

Abstract
Chondroitin sulfates are the glycosaminoglycan chains of proteoglycans critical in the normal development and pathophysiology of all animals. Chondroitinase ACII, a polysaccharide lyase originally isolated from Arthrobacter aurescens IAM 110 65, which is widely used in the analysis and study of chondroitin structure, is no longer commercially available. The aim of the current study was to prepare recombinant versions of this critical enzyme for the glycobiology research community. Two versions of recombinant chondroitinase ACII were prepared in Escherichia coli, and their activity, stability, specificity and action pattern were examined, along with a non-recombinant version secreted by an Arthrobacter strain. The recombinant enzymes were similar to the enzyme obtained from Arthrobacter for all examined properties, except for some subtle specificity differences towards uncommon chondroitin sulfate substrates. These differences are believed to be due to either post-translational modification of the Arthrobacter-secreted enzyme or other subtle structural differences between the recombinant and natural enzymes. The secreted chondroitinase can serve as a suitable replacement for the original enzyme that is currently unavailable, while the recombinant ones can be applied generally in the structural determination of most standard chondroitin sulfates.
Keywords: chondroitinase, lyase, chondroitin sulfate, recombinant expression, specificity
Graphical Abstract
Chondroitinase ACII, a polysaccharide lyase originally isolated from Arthrobacter aurescens IAM 110 65, is widely used in the analysis and study of chondroitin structure, but is no longer commercially available from its sole supplier, Seikagaku Corporation. In this study we prepared two versions of recombinant chondroitinase ACII in Escherichia coli, along with a secreted, non-recombinant version from an Arthrobacter strain, and examined their activity, stability, specificity and action pattern. Based on its similarity in properties and characteristics, we believe that our Arthrobacter-secreted enzyme can serve as a suitable replacement for the original inaccessible enzyme, while the recombinant E. coli expressed versions are suitable for use in the structural determination of most standard chondroitin sulfates.
1 Introduction
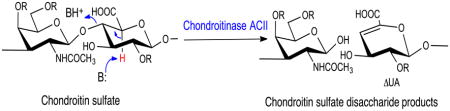
Chondroitinase AC II (chondroitin lyase ACII, EC 4.2.2.5) from Arthrobacter aurescens IAM 110 65 was first purified and characterized in 1975 [1]. It is an eliminase, which cleaves the (1→4) glycosidic linkage between N-acetyl-β-D-galactosamine (GalNAc) and β-D-glucuronic acid (GlcA) residues in chondroitin, chondroitin sulfate-A and chondroitin sulfate-C (CS-A and CS-C), yielding oligosaccharides product, mainly unsaturated disaccharides ΔDi-0S, ΔDi-4S, and ΔDi-6S, respectively [2,3] (Figure 1). While chondroitinase ACII is also active on hyaluronic acid, cleaving the (1→4) glycosidic linkage between N-acetylglucosamine (β-D-GlcNAc) and GlcA residues, it shows no activity towards chondroitin sulfate-B (CS-B or dermatan sulfate, →3) GalNAc(1→4) α-L-iduronic acid (IdoA) (1→). Chondroitinase ACII displays an exolytic action pattern in which the enzyme cleaves one disaccharide at a time from the non-reducing end of the polysaccharide chain, while a second enzyme, chondroitinase ACI from Flavobacterium heparinum (chondroitin lyase ACI), acts in a random endolytic action pattern, cleaving the polysaccharide substrate at randomly selected β-D-GlcNAc (1→4) GlcA linkages [4,5]. The elimination reaction catalyzed by the chondroitinase AC enzymes generates an unsaturated uronic acid residue (ΔUA 4-deoxy-α-L-threo-hexenopyranosyluronic acid) (Figure 1), which can be detected by UV spectroscopy at 232 nm, while leaving the chemical structure of glucosamine reducing end unaltered [2,3,6].
Figure 1.

The action of chondroitinase ACII on GAGs. CS and HA are substrates for chondroitinase ACII which acts to abstract the acidic C-5 proton (shown in red) of GlcA α to the carboxyl group. This results in cleavage of the adjacent glycosidic linkage and the formation of a ΔUA containing product and a second disaccharide product with a reducing end GalNAc residue (or GlcNAc residue in the case of HA) attached to an uronic acid residue. Since chondroitinase ACII is an exolytic enzyme it continues to cut one disaccharide at a time from the non-reducing terminus of the GAG chain. DS, containing an IdoA residue, incorrectly positions its acidic C-5 proton (shown in red) and, hence is not a substrate for chondroitinase ACII. In CS-A the R at position 4 is commonly SO3− and the R at positions 2 and 6 is H. In CS-C the R at position 4 is commonly SO3− and the R at positions 2 and 6 is H. In CS-D the R at positions 2 and 6 are commonly SO3− and the R at positions 4 is H. In CS-E the R at position 4 and 6 are commonly SO3− and the R at positions 2 is H. In DS the R at position 4 is commonly SO3− and the R at positions 2 and 6 is H.
Chondroitinase AC II is widely used for the analysis of CSs. Its unique enzymatic specificity causes it to act at the non-reducing end of most CSs to release disaccharide products, while leaving dermatan sulfate (DS) intact. Thus, it is useful in distinguishing CS from DS and can aid in the study of structural and sequence motifs of GAGs [7]. Seikagaku Corporation in Japan, the sole supplier of chondroitinase ACII, discontinued its production in 2011, and subsequently the glycobiology research community has had very limited access to this enzyme for the in-depth study and analysis of CS. Recently, an ortholog from Arthrobacter sp. GAG sharing moderate sequence similarity (65%) to the original A. aurescens IAM 110 65 chondroitinase ACII was characterized after heterologous expression in E. coli, and it was found to share similar enzymatic properties with the original enzyme when tested against a narrow panel of non-sulfated chondroitin oligosaccharides [8]. However, there remains an unmet need for the original enzyme that has well-defined substrate specificity and that is already established as a reagent in many labs and even in pharmaceutical quality control pipelines. Thus, in this study, we expressed and characterized chondroitinase ACII as a recombinant enzyme in Escherichia coli. Using as a template the protein sequence derived from the solved high-resolution crystal structure of the Seikagaku chondroitinase ACII [9], we located a nearly identical chondroitinase ACII encoded in the genome of Arthrobacter sp. 161MFSha2.1. This sequence was used to prepare two recombinant enzymes in E. coli. Similar activity was observed between the A. sp. 161MFSha2.1 wild-type sequence deduced from the crystal structure, and a point mutant reverting the enzyme to what we believe is the original A. aurescens sequence, by changing a single dissimilar side chain. The wild-type A. sp. 161MFSha2.1 strain was also obtained and utilized to prepare a secreted, non-recombinant version of chondroitinase ACII. The activity, stability, and substrate specificity of natural and recombinant chondroitinase ACII enzymes were compared to the original commercial product.
2 Materials and methods
2.1 Media and Chemicals
Luria-Bertani (LB, Sigma) medium with or without kanamycin (50 μg/mL) was used for the cell growth and transformation screening for the recombinant chondroitinases. LB medium supplemented with 0.2% CS-A or 0.2% CS-C was used for growth of the Arthrobacter strain and secretion of chondroitinase. Super optimal broth with catabolite repression (SOC) was used for cell recovery after heat shock or electroporation during the transformation experiments. Plasmid maintenance and propagation were performed using E. coli DH5α ™ strain (Invitrogen). E. coli BL21 Star™ (DE3) strain (Invitrogen) was used as the production strain with the expression of the plasmid pET28a(+)-tA16ACII. All other nutrients and chemicals for medium preparation were from Sigma Chemical Co. (St. Louis, MO). CS disaccharide standards were purchased from Iduron (Manchester, U.K). Sodium cyanoborohydride, 1,9-dimethylmethylene blue (DMMB), 2-aminoacridone (AMAC), and acetic acid were purchased from Sigma–Aldrich (St. Louis, MO), and methanol (high performance liquid chromatography (HPLC) grade), ammonium acetate (HPLC grade), and dimethyl sulfoxide (DMSO) were purchased from Fisher Scientific (Springfield, NJ).
E. coli expression and purification of the recombinant Proteus vulgaris chondroitin lyase ABC (EC No. 4.2.2.20) was performed in our laboratory as previously described [10]. CS-A (6.3% of ΔDi-0S, 74.2% of ΔDi-4S, 19.5% of ΔDi-6S, 0.3% of ΔDi-diSE) (see Figure S1 in Supporting Information for structures of disaccharides) from whale cartilage, CS-C (1.8% of ΔDi-0S, 13.7% of ΔDi-4S, 70.8% of ΔDi-6S, 2.9% of ΔDi-diSE, 10.8% of ΔDi-diSD) from shark cartilage, CS-E (10.0% of ΔDi-0S, 17.0% of ΔDi-4S, 8.31% of ΔDi-6S, 63.6% of ΔDi-diSE, 1.08% of ΔDi-diSD) from squid cartilage, DS (6.5% of Δdi-0S, 83.2% of ΔDi-4S, 1.2% of ΔDi-6S, 9.1% of ΔDi-diSB) from porcine intestine were purchased from Seikagaku Corp., Tokyo, Japan. The preparations of CS-E from M. chinensis [11] and CS-K from Enteroctopus dofleini (octopus) cartilage [12] were previously described.
2.2 Plasmid construction
The putative chondroitinase ACII from Arthrobacter sp. 161MFSha2.1, hereafter referred to as tA16ACII, was synthesized without its N-terminal signal peptide and cloned into the NdeI and XhoI sites of pET28a(+) (GenScript) in frame with the N-terminal 6x-His tag for purification (the strains and plasmids used in this study are provided in Table S1). The plasmid pET28a(+)-tA16ACII was then transformed into E. coli BL21 Star™ (DE3) by electroporation using a Bio-Rad Gene Pulser Xcell™ transformation system (2 mm cuvettes, 2.5 kV, 25 μF and 200 Ω). Cells were recovered in SOC medium for 50 min and plated on LB agar, supplemented with 50 μg/mL of kanamycin. Primers from IDT were synthesized to perform site-directed mutagenesis on amino acid number 236 of the pET28a(+)-tA16ACII construct, converting an isoleucine codon (ATT) to a threonine codon (ACC). The QuikChange Site-Directed Mutagenesis Kit from Agilent was used according to the manufacturer’s protocol, followed by DpnI digestion to remove the original template before transformation into E. coli DH5α. Restriction digestion and Sanger sequencing through Genewiz verified the final construct. The new construct with the point mutation was transformed into E. coli BL21 Star™ (DE3) and plated as previously described. The enzyme produced from this construct will hereafter be referred to as tA16ACII(I236T).
The primers used for site-directed mutagenesis were:
| Primer name | Primer Sequence (5′ → 3′) |
|---|---|
| AC_Lyase_Mut_F | CGTTCATCCAACACAGCACCACTCCGTACACCGGTTC |
| AC_Lyase_Mut_R | GAACCGGTGTACGGAGTGGTGCTGTGTTGGATGAACG |
2.3 Expression and Purification of enzymes
Single colonies of E. coli BL21 Star™ (DE3) harboring the plasmids for the recombinant tA16ACII and tA16ACII(I236T) constructs were picked from each plate and separately inoculated in 1 L of LB supplemented with 50 μg/mL kanamycin in Pyrex™ Fernbach culture flasks (Corning Life Sciences). The cell cultures were incubated in a rotary air shaker (New Brunswick Scientific Innova 44R) at 37 °C, 220 rpm until the optical density (OD) A600 reached ~0.8–1.0. At this point, gene expression was initiated by inducing with 0.2 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG). The cultures were then incubated for 16–20 h at 22 °C. Cells were harvested by centrifugation at 4°C (5,000 × g for 10 min) and stored at −20 °C until ready for purification. Pelleted E. coli cells were re-suspended in 20 mL of loading buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 25 mM imidazole) followed by sonication, with intermittent cooling on ice. The cell debris was removed by a centrifugation step (16,000 × g for 30 min) at 4 °C. The resulting cell lysate was filtered (0.45 μm) and the supernatant was applied to a Ni Sepharose 6 Fast Flow (GE Health) column. The columns were rinsed with washing buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 25 mM imidazole), and the bound proteins were eluted with elution buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 250 mM imidazole). The imidazole was removed by carrying out a buffer exchange against storage buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 100 mM lactose), after which the proteins were freeze-dried and stored at −80 °C until needed. The purity of both recombinant chondroitinase ACII enzymes was determined using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
A third form of the chondroitinase ACII, hereafter referred to as A16ACII, was produced and purified from Arthrobacter sp. 161MFSha2.1, generously provided by Jeff Dangl (University of North Carolina, Chapel Hill, NC, USA). A colony was picked from an agar plate streaked with cells from A. sp. 161MFSha2.1 and a 5 mL pre-culture was grown in LB medium overnight at 30 °C. One milliliter of this pre-culture was transferred to each of three 500 mL flasks containing 100 mL of LB supplemented with 0.2% CS-A, 100 mL of LB supplemented with 0.2% CS-C, and 100 mL of plain LB, respectively. After shaking for 2 days at 30 °C and 250 rpm, cells were removed by centrifugation at 15,000 × g for 20 min. The supernatant fluid was collected and concentrated to 10 mL by tangential flow filtration using a Vivaflow 200 cassette (Sartorius) with a 10 kDa polyethersulfone (PES) membrane. The concentrated supernatant containing the secreted enzyme was then transferred to a pre-washed 10 kDa spin column (Sigma-Aldrich) and buffer exchanged several times against storage buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 100 mM trehalose), until residual medium was removed. The A16ACII enzyme was then freeze-dried and stored at −80 °C until needed. The purity of the A16ACII enzyme was assessed by SDS-PAGE. Table S2 tabulates differences between all enzymes utilized in this study.
2.4 Activity assays
2.4.1 Initial tA16ACII activity
The activity of the tA16ACII was measured by detecting the increase in ultraviolet absorption at 232 nm, according to a previously reported procedure [13]. Initial activity assays were carried out on tA16ACII in a quartz crystal cuvette. CS-A (50 μL of 20 mg/mL solution) was added to 640 μL of digestion buffer (50 mM ammonium acetate, pH 7.4) and pre-heated to 37 °C. Purified tA16ACII was diluted appropriately and 10 μL of the enzyme was added to the mixture. Within 30 s of adding enzyme the absorbance was measured continuously for approximately 2 min until the reading reached a plateau (Figure 2). All absorbance measurements were done using a Synergy 2 Multi-Mode Reader (BioTek). The enzyme activity was evaluated by the following equation (1):
| (1) |
where 1 U = 1 μmol product formed per minute, ΔA232 is the change in absorbance over the Δtime, Vtotal is the total volume of the solution, Venzyme is the volume of the enzyme added to the solution and ε is the product disaccharide extinction coefficient (5260 M−1 cm−1) [6]. The optimal temperature for effective elimination reactions was also investigated by recording and comparing the enzyme reaction rate at various temperatures (30 °C, 37 °C, 50 °C, and 70 °C). Additionally, to determine the best storage conditions for recovery of enzyme activity, the purified tA16ACII enzyme was buffer exchanged against three different buffers: Tris buffer, Tris buffer with the addition of 100 mM trehalose, and Tris buffer with the addition of 100 mM lactose. The activities of the freeze-dried enzymes were then calculated and compared to those of the original enzymes.
Figure 2.

The activity assays for tA16ACII and CS ABC. CS-A and CS-B (DS) were used as substrates for the same protein concentration of tA16ACII and CS ABC expressed in E. coli. Assay were carried out in a quartz crystal cuvette at 37 °C and each consisted of 50 μL of 20 mg/mL CS-A or CS-B, 640 μL of digestion buffer (50 mM ammonium acetate, pH 7.4), and 10 μL of the same concentration of tA16ACII or CS ABC. Absorbance was measured at 232 nm.
2.4.2 Effect of point mutation on stability and activity of recombinant chondroitinase ACII
Activity assays were carried out on both the tA16ACII and tA16ACII(I236T) enzymes directly after purification to determine the effect of the point mutation on activity and stability. Each assay consisted of 3 μL of purified enzyme normalized to a concentration of ~25 μg/mL, 14 μL of 20 mg/mL CS-A, and 183 μL of digestion buffer (50 mM ammonium acetate, pH 7.4). The CS-A and buffer were mixed well and preheated to 37 °C in a Quartz Microplate (Hellma Analytics) before the enzyme was added. Subsequent activity assays were carried out on both enzymes after leaving them in Tris buffer (with 100 mM lactose) at room temperature for 30 days.
2.4.3 A16ACII activity in LB supplemented with CS-A or CS-C
Activity assays for the A16ACII enzyme secreted from A. sp. 161MFSha2.1 were carried out in a similar fashion, comparing the activity of the enzyme produced from growth in LB only and the enzymes produced from growth in LB supplemented with 0.2% CS-A or 0.2% CS-C.
2.4.4 Chondroitinase ACII activities on standard and uncommon CS substrates
Next, the active A16ACII enzyme was tested alongside tA16ACII, tA16ACII(I236T), and CS ABC on a variety of CS substrates. These activity assays consisted of 187 μL of digestion buffer, 10 μL of 1 mg/mL CS substrate, and 3 μL of ~5 μg/mL enzyme. The seven CS substrates used in these assays were commercially acquired CS-A, DS (CS-B), CS-C, CS-D, and CS-E, along with CS-E from M. chinensis and CS-K from E. dofleini cartilage. Reactions were run overnight at 37°C, allowing them to reach completion before processing for disaccharide analysis. Each reaction consisted of two independent replicates that were pooled for LC-MS analysis.
2.4.5 Enzymatic Digestions
A final set of assays was carried out on the three enzymes to determine their action patterns. CS-A (5 μL at 1 mg/mL) and digestion buffer (93 μL) were added to several PCR strip tubes (VWR) and incubated at 37°C for 5 min. Concentrations of the tA16ACII, tA16ACII(I236T), and A16ACII enzymes were normalized to ~5 μg/mL with digestion buffer and 2 μL of enzyme were added to a new pre-heated reaction tube every 5 min after the first 2 min, so that reactions were running for 0, 2, 5, 10, 15, 20 min, etc., for up to 40 min when a plateau was reached. Enzymatic reactions were terminated by heating for 10 min at 100 °C using a Bio-Rad C1000 Thermal Cycler. The enzymes were removed from the reaction mixtures by centrifugation and the supernatant was freeze-dried for disaccharide analysis.
2.5 Disaccharide analysis using LC-MS
Reaction mixtures for disaccharide analysis were collected in the flow through of 3 kDa spin columns. The filter units were washed twice with 100 μL of distilled water and the filtrates containing the disaccharide products were freeze-dried. The freeze-dried samples containing chondroitin disaccharides or chondroitin disaccharide standards from Iduron (Manchester, U.K.) were added to 10 μL of 0.1 M AMAC solution in acetic acid (AcOH)/DMSO (3:17, v/v) and mixed by vortexing for 5 min. Next, 10 μL of 1 M sodium cyanoborohydride was added to the reaction mixture and incubated at 45 °C for 1 h. After the AMAC-labeling reaction, the samples were centrifuged at 13,000 × g for 10 min and the supernatants were recovered. The AMAC-tagged disaccharide was diluted to different concentrations using 50% (v/v) aqueous DMSO and LC-MS analysis was performed.
Liquid chromatography-mass spectrometry (LC-MS) analyses were performed on an Agilent 1200 LC/MSD instrument (Agilent Technologies, Inc. Wilmington, DE) equipped with a 6300 ion-trap and a binary pump. The column used was a Poroshell 120 EC-C18 column (3.0 × 100 mm, 2.7 μm, Agilent, USA) at 45° C. Eluent A was 50 mM ammonium acetate solution and eluent B was methanol. Solution A and 5% solution B was flown (150 μL/min) through the column for 20 min followed by linear gradients 40% solution B from 20 to 30 min. The column effluent entered the electrospray ionization-MS source for continuous detection by MS. The electrospray interface was set in negative ionization mode with a skimmer potential of −40.0 V, a capillary exit of −40.0 V, and a source temperature of 350° C, to obtain the maximum abundance of the ions in a full-scan spectrum (300–1200 Da). Nitrogen (8 L/min, 40 psi) was used as a drying and nebulizing gas.
Samples for investigating substrate specificity were similarly AMAC labeled, then mass spectrometry analysis was carried out as described in our previous work [14]. The data acquired was analyzed using Thermo Xcalibur software, and disaccharides were quantified by comparing peak areas to those of an external standard.
3 Results
3.1 Expression, purification, and initial activity of recombinant chondroitinases
The tA16ACII and tA16ACII(I236T) enzymes were successfully overexpressed in E. coli in shake flasks. The peptide sequence of the tA16ACII(I236T) construct was presumed to be identical to AaACII from Arthrobacter aurescens IAM 110 65, originally produced by Seikagaku. The calculated molecular weights of the recombinant enzymes were both ~83.3 kDa and theoretical pI values were 11.95, based on the online bioinformatics tool ExPASy. The proteins of interest expressed in E. coli showed a molecular weight based on sodium dodecyl sulfate –polyacrylamide gel electrophoresis (SDS-PAGE) of ~75 kDa (Figure 3). The expressed proteins were primarily found in the soluble fraction and then were purified using a Ni-Sepharose column (Figure 3). The measured molecular weight of chondroitinase ACII enzymes purified from E. coli of 75 kDa is slightly lower than the calculated molecular weight. The final yields of the recombinant chondroitinase ACII enzymes were ~250 mg/L cell culture. The catalytic activities of these enzymes were calculated using equation (1) to be ~107 U/mL with a specific activity of ~4 units/mg protein.
Figure 3.

The SDS-PAGE analysis of chondroitinase ACII enzymes with the ladder labeled L for each gel. Panel A gel result shows the induced and uninduced fractions of the tA16ACII and tA16ACII(I236T) enzymes expressed in E. coli BL21. The theoretical molecular weight was predicted to be 83.5 kDa and in the gel, the approximate molecular weight is ~75 kDa. The lanes are - induced tA16ACII(I236T): 1a, uninduced tA16ACII(I236T): 2a, induced tA16ACII: 3a, uninduced tA16ACII: 4a. Panel B gel result shows the tA16ACII and tA16ACII(I236T) enzymes expressed in E. coli BL21 and purified from Nickel column, with the tA16ACII(I236T) enzyme in the lanes on the left side of the gel and tA16ACII on the right side. The lanes are – 1b & 5b: soluble fraction, 2b & 6b: insoluble fraction, 3b & 7b: flow through, 4b & 8b: first wash. Panel C shows the gel result of the purified recombinant enzymes after buffer exchange in a 10 kDa spin column, with the lanes labeled – 1c: tA16ACII, 2c: tA16ACII(I236T). Panel D gel result shows the A16ACII enzyme (which has a theoretical molecular weight of ~76 kDa), among other proteins in the crude supernatant. The lanes are labeled – 1d: A16ACII grown in LB supplemented with 0.2% CS-A: A and 2d: A16ACII grown in LB supplemented with 0.2% CS-C.
3.2 Effect of temperature and storage buffer on enzymatic activity and stability
Enzyme stability was measured under the conditions where the enzyme was saturated with substrate. The reaction rate catalyzed by the enzyme increased with increasing temperature, 68% from 30 °C to 37 °C, affording an optimal temperature at 37 °C. Over 90% of enzyme activity was lost above 50 °C and recombinant enzyme tA16ACII was completely inactivated at 70 °C (Figure S2). Purified tA16ACII enzyme was buffer exchanged against three different buffers, Tris buffer, Tris buffer containing 100 mM trehalose, and Tris buffer containing 100 mM lactose, to examine optimal enzyme storage conditions, since disaccharides have been shown to provide effective stabilization of proteins during freeze-drying [15]. The enzyme was then freeze-dried at −40 °C at a protein concentration of 0.2–0.3 mg/mL. After testing lactose and trehalose as excipients for freeze-drying, we found that the best storage condition involved buffer exchange in the presence of either lactose or trehalose, followed by freeze-drying. Although both reducing and non-reducing disaccharides are effective for protection during the freeze-drying cycle, trehalose was subsequently selected for use in the storage buffer, since reducing sugars like lactose have the potential to degrade proteins during storage [15]. While nearly 100% of enzymatic activity could be recovered following freeze-drying from these buffer systems and long-term storage (Figures S3 and S4), ~60% of enzymatic activity was lost on long-term storage in the absence of both trehalose and lactose. The Arthrobacter enzyme, A16ACII even showed a slight increase in activity after being rehydrated with deionized water (Figure S4).
3.2 Effect of point mutation on recombinant enzyme activity and stability
Initial activity assays were carried out on tA16ACII and tA16ACII(I236T), under identical conditions with normalized enzyme concentrations, to understand how activity and stability were impacted by the point mutation. Activity assays were subsequently repeated on both enzymes after being left in storage buffer at room temperature (~23 °C) for 30 days. When compared to initial results, both enzymes displayed similar activity and stability, maintaining an average activity of ~30 U/mL (Figure S8) as calculated using equation (1). Thus, this point mutation had no observable impact on stability and activity against CS-A substrate.
3.3 Effect of CS supplementation on Arthrobacter chondroitinase secretion
A. sp. 161MFSha2.1 secretes a substrate-inducible chondroitinase ACII (A16ACII), having a molecular weight of ~76 kDa (Figure 3), into the culture broth. Initial experiments relied on media supplemented with CS-C as suggested in previous papers describing protocols for chondroitinase ACII production by Arthrobacter [1]. CS-A was ultimately used as an alternative supplement as it was more available to us as a reagent than CS-C. The enzymatic activity observed in plain LB medium and LB medium supplemented with 0.2% CS-A or CS-C is shown in Figure S3. The absorbance profiles for the enzyme produced in unsupplemented LB medium were identical to those for the assay where DS was used as the substrate.
3.4 Difference in action of recombinant and secreted chondroitinases on various CS substrates
Substrate specificity of the chondroitinase ACII enzymes was determined by analyzing the disaccharides produced after digesting several CS substrates with each enzyme. The percentage composition of various disaccharide products formed through the action of each chondroitinase ACII on different CS substrates is shown in Figure 4. The data clearly illustrates that tA16ACII and tA16ACII(I236T) exhibit comparable substrate specificities, producing similar product profiles for all seven substrates. In contrast, while the A16ACII enzyme matched the product profiles of tA16ACII and tA16ACII(I236T) acting on CS- A, B, C, D and E substrates, the more unusual CS substrates—CS-E from M. chinensis and CS-K from octopus cartilage—showed increased product complexity affording additional 2S, 2S4S, and 2S6S disaccharide products. Although each CS contains a primary disaccharide unit consistent with its structure, all CS types are heterogeneous polysaccharides, containing multiple types of disaccharide units and thus affording multiple disaccharide products on digestion. The major disaccharide unit contained in each CS type corresponded to one of the more dominant disaccharides in the observed digestion products (Figure 4).
Figure 4.

The enzymes discussed in this study are 1: tA16ACII(I236T), 2: tA16ACII, 3: A16ACII, and 4: CS ABC) were tested against a panel of various CS substrates. Each bar chart shows the percent composition of disaccharide on the x-axis while the y-axis illustrates which enzyme is involved in the reaction. Each reaction consisted of two independent replicates that were pooled for LC-MS disaccharide analysis. Above each bar chart is the characteristic repeating disaccharide structure found in each substrate, comprising anywhere from 10% to 90% of each structure. The structure of CS-E from M. chinensis is based on the work of Higashi et al. [11]. The structure of octopus CS-K is currently unavailable.
3.5 Exolytic action pattern of chondroitinase ACII enzymes
Enzymes were sufficiently diluted so that each assay contained the same concentration of enzyme and the reaction rate was slow enough to allow for the reaction to be quenched by thermal inactivation of chondroitinase ACII at various time points, to recover aliquots for disaccharide analysis. The change in the mass of disaccharide released (ng) per mg of enzyme in the reaction, versus time is plotted in Figures 5 and is also illustrated in Figure S5. Each reaction was run induplicate and the average disaccharide content at each time point was plotted on the y-axis. The r2 value of each line indicates that the data correlated to a linear fit, thus, corresponding to a characteristic of an exolytic action pattern, where disaccharide is continuously formed throughout the entire reaction. The linear equations derived from these plots were: y = 0.0286x+ 0.0028 (for tA16ACII), y = 0.029x – 0.0027 (for tA16ACII(I236T)), and y = 0.0314x + 0.0353 (for A16ACII). The major digestion product of these enzymes was the monosulfated unsaturated disaccharide.
Figure 5.

This figure graphically shows the change in mass of disaccharide released (ng) per mg of enzyme in reactions consisting of 5 μL of 1 mg/mL CS-A, 93 μL of digestion buffer, and 2 μL of ~5 μg/mL of tA16ACII, tA16ACII(I236T), or A16ACII enzyme. The reactions were thermally inactivated and processed for disaccharide analysis after for 0, 2, 5, 10, 15, 20 min, etc., for up to 40 min, as shown on the x-axis labels. Each reaction was run in duplicate and the average disaccharide content at each time point was plotted on the y-axis. The linear equations derived from these plots are: y = 0.0286x +0.0028 (for tA16ACII); y = 0.029x – 0.0027 (for tA16ACII(I236T)); y = 0.0314 + 0.0353 (for A16ACII).
4 Discussion
4.1 Identification and synthesis of A. aurescens CS AC II ortholog
The gene sequence of the chondroitin AC lyase II (referred to as AaACII in this paper), widely available from 1980s to 2011 and produced from a commercial strain of A. aurescens by Seikagaku Corporation in Japan, has not been reported [1]. Fortunately, a high resolution X-ray crystal structure of the commercial enzyme had been published, from which a partial sequence could be deduced [9]. BLASTP 2.3.1 [16,17] was utilized to search the NCBI non-redundant protein sequences (nr) database for sequences similar to the deduced sequence of AaACII [9] and a putative ortholog possessing the highest similarity was identified in Arthrobacter sp. 161MFSha2.1 (RefSeq WP_018778839.1, annotated as a hypothetical protein), with 747/757 amino acid identities (98%) and 755/757 positives (99%). Notably, the putative chondroitinase AC II possessed 33 additional N-terminal amino acids, which were predicted by SignalP 4.1 [18] to be a Gram-positive signal peptide. This finding is consistent with the original isolation of AaACII as a secreted enzyme in the supernatant of A. aurescens culture medium [1]. Furthermore, the first amino acid following the predicted cleavage site (between residues 33 and 34) was the same as that identified in the original AaACII crystal structure. The Clustal Omega 1.2.1 amino acid alignment [19,20,21] of AaACII and the putative ortholog from Arthrobacter sp. 161MFSha2.1 (A16ACII) is presented in Figure S6. Interestingly, most of the unconserved residues between the two sequences are asparagine/aspartic acid or glutamine/glutamic acid, suggesting that the residues were miscalled during side-chain assignment from crystal structure electron density interpretation, and only one dissimilar side-chain (threonine or isoleucine at residue 236 in the signal peptide-truncated protein) exists between the two orthologs.
The nucleotide sequence of the putative ortholog was obtained using TBLASTN 2.3.1 [17] to search the NCBI refseq_genomic database for the previously identified protein sequence as a translated nucleotide sequence, constrained by the organism to Arthrobacter sp. 161MFSha2.1 (taxid:1151118). A perfect match was found in Arthrobacter sp. 161MFSha2.1 genomic scaffold C567DRAFT_scaffold00008.8, whole genome shotgun sequence (NCBI Reference Sequence NZ_KB895790.1; locus tag C567_RS22185). The nucleotide sequence, excluding the 33 N-terminal residues forming the predicted signal peptide, was synthesized (express cloning option) into NdeI and XhoI restriction sites of pET-28a(+) by GenScript (Piscataway, NJ), creating an N-terminal 6xHis-tag fusion for expression in E. coli as shown in Figure S7. The enzyme produced from this construct (tA16ACII) showed similar specificity to the AaACII enzyme manufactured by Seikagaku, as illustrated in Figure 2, with a higher activity towards CS-A compared to the same concentration of chondroitinase ABC. For DS, tA16ACII showed significantly lower activity than chondroitinase ABC, closely corresponding to the enzyme characterization description of chondroitinase ACII [1]. The slight increase in absorbance at 232 nm observed for tA16ACII may be due to impurities of glucuronic acid in the DS substrate.
4.2 Comparison of activity and stability of recombinant chondroitinases
We next turned our attention to a wide array of Arthrobacter chondroitinase ACII enzymes in order to make improvements to the tA16ACII enzyme. The single residue mutation between tA16ACII (I236) and the AaACII crystal structure sequence (T236, signal peptide-truncated) is within close proximity to the enzyme’s putative active site, and the orthologs with highest overall similarity to both enzymes maintained a threonine at this position. Thus, site-directed mutagenesis was used to revert this divergent side-chain in recombinant tA16ACII from isoleucine to threonine at residue 236 of the signal peptide-truncated protein, thereby producing the recombinant enzyme tA16ACII(I236T). This peptide sequence was assumed to be identical to the natural secreted Arthrobacter aurescens enzyme (AaACII) manufactured by Seikagaku. The point mutation did not cause any significant change in the physical properties of tA16ACII, as both versions of the recombinant enzyme possessed similar activity and stability (Figures S4 and S8). Additionally, Figure 4 reveals that both versions of the recombinant enzyme displayed almost identical substrate specificities and disaccharide product profiles when tested on several types of CS substrates.
4.3 Unique activity and specificity of Arthrobacter-expressed chondroitinase ACII
We next adjusted our approach, searching for and locating a similar Arthrobacter strain from which the tA16ACII sequence was based. This would permit the direct isolation of a non-recombinant version of A16ACII from Arthrobacter (as it might have been prepared by Seikagaku). This enzyme, secreted by Arthrobacter sp. 161MFSha2.1, had comparable activity and stability to the two recombinant versions of the enzyme from E. coli (Figures S4). However, when we screened the secreted enzyme A16ACII against a battery of substrates we found subtle differences between it and the recombinant enzymes, particularly among highly unusually CS substrates, CS-E from M. chinensis and CS-K from octopus (Figure 4). On standard CS substrates (CS-A, CS-B, CS-C, CS-D, and CS-E), the recombinant enzymes (tA16ACII and tA16ACII(I236T)) showed comparable behavior and product profiles to the enzyme secreted from Arthobacter (A16ACII). However, the Arthobacter-produced A16ACII enzyme afforded a more complex product mixture than the recombinant ones when applied to CS-E from M. chinensis and CS-K from octopus substrates, even though Arthobacter A16ACII possesses the same peptide sequence as the recombinant enzyme tA16ACII. To further understand this small but enigmatic difference between recombinant tA16ACII and Arthrobacter-isolated A16ACII, we hypothesized that Arthrobacter-expressed AaACII might also show additional product complexity that is absent from recombinant tAaACII. Thus, a small amount of available authentic Seikagaku AaACII product was tested and showed similar product complexity to A16ACII on CS-E from M. chinensis and CS-K from octopus [11,12]. These data support our conclusion that A16ACII from Arthobacter is a closer match to the original commercial Seikagaku AaACII enzyme than either of the recombinant chondroitinases produced in E. coli, underscoring that the expression system and production host might lead to subtle differences in evaluated enzyme properties, while the I236T point mutation does not (since A16ACII and recombinant tA16ACII peptide sequences are identical). This suggests that the Arthrobacter host alters the activity of the enzyme, possibly through mechanisms of indeterminate post-translational modification, glycosylation, and/or improved protein folding, endowing it with the ability to break down otherwise resistant domains. This explanation is further supported as Arthrobacter is historically known to glycosylate its proteins and has been shown to produce three distinct isoforms of chondroitinase ACII with identical peptide composition but different carbohydrate content [22]. Further post-translational modification and alteration at the peptide level, including side-chain modification, cleavage during secretion, or capping of the N-terminus, might also account for the difference in activity. Finally, the presence of the His-6x tag on the N-terminus of the E. coli BL21 expressed enzymes could potentially have an effect on the activity of the recombinant enzyme.
4.4 Activities of chondroitinase ACII enzymes and chondroitinase ABC on various substrates
The disaccharides produced from the action of tA16ACII, tA16ACII(I236T), A16ACII, and CS ABC on commercially available CS, including CS-A (50–80% of A-type unit), CS-C (50–70% of C-type unit), CS-D (20–40% of D-type unit) and CS-E (63.6% of E-type unit) are presented in Figure 4. Seikugaku AaACII was not included in this and further studies due to limited availability of the reagent and inability to obtain Arthrobacter aurescens IAM 110 65 for in-house preparation. The repeat region of CS chains is a repeating disaccharide composed of [-4)GlcAβ(1–3)GalNAcβ(1-]n (which may be sulfated on the C4 and/or C6 of GalNAc and C2 of GlcA) [23]. Structure, sulfation pattern, and composition of CS chains are also known to differ between tissue sources as well as within the chain [24]. Since dermatan sulfates contains IdoA instead of GlcA and have a very low occurrence of 6-sulfation [23], the digestion products shown for the DS substrate in Figure 4 are possibly due to other CS contaminants in the substrate or CS domains within the DS chain. The average total mass of disaccharides produced from each DS digestion with the chondroitinase ACII enzymes was approximately 5% of the mass of disaccharides released from a similar concentration of chondroitinase ABC acting on the same amount of substrate (Table S3). This negligible conversion is consistent with the resistance of DS to chondroitinase ACII [25].
The novel CS-E from the clam, Mactra chinensis, contains the KS disaccharide unit [D-GlcNAc6S-(1→3)-β-D-Gal-(1→] at the C-3 position of GlcA [11]. The proposed structure of KS disaccharide branched CS is shown in Figure 4, as deduced in the work of Higashi et al. [11]. This CS from M. chinensis contains unknown consecutive repeating structures that show distinct chondroitinase susceptibilities in comparison to other CS structures. KS-branched CS has been found to be resistant to degradation by both chondroitinase ABC and chondroitinase ACII at very low concentrations, but when treated with Seikagaku’s chondroitinase ACII at a concentration of 12.5 mU, several unidentified peaks were observed on the reaction’s anion-exchange chromatogram [11].
Considerable levels of K-type units have been discovered in CS isolated from octopus cartilage (E. dofleini), with the disaccharide composition reported to be 4.9% of ΔDi-0S, 76.9% of ΔDi-4S, 4.22% of ΔDi-diSE and 13.9% of GalNAc (4S) (K-type units) [12]. Upon treating crude glycosaminoglycan (GAG) containing octopus cartilage with Seikagaku’s chondroitinase ACII, peaks were seen on the disaccharide chromatogram corresponding to ΔDi-0S, ΔDi-4S and small amounts of ΔDi-diSE, along with another unidentified peak. However, when the same crude GAG containing CS-K was treated with chondroitinase ABC, the unidentified peak completely disappeared [12]. These unidentified peaks obtained by treating M. chinensis CS-E and octopus CS-K substrates with AaACII are analogous to the additional disaccharide products obtained from these same substrates when treated with the A16ACII enzyme secreted by Arthrobacter (Figure 4).
All three enzymes exhibited an exolytic action pattern as is typical of chondroitinase ACII enzymes [4]. All three enzymes showed no activity on KS as a substrate but were active on hyaluronan (HA), producing mainly 0S disaccharides (data not shown). This behavior is characteristic of all chondroitinases, which are members of polysaccharide lyase family 8 (including hyaluronate lyases), a group that shares significant sequence, structural, and mechanistic homology [26].
4.5 Activity of A16ACII induced with CS-C and CS-A
Arthrobacter produced active enzyme when the medium was supplemented with CS-A or CS-C as inducer, but not in the absence of CS (Figure S3). Based on our testing of these two types of CS, it appears that sulfation pattern does not have a significant impact on the induction of A16ACII production in Arthrobacter, as both preparation methods yielded enzyme displaying comparable activity and stability (Figures S3 and S4).
4.6 Action pattern of chondroitinase ACII enzymes
We also examined the action pattern of all three chondroitinase ACII enzymes (excluding Seikagaku AaACII) using a single substrate type, CS-A. The equations obtained for the change in the mass of disaccharide released as a function of reaction time, illustrates a linear increase in disaccharide content (Figure 5), consistent with an exolytic action pattern on CS-A substrate. This matches the reported exolytic action pattern previously reported for AaACII from Seikagaku [4], which is no longer available for testing. As expected, the major digestion product of these enzymes was the monosulfated unsaturated disaccharide, and their exolytic action pattern was easily established by the immediate appearance of disaccharides at steadily increasing quantities over time.
Based on the similarity in properties and characteristics between the AaACII enzyme from Seikagaku and the A16ACII enzyme from the Arthrobacter strain discussed in this study, we believe that A16ACII can serve as a suitable replacement for the original enzyme, which is no longer commercially available from Seikagaku. Notably, it displays a distinguishing product complexity on octopus CS-K and M. chinensis CS-E substrates, as does the Seikagaku preparation. This enzyme will fill a great need within the glycobiology research community, which has had limited access to AaACII since 2011. It has been widely used for the analysis of CS/DS composition in GAGs, in studies generating crucial data allowing an understanding of species, tissue, age and pathology related differences across materials from diverse sources [7]. Furthermore, the recombinant E. coli expressed versions of chondroitinase ACII, tA16ACII and tA16ACII(I236T), developed and characterized herein, are suitable for general application in the structural determination of most standard chondroitin sulfates. The capability for efficient recombinant expression of these chondroitinases will facilitate the structure-function characterization of these enzymes and allow for the advancement of chondroitinases as enzymatic tools for the characterization and sequencing of CS/DS [27].
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL096972) and the National Science Foundation (MCB-1448657, CBET-1604547).
Abbreviations
- AcOH
acetic acid
- AMAC
2-aminoacridone
- CS
chondroitin sulfate
- ΔUA
4-deoxy-α-L-threo-hexenopyranosyluronic acid
- DS
dermatan sulfate
- DMMB
1,9-dimethylene blue
- DMSO
dimethylsulfoxide
- GAG
glycosaminoglycan
- GlcA
glucuronic acid
- GalNAc
N-acetylgalactosamine
- GlcNAc
N-acetylglucosamine
- HPLC
high performance liquid chromatography
- HA
hyaluronan
- IdoA
iduronic acid
- KS
keratan sulfate
- LB
Luria-Bertani
- LC-MS
liquid chromatography-mass spectrometry
- OD
optical density
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SOC
super optimal broth with catabolite repression
Footnotes
The authors declare no financial or commercial conflict of interest.
ORCIDs: 0000-0002-2948-2846
References
- 1.Hiyama K, Okada S. J Biol Chem. 1975;250:1824. [PubMed] [Google Scholar]
- 2.Linhardt RJ, Galliher PM, Cooney CL. Appl Biochem Biotechnol. 1986;12:135. doi: 10.1007/BF02798420. [DOI] [PubMed] [Google Scholar]
- 3.Linhardt RJ. Curr Protoc Mol Biol. 2001;Chapter 17(Unit17):13B. doi: 10.1002/0471142727.mb1713bs48. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z, Park Y, Kemp MM, Zhao W, Im AR, Shaya D, Cygler M, Kim YS, Linhardt RJ. Anal Biochem. 2009;385:57. doi: 10.1016/j.ab.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jandik KA, Gu K, Linhardt RJ. Glycobiology. 1994;4:289. doi: 10.1093/glycob/4.3.289. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki S. J Biol Chem. 1960;235:3580. [PubMed] [Google Scholar]
- 7.Huckerby TN, Nieduszynski IA, Giannopoulos M, Weeks SD, Sadler IH, Lauder RM. FEBS J. 2005;272:6276. doi: 10.1111/j.1742-4658.2005.05009.x. [DOI] [PubMed] [Google Scholar]
- 8.Yin FX, Wang FS, Sheng JZ. J Biol Chem. 2016;291:4399. doi: 10.1074/jbc.C115.708396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lunin VV, Li Y, Linhardt RJ, Miyazono H, Kyogashima M, Kaneko T, Bell AW, Cygler M. J Mol Biol. 2004;337:367. doi: 10.1016/j.jmb.2003.12.071. [DOI] [PubMed] [Google Scholar]
- 10.Prabhakar V, Capila I, Soundararajan V, Raman R, Sasisekharan R. J Biol Chem. 2009;284:974. doi: 10.1074/jbc.M806630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higashi K, Takeda K, Mukuno A, Okamoto Y, Masuko S, Linhardt RJ, Toida T. Biochem J. 2016;473:4145. doi: 10.1042/BCJ20160655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higashi K, Okamoto Y, Mukuno A, Wakai J, Hosoyama S, Linhardt RJ, Toida T. Carbohydr Polym. 2015;134:557. doi: 10.1016/j.carbpol.2015.07.082. [DOI] [PubMed] [Google Scholar]
- 13.Hernaiz MJ, Linhardt RJ. Methods Mol Biol. 2001;171:363. doi: 10.1385/1-59259-209-0:363. [DOI] [PubMed] [Google Scholar]
- 14.Sun X, Li L, Overdier KH, Ammons LA, Douglas IS, Burlew CC, Zhang F, Schmidt EP, Chi L, Linhardt RJ. Anal Chem. 2015;87:6220. doi: 10.1021/acs.analchem.5b00913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carpenter JF, Chang BS, Garzon-Rodriguez W, Randolph TW. Pharm Biotechnol. 2002;13:109. doi: 10.1007/978-1-4615-0557-0_5. [DOI] [PubMed] [Google Scholar]
- 16.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK. FEBS J. 2005;272:5101. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petersen TN, Brunak S, von Heijne G, Nielsen H. Nat Methods. 2011;8:785. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 19.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. Nucleic Acids Res. 2010;38:W695. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Nucleic Acids Res. 2013;41:W597. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hiyama K, Okada S. Agricultural and Biological Chemistry. 1977;41:1279. [Google Scholar]
- 23.Lauder RM, Huckerby TN, Nieduszynski IA. Glycobiology. 2000;10:393. doi: 10.1093/glycob/10.4.393. [DOI] [PubMed] [Google Scholar]
- 24.Lauder RM. Complement Ther Med. 2009;17:56. doi: 10.1016/j.ctim.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Yamagata T, Saito H, Habuchi O, Suzuki S. J Biol Chem. 1968;243:1523. [PubMed] [Google Scholar]
- 26.Stern R, Jedrzejas MJ. Chem Rev. 2006;106:818. doi: 10.1021/cr050247k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pojasek K, Shriver Z, Kiley P, Venkataraman G, Sasisekharan R. Biochem Biophys Res Commun. 2001;286:343. doi: 10.1006/bbrc.2001.5380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.