

Abstract
Aims
Chloride (Cl) is an established key electrolyte for the activation of the renin–angiotensin–aldosterone system. Recent studies have shown the serum Cl as a key electrolyte for the regulation of body fluid distribution in heart failure (HF) patients. The clinical differences of worsening HF status according to the changes in serum Cl concentration are unclear.
Methods and results
Data from 47 chronic HF patients were analysed. Upon worsening HF, each patient exhibited at least two HF‐related signs. Blood tests included haemoglobin (Hb), haematocrit (Ht), mean red blood cell volume (MCV), albumin, serum solutes, and b‐type natriuretic peptide. The relative change in the plasma volume (%PV) from stable to worsening HF was estimated as follows: 100 × {Hb (stable) × [1 − Ht (worse)]}/{Hb (worse) × [1 − Ht (stable)]} − 100. When patients were divided into two groups based on changes in serum Cl concentration from stable to worsening HF, the pathophysiologic features of the patients with increased Cl (range 1–23 mEq/L; n = 31) included a greater increase in serum sodium (2.94 ± 4.15 vs. −0.69 ± 3.75 mEq/L, P = 0.005), higher vascular expansion (12 ± 11.1 vs. 4.81 ± 7.94%, P = 0.026), a tendency towards a greater MCV (1.23 ± 2.36 vs. −0.06 ± 1.88 fL, P = 0.065), and preserved renal function defined by the absence of an increase of serum creatinine (−0.24 ± 0.39 vs. −0.05 ± 0.12 mg/dL, P = 0.057) compared to patients with non‐increased Cl (range −9 to 0 mEq/L; n = 16). Clinically, the increased Cl group had fewer HF signs (2.65 ± 0.71 vs. 3.31 ± 0.79, P = 0.005) although the change in symptoms did not differ between groups (48% vs. 63%, P = 0.54).
Conclusions
The present study suggests a new clinical entity of worsening HF status, that is, HF with increased vs. non‐increased serum Cl concentration from clinical stability to worsening HF.
Keywords: Heart failure, Chloride, Body fluid, Vascular volume, Red blood cell volume, Renal function
Introduction
Until recently, most studies focused on the body fluid dynamics in heart failure (HF) status through the control of sodium, potassium, and water balance in the body,1, 2 and through the regulation of solutes and water by the renin–angiotensin–aldosterone system (RAAS) and antidiuretic hormone (ADH).3, 4, 5 Thus, the proposed hypothesis of body fluid regulation in health and disease is that the arterial circulation is the primary body fluid compartment modulating renal sodium and water excretion.3, 4 Chloride, despite flanking sodium as its anionic counterpart in salt, has remained largely ignored, presenting in the medical literature and in clinical practice as an afterthought to the more popular electrolytes sodium and potassium, or simply as a substitute for bicarbonate to preserve electroneutrality.6 I recently reported that serum chloride is a key electrolyte for the regulation of intravascular7 and intracellular body fluid8 in HF patients. Considering both my recent observations7, 8 and established finding that chloride is a key electrolyte for RAAS activation and for tubuloglomerular feedback in the kidney,9, 10, 11, 12 I tested the hypothesis that there is a different clinical presentation of worsening HF status according to the changes in serum chloride concentration from clinical stability to worsening HF.
Methods
Study population
The present study was a sub‐study of my follow‐up study13, 14 focusing on monitoring body composition changes in established HF patients performed at the cardiology clinic of Nishida Hospital between June 2003 and March 2009. Eligible patients included into this follow‐up study had at least one decompensated HF episode resulting in hospitalization or outpatient treatment with conventional diuretics. In the present analysis, HF patients with severe renal failure (serum creatinine concentration >3.5 mg/dL at stable HF status) were excluded. Informed consent was obtained from all patients before study enrolment.
Study protocol
At study entry, patient characteristics, history, and primary aetiology were recorded. Patients enrolled in this study were interviewed regarding changes in symptoms and examined for the appearance of physical signs of fluid retention upon each visit to the clinic by a clinician (H.K.). Additional routine tests included conducting ultrasound examination to search for pleural effusion,14, 15 monitoring changes in the fluid status using a digital body weight scale incorporating a bioelectrical impedance analyser (HBF‐352‐W, Omron Healthcare Co., Kyoto, Japan),13, 14, 16 and measuring b‐type natriuretic peptide (BNP) levels.17 Peripheral blood tests, chest X‐ray, standard 12‐lead electrocardiography, and echocardiography were performed at study entry and a clinic visit during follow‐up after an appropriate interval.
Data collection
Blood tests
Peripheral haematologic and biochemical tests were performed by standard laboratory techniques. Blood tests included measurements of red blood cell count, mean red blood cell volume (MCV), haemoglobin (Hb), haematocrit (Ht), total protein, albumin, serum electrolytes (sodium, potassium, and chloride), blood urea nitrogen, and creatinine. The percentage shift in the plasma volume (%PV) during a change in HF status was estimated from serial concomitant Hb and Ht concentrations according to the following formula: 100 × {Hb (stable) × [1 − Ht (worse)]}/{Hb (worse) × [1 − Ht (stable)]} − 100.18
Thoracic ultrasound
Thoracic ultrasound was performed on each patient using a commercially available real‐time, wide‐angle phased‐array system.15 The patient was placed in a sitting position on a bed or chair, and chest ultrasound was performed on each hemithorax using a 3.5‐MHz sector transducer through the intercostal space and scanning along the paravertebral, scapular, and posterior axillary lines. The presence of pleural effusion was diagnosed by the appearance of an anechoic space between the parietal pleura and the highly reflective visceral pleura–lung interface.
Monitoring of weight gain
Evaluation of fluid body weight gain by measuring body weight and percentage body fat was described in detail previously.13, 16 Briefly, for monitoring definite HF patient, body weight and percentage body fat were measured twice and averaged. Patients wore light clothes and were barefoot during weighing. Based on prior experience,13, 16 the criterion of significant fluid weight gain was defined as a body weight gain of at least 1.5 kg with a concomitant decrease in percentage body fat from the most recent visit with clinical stability to the time of target weight gain. Adjustments of reference dry weight, as determined by the evaluation of symptoms, physical exam and chest ultrasound, were made at each clinic visit.
Definition of worsening HF
Criteria for selecting the event of worsening HF consisted of the appearance of at least two of the following HF‐related signs, irrespective of the presence of symptomatic changes: physical signs (third heart sound, pulmonary crackles, and leg oedema), fluid weight gain (≥1.5 kg), and pleural effusion on ultrasound. Worsening HF was treated with conventional therapy using a combination of loop diuretics, aldosterone blockade, thiazide diuretics, and/or inotropic drugs by oral and/or intravenous routes in the hospital or outpatient clinic. After returning to stable HF status from acutely worsening HF status, oral HF therapy was adjusted and maintained during the following clinical course at the outpatient clinic. In the event of multiple episodes of worsening HF in a single patient, the first episode was selected for the present analysis.
Statistical analysis
All statistical analyses were performed using commercially available statistical software (GraphPad Prism 4, San Diego, CA). All data are expressed as a mean ± SD for continuous data and percentage for categorical data. Paired and unpaired t‐tests for continuous data and Fisher's exact test for categorical data were used for two‐group comparisons. A P‐value of less than 0.05 was considered statistically significant.
Results
Ambulatory patients with HF (n = 83) were enrolled and followed up at the outpatient clinic of Nishida Hospital. Peripheral haematologic and biochemical tests, except BNP measurement, were not routinely obtained for all patients in the original study because that study did not focus on the correlation between HF status and blood chemistry. Thus, the patients and blood data of the present study were not selected consecutively but were included based on the availability of complete blood data as well as simultaneous HF‐related clinical data during clinically stable HF, worsening HF, and resolution of worsening HF after therapy. Of 83 HF patients enrolled in the study at our hospital, 47 had available data of worsening HF event(s) for the analysis in the present study. After HF therapy using conventional diuretics and adjunctive drugs, all the study patients returned to clinical stability from worsening HF.
The demographic features of the 47 patients with clinical stability at study entry are summarized in Table 1. The interval between the clinical stability to worsening HF was 37.5 ± 16.3 days (range: 14–67 days). Diagnosis and precipitating factors of worsening HF status in the 47 HF patients are reported in the Supporting Information (Table S1 ). The cumulative number of HF‐related signs/tests was 2.87 ± 1.52 (range: 2–5).
Table 1.
Clinical characteristics of the study patients
| Characteristics | N = 47 |
|---|---|
| Age (years) | 78.2 ± 9.7 (29–93) |
| Male | 15 (32) |
| Primary cause of heart failure | |
| Hypertension | 25 (53) |
| Valvular | 8 (17) |
| Cardiomyopathy | 6 (13) |
| Ischaemic | 3 (6) |
| Arrhythmia | 3 (6) |
| Congenital | 2 (4) |
| Left ventricular EF (%) | 56 ± 14 |
| Left ventricular EF > 50% | 25 (53) |
| Atrial fibrillation | 16 (34) |
| NYHA‐FC at stable period | |
| II/III | 34 (72)/13 (28) |
| Medication | |
| Diuretics | 46 (98) |
| Loop diuretics | 31 (66) |
| Thiazide diuretics | 24 (51) |
| Potassium‐sparing diuretics | 38 (81) |
| ACE inhibitors/ARB | 30 (64) |
| Calcium antagonists | 21 (45) |
| Beta‐blockers | 19 (40) |
| Digitalis | 5 (11) |
| Nitrates | 3 (6) |
ACE, angiotensin‐converting enzyme; ARB, angiotensin II receptor blocker; EF, ejection fraction; NYHA‐FC, New York Heart Failure functional class.
Data presented as number (%) of patients unless otherwise specified.
A histogram of the magnitude in changes in serum chloride concentration from stable to worsening HF is shown in Figure 1 . When participants were divided into two groups based on changes in the serum chloride concentration from clinical stability to worsening HF (Table 2 ), the increased chloride group (range 1–23 mEq/L; n = 31) had fewer HF signs (2.65 ± 0.71 vs. 3.31 ± 0.79, P = 0.005) and a lower incidence of pulmonary rales compared to the non‐increased chloride group (range −9 to 0 mEq/L; n = 16). The change in symptoms did not differ between groups (48% vs. 63%, P = 0.54).
Figure 1.
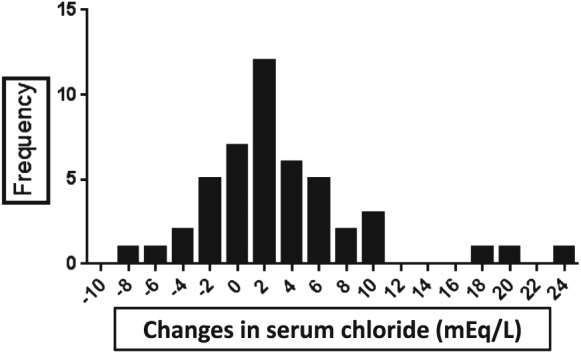
Histogram of the magnitude in changes in serum chloride concentration from stable to worsening heart failure.
Table 2.
Comparison of symptoms and physical signs between groups of increased vs. non‐increased serum chloride concentration
| Increased Cl group | Non‐increased Cl group | P‐value | |
|---|---|---|---|
| (n = 31) | (n = 16) | ||
| Worsening of dyspnoea | 15 (48%) | 10 (63%) | 0.54 |
| Cumulative number of HF‐related signs | 2.65 ± 0.71 | 3.31 ± 0.79 | 0.005a |
| Body weight gain ≥ 1.5kg | 27 (87%) | 15 (94%) | 1 |
| Bilateral pulmonary rales | 5 (16%) | 10 (63%) | 0.0024a |
| Bilateral leg oedema above ankle | 21 (68%) | 12 (75%) | 0.742 |
| Third heart sound (S3) | 7 (23%) | 4 (67%) | 1 |
| Ultrasound pleural effusion | 25 (81%) | 12 (75%) | 0.716 |
Cl, chloride; HF, heart failure.
Significant.
Laboratory data for the two groups are shown in Table 3. Patients with increased serum chloride concentrations had increased serum sodium (2.94 ± 4.15 mEq/L, P = 0.0004), greater MCV enlargement (1.23 ± 2.36 fL, P = 0.007), and preserved renal function defined as a decrease in serum creatinine (−0.24 ± 0.39 mg/dL, P = 0.0015) and blood urea nitrogen (−9.47 ± 15.4 mg/dL, P = 0.0018). In contrast, patients without an increased serum chloride concentration did not have the accompanying changes in serum sodium, MCV, serum creatinine, and blood urea nitrogen from clinical stability to worsening HF. When compared to patients without an increase in serum chloride, patients with increased chloride also exhibited a greater increase in serum sodium (2.94 ± 4.15 vs. −0.69 ± 3.75 mEq/L, P = 0.005), higher vascular expansion determined by %PV (12 ± 11.1 vs. 4.81 ± 7.94%, P = 0.026), a tendency towards a greater MCV (1.23 ± 2.36 vs. −0.06 ± 1.88 fL, P = 0.065), and preserved renal function defined by the absence of an increase of serum creatinine (−0.24 ± 0.39 vs. −0.05 ± 0.12mg/dL, P = 0.057) from stable to worsening HF, although the body weight increase (P = 0.57) and log BNP (P = 0.126) did not differ between groups.
Table 3.
Comparison of changes in blood examination between groups of increased vs. non‐increased serum chloride concentration from clinical stability to worsening of heart failure
| Variables | Total | Changes in serum chloride | P‐value | |
|---|---|---|---|---|
| Increase | Non‐increase | |||
| (n = 47) | (n = 31) | (n = 16) | ||
| Serum chloride (mEq/L) | ||||
| Stability | 101 ± 5.36 | 100 ± 5.29 | 103 ± 4.94 | 0.044a |
| Worsening | 104 ± 5.44 | 106 ± 4.22 | 101 ± 6.3 | 0.0038a |
| Δstability to worsening | 2.72 ± 6.02 | 5.45 ± 5.42 | −2.6 ± 2.73 | <0.0001a |
| P‐value | 0.0033a | <0.0001a | 0.0019a | |
| Body weight (kg) | ||||
| Stability | 49.8 ± 11.8 | 49.3 ± 13.5 | 50.8 ± 7.73 | 0.691 |
| Worsening | 52.3 ± 12.0 | 51.9 ± 13.9 | 53.1 ± 7.6 | 0.747 |
| Δstability to worsening | 2.51 ± 1.42 | 2.59 ± 1.56 | 2.34 ± 1.12 | 0.572 |
| P‐value | <0.0001a | <0.0001a | <0.0001a | |
| Serum log BNP (pg/mL) | ||||
| Stability | 2.08 ± 0.39 | 2.06 ± 0.43 | 2.12 ± 0.33 | 0.621 |
| Worsening | 2.57 ± 0.34 | 2.60 ± 0.36 | 2.51 ± 0.28 | 0.405 |
| Δstability to worsening | 0.488 ± 0.26 | 0.54 ± 0.31 | 0.39 ± 0.30 | 0.126 |
| P‐value | <0.0001a | <0.0001a | 0.0001a | |
| ΔChange in %plasma volume | ||||
| Mean ± SD | 9.54 ± 10.6 | 12.0 ± 11.1 | 4.81 ± 7.94 | 0.0026a |
| Range | −19~35.8 | −19 ~ 35.8 | −10.8 ~ 14.9 | |
| MCV (fL) | ||||
| Stability | 96 ± 5.16 | 96.3 ± 4.65 | 95.4 ± 6.15 | 0.597 |
| Worsening | 96.8 ± 6.0 | 97.5 ± 5.08 | 95.4 ± 7.44 | 0.25 |
| Δstability to worsening | 0.79 ± 2.27 | 1.23 ± 2.36 | −0.06 ± 1.88 | 0.065 |
| P‐value | NS | 0.007a | 0.896 | |
| Albumin (g/dL) | ||||
| Stability | 3.88 ± 0.35 | 3.83 ± 0.34 | 3.98 ± 0.35 | 0.15 |
| Worsening | 3.6 ± 0.38 | 3.51 ± 0.38 | 3.78 ± 0.29 | 0.016a |
| Δstability to worsening | −0.28 ± 0.31 | −0.32 ± 0.32 | −0.20 ± 0.28 | 0.215 |
| P‐value | <0.0001a | <0.0001a | 0.0012a | |
| Serum sodium (mEq/L) | ||||
| Stability | 139 ± 4.1 | 139 ± 4.05 | 140 ± 4.31 | 0.678 |
| Worsening | 141 ± 5.1 | 142 ± 3.55 | 139 ± 6.84 | 0.046a |
| Δstability to worsening | 1.7 ± 4.3 | 2.94 ± 4.15 | −0.69 ± 3.75 | 0.0053a |
| P‐value | 0.01a | 0.0004a | 0.475 | |
| Serum potassium (mEq/L) | ||||
| Stability | 4.23 ± 0.58 | 4.11 ± 0.55 | 4.46 ± 0.60 | 0.047a |
| Worsening | 4.1 ± 0.69 | 3.98 ± 0.65 | 4.34 ± 0.73 | 0.098 |
| Δstability to worsening | −0.12 ± 0.65 | −0.12 ± 0.73 | −0.13 ± 0.47 | 0.991 |
| P‐value | 0.2 | 0.36 | 0.305 | |
| Blood urea nitrogen (mg/dL) | ||||
| Stability | 27.1 ± 14.4 | 29.2 ± 16.4 | 23.1 ± 8.26 | 0.166 |
| Worsening | 20.4 ± 7.97 | 19.7 ± 8.59 | 21.7 ± 6.69 | 0.166 |
| Δstability to worsening | −6.7 ± 13.3 | −9.47 ± 15.4 | −1.37 ± 4.85 | 0.047a |
| P‐value | 0.0012a | 0.0018a | 0.277 | |
| Serum creatinine (mg/dL) | ||||
| Stability | 1.19 ± 1.64 | 1.21 ± 0.5 | 1.17 ± 0.41 | 0.805 |
| Worsening | 1.02 ± 0.39 | 0.96 ± 0.35 | 1.12 ± 0.45 | 0.19 |
| Δstability to worsening | −0.176 ± 0.33 | −0.24 ± 0.39 | −0.05 ± 0.12 | 0.057 |
| P‐value | 0.0007a | 0.0015a | 0.121 | |
| Serum uric acid (mg/dL) | ||||
| Stability | 7.16 ± 2.79 | 7.38 ± 3.11 | 6.73 ± 2.05 | 0.458 |
| Worsening | 6.07 ± 2.15 | 6.02 ± 2.32 | 6.16 ± 1.85 | 0.843 |
| Δstability to worsening | −1.09 ± 1.87 | −1.35 ± 2.05 | −0.58 ± 1.39 | 0.178 |
| P‐value | 0.0002a | 0.0009a | 0.118 | |
BNP, b‐type natriuretic peptide; MCV, mean red blood cell volume.
Significant.
Discussion
The present study has shown that there are two distinct modes of acute on chronic HF progression with different clinical characteristics based on the chloride accumulation pattern in relation to water retention, that is, worsening HF with increased vs. non‐increased serum chloride concentration from clinical stability to worsening HF. Pathophysiologically important alterations in the sympathetic nervous system,19 the RAAS and ADH systems,3, 4, 5, 20 and vasodilatory/natriuretic pathways21 are present in HF patients. These disturbances are translated at the renal circulation and tubular level in such a way that sodium and water retention are increased.3, 4, 5, 20 Surprisingly, the role of chloride in HF pathophysiology has remained largely ignored and has not been investigated for several decades.6
Taking together the established central role of chloride in the RAAS,9, 10, 11, 12 my recent observations revealing the important role of chloride for regulation of intravascular7 and intracellular body fluid8 in HF patients and the present study, I propose a hypothesis for the ‘chloride theory’ for worsening HF. This hypothesis states that changes in the serum chloride concentration are the primary determinant of changes in the plasma volume, RAAS, and ADH systems under worsening HF, as shown in Figure 2 , which depicts the two types of acute on chronic HF worsening according to the directional movement of serum chloride, that is, worsening HF type with increased (Figure 2 , left half) vs. decreased (Figure 2 , right half) serum chloride concentration. Each type of worsening HF might reflect differences in the RAAS and ADH activation and effectiveness, and subsequent movement of serum electrolytes and water, in which body fluid is re‐distributed to each body fluid compartment comprising extracellular (intravascular and interstitial space) and intracellular spaces.
Figure 2.
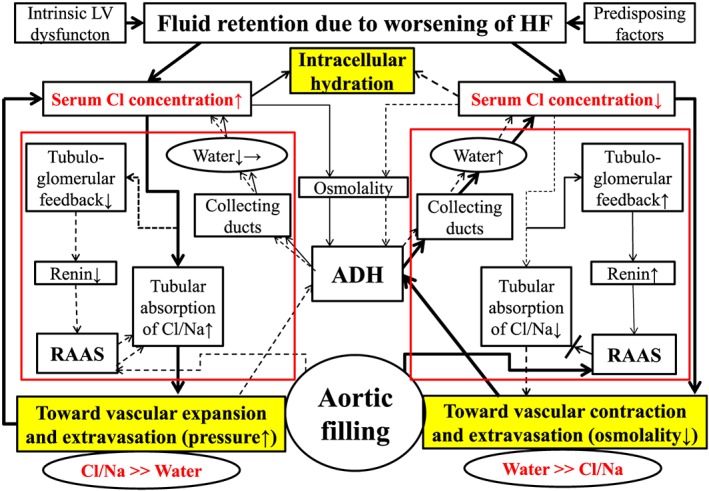
The ‘chloride theory’ for explaining fluid dynamics in the course of worsening heart failure. Solid line indicates enhanced supply or excitatory effect, and dotted line, reduced supply or inhibitory effect. Different effect strengths are expressed by the thickness of each line. The pathway of the RAAS, tubuloglomerular feedback, and ADH in the kidney is outlined in red. ADH, antidiuretic hormone; Cl, chloride; HF, heart failure; LV, left ventricular; Na, sodium; RAAS, renin‐angiotensin‐aldosterone system.
Role of chloride in the kidney and distribution of body fluid
Chloride is a crucial electrolyte that enables reabsorption of the filtered sodium from the urinary tubules into the extracellular body spaces and maintains arterial pressure.9, 22, 23, 24 Chloride also plays a central role in the regulation of renin release from the juxtaglomerular apparatus (i.e. tubuloglomerular feedback), which depends on macula densa chloride transport.9, 10, 11, 12
I recently demonstrated the heterogeneous distribution of plasma volume (from −19% to 36%) and an independent positive association between the changes in %PV and serum chloride concentration, which is not dependent on serum sodium, from stability to worsening HF.7 An accumulation of serum chloride would increase or maintain the plasma volume and probably tend to inhibit a fluid shift from the blood vessels to the interstitial space25, 26 owing to an increase in serum osmolality or tonicity, which is mainly induced by serum chloride,7 not solely by serum sodium,27 and vice versa. In addition, I recently observed that MCV serially changes according to the transition of the HF status, that is, worsening and recovery of HF, and that serum chloride is a key electrolyte for the regulation of MCV, presumably reflecting the intracellular fluid status.8 As shown by my recent observations7, 8 and the findings of the present study, chloride in the body fluid appears to have an important role in regulating the reabsorption of filtered solutes and water in the kidney, for distributing the body fluid between the intracellular and extracellular spaces, and between the intravascular and interstitial spaces according to the changes in various HF states. Thus, it is quite reasonable to organize newly the pathophysiologic background of acute or chronic HF according to the dynamics of chloride in the human body.
Worsening heart failure with increased vs. decreased serum chloride concentration
In patients with worsening HF and increased serum chloride concentration from clinical stability to worsening HF (Figure 2 , left half), the greater accumulation of serum chloride acts to maintain or increase intravascular volume, which places a greater burden on the failing heart. It might be that maintaining plasma volume is beneficial for supplying blood to the kidney and preserving its function,27 as shown in the group with an increased chloride concentration compared with the group with a decreased chloride concentration. As for the lung, I observed a lower incidence of audible bilateral pulmonary rales in this subtype of worsening HF group, which might be due not only to differences in the levels of accumulated serum chloride but also to differences in the changes in serum albumin levels. A decrease in serum albumin would accompany a decrease in the interstitial albumin concentration,28 which would promote drainage of interstitial fluid into the lymphatics.29, 30 This subtype of worsening HF would show signs of remarkable body fluid retention in the interstitial or third spaces when the venous and lymphatic systems can no longer accommodate extra body fluid from the interstitial space through the lymphatic drainage system.
In patients with worsening HF and no increase in the serum chloride concentration from clinical stability to worsening HF (Figure 2 , right half), a decreased supply of chloride to the urinary tubules would result in decreased reabsorption of filtered sodium from the tubules into the extracellular space,9, 22, 23, 24 resulting in reduced blood vessel expansion or even blood vessel contraction. As a result, this subtype of worsening HF might accompany enhanced ADH activation and increased water retention in relation to serum chloride (and mostly sodium) to correct arterial under‐filling.3 In this subtype of worsening HF, the RAAS might be overactivated owing to positive tubuloglomerular feedback because of a decreased supply of filtered chloride to the macula densa, but RAAS activation to increase reabsorption of filtered solutes (chloride, sodium, or both) would be ineffective because the supply of these solutes in itself is reduced in the urinary tubules in HF patients with no increase in the serum chloride concentration. Ineffective reabsorption of filtered chloride and sodium despite enhanced RAAS activation, probably also including activation of the sympathetic nervous system,19 contributes to the deficits in the intravascular accumulation of serum chloride and the resultant loss of plasma volume. Thus, this subtype of worsening of HF would have a more serious clinical presentation3, 31, 32 with vascular contraction, extravasation of vascular fluid into the interstitial space, enhanced peripheral arterial resistance, and worsening of organ perfusion, as is partly reported here. A substantial part of the clinical features of this type of worsening of HF would overlap with that of the HF patients with overhydrated hyponatraemia.33, 34, 35
The applicability of this proposed hypothesis, the ‘chloride theory’, to HF pathophysiology is well supported by the fact that ‘maintenance of arterial circulatory integrity’ as the conceptual core of the established ‘arterial under‐filling theory’ for HF pathophysiology3, 4 depends deeply on the chloride itself because this electrolyte holds the central role for maintaining the arterial circulatory integrity.9, 22, 23, 24 Additionally, interactions between dynamic changes in the serum chloride concentration and haemodynamics are demonstrated through the changes in plasma volume as shown in Figure 3 (blue arrows). Namely, changes in plasma volume affect venous return to the heart and changes in cardiac output by the Frank–Starling mechanism,36, 37 but the failing heart may have a maladaptive response to the altered plasma volume owing to limited preload reserve and afterload mismatch.37, 38, 39, 40 Importantly, a recent study by Grodin et al.41 reported strong positive relationship between the cardiac index and the serum chloride concentration in 436 patients with advanced chronic HF, confirming the link between changes in the serum chloride concentration and haemodynamics (Figure 3 , red arrows).
Figure 3.
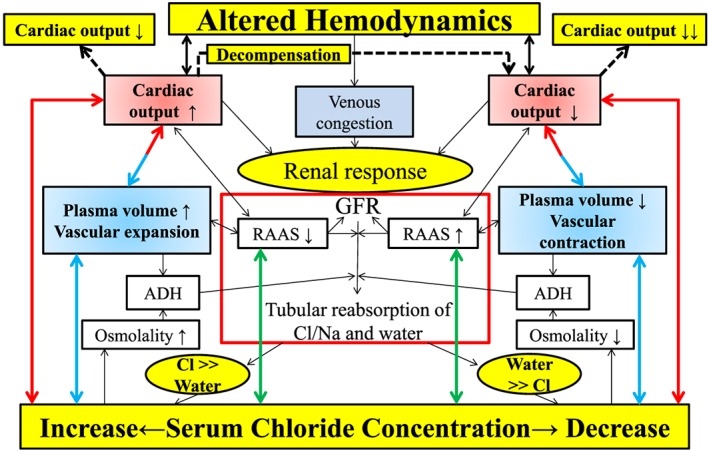
Interactions between changes in the RAAS activity (green arrows), plasma volume (blue arrows), and haemodynamics (red arrows) through changes in the serum chloride concentration in heart failure pathophysiology. ADH, antidiuretic hormone; Cl, chloride; GFR, glomerular filtration rate; RAAS, renin–angiotensin–aldosterone system.
As summarized in Figure 3 , changes in the serum chloride concentration are related to changes in plasma volume (blue arrows), RAAS activation in the kidney (green arrows), and alterations of the haemodynamic status (red arrows) in each case. Thus, the ‘chloride theory’ proposed here incorporates qualitatively different but important main pathways of HF pathophysiology, including biochemical, neurohormonal, and haemodynamic pathways, solely via the chloride electrolyte.
Pathophysiologic differences in HF subtypes stratified by changes in serum sodium vs. chloride concentration
The present study characterized and proposed two distinct types of worsening chronic HF progression. In addition to my recent observations, the proposed HF pathophysiology defined by the dynamics of serum chloride reported here would be rational when considering the central role of chloride in the RAAS and for the maintenance of aortic filling. How, then, can we expect the differences in the clinical pictures and blood examination results when patients with worsening HF are stratified by changes in serum sodium or chloride concentration? The change in serum chloride under worsening HF in the present HF patients correlated moderately with the change in serum sodium (r = 0.762, P < 0.0001; see Supporting Information, Figure S1 ), but discordant movement of chloride and sodium was observed in as many as 9 of 47 patients (19% of study patients); two patients showed increased sodium and decreased chloride, and seven patients showed decreased sodium and increased chloride. Accordingly, stratification of worsening HF patients in the present study (n = 47) into two groups based on changes in serum sodium (26 patients with and 21 without increased serum sodium concentration) or serum chloride (31 patients with and 16 without increased serum chloride concentration) revealed some intra‐group or inter‐group differences in the clinical picture and laboratory test results, such as differences in body weight and pulmonary crackles at baseline (intra‐group; see Supporting Information, Table S2 ) and change in the serum potassium concentration upon worsening HF (inter‐group; see Supporting Information, Table S3 ). The number of subjects in the present study was too small, however, to draw conclusions regarding the pathophysiologic differences in HF patients stratified by the dynamics of each type of electrolyte. Further studies with a large population of HF patients are needed to resolve this problem.
Future perspectives
Adding to the present observations, my recent study demonstrated a novel finding of a positive and independent association between vascular volume and the serum chloride concentration after adequate decongestive therapy for worsening HF,42 indicating that the proposed ‘chloride theory’ hypothesis for worsening HF reported here could be analogically applicable to HF pathophysiology during the resolution of worsening HF after diuretic therapy. Although the targeting level of the serum chloride concentration is not yet clear, modulation of the serum chloride concentration could become an attractive therapeutic target for HF treatment, such as for reducing serum chloride concentrations using conventional diuretics43 and for enhancing serum chloride concentration using a V2 receptor antagonist5, 20, 27 and hyperosmotic saline infusion.44 Our recent case report45 demonstrated the importance of modulating serum chloride concentration in an HF patient undergoing HF therapy using a vasopressin antagonist. To date, the pathophysiologic role of chloride in HF has been ignored because of the long‐term lack of awareness of its role in HF status,6 except for several very recent studies.7, 8, 41, 42, 46 but it is now clearly time to explore the clinical significance, features, and pathophysiologic roles of this electrolyte in HF syndrome.
Study limitations
First, this study is a cross‐sectional observational study and should be considered to be hypothesis generating and to have some limitations. Second, this study was a small‐sized retrospective observational study with a selection bias due to data availability. Thus, a study including a larger number of HF patients is needed to better understand the role of chloride in the pathophysiology of HF patients. Finally, the pathophysiologic role of chloride in relation to the RAAS and the ADH axis was not clarified in the present study, and thus, additional studies should be performed.
Conclusions
My findings suggest a new clinical entity of worsening HF status, that is, HF with increased vs. non‐increased serum chloride concentration (‘chloride theory’). Differential activation of the RAAS and ADH systems might lead to an imbalance in the chloride‐to‐water ratio, and thus different pathophysiologic and clinical signs of worsening HF.
Conflict of interest
None declared.
Supporting information
Figure S1. Relationship of the changes between sodium (Na) and chloride (Cl) from stability to worsening of HF.
Table S1. Diagnosis and precipitating factors of worsening HF status in 47 HF patients.
Table S2. Comparison of symptoms and physical signs between increased vs. non‐increased serum chloride or sodium concentration.
Table S3. Comparison of changes in blood examination between groups of increased vs. non‐increased serum chloride or sodium concentration from clinical stability to worsening of HF.
Kataoka, H. (2017) Proposal for heart failure progression based on the ‘chloride theory’: worsening heart failure with increased vs. non‐increased serum chloride concentration. ESC Heart Failure, 4: 623–631. doi: 10.1002/ehf2.12191.
References
- 1. Cody RJ, Covit AB, Schaer GL, Laragh JH, Sealey JE, Feldschuh J. Sodium and water balance in chronic congestive heart failure. J Clin Invest 1986; 77: 1441–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Volpe M, Tritto C, DeLuca N, Rubattu S, Rao MAE, Lamenza F, Mirante A, Enea I, Rendina V, Mele AF, Trimarco B, Condorelli M. Abnormalities of sodium handling and of cardiovascular adaptations during high salt diet in patients with mild heart failure. Circulation 1993; 88(part 1): 1620–1627. [DOI] [PubMed] [Google Scholar]
- 3. Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med 1999; 341: 577–585. [DOI] [PubMed] [Google Scholar]
- 4. Sica DA. Sodium and water retention in heart failure and diuretic therapy: basic mechanisms. Cleve Clin J Med 2006; 73(suppl. 2): S2–S7. [DOI] [PubMed] [Google Scholar]
- 5. Goldsmith SR, Gheorghiade M. Vasopressin antagonism in heart failure. J Am Coll Cardiol 2005; 46: 1785–1791. [DOI] [PubMed] [Google Scholar]
- 6. O'Connor CM, Ahmad T. The role of sodium and chloride in heart failure: does it take two to tango? J Am Coll Cardiol 2015; 66: 667–669. [DOI] [PubMed] [Google Scholar]
- 7. Kataoka H. Vascular expansion during worsening of heart failure: effects on clinical features and its determinants. Int J Cardiol 2017; 230: 556–561. [DOI] [PubMed] [Google Scholar]
- 8. Kataoka H. Serial changes in red blood cell volume during transition of heart failure status: a reflection of cellular hydration status? (Abstract). Eur Heart J 2015; 36(suppl. 1): 1002–1003. [DOI] [PubMed] [Google Scholar]
- 9. Kotchen TA, Luke RG, Ott CE, Galla JH, Whitescarver S. Effect of chloride on renin and blood pressure responses to sodium chloride. Ann Intern Med 1983; 98: 817–822. [DOI] [PubMed] [Google Scholar]
- 10. Lapointe J‐Y, Bell PD, Cardinal J. Direct evidence for apical Na+ : 2Cl− : K+ cotransport in macula densa cells. Am J Physiol 1990; 258: F1466–F1469. [DOI] [PubMed] [Google Scholar]
- 11. Lorenz JN, Weihprecht H, Schnermann J, Skøtt O, Briggs JP. Renin release from isolated juxtaglomerular apparatus depends on macula densa chloride transport. Am J Physiol 1991; 260: F486–F493. [DOI] [PubMed] [Google Scholar]
- 12. Schnermann J. Juxtaglomerular cell complex in the regulation of renal salt excretion. Am J Physiol 1998; 274: R263–R279. [DOI] [PubMed] [Google Scholar]
- 13. Kataoka H. Detection of preclinical body fluid retention in established heart failure patients during follow‐up by a digital weight scale incorporating a bioelectrical impedance analyzer. Congest Heart Fail 2012; 18: 37–42. [DOI] [PubMed] [Google Scholar]
- 14. Kataoka H. Clinical significance of bilateral leg edema and added value of monitoring weight gain during follow‐up of patients with established heart failure. ESC Heart Fail 2015; 2: 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kataoka H, Takada S. The role of thoracic ultrasonography for evaluation of patients with decompensated chronic heart failure. J Am Coll Cardiol 2000; 35: 1638–1646. [DOI] [PubMed] [Google Scholar]
- 16. Kataoka H. A new monitoring method for the estimation of body fluid status by digital weight scale incorporating bioelectrical impedance analyzer in definite heart failure patients. J Card Fail 2009; 15: 410–418. [DOI] [PubMed] [Google Scholar]
- 17. Kataoka H. Relation of body fluid status to b‐type natriuretic peptide levels in patients with chronic heart failure during long‐term follow‐up. Clin Cardiol 2006; 29: 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalra PR, Anagnostopoulos C, Bolger AP, Coats AJS, Anker SD. The regulation and measurement of plasma volume in heart failure. J Am Coll Cardiol 2002; 39: 1901–1908. [DOI] [PubMed] [Google Scholar]
- 19. Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure. J Am Coll Cardiol 2009; 54: 1747–1762. [DOI] [PubMed] [Google Scholar]
- 20. Lanfear DE, Sabbah HN, Goldsmith SR, Greene SJ, Ambrosy AP, Fought AJ, Kwasny MJ, Swedberg K, Yancy CW, Konstam MA, Maggioni AP, Zannad F, Gheorghiade M, on behalf of the EVEREST trial investigators . Association of arginine vasopressin levels with outcomes and the effect of V2 blockade in patients hospitalized for heart failure with reduced ejection fraction: insights from the EVEREST trial. Circ Heart Fail 2013; 6: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gupta DK, Wang TJ. Natriuretic peptides and cardiometabolic health. Circ J 2015; 79: 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamlyn JM, Blaustein MP. Sodium chloride, extracellular fluid volume, and blood pressure regulation. Am J Physiol 1986; 251: F563–F575. [DOI] [PubMed] [Google Scholar]
- 23. Shore AC, Markandu ND, MacGregor GA. A randomized crossover study to compare the blood pressure response to sodium loading with and without chloride in patients with essential hypertension. J Hypertension 1988; 6: 613–617. [DOI] [PubMed] [Google Scholar]
- 24. Boegehold MA, Kotchen TA. Importance of dietary chloride for salt sensitivity of blood pressure. Hypertension 1991; 17(suppl. I): I‐158–I‐161. [DOI] [PubMed] [Google Scholar]
- 25. Figueras J, Weil MH. Blood volume prior to and following treatment of acute cardiogenic pulmonary edema. Circulation 1978; 57: 349–355. [DOI] [PubMed] [Google Scholar]
- 26. Kreimeier U. Pathophysiology of fluid imbalance. Crit Care 2000; 4(suppl. 2): S3–S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goldsmith SR, Bart BA, Burnett J. Decongestive therapy and renal function in acute heart failure: time for a new approach? Circ Heart Fail 2014; 7: 531–535. [DOI] [PubMed] [Google Scholar]
- 28. Noddeland H, Riisnes SM, Fadnes HO. Interstitial fluid colloid osmotic and hydrostatic pressures in subcutaneous tissue of patients with nephrotic syndrome. Scand J Clin Lab Invest 1982; 42: 139–146. [PubMed] [Google Scholar]
- 29. Davies SW, Bailey J, Keegan J, Balcon R, Rudd RM, Lipkin DP. Reduced pulmonary microvascular permeability in severe chronic left heart failure. Am Heart J 1992; 124: 137–142. [DOI] [PubMed] [Google Scholar]
- 30. Onwuanyi A, Taylor M. Acute decompensated heart failure: pathophysiology and treatment. Am J Cardiol 2007; 99(suppl. D): 25D–30D. [DOI] [PubMed] [Google Scholar]
- 31. Levine TB, Francis GS, Goldsmith SR, Simon AB, Cohn JN. Activity of the sympathetic nervous system and renin–angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol 1982; 49: 1659–1666. [DOI] [PubMed] [Google Scholar]
- 32. Benedict CR, Johnstone DE, Weiner DH, Bourassa MG, Bittner V, Kay R, Kirlin P, Greenberg B, Kohn RM, Nicklas JM, McIntyre K, Quinones MA, Yusuf S, for the SOLVD Investigators . Relation of neurohumoral activation to clinical variables and degree of ventricular dysfunction: a report from the registry of Studies of Left Ventricular Dysfunction. J Am Coll Cardiol 1994; 23: 1410–1420. [DOI] [PubMed] [Google Scholar]
- 33. Sica DA. Hyponatremia and heart failure: pathophysiology and implications. Cong Heart Fail 2005; 11: 274–277. [DOI] [PubMed] [Google Scholar]
- 34. Gheorghiade M, Rossi JS, Cotts W, Shin DD, Hellkamp AS, Piña IL, Fonarow GC, DeMarco T, Pauly DF, Rogers J, DiSalvo TG, Butler J, Hare JM, Francis GS, Stough WG, O'Connor CM. Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE trial. Arch Intern Med 2007; 167: 1998–2005. [DOI] [PubMed] [Google Scholar]
- 35. Ghali JK, Tam SW. The critical link of hypervolemia and hyponatremia in heart failure and the potential role of arginine vasopressin antagonists. J Card Fail 2010; 16: 419–431. [DOI] [PubMed] [Google Scholar]
- 36. Guyton AC. Determination of cardiac output by equating venous return curves with cardiac response curves. Physiol Review 1955; 35: 123–129. [DOI] [PubMed] [Google Scholar]
- 37. Henderson WR, Griesdale DEG, Walley KR, Sheel AW. Clinical review: Guyton—the role of mean circulatory filling pressure and right atrial pressure in controlling cardiac output. Crit Care 2010; 14: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Braunwald E. Heart failure. J Am Coll Cardiol HF 2013; 1: 1–20. [DOI] [PubMed] [Google Scholar]
- 39. Mangano DT, van Dyke DC, Ellis RJ. The effect of increasing preload on ventricular output and ejection in man: limitations of the Frank–Starling mechanism. Circulation 1980; 62: 535–541. [DOI] [PubMed] [Google Scholar]
- 40. Ross J Jr. Cardiac function and myocardial contractility: a perspective. J Am Coll Cardiol 1983; 1: 52–62. [DOI] [PubMed] [Google Scholar]
- 41. Grodin JL, Mullens W, Dupont M, Taylor D, McKie P, Starling R, Testani J, Tang WH. Hemodynamic determinants of serum chloride in ambulatory patients with advanced heart failure. (Abstract) J Am Coll Cardiol 2016; 67 (suppl. 5):1322. [Google Scholar]
- 42. Kataoka H. Determinants of changes in plasma volume after decongestion therapy for worsening heart failure. (Abstract) Eur Heart J 2016; 37 (Abstract Supplement): 915. [DOI] [PubMed] [Google Scholar]
- 43. Jentzer JC, DeWald TA, Hernandez AF. Combination of loop diuretics with thiazide‐type diuretics in heart failure. J Am Coll Cardiol 2010; 56: 1527–1534. [DOI] [PubMed] [Google Scholar]
- 44. Hirotani S, Masuyama T. When to increase or reduce sodium loading in the management of fluid volume status during acute decompensated heart failure. ESC Heart Fail 2014; 1: 75–81. [DOI] [PubMed] [Google Scholar]
- 45. Kataoka H, Yamasaki Y. Strategy for monitoring decompensated heart failure treated by an oral vasopressin antagonist with special reference to the role of serum chloride: a case report. J Card Cases 2016; 14: 185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grodin JL, Simon J, Hachamovitch R, Wu Y, Jackson G, Halkar M, Starling RC, Testani JM, Tang WHW. Prognostic role of serum chloride levels in acute decompensated heart failure. J Am Coll Cardiol 2015; 66: 659–666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Relationship of the changes between sodium (Na) and chloride (Cl) from stability to worsening of HF.
Table S1. Diagnosis and precipitating factors of worsening HF status in 47 HF patients.
Table S2. Comparison of symptoms and physical signs between increased vs. non‐increased serum chloride or sodium concentration.
Table S3. Comparison of changes in blood examination between groups of increased vs. non‐increased serum chloride or sodium concentration from clinical stability to worsening of HF.