, the constructs were prepped under endotoxin-free conditions with Zyppy plasmid miniprep and maxiprep kits (Zymo Research) and QIAfilter plasmid and EndoFree plasmid maxi kits (Qiagen).
Proximity labeling.
Proximity labeling using the BioID approach was accomplished as described previously (96), with modifications. HeLa cells alone or expressing P2-BirA or AHNAK-BirA were cultivated in the presence of 50 μM biotin for 15 h and then infected with WT Chlamydia at an MOI of 20 in the presence of biotin. Cultures were maintained in 50 μM biotin (Sigma) for an additional 5 h, washed with HBSS, and disrupted in lysis buffer (50 mM Tris, pH 7.4, 500 mM NaCl, 0.2% SDS) for processing as described previously (96). Material from cleared lysates was probed in immunoblots with NeutrAvidin-horseradish peroxidase (HRP) (Molecular Probes) and anti-actin (Sigma) or combined with NeutrAvidin agarose (Molecular Probes) beads to specifically purify biotinylated proteins. Immunoprecipitated material was resolved via SDS-PAGE and probed with antibodies specific for Chlamydia Hsp60, TarP (38), TmeA, or host annexin A2 (Cell Signaling).
Recombinant protein purification.
C. trachomatis TmeA and human AHNAK genes were cloned using GATEWAY technology (Invitrogen) according to the manufacturer's instructions. For ahnak, the final 1,000 codons were amplified from HeLa cell cDNA isolated from the ProQuest premade HeLa cDNA library (Invitrogen) using Q5 high-fidelity (New England BioLabs) DNA polymerase and custom oligonucleotide primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCCGTCTGGATTTCGAAGGCCC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTACTCTTTCTTTGTGGAAACTGACAG-3′. Coding sequence for TmeA was similarly amplified from genomic C. trachomatis L2 DNA using primers SHCT694-4A and SHCT694-4B (49). PCR products were mobilized into the pDNR221 (Invitrogen) plasmid and subsequently into pDEST15 and pDEST17 to generate GST- and His-tagged proteins, respectively. Constructs were verified via DNA sequencing (GENEWIZ).
Tagged constructs were transformed into E. coli BL21-Al, and expression was induced by the addition of 0.2% (vol/vol) l-arabinose (Sigma-Aldrich). Cell pellets containing GST-tagged proteins were lysed with BugBuster protein extraction reagent (Novagen) or by passage of suspended pellet in PBS through a French pressure cell. His-tagged cell pellets were lysed with CelLytic B cell lysis reagent (Sigma-Aldrich). Affinity chromatography was used to purify the proteins. His-tagged proteins and GST-tagged proteins were purified using a PrepEase His-tagged protein purification kit (USB Corporation) or GST-Bind resin (Novagen), respectively, according to the manufacturer's protocols. All proteins were dialyzed into PBS, assessed for purity by Coomassie brilliant blue R-250 staining of SDS-PAGE resolved material, and quantified via Bradford assay (Sigma-Aldrich).
Actin bundling and interactions assays.
An actin binding protein biochemistry kit (Cytoskeleton) was employed utilizing method 3, detecting F-actin bundling activity, with modifications to the manufacturer's protocol (40). F-actin was filamented overnight at 4°C in general actin buffer supplemented with actin polymerization buffer (5 mM KCl, 200 μM MgCl2, and 10 μM ATP) to 9% (vol/vol), resulting in a 21 μM F-actin stock solution. Concentrations (20 μM) of purified GST-tagged TmeA and AHNAK underwent a clarification spin at 12,000 × g at 4°C for 1 h. Twenty microliters of 21 μM F-actin or F-actin buffer (10% [vol/vol] actin polymerization buffer in general actin buffer) was combined on ice with 10 μl of 20 μM clarified test proteins at a 1:1 or 1:1:1 molar ratio of 4 μM plus an appropriate amount of PBS to keep reaction volumes equal between samples. The positive (α-actinin) and negative (BSA and GST) controls were prepared and combined with 20 μl of F-actin or F-actin buffer as indicated by the manufacturer. The protein combinations were incubated at room temperature for 90 min and sedimented by a 12,000 × g centrifugation for 15 min at 21°C. Samples were separated into insoluble pellet and supernatant fractions. Proteins were visualized in material resolved by SDS-PAGE by either Coomassie staining or immunoblotting with AHNAK-specific or TmeA-specific antibodies (49). Relative quantification of the amount of F-actin in the supernatant and pellet fractions was conducted using Quantity One V.4.6 (Bio-Rad).
Actin structure was visualized directly by transmission electron microscopy (TEM). F-actin was prepared as previously noted and combined with GST-tagged TmeA and AHNAK C1000 in a 1:1 or 1:1:1 molar ratio of 4 μM each protein in general actin buffer and processed for visualization by TEM. Samples were postfixed in 2% osmium tetroxide in 0.1 M phosphate buffer, dehydrated through a series of graded ethanols, and embedded in EM-bed (Electron Microscopy Sciences, Fort Washington, PA). Eighty-nanometer sections were cut on a Leica Ultracut-R ultramicrotome (Leica Microsystems Inc., Buffalo Grove, IL) and stained with uranyl acetate and lead citrate. The grids were viewed at 80 kV on a Philips CM-10 transmission electron microscope (Koninklijke Philips) and images captured by a Gatan ES1000W digital camera (Gatan, Inc., Pleasanton, CA). All images were acquired utilizing Gatan Microscopy Suite (GMS; Gatan) software. Images were processed using Adobe Photoshop 6.0 (Adobe Systems).
Statistical analysis.
All presented data are representative of a minimum of three experiments. Unless otherwise noted, quantitative data were generated from experiments containing triplicate biological samples and duplicate technical replicates. Calculation of standard deviations of the means (SD) and assessment via analysis of variance (ANOVA) or Student's t test statistical analyses were performed using GraphPad Prism 6, version 6.04 (GraphPad Software Inc., La Jolla, CA).
Supplementary Material
ACKNOWLEDGMENTS
Transmission electron microscopy was accomplished at the University of Miami Miller School of Medicine Electron Microscopy Core Facility. We thank Margaret Bates and Vania Almeida for their support in preparing samples and performing TEM imaging. We thank R. Hayman, G. Kebb, M. Clouse, K. Wolf, and M. Rahnama for critical reading of the manuscript.
This work was supported by Public Health Service grants from the National Institutes of Health, NIAID (AI065530 and AI124649), to K.A.F.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00640-17.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2013. Sexually transmitted disease surveillance 2012. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 2.Baneke A. 2012. Review: targeting trachoma: strategies to reduce the leading infectious cause of blindness. Travel Med Infect Dis 10:92–96. doi: 10.1016/j.tmaid.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Abdelrahman YM, Belland RJ. 2005. The chlamydial developmental cycle. FEMS Microbiol Rev 29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Binet R, Maurelli AT. 2009. Transformation and isolation of allelic exchange mutants of Chlamydia psittaci using recombinant DNA introduced by electroporation. Proc Natl Acad Sci U S A 106:292–297. doi: 10.1073/pnas.0806768106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agaisse H, Derré I. 2014. Expression of the effector protein IncD in Chlamydia trachomatis mediates recruitment of the lipid transfer protein CERT and the endoplasmic reticulum-resident protein VAPB to the inclusion membrane. Infect Immun 82:2037–2047. doi: 10.1128/IAI.01530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wickstrum J, Sammons LR, Restivo KN, Hefty PS. 2013. Conditional gene expression in Chlamydia trachomatis using the Tet system. PLoS One 8:e76743. doi: 10.1371/journal.pone.0076743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller KE, Fields KA. 2015. Application of β-lactamase reporter fusions as an indicator of protein secretion during infections with the obligate intracellular pathogen Chlamydia trachomatis. PLoS One 10:e0135295. doi: 10.1371/journal.pone.0135295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauler LD, Hackstadt T. 2014. Expression and targeting of secreted proteins from Chlamydia trachomatis. J Bacteriol 196:1325–1334. doi: 10.1128/JB.01290-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kokes M, Dunn Joe D, Granek Joshua A, Nguyen Bidong D, Barker Jeffrey R, Valdivia Raphael H, Bastidas Robert J. 2015. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17:716–725. doi: 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci U S A 108:7189–7193. doi: 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson CM, Fisher DJ. 2013. Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PLoS One 8:e83989. doi: 10.1371/journal.pone.0083989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mueller KE, Wolf K, Fields KA. 2016. Gene deletion by fluorescence-reported allelic exchange mutagenesis (FRAEM) in Chlamydia trachomatis. mBio 7:e01817-15. doi: 10.1128/mBio.01817-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elwell C, Mirrashidi K, Engel J. 2016. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14:385–400. doi: 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dautry-Varsat A, Subtil A, Hackstadt T. 2005. Recent insights into the mechanisms of Chlamydia entry. Cell Microbiol 7:1714–1722. [DOI] [PubMed] [Google Scholar]
- 17.Scidmore MA, Rockey DD, Fischer ER, Heinzen RA, Hackstadt T. 1996. Vesicular interactions of the Chlamydia trachomatis inclusion are determined by chlamydial early protein synthesis rather than route of entry. Infect Immun 64:5366–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saka HA, Thompson JW, Chen Y-S, Kumar Y, Dubois LG, Moseley MA, Valdivia RH. 2011. Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol Microbiol 82:1185–1203. doi: 10.1111/j.1365-2958.2011.07877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen JC, Zhang JP, Stephens RS. 1996. Structural requirements of heparin binding to Chlamydia trachomatis. J Biol Chem 271:11134–11140. doi: 10.1074/jbc.271.19.11134. [DOI] [PubMed] [Google Scholar]
- 20.Stephens RS, Koshiyama K, Lewis E, Kubo A. 2001. Heparin-binding outer membrane protein of chlamydiae. Mol Microbiol 40:691–699. doi: 10.1046/j.1365-2958.2001.02418.x. [DOI] [PubMed] [Google Scholar]
- 21.Yabushita H, Noguchi Y, Habuchi H, Ashikari S, Nakabe K, Fujita M, Noguchi M, Esko JD, Kimata K. 2002. Effects of chemically modified heparin on Chlamydia trachomatis serovar L2 infection of eukaryotic cells in culture. Glycobiology 12:345–351. doi: 10.1093/glycob/12.5.345. [DOI] [PubMed] [Google Scholar]
- 22.Zhang JP, Stephens RS. 1992. Mechanism of C. trachomatis attachment to eukaryotic host cells. Cell 69:861–869. doi: 10.1016/0092-8674(92)90296-O. [DOI] [PubMed] [Google Scholar]
- 23.Wuppermann FN, Hegemann JH, Jantos CA. 2001. Heparan sulfate-like glycosaminoglycan is a cellular receptor for Chlamydia pneumoniae. J Infect Dis 184:181–187. doi: 10.1086/322009. [DOI] [PubMed] [Google Scholar]
- 24.Carabeo R, Hackstadt T. 2001. Isolation and characterization of a mutant Chinese hamster ovary cell line that is resistant to Chlamydia trachomatis infection at a novel step in the attachment process. Infect Immun 69:5899–5904. doi: 10.1128/IAI.69.9.5899-5904.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fudyk T, Olinger L, Stephens RS. 2002. Selection of mutant cell lines resistant to infection by Chlamydia spp. Infect Immun 70:6444–6447. doi: 10.1128/IAI.70.11.6444-6447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carabeo R. 2011. Bacterial subversion of host actin dynamics at the plasma membrane. Cell Microbiol 13:1460–1469. doi: 10.1111/j.1462-5822.2011.01651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carabeo RA, Grieshaber SS, Fischer E, Hackstadt T. 2002. Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells. Infect Immun 70:3793–3803. doi: 10.1128/IAI.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynolds DJ, Pearce JH. 1991. Endocytic mechanisms utilized by chlamydiae and their influence on induction of productive infection. Infect Immun 59:3033–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiwani S, Ohr RJ, Fischer ER, Hackstadt T, Alvarado S, Romero A, Jewett TJ. 2012. Chlamydia trachomatis Tarp cooperates with the Arp2/3 complex to increase the rate of actin polymerization. Biochem Biophys Res Commun 420:816–821. doi: 10.1016/j.bbrc.2012.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subtil A, Wyplosz B, Balana ME, Dautry-Varsat A. 2004. Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity. J Cell Sci 117:3923–3933. doi: 10.1242/jcs.01247. [DOI] [PubMed] [Google Scholar]
- 31.Carabeo RA, Grieshaber SS, Hasenkrug A, Dooley C, Hackstadt T. 2004. Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic 5:418–425. doi: 10.1111/j.1398-9219.2004.00184.x. [DOI] [PubMed] [Google Scholar]
- 32.Damiani MT, Gambarte Tudela J, Capmany A. 2014. Targeting eukaryotic Rab proteins: a smart strategy for chlamydial survival and replication. Cell Microbiol 16:1329–1338. doi: 10.1111/cmi.12325. [DOI] [PubMed] [Google Scholar]
- 33.Bastidas RJ, Elwell CA, Engel JN, Valdivia RH. 2013. Chlamydial intracellular survival strategies. Cold Spring Harb Perspect Med 3:a010256. doi: 10.1101/cshperspect.a010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He SY, Nomura K, Whittam TS. 2004. Type III protein secretion mechanism in mammalian and plant pathogens. Biochim Biophys Acta 1694:181–206. doi: 10.1016/j.bbamcr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Mueller KE, Plano GV, Fields KA. 2014. New frontiers in type III secretion biology: the Chlamydia perspective. Infect Immun 82:2–9. doi: 10.1128/IAI.00917-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jamison WP, Hackstadt T. 2008. Induction of type III secretion by cell-free Chlamydia trachomatis elementary bodies. Microb Pathog 45:435–440. doi: 10.1016/j.micpath.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fields K, Mead D, Dooley C, Hackstadt T. 2003. Chlamydia trachomatis type III secretion: evidence for a functional apparatus during early-cycle development. Mol Microbiol 48:671–683. doi: 10.1046/j.1365-2958.2003.03462.x. [DOI] [PubMed] [Google Scholar]
- 38.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. 2004. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci U S A 101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel J. 2004. Tarp and Arp: how Chlamydia induces its own entry. Proc Natl Acad Sci U S A 101:9947–9948. doi: 10.1073/pnas.0403633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. 2006. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci U S A 103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jewett TJ, Miller NJ, Dooley CA, Hackstadt T. 2010. The conserved Tarp actin binding domain is important for chlamydial invasion. PLoS Pathog 6:e1000997. doi: 10.1371/journal.ppat.1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehlitz A, Banhart S, Maurer AP, Kaushansky A, Gordus AG, Zielecki J, Macbeath G, Meyer TF. 2010. Tarp regulates early Chlamydia-induced host cell survival through interactions with the human adaptor protein SHC1. J Cell Biol 190:143–157. doi: 10.1083/jcb.200909095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA. 2008. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog 4:e1000014. doi: 10.1371/journal.ppat.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brinkworth AJ, Malcolm DS, Pedrosa AT, Roguska K, Shahbazian S, Graham JE, Hayward RD, Carabeo RA. 2011. Chlamydia trachomatis Slc1 is a type III secretion chaperone that enhances the translocation of its invasion effector substrate TARP. Mol Microbiol 82:131–144. doi: 10.1111/j.1365-2958.2011.07802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pais SV, Milho C, Almeida F, Mota LJ. 2013. Identification of novel type III secretion chaperone-substrate complexes of Chlamydia trachomatis. PLoS One 8:e56292. doi: 10.1371/journal.pone.0056292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y-S, Bastidas RJ, Saka HA, Carpenter VK, Richards KL, Plano GV, Valdivia RH. 2014. The Chlamydia trachomatis type III secretion chaperone Slc1 engages multiple early effectors, including TepP, a tyrosine-phosphorylated protein required for the recruitment of CrkI-II to nascent inclusions and innate immune signaling. PLoS Pathog 10:e1003954. doi: 10.1371/journal.ppat.1003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carpenter V, Chen Y-S, Dolat L, Valdivia RH. 2017. The effector TepP mediates recruitment and activation of phosphoinositide 3-kinase on early Chlamydia trachomatis vacuoles. mSphere 2:e00207-17. doi: 10.1128/mSphere.00207-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 49.Hower S, Wolf K, Fields KA. 2009. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol 72:1423–1437. doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bullock HD, Hower S, Fields KA. 2012. Domain analyses reveal that C. trachomatis CT694 belongs to the membrane-localized family of type III effector proteins. J Biol Chem 287:28078–28086. doi: 10.1074/jbc.M112.386904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masunaga T, Shimizu H, Ishiko A, Fujiwara T, Hashimoto T, Nishikawa T. 1995. Desmoyokin/AHNAK protein localizes to the non-desmosomal keratinocyte cell surface of human epidermis. J Investig Dermatol 104:941–945. doi: 10.1111/1523-1747.ep12606213. [DOI] [PubMed] [Google Scholar]
- 52.Shtivelman E, Bishop JM. 1993. The human gene AHNAK encodes a large phosphoprotein located primarily in the nucleus. J Cell Biol 120:625–630. doi: 10.1083/jcb.120.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sussman J, Stokoe D, Ossina N, Shtivelman E. 2001. Protein kinase B phosphorylates AHNAK and regulates its subcellular localization. J Cell Biol 154:1019–1030. doi: 10.1083/jcb.200105121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gentil BJ, Benaud C, Delphin C, Remy C, Berezowski V, Cecchelli R, Feraud O, Vittet D, Baudier J. 2005. Specific AHNAK expression in brain endothelial cells with barrier properties. J Cell Physiol 203:362–371. doi: 10.1002/jcp.20232. [DOI] [PubMed] [Google Scholar]
- 55.Gentil BJ, Delphin C, Benaud C, Baudier J. 2003. Expression of the giant protein AHNAK (desmoyokin) in muscle and lining epithelial cells. J Histochem Cytochem 51:339–348. doi: 10.1177/002215540305100309. [DOI] [PubMed] [Google Scholar]
- 56.Nie Z, Ning W, Amagai M, Hashimoto T. 2000. C-terminus of desmoyokin/AHNAK protein is responsible for its translocation between the nucleus and cytoplasm. J Investig Dermatol 114:1044–1049. doi: 10.1046/j.1523-1747.2000.00949.x. [DOI] [PubMed] [Google Scholar]
- 57.Benaud C, Gentil BJ, Assard N, Court M, Garin J, Delphin C, Baudier J. 2004. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J Cell Biol 164:133–144. doi: 10.1083/jcb.200307098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shtivelman E, Cohen FE, Bishop JM. 1992. A human gene (AHNAK) encoding an unusually large protein with a 1.2-micron polyionic rod structure. Proc Natl Acad Sci U S A 89:5472–5476. doi: 10.1073/pnas.89.12.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Komuro A, Masuda Y, Kobayashi K, Babbitt R, Gunel M, Flavell RA, Marchesi VT. 2004. The AHNAKs are a class of giant propeller-like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc Natl Acad Sci U S A 101:4053–4058. doi: 10.1073/pnas.0308619101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee IH, Lim HJ, Yoon S, Seong JK, Bae DS, Rhee SG, Bae YS. 2008. Ahnak protein activates protein kinase C (PKC) through dissociation of the PKC-protein phosphatase 2A complex. J Biol Chem 283:6312–6320. doi: 10.1074/jbc.M706878200. [DOI] [PubMed] [Google Scholar]
- 61.Sekiya F, Bae YS, Jhon DY, Hwang SC, Rhee SG. 1999. AHNAK, a protein that binds and activates phospholipase C-γ1 in the presence of arachidonic acid. J Biol Chem 274:13900–13907. doi: 10.1074/jbc.274.20.13900. [DOI] [PubMed] [Google Scholar]
- 62.Lee IH, Sohn M, Lim HJ, Yoon S, Oh H, Shin S, Shin JH, Oh SH, Kim J, Lee DK, Noh DY, Bae DS, Seong JK, Bae YS. 2014. Ahnak functions as a tumor suppressor via modulation of TGFbeta/Smad signaling pathway. Oncogene 33:4675–4684. doi: 10.1038/onc.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee IH, You JO, Ha KS, Bae DS, Suh PG, Rhee SG, Bae YS. 2004. AHNAK-mediated activation of phospholipase C-gamma1 through protein kinase C. J Biol Chem 279:26645–26653. doi: 10.1074/jbc.M311525200. [DOI] [PubMed] [Google Scholar]
- 64.Haase H, Pagel I, Khalina Y, Zacharzowsky U, Person V, Lutsch G, Petzhold D, Kott M, Schaper J, Morano I. 2004. The carboxyl-terminal ahnak domain induces actin bundling and stabilizes muscle contraction. FASEB J 18:839–841. [DOI] [PubMed] [Google Scholar]
- 65.Hohaus A, Person V, Behlke J, Schaper J, Morano I, Haase H. 2002. The carboxyl-terminal region of ahnak provides a link between cardiac L-type Ca2+ channel and the actin-based cytoskeleton. FASEB J 16:1205–1216. doi: 10.1096/fj.01-0855com. [DOI] [PubMed] [Google Scholar]
- 66.De Seranno S, Benaud C, Assard N, Khediri S, Gerke V, Baudier J, Delphin C. 2006. Identification of an AHNAK binding motif specific for the Annexin2/S100A10 tetramer. J Biol Chem 281:35030–35038. doi: 10.1074/jbc.M606545200. [DOI] [PubMed] [Google Scholar]
- 67.Dempsey BR, Rezvanpour A, Lee TW, Barber KR, Junop MS, Shaw GS. 2012. Structure of an asymmetric ternary protein complex provides insight for membrane interaction. Structure 20:1737–1745. doi: 10.1016/j.str.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 68.Rezvanpour A, Santamaria-Kisiel L, Shaw GS. 2011. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J Biol Chem 286:40174–40183. doi: 10.1074/jbc.M111.244038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rucks EA, Olson MG, Jorgenson LM, Srinivasan RR, Ouellette SP. 2017. Development of a proximity labeling system to map the Chlamydia trachomatis inclusion membrane. Front Cell Infect Microbiol 7:40. doi: 10.3389/fcimb.2017.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fields KA, McCormack R, de Armas LR, Podack ER. 2013. Perforin-2 restricts growth of Chlamydia trachomatis in macrophages. Infect Immun 81:3045–3054. doi: 10.1128/IAI.00497-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hueck CJ. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 62:379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.da Cunha M, Milho C, Almeida F, Pais S, Borges V, Mauricio R, Borrego M, Gomes J, Mota L. 2014. Identification of type III secretion substrates of Chlamydia trachomatis using Yersinia enterocolitica as a heterologous system. BMC Microbiol 14:40. doi: 10.1186/1471-2180-14-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38:868–877. doi: 10.1093/nar/gkp1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.So EC, Mattheis C, Tate EW, Frankel G, Schroeder GN. 2015. Creating a customized intracellular niche: subversion of host cell signaling by Legionella type IV secretion system effectors. Can J Microbiol 61:617–635. doi: 10.1139/cjm-2015-0166. [DOI] [PubMed] [Google Scholar]
- 76.Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN. 2008. RNA interference screen identifies Able Kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog 4:e1000021. doi: 10.1371/journal.ppat.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abromaitis S, Stephens RS. 2009. Attachment and entry of Chlamydia have distinct requirements for host protein disulfide isomerase. PLoS Pathog 5:e1000357. doi: 10.1371/journal.ppat.1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stallmann S, Hegemann JH. 2016. The Chlamydia trachomatis Ctad1 invasin exploits the human integrin β1 receptor for host cell entry. Cell Microbiol 18:761–775. doi: 10.1111/cmi.12549. [DOI] [PubMed] [Google Scholar]
- 79.Patel AL, Chen X, Wood ST, Stuart ES, Arcaro KF, Molina DP, Petrovic S, Furdui CM, Tsang AW. 2014. Activation of epidermal growth factor receptor is required for Chlamydia trachomatis development. BMC Microbiol 14:1–20. doi: 10.1186/1471-2180-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Subbarayal P, Karunakaran K, Winkler A-C, Rother M, Gonzalez E, Meyer TF, Rudel T. 2015. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog 11:e1004846. doi: 10.1371/journal.ppat.1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kari L, Southern TR, Downey CJ, Watkins HS, Randall LB, Taylor LD, Sturdevant GL, Whitmire WM, Caldwell HD. 2014. Chlamydia trachomatis polymorphic membrane protein D is a virulence factor involved in early host-cell interactions. Infect Immun 82:2756–2762. doi: 10.1128/IAI.01686-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rogers LD, Brown NF, Fang Y, Pelech S, Foster LJ. 2011. Phosphoproteomic analysis of Salmonella-infected cells identifies key kinase regulators and SopB-dependent host phosphorylation events. Sci Signal 4:rs9. doi: 10.1126/scisignal.2001668. [DOI] [PubMed] [Google Scholar]
- 83.Jolly C, Winfree S, Hansen B, Steele-Mortimer O. 2014. The Annexin A2/p11 complex is required for efficient invasion of Salmonella Typhimurium in epithelial cells. Cell Microbiol 16:64–77. doi: 10.1111/cmi.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davis TA, Loos B, Engelbrecht A-M. 2014. AHNAK: the giant jack of all trades. J Cell Signal 26:2683–2693. doi: 10.1016/j.cellsig.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 85.Khurana S, George S. 2011. The role of actin bundling proteins in the assembly of filopodia in epithelial cells. Cell Adh Migr 5:409–420. doi: 10.4161/cam.5.5.17644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shaw EI, Dooley CA, Fischer ER, Scidmore MA, Fields KA, Hackstadt T. 2000. Three temporal classes of gene expression during the Chlamydia trachomatis developmental cycle. Mol Microbiol 37:913–925. doi: 10.1046/j.1365-2958.2000.02057.x. [DOI] [PubMed] [Google Scholar]
- 87.Jiwani S, Alvarado S, Ohr RJ, Romero A, Nguyen B, Jewett TJ. 2013. Chlamydia trachomatis tarp harbors distinct G and F actin binding domains that bundle actin filaments. J Bacteriol 195:708–716. doi: 10.1128/JB.01768-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mueller KE, Wolf K, Fields KA. 2017. Chlamydia trachomatis transformation and allelic exchange mutagenesis. Curr Protocol Microbiol 45:11A.3.1–11A.3.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caldwell HD, Kromhout J, Schachter J. 1981. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun 31:1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA. 1996. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J 15:964–977. [PMC free article] [PubMed] [Google Scholar]
- 91.Furness G, Graham DM, Reeve P. 1960. The titration of trachoma and inclusion blennorrhoea viruses in cell cultures. J Gen Microbiol 23:613–619. [DOI] [PubMed] [Google Scholar]
- 92.Scidmore MA, Hackstadt T. 2001. Mammalian 14-3-3 beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol 39:1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- 93.Darville T, Andrews CW, Laffoon KK, Shymasani W, Kishen LR, Rank RG. 1997. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun 65:3065–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maniatis T, Sambrook J, Fritsch EF. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 95.McCormack R, de Armas LR, Shiratsuchi M, Ramos JE, Podack ER. 2012. Inhibition of intracellular bacterial replication in fibroblasts is dependent on the Perforin-like protein (Perforin-2) encoded by macrophage-expressed gene 1. J Innate Immun 5:185–194. doi: 10.1159/000345249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roux KJ, Kim DI, Burke B. 2001. BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci 74:Unit 19.23. doi: 10.1002/0471140864.ps1923s74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.