

Abstract
Our understanding of the structure of DNA has helped pave the way for tremendous advancements in understanding the mechanisms of DNA replication. Semiconservative DNA replication has provided an elegant solution to the fundamental problem of how life is able to proliferate in a way that allows cells, organisms, and populations to survive and replicate many times over. Somewhat lost, however, in our admiration for this elegant mechanism is an appreciation for the asymmetries that inevitably occur in the process of DNA replication. As we will discuss in this review, these asymmetries arise as a consequence of the structure of the DNA molecule and the enzymatic mechanism DNA synthesis. Furthermore, increasing evidence suggests that these asymmetries are utilized as mechanisms to drive diverse processes ranging from adaptation and evolution, to cell fate decisions related to patterning and development.
Keywords: DNA replication, asymmetric, histones, epigenetic inheritance, chromatin
1. INTRODUCTION: A HISTORY OF DNA REPLICATION
The combined efforts of Rosalind Franklin, Francis Crick, James Watson, and Maurice Wilkins provided the scientific community at large with an understanding of how two antiparallel strands of DNA are able to pair together in the double-helix structure (Watson and Crick, 1953b, Wilkins et al., 1953, Franklin and Gosling, 1953b, Watson and Crick, 1953a, Franklin and Gosling, 1953a, Avery et al., 1944). Following this groundbreaking discovery, those in the field of DNA research immediately sought to understand the mechanisms by which DNA is able to copy itself to allow for transmission of genetic information from parents to subsequent generations. Indeed, at the time of this discovery, the structure of the double helix itself seemed to suggest an elegant copying mechanism by which both strands, once separated, could simultaneously serve as templates for the copying of duplicate strands. Subsequent experiments by Meselson & Stahl (Meselson and Stahl, 1958) further demonstrated that this model of DNA replication was indeed the primary method utilized by cells to replicate their genomic material. This model, termed semiconservative DNA replication, suggests that both existing strands are used as templates from which two identical strands of DNA, termed sister chromatids, can be produced and eventually segregated during mitosis.
However, the two strands that result from semiconservative DNA replication are not always precise duplicates of one another. Asymmetries between sister chromatids arising from the process of DNA replication can, in large part, be understood by revisiting the basic physical structure of the DNA fiber. The antiparallel structure of the DNA double helix is extremely important in the context of DNA replication, specifically because of the enzymatic activity of DNA polymerase. Following the discovery of the structure of DNA, Arthur Kornberg discovered that DNA polymerases, the molecules that can elongate DNA from a single-stranded DNA (ssDNA) template and a primer, can only add new bases in the 5′-to-3′ direction. This directionality is due to the fact that DNA polymerase requires a free 3′ hydroxyl (–OH) group to catalyze the formation of phosphodiester bonds during the process of strand elongation. Consequently, the template strand with its 5′ end oriented in the same direction as the movement of the helicase—termed the leading strand—will be able to be read and synthesized in a continuous fashion (Bessman et al., 1956, Bessman et al., 1958, Kornberg et al., 1989, Lehman et al., 1958). Conversely, the lagging strand, whose template strand is oriented with its 5′ end opposite to the direction of the helicase movement, is synthesized in short, discontinuous segments. These segments, termed Okazaki fragments after their discoverers, are synthesized in the opposite direction of fork movement and are typically sized between 100 and 200 base pairs (bp) in eukaryotes and between 1,000 and 2,000 bp in prokaryotes (Balakrishnan and Bambara, 2013, Sakabe and Okazaki, 1966, Okazaki et al., 1968). In summary, by virtue of the antiparallel organization of the DNA double helix in conjunction with the unidirectional enzymatic mechanism of DNA polymerase, DNA replication is inherently asymmetric. As a result, DNA synthesis occurs in two distinct mechanisms: continuous (leading-strand) synthesis and discontinuous (lagging-strand) synthesis. In this review, we elaborate on new and exciting research demonstrating the important biological functions that these asymmetries have in roles ranging from adaptation and evolution to cell fate decisions during development.
2. DNA REPLICATION
2.1. Initiation and Elongation of DNA Replication
Before discussing the molecular details of how semiconservative DNA replication can produce asymmetric strands of DNA, it is useful to briefly discuss the basics of DNA replication. In doing so below, we will elaborate on the fundamental reasons why the process of DNA replication is an inherently asymmetric event. Generally, in this review we discuss what is known about DNA replication and chromatin establishment in eukaryotes. When possible, we address findings in metazoans, although a large portion of DNA and chromatin replication studies have been conducted in unicellular organisms and viruses.
2.1.1. Early steps and fundamentals of DNA replication
Generally, the goal of DNA replication in dividing cells is to produce two identical daughter DNA molecules from one parent DNA molecule with as high fidelity as possible without under-replicating or re-replicating any regions of the genome. To propagate their genome, every species must be able to efficiently perform this fundamental process. The basic units of DNA replication are the replicon and the replisome. The replisome refers to the protein complexes used to replicate DNA, and the replicon is the stretch of DNA replicated by a single replisome. The first step of DNA replication occurs in G1 phase immediately after the separation of sister chromatids, where the origin of replication complex, specifically Orc 1–6, binds DNA (Bell and Stillman, 1992) (Figure 1). In most prokaryotes and some eukaryotes, ORC binds a specific DNA sequence denoted as ORI (Fuller et al., 1984). In higher-order eukaryotes, however, a multitude of factors help specify sites of ORC binding, but there are few clear ORI sequences as in prokaryotes and lower eukaryotes (Vashee et al., 2003). However, it is becoming clear that in metazoans a wide variety of features help specify flexible, but non-random origin localization (Cayrou et al., 2011).
Figure 1. Overview of DNA replication.
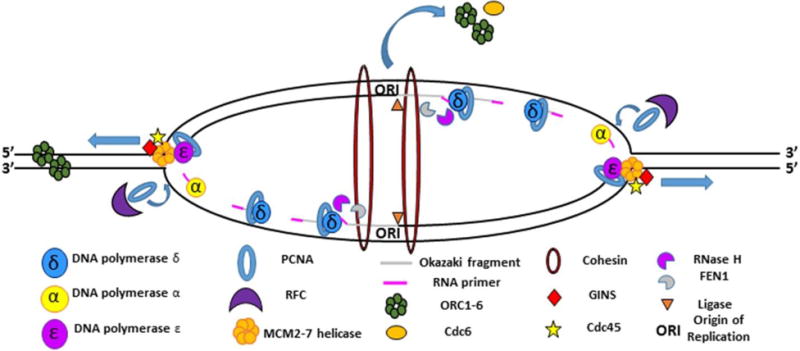
The fork fires as the MCM2-7 helicase proceeds out in a bidirectional fashion from the origin of replication (ORI), unwinding double-stranded DNA as it goes. ORC proteins and cdc6 dissociated from the origin, where they were initially bound to facilitate helicase loading. CDC45 and GINS travel with the MCM2-7 helicase to create the CMG replication complex. The leading strand is synthesized primarily through the actions of DNA polymerase ε. The lagging strand is discontinuously synthesized primarily through the actions of DNA polymerase α and δ. Pol α synthesizes the RNA primer needed to initiate DNA synthesis. The lagging strand is additionally processed by FEN1 and ligase to complete synthesis. The RFC complex is responsible for loading PCNA, which acts as a processivity factor for DNA pol ε and DNA pol δ. Cohesins link the two sister chromatids following passage of the replication fork.
ORC operates by recruiting Cdc6 to a subset of origins (Hateboer et al., 1998), which in turn helps recruit the MCM2-7 and cdt1 complex (Tsakraklides and Bell, 2010). MCM2–7 is the eukaryotic helicase used in DNA replication to unwind the DNA double helix (Labib et al., 2000, Nishitani et al., 2000). After MCM2–7 is loaded, the next major step in DNA replication is firing the replication forks at the G1/S transition upon signaling from Ddk and Cdk, which among other roles phosphorylate MCM (Nougarede et al., 2000). The phosphorylated MCM2-7 is bound by Cdc45, the limiting reagent of DNA replication, along with MCM10 (Zou et al., 1997, Moyer et al., 2006, Wohlschlegel et al., 2002). GINS and DNA polymerase ε (Pol ε) are additionally loaded at this point (Muramatsu et al., 2010). The Cdc45, MCM2-7, and GINS complex form the CMG complex which drives the progression of the replisome (Gambus et al., 2006). Upon further binding of additional DNA polymerases and additional DNA replication factors, the fork will fire.
2.1.2. Bidirectional firing and progression of the replication fork
In metazoan genomes, the entire genome does not replicate at the same time. Rather, there is a very complex and highly studied temporal order of replication events. The order of replication events is neither truly stochastic nor truly sequential in most eukaryotic cells, as there are not defined origins that fire in a predetermined sequence in most metazoans; there are also clearly early firing and late firing regions wherein euchromatin tends to fire early and heterochromatin fires late, but a slew of other factors also bias timing (Rhind, 2006, Ryba et al., 2010). The firing of replication forks is spatially organized into replication factories where active origins are clustered and fire coordinately as a means to localize key replication factors predominantly to actively replicating sites (Leonhardt et al., 2000).
Upon firing, a pair of CMG complexes progress bi-directionally in the 3′-to-5′ direction (on the leading strand) in eukaryotes and in the 5′-to-3′ direction (on the lagging strand) in bacteria, separating the DNA strands as it progresses and creating the characteristic DNA replication bubble (Figure 1). At each helicase, one strand is opened as ssDNA in the 5′-to-3′direction and the other strand opened in the 3′-to-5′ direction (O’Donnell et al., 2013). The synthesis of DNA on these two strands is a hugely different biochemical process. The physical firing of the fork and the process of synthesizing new DNA behind the advancing helicase require the recruitment of a variety of components needed for the completion of the leading and lagging strands. Behind the fork, DNA is quickly synthesized on the two template ssDNA molecules by the addition of dNTPs by DNA polymerases.
2.2. Synthesis and Processing of the Leading and Lagging Strands
The leading strand is synthesized continuously in the 5′-to-3′ direction while the lagging strand is synthesized in the 3′-to-5′irection in the form of Okazaki fragments which must be processed and ligated to create a continuous strand. These two divergent processes underlie the asymmetry of DNA replication and will be discussed in molecular detail in this section.
2.2.1. Leading-strand synthesis and essential DNA replication components
The biochemical properties of the DNA polymerases underlie the physical systems of DNA replication. As discussed above, DNA polymerase can synthesize new DNA only in the 5′to-3′direction because the polymerase catalyzes the addition of the 5′ end of a nucleotide base to the 3′ end of another nucleotide base. Having to add to the 3′ end of an existing base requires that before DNA polymerases can synthesize new DNA, an RNA primer must be synthesized. In eukaryotes, this is a short 8−10-nt (nucleotide) RNA primer made by primase (Lark, 1972b, Lark, 1972a). The leading strand requires only a single RNA primer, as DNA can be continuously synthesized in the 5′-to-3′ direction during leading strand synthesis. During synthesis, DNA polymerase processivity is greatly enhanced by through association with DNA clamps. In eukaryotes, the main DNA clamp is Proliferating Cell Nuclear Antigen (PCNA), which is loaded by the clamp loader RFC behind the fork. PCNA, a trimeric complex that binds DNA, has a variety of other interacting domains that serve a host of different functions, including maintaining polymerase association with DNA while also recruiting other DNA replication factors. Such factors include FEN1, a key lagging-strand component; CAF1, a histone chaperone; and cohesin, a multiprotein complex involved in chromatin organization (Moldovan et al., 2006). Due to the variety of processes with which PCNA is associated, it is generally referred to as the tool belt of DNA replication. It has long been acknowledged that Pol ε is a leading-strand-specific polymerase (Hubscher et al., 2002). However, although it is largely accepted that Pol ε operates on the leading strand, there is an emerging debate about whether Pol ε is the main DNA polymerase on the leading strand or whether Pol δ is the major polymerase on the leading strand, as is true on the lagging strand (Johnson et al., 2015, Pursell et al., 2007). If the latter is the case, it would stand to reason that the primary role of Pol ε would be as a proofreader on the leading strand as opposed to as a synthesizer of the bulk of DNA (Albertson et al., 2009). Both Pol δ and Pol ε have very high processivity when interacting with PCNA and have high fidelity due to their proofreading activity (Korona et al., 2011) (Figure 1).
2.2.2. Lagging-strand synthesis
The lagging strand has a far more complicated synthesis relative to the leading strand. As the overall synthesis of the lagging strand is in the 3′-to-5′ direction, DNA polymerases working on the lagging strand must add new nucleotides in a direction opposite to that of the advancing helicase (Figure 1). A consequence of this mode of synthesis is that new DNA cannot be added continuously at the edge of the fork. Rather, new DNA must be synthesized discontinuously in small fragments, termed Okazaki fragments, opposite the direction of the movement of the fork (Sakabe and Okazaki, 1966). As the polymerase is not acting directly behind the fork, the lagging-strand DNA remains single stranded for a stretch and is bound by ssDNA-binding proteins, such as RPA, that stabilize this ssDNA and recruit the Pol α/primase complex (Ryba et al., 2010, Leonhardt et al., 2000).
The lagging strand is first replicated by a Pol α/primase complex that lays down an RNA primer as well as an additional short fragment of DNA of approximately 20 nucleotides in length (Masai et al., 2010, O’Donnell et al., 2013). Although Pol α was the first discovered DNA polymerase and was originally thought to replicate the entire genome, it is now recognized that the primary role of Pol α is simply to start the synthesis of the lagging strand as opposed to serving as the main polymerase for the lagging strand, let alone the entire genome (Hubscher and Seo, 2001). Pol α has low processivity and, very importantly, has far lower fidelity than the other eukaryotic polymerases (O’Donnell et al., 2013). Unsurprisingly, most of the DNA synthesized by Pol α is replaced by Pol δ, which as mentioned above is considered the main polymerase on the lagging strand. Pol δ is loaded onto the lagging strand by PCNA and RFC and, using the Pol α DNA as the primer, synthesizes the Okazaki fragments (Leonhardt et al., 2000). The length of these fragments varies among species, but generally bacteria have relatively long fragments of approximately 1,200 nt, whereas eukaryotes have approximately 200-nt fragments sized according to chromatin repeats (Ogawa and Okazaki, 1980, Smith and Whitehouse, 2012). Pol δ synthesizes under and displaces much of the Pol α–synthesized RNA/DNA, generating a 5′ flap which will be subject to further processing (discussed hereafter). Pol δ has higher processivity than Pol α, as it is associated with PCNA, and has higher fidelity, as it has 3′-to-5′ exonuclease activity, allowing for proofreading activity (Prindle and Loeb, 2012).
2.2.3. Lagging-strand processing
After the synthesis of the lagging strand, the Pol α–synthesized 5′ flaps have to be cleaved, the RNA primer has to be removed, and the nicks between the Okazaki fragments must be ligated. RNase H is responsible for removing the RNA primers (Cerritelli and Crouch, 2009). There are at least two pathways by which the flap is metabolized. The simplest pathway is the short flap pathway, in which FEN1, an exonuclease, is attached to the Pol δ complex and cleaves the RNA/DNA fragment at a single site at the base of the flap, leaving just a nick between Okazaki fragments that is ligated by DNA ligase (Bambara et al., 1997). If FEN1 is not associated with the fragment and does not remove the flap before Pol δ displaces a large chunk of DNA, then a long ssDNA flap is formed to which RPA is able to bind. Once RPA binds the long flap, FEN1 is unable to process the long flap. To dissociate RPA, a different exonuclease, Dna2, cleaves the long flap, leaving a short flap behind which FEN1 is now able to efficiently process. Interestingly, the helicase Pif1 biases the strands that are normally processed by the short flap pathway to instead be processed via the long flap pathway (Rossi et al., 2008).
2.3. The Functional Relevance of the Leading- and Lagging-Strand Asymmetries
More recently, studies have focused on not only the mechanisms by which the leading and lagging strands are replicated, but also the effects of the asymmetries generated by these two different divergent replication processes. This section will discuss the impact that differences between continuous (leading strand) replication and discontinuous (lagging-strand) replication have on patterns of mutagenesis as well as the role that replication asymmetry plays in the process of yeast mating type switching.
2.3.1. The leading and lagging strands shape the mutagenic landscape of DNA
Random mutations created during DNA replication represent a double-edged sword in terms of their effects on the survival and stability of an individual organism as well as the population as a whole. On the one hand, random mutations represent an indispensable tool for evolutionary change, without which rapid adaptation to severe changes in environment would be nearly impossible. On the other hand, mutations also represent a great source of chaos in an organism’s genome that, if allowed to proliferate in an unchecked manner, could spell doom for both the organism and the population (Furusawa and Doi, 1992, Furusawa and Doi, 1998). Therefore, as with many aspects of biological systems, a balance is required to achieve the desired degree of both evolutionary flexibility and genomic stability. Interestingly, the asymmetric structure and replication of the DNA molecule may provide an elegant solution to the problem of balancing flexibility and stability by impacting the distribution of mutations within the genome.
On the basis of the fact that the discontinuous synthesis of the lagging strand would require the coordinated effort of a greater number of proteins to proceed efficiently, researchers initially hypothesized that lagging-strand synthesis could generate mutations at a higher rate than leading-strand synthesis at sister genomic regions. This type of strand-based mutational imbalance was termed disparity mutagenesis by researchers studying evolutionary adaptations, and was initially proposed as a mechanism to allow organisms to achieve a high mutational load without severely compromising an organism’s fitness through loss of genome stability (Fujihara and Furusawa, 2016). The evolutionary logic of disparity mutagenesis is as follows: A majority of random mutations are concentrated upon the lagging strand as a result of discontinuous lagging-strand synthesis. The leading strand, by virtue of continuous synthesis, remains essentially error free. Inside both an organism (diploid) and a population (haploid), this system creates a mixture of stable, leading-strand-synthesized genes and more plastic, flexible lagging-strand-derived genes. The leading-strand genes reliably propagate the parental genetic state, providing stability for the organism and allowing for continued growth and survival in stable environmental conditions with low selective pressure (Furusawa, 2014). However, if the environment suddenly changes to much harsher conditions with more competition and higher selective pressure, now the lagging-strand-derived genes provide an opportunity for rapid adaptation to changing environmental conditions.
Researchers demonstrated via modeling that by directing a majority of the mutational load to one side of the replication fork, organisms could survive under a significantly higher mutation rate than if those same mutational rates were randomly distributed throughout the genome. Researchers further demonstrated the evolutionary value of disparity mutagenesis by using genetic tools to exacerbate the degree to which the lagging strand acquires novel mutations. Using polymerase δ mutants that preferentially increase mutagenesis rates on the lagging strand, researcher found that they could culture yeast in extremely harsh environmental conditions in which wild-type yeast would be unable to adapt and grow (Tanabe et al., 1999, Shimoda et al., 2006). Although early studies into disparity mutagenesis were largely theoretical in nature, they suggested that if certain mechanisms could bias mutation distribution in nature, such a process would be extremely beneficial to an organism’s survival and adaptability in a diverse range of climates (Furusawa, 2012). Interestingly, research discussed below has revealed evidence for molecular mechanisms that could generate asymmetries in strand mutational rates.
The divergent synthesis of the leading strand and the lagging strand allows for the intriguing possibility that the two strands could have different rates or types of mutagenesis. Early studies on the topic primarily relied on using the Kunkel mutagenesis method (Kunkel, 1985) to introduce individual site specific mutation in a wide array of genes in both eukaryotic and prokaryotic unicellular organisms and in vitro. As the mutations are introduced into sites that can be screened by phenotype or survival, reversion rates of the genes can be measured. The overwhelming conclusions from this body of work were that reversions were more common in a wide variety of circumstances during lagging strand replication than leading strand replication (Francino and Ochman, 1997). Some groups have documented that in particular conditions, the leading strand can be more error prone (Fijalkowska et al., 1998), but it is possible that transcription could be playing a larger role in these cases than originally thought. Overall, the generality is that the lagging strand has higher overall rates of mutagenesis. One reason for the different rates of mutations between the two strands could be the different fidelities of DNA polymerases. A recent study found that in both yeast and human cells, approximately 1.5% of the genome after DNA replication was Pol α–synthesized DNA that is retained near Okazaki junctions at the end of Okazaki fragments. Importantly, this retained Pol α–synthesized DNA underlies the higher mutagenesis on the lagging strand due to the lower fidelity of Pol α (Reijns, 2015). Pol α synthesized DNA may additionally be retained at higher rates near transcription factor binding sites, creating hot spots of lagging strand derived mutations. Together, these studies indicate that the lagging strand shapes the mutational landscape of cells (Reijns, 2015). Furthermore, these findings support the disparity mutagenesis model across multiple domains of life.
One interesting finding originating from studies of mutation distribution is that actively transcribed genes, in particular, have increased asymmetries in mutation rates and therefore gene evolution, depending on whether the genes are on the leading or the lagging strand (Liu and Alberts, 1995). Leading-strand genes are transcribed in the same direction as the movement of the fork, whereas the lagging-strand genes are transcribed in the opposite direction of the fork’s progression. Genes on the lagging strand can be referred to as head-on genes, as when the genes are highly transcribed, the RNA polymerase and DNA replication fork can collide head on. This process can be highly mutagenic, resulting in high rates of insertions and deletions in the gene body and promotor, as well as high rates of base substitutions specifically enriched in promoter regions (Sankar et al., 2016) (Figure 2). When studied in the context of the ThyP3 locus, this results in a 60% increase in the rate of mutagenesis in head on genes compared to co-directional gene orientation at the ThyP3 locus. It is thought that the increased rate of insertions and deletions is due to the replication and transcription machinery collisions, as these collisions induce replication fork stalling which can lead to fork slippage, the result of which can often be insertions and deletions. The base substitution rate is significantly increased in the promoter region, specifically at the binding site of the sigma factor on the lagging strand. It is currently debated whether increased base substitution is due to the higher baseline rate of base substitution on the lagging strand or due to replication-transcription collisions (Sankar et al., 2016). Other groups have demonstrated that the increased rate in lagging strand mutagenesis in the head on orientation is transcriptionally dependent, which does provide evidence that base substitution may also be higher in genes on the lagging strand when transcribed with head on orientation (Paul et al., 2013). Hence, it is likely that mutation rates on the lagging strand are significantly higher than on the leading strand due to both incorporation of Pol α synthesized DNA on the lagging strand and head on replication-transcription collisions on lagging strand transcribed genes.
Figure 2. Mutational Imbalance during DNA replication.
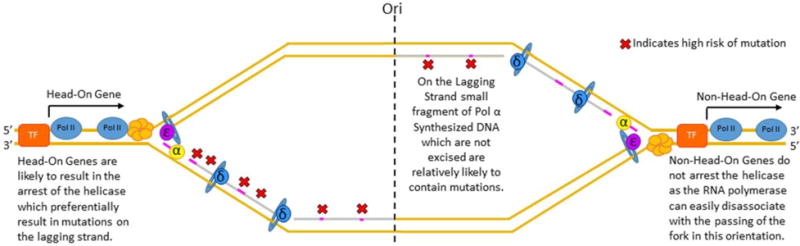
Mutagenesis occurs asymmetrically on the two sisters during DNA replication. The lagging strand may have a higher rate of mutagenesis than the leading strand due to both incorporation of low fidelity DNA Polymerase α and the strands increased susceptibility to mutagenesis with head on transcription and DNA replication fork collisions.
2.3.2. Asymmetries in mutagenesis could play a key evolutionary role
If we consider that actively transcribed genes on the lagging strand have higher rates of mutations when compared to activley transcribed genes on the leading strand, it would stand to reason that the asymmetric nature of DNA replication plays a key evolutionary role. For key housekeeping genes, where high rates of mutations resulting in changes to amino acid composition would be deleterious, it may be favorable to transcribe these genes on the leading strand to avoid head on mutagenic replication transcription collisions. Strikingly, 94% of the essential genes in the genome of Bacillus subtilis are, in fact, transcribed on the leading strand, avoiding head on transcription-replication collisions and elevated rates of mutagenesis (Rocha and Danchin, 2003). This suggests that for essential genes, inversions that orient these genes to be transcribed away from the ORI sequence, which are thereby transcribed codirectionally to DNA replication on the leading strand, are selected for in order to avoid deleterious mutations from replication-transcription head on collisions. While it is debated whether there is selection for orienting non-essential genes to be transcribed on the lagging strand, it does appear that no matter the reason for their orientation on the lagging strand, these genes do have elevated rates of mutagenesis and undergo accelerated gene evolution. This serves to rapidly create heterogeneity in populations of unicellular organisms, which could help populations of unicellular organisms survive changing environmental conditions as postulated by the disparity mutagenesis hypothesis.
The response to transcription-replication collisions has also been studied in metazoans. In metazoans, where origin specification is flexible, and where genetic heterogeneity among a population of cells is more deleterious than beneficial, transcription replication collisions are avoided far more dynamically than in unicellular organisms. While unicellular organisms orient their genomes to passively promote codirectionallity, metazoans tend to avoid replication-transcription collisions altogether by spatially and temporally separating transcription and replication (Helmrich et al., 2013), which is accomplished in a variety of different ways for different genes and in different cells. Interestingly, genes that can’t avoid transcription-replication collisions, such as the genes FHIT, WWOX, and IMMP2L, which in human cells are reported to take longer than one cell cycle to transcribe due to their enormous size, cannot easily temporally separate replication-transcription collisions. Resultingly, these genes are common fragile sites, particularly susceptible to DNA double strand breaks and R-loop formation in response to replication-transcription collisions, suggesting that in metazoans replication-transcription collisions can result in major genome instability when not actively avoided (Helmrich et al., 2011). While many S-phase transcribed genes have active mechanisms preventing replication-transcription collisions altogether, we hypothesize that flexible origin specification may serve as an additional protection against creating hot spots for mutagenesis due to replication-transcription collisions. Dynamic origin specfication could allow the metazoan cell to minimizes the chances that a gene would be consistently replicated in a head-on fashion (and thus highly mutagenized) by varying the location of that genes’ distal-most origin of replication. Furthermore, many promoters additionally serve as sites of preferential origin binding and activity due to their open chromatin state (Sequeira-Mendes et al., 2009). Localizing origins to promoters for actively transcribed genes is likely to ensure that even if replication-transcription collisions occur, the origin is more likely to be downstream of the gene and therefore any transcription-replication collisions are likely to be codirectional, minimizing mutagenicity. Overall, it appears that metazoans use a wide variety of techniques to actively avoid replication-transcription collisions, while bacteria appear to minimize fitness reducing mutations by orienting essential genes to be transcribed codirectionally to replication, while potentially using head on lagging strand transcription to promote genetic heterogeneity in non-essential genes.
2.3.3. The leading and lagging strands contribute to yeast mating-type switches
Asymmetries in DNA replication have the potential to generate a variety of strand-based differences capable of effecting important cell fate decisions. The mating-type switching behavior exhibited by yeast has long served as an excellent model system to study the underlying mechanisms of asymmetric cell division (ACD): the phenomena by which two daughter cells acquire different fates following division (Haber, 2012, Hanson and Wolfe, 2017, Holmes et al., 2005). In yeast, mating type refers to a cell’s competency to mate with other haploid yeast cells. For instance, in the yeast Saccharomyces cerevisiae, haploid mating-type a cells are competent to mate with mating type α cells, but cannot mate amongst the same typed cells. A similar principle is true for Saccharomyces pombe, where yeast of mating type plus (P) cannot mate amongst themselves but are competent to mate with S. pombe of mating type minus (M). This mating competency reflects the function output of differential transcription of mating-type related genes in each of the two different mating yeast groups (a vs. α; P vs. M) (Haber, 2012). Interestingly, mating type is not a fixed identity in yeast. Rather, a yeast cell can alter its mating type over the course of several cell divisions in a process known as mating type switching. Several decades of study in the fission yeast Saccharomyces pombe have revealed that mating-type switching is driven by the asymmetries inherent in the process of DNA replication (Dalgaard and Klar, 2001). More specifically, the process of discontinuous lagging strand synthesis of the mating type locus (MAT1) marks one sister chromatid with a molecular lesion which functions directly in the process of mating-type switching. The lesion is composed of two RNA nucleotides, likely the result of imprecise excision of an RNA primer (Klar, 1987, Klar, 1990, Klar, 2010). The function of this lesion does not become obvious until the next S-phase, when its acts as an impediment to the motion of the replication fork as it is moving through the MAT1 locus (Klar et al., 1991, Nakayama et al., 2001). When the fork collides with the lesion it causes a stalling event (Yamada-Inagawa et al., 2007). To rescue the stalled fork, the yeast cell undergoes a recombination event similar to synthesis dependent strand annealing. During this process, DNA from a nearby donor locus (MAT2 or MAT3) is copied into the MAT1 locus, replacing the previous genetic material the result of which is a switch in cellular mating type (Arcangioli and de Lahondes, 2000, Egel, 2004) (Figure 3). Further study into the molecular mechanism responsible for regulating the process of mating type switching have revealed that the cell actively works to coordinate replication forks in the MAT1 region as a means to ensure that the gene locus is always replicated in a uni-direction manner (Dalgaard and Klar, 1999). The primary tool the cell uses to control fork progression is a cis-acting polar terminator of replication (RTS1). RTS1 acts to control fork progression in the MAT1 locus by halting fork progression in the centromere-distal direction. These findings demonstrate that beyond simply duplicating the genetic material, the asymmetries inherent in the process of DNA replication can be controlled and utilized to ensure a robust developmental outcome during the process of ACD.
Figure 3. Asymmetries in DNA replication underlie mating-type switching in S. pombe.
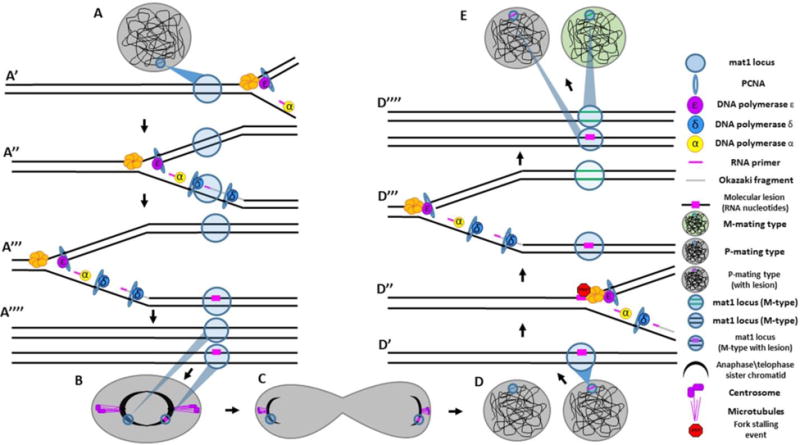
(A) A P-type mating cell entering DNA replication with the mat1 (mating type) locus labeled with blue circle. (A′) The replication fork approaches the mat1 locus. (A″) As replication proceeds, one strand is replicated by continuous (leading strand) synthesis whereas the other strand is replicated by discontinuous (lagging-strand) synthesis, characterized by numerous RNA primers. (A‴) One of these primers fails to get properly removed during lagging strand processing, leaving a molecular lesion (two RNA nucleotides) necessary for mating-type switching to occur. (A‴′) Two sister chromatids now exist: one with a molecular lesion and one which is unmarked. (B–C) The two sister chromatids are segregated to distinct daughter cells during mitosis. (D) Two daughter cells now enter S-phase; one with the molecular lesion and one without. (D′) In the daughter cell with the molecular lesion, the replication fork approaches the mat1 locus. (D″) The replication fork stalls when it hits the lesion. (D‴) The fork is rescued in DNA repair mechanism resembling synthesis dependent strand annealing. The result of this process is that the strand where the collision occurred has new DNA copied into the mat1 locus, resulting in a switch in mating type from P ➔ M. (D‴′) Two sister chromatids are produced; one which has switched mating type, the other which now contains a molecular lesion. (E) Two daughter cells are produced: one which has switch mating type, the other of which has not.
3. REPLICATION AND THE CHROMATIN LANDSCAPE
In addition to having impacts on DNA sequence, the asymmetries inherent in the process of DNA replication have important impacts on epigenetic phenomena (Probst et al., 2009). Epigenetic phenomena refer to modifications made to DNA or DNA-associated proteins that do not alter DNA sequences but lead to inheritable changes in gene expression. Many epigenetic phenomena affect changes in gene expression via altering DNA structure/packaging. By regulating DNA structure/accessibility, epigenetic phenomena play an essential role in regulating cell fate decisions and are involved in helping to establish and maintain proper cell identity throughout the life of the cell and the organism as a whole (Allis and Jenuwein, 2016). In this section, we discuss prominent epigenetic mechanisms and explore ways in which asymmetric DNA replication may impact the propagation of these molecular marks.
3.1. DNA Methylation
The addition of a single methyl (–CH3) group to the fifth carbon in the cytosine base (abbreviated 5mC) is one of the best-studied and fully characterized epigenetic signatures known to modulate gene expression. Cytosine methylation has a repressive effect on DNA bearing the epigenetic mark, an effect with a variety of important roles in regulating the chromatin landscape (Kulis and Esteller, 2010, Smith and Meissner, 2013). For many years, cytosine methylation was believed to be the only DNA modification known to have a specific role in regulating gene expression in eukaryotic genomes (Bird, 2002). However, recent work in Caenorhabditis elegans and Drosophila melanogaster as well as in human embryonic stem cells has demonstrated the presence of another DNA modification, methylation of the sixth carbon in the adenine base (6mA), which has been shown to have functional consequences in epigenetic regulation (Greer et al., 2015, Zhang et al., 2015, Wu et al., 2016). 6mA has initially been characterized in bacteria, in which it plays important roles in regulating gene expression (Low et al., 2001). Interestingly, deposition of new adenine methylation following replication fork passage appears to have a marked asymmetry in bacterial genomes. Although marks are fully restored on both sister chromatids following DNA replication, the kinetics of mark restoration differ significantly between sisters built via leading-strand synthesis and sisters built via lagging-strand synthesis. Leading strands re-methylate nascent DNA strands as much as two seconds before the lagging strand, leading to a spatial separation of ~700 nm. Researchers hypothesize that the temporal and spatial asymmetries observed between the leading strand and the lagging strand in terms of the recovery of adenine methylation reflect the additional amount of processing required to complete synthesis of lagging-strand DNA. More specifically, researchers conclude that the Okazaki fragments formed during lagging-strand synthesis must be fully ligated before DNA methylation can be fully restored (Stancheva et al., 1999). Though a clear biological role for this phenomenon has yet to be demonstrated, many researchers have speculated that asymmetric DNA methylation may play an important role in the process of nonrandom sister chromatid segregation, a phenomenon discussed below (Tajbakhsh and Gonzalez, 2009, Yamashita, 2013, Lopez-Vernaza and Leach, 2013).
3.2. Transcription and Chromatin Replication
Transcription factors are an essential class of DNA-binding proteins that recognize and bind to a consensus DNA sequence to help regulate the expression of a specific gene or genes in certain cases (Spitz and Furlong, 2012). During DNA replication, transcription factors must be removed to allow for the progression of the replication fork. After dissociating, transcription factors must rebind their target sites to maintain proper gene expression. When rebinding, recently dissociated transcription factors are faced with a choice of whether to bind the sister chromatid synthesized via continuous leading-strand synthesis or the sister chromatid synthesized via discontinuous lagging-strand synthesis. Interestingly, recent work from a variety of sources has revealed not only that this decision may not be random, but that this choice may have important functional consequences for gene regulation and epigenetic inheritance (Alabert and Groth, 2012).
Recent studies on chromatin maturation following DNA replication have revealed that the replicative history of leading vs. lagging sister chromatids plays an important role in the relative rates of chromatin maturation and resumption of transcription following passage of the replication fork. Interestingly, as described above (Figure 2), transcription ahead of the fork also appears to have an important role in regulating fork progression/speed. In cases in which transcription ahead of the fork runs co-directionally in the same direction as fork progression, chromatin matures faster on the leading strand than it does on the complementary lagging strand following passage of the replication fork. Interestingly, this faster recovery is dependent on transcription, as the leading and lagging strands mature at comparable rates when transcription is inhibited. Furthermore, when transcription runs counter to the direction of fork progression, chromatin matures more quickly on the lagging strand than on the leading strand. It has been interpreted as follows (Vasseur et al., 2016): When transcription progresses in the same direction as the replication fork, the replication fork will be allowed to move forward, uninhibited by collisions with transcription bubbles. The leading strand, due to the continuous nature of its synthesis, acts as a more capable binding target for the recently dissociated transcription factor. Consequently, the leading strand resumes transcription prior to the lagging strand (Figure 4). Conversely, when transcription is oriented counter to the direction of the replication fork, the progression of the replication fork is slowed, thus allowing lagging-strand processing to catch up to the leading strand. Although this model does not clarify why the lagging strand would be favored over the leading strand, it has been proposed that in the immediate aftermath of fork passage, transcription frequently resumes in an asymmetric manner, possibly as a mechanism to buffer gene dosage increases caused by genome duplication during S phase (Vasseur et al., 2016).
Figure 4. Asymmetries in DNA replication effect chromatin maturation following fork passage.
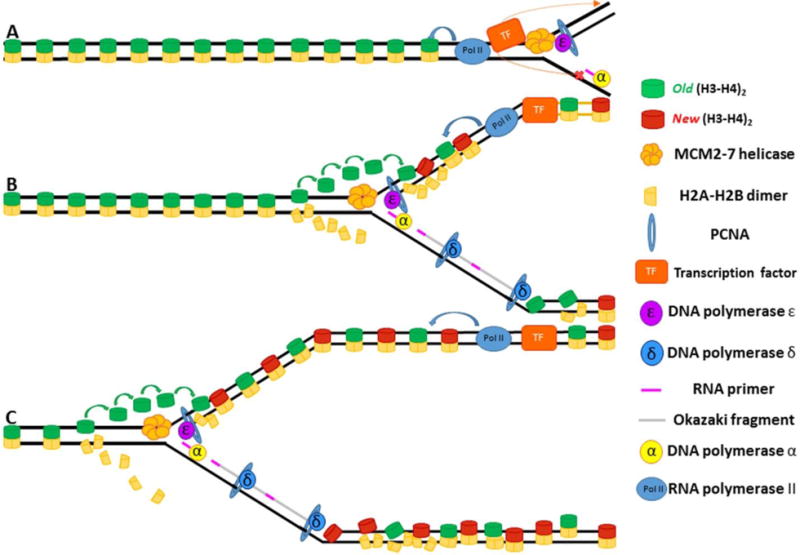
(A) The replisome approaches the bound transcription factor with Pol II transcribing in the same direction as the fork. The approaching helicase causes the transcription factor to dissociate. Due to the fact that the lagging strand is still being processed, the transcription factor will preferentially bind the leading strand. (B) Once bound, the transcription factor can initiate transcription, which helps structure and order the nucleosomes on the leading strand. (C) The leading strand now has ordered, phased nucleosomes whereas the lagging strand, due to lack of transcription, still has disordered nucleosomes.
Interestingly, recent studies on the inheritance of transcriptional memory in Drosophila have revealed that differences in transcription in sister chromatids may be reliably inherited throughout the process of cell division. Using a transgene containing a suboptimal Snail enhancer element driving synthesis of an MS2-hairpin reporter, researchers observed that following cell division, daughter cells derived from an actively transcribing mother cell were able to initiate transgene transcription significantly earlier than daughter cells derived from a non-transcribing mother. Furthermore, one of the two daughters of the transcribing mother showed transcription initiation significantly earlier than the other daughter, a finding that suggests that memory of transcriptional activity is preserved and inherited on only one of the two sister chromatids containing the transgene reporter element. Researchers went on to demonstrate that differences in epigenetic information on sister chromatids are likely the source of the observed asymmetries in resumption of transcription following mitotic division (Ferraro et al., 2016).
What is the source of epigenetic asymmetries between sister chromatids that regulate asymmetric transcription initiation following mitosis? Based on previously discussed findings (Vasseur et al., 2016), we hypothesize that DNA replication may play a role in establishing transcriptional asymmetries between sister chromatids. Due to the role that replicative history can have on the resumption of transcription following replication fork passage (described above), asymmetries present during sister chromatid synthesis may play an important role in establishing asymmetric epigenetic states during DNA replication. To explore this intriguing possibility, we would like to propose the following hypothetical example:
Following replication fork passage, a co-directional gene reestablishes TF binding and initiates transcription on the leading strand-synthesized sister chromatid while the complementary lagging strand-synthesized sister chromatid continues to undergo chromatin maturation (Vasseur et al., 2016). If no transcription factor binds the lagging strand before chromatin fully matures, it is conceivable that nucleosomes could invade enhancer/promotor binding sites, thereby reducing the likelihood of initiation of transcription (Thurman et al., 2012, Luebben et al., 2010). Furthermore, active transcription on the leading strand could introduce chromatin modification capable of reinforcing transcriptional activity. Such chromatin modifications could include deposition of histone marks associated with gene activation (H3K4me3) as well as replacement of histone H3 with the replication-associated variant (H3.3) (Buratowski and Kim, 2010, Chen et al., 2013, Ahmad and Henikoff, 2002). If the lagging strand-synthesized sister chromatid fails to reinitiate transcription (or does so at a reduced rate), it is conceivable that these chromatin modifications would never be deposited, leading to a relative enrichment of these modifications on the leading strand-synthesized sister (Figure 5). As H3K4me3 and H3.3 can be maintained and inherited through the process of mitosis (Muramoto et al., 2010), differences on both of these heritable modifications (H3K4me3 and H3.3) between leading and lagging strand synthesized sister chromatids could serve as a means to bias transcriptional activity in daughter nuclei following mitosis. This hypothesis is supported by observations suggesting that once asymmetries between sister chromatids are established, they can be reliably inherited through mitosis in a manner that impacts daughter cell gene regulation (Ferraro et al., 2016). These findings hint at the intriguing possibility that cells might be able to use asymmetries present during DNA replication to ensure asymmetric transcriptional outcomes in daughter cell genomes.
Figure 5. Asymmetric chromatin maturation leads to epigenetic asymmetries on sister chromatids.
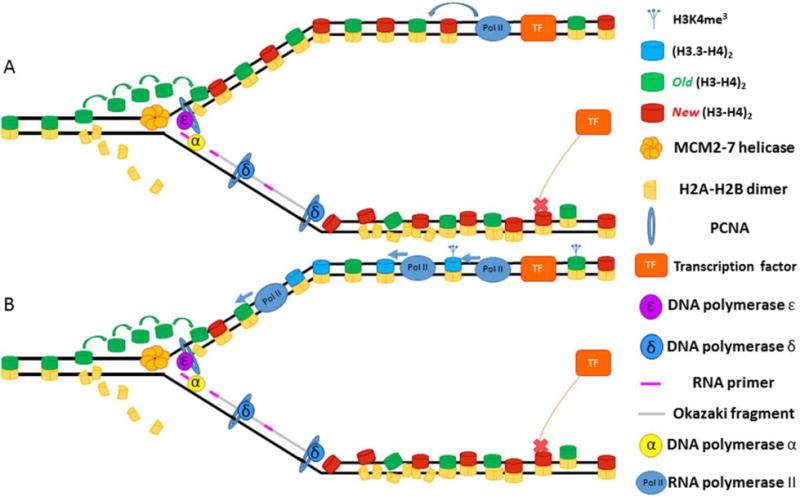
(A) One the leading strand, the transcription factor (TF) binds and initiates transcription, which helps structure and order the nucleosomes on the leading strand. On the lagging strand, TF binding fails to occur in a timely manner, allowing nucleosomes to encroach upon binding site. (B) Transcription on the leading strand leads to deposition of marks associated with transcription such as H3 variant H3.3 and tri-methylation of the 4th lysine on histone H3 (H3K4me3). The transcriptionally-inert lagging strand receives none of these chromatin modifications.
While such a mechanism would be ideal for patterning gene expression during ACD, a global epigenetic asymmetry between sister chromatids seems ill-suited for the stable propagation of the parental epigenetic states required for symmetric cell division. More specifically, if the asymmetries inherent in the replication fork can be stably propagated and inherited throughout cell division, how then does a cell ensure that key genetic regions that should be expressed at similar levels in daughter cells achieve a more similar level of activity post–S phase and post-mitotically? One potential answer lies in the role that cohesins may play during S phase to ensure that nucleosome-free regions (NFRs) are not lost in the process of genome duplication (Figure 6). As transcription factors must compete with histones for binding sites in the eukaryotic genome, a mechanism must be present to ensure that regions normally occupied by transcription factors do not disappear as a result of promiscuous nucleosome incorporation during DNA replication. Recent studies have found evidence that, in regions in which a host of transcription factors are reported to bind, cohesins also bind after replication fork passage to maintain NFRs and ensure nucleosome organization proximal to transcription factor hot spots (Yan et al., 2013). As cohesins encircle both sister chromatids following fork passage, we hypothesize that cohesin binding could serve as a mechanism to buffer any molecular/temporal differences present in the construction of the two sister chromatids to ensure that both sisters maintain NFRs at key genomic regions. As cohesin binding sites differ among different cell types, different cells may utilize cohesin binding as a way to ensure uniform expression patterns among housekeeping genes as well as more lineage-specific genes needed for cell type specification. While it is unclear whether cohesin plays a direct role in buffering strand asymmetries created in the process of DNA replication, studies of cohesin mutants have revealed that loss of cohesin severely disrupts the chromatin landscape in a manner which leads to gene misregulation. Furthermore, gene misregulation resulting from loss of cohesin function has been linked to several human disorders including Cornelia de Lange syndrome and Coffin-Siris syndrome; both of which have been shown to have significant alternations to chromatin landscape (Lopez-Vernaza and Leach, 2013).
Figure 6. Cohesins maintain nucleosome free regions (NFRs) in the wake of the replication fork.
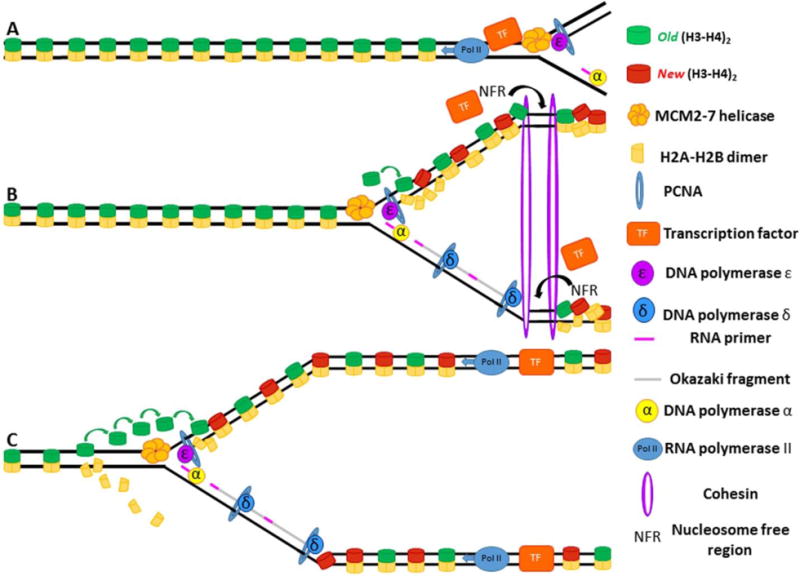
(A) The replisome approaches a bound transcription factor, causing the transcription factor to dissociate. (B) Cohesin binds immediately after the passage of the replication fork and prevents histones from invading transcription factor binding sites, maintaining them as nucleosome free regions. (C) Transcription factors can now bind leading and lagging strands and initiate synthesis in the wake of replication fork passage.
3.3. Nucleosomes in DNA Replication
As with other components of chromatin, the process of DNA replication presents a challenge for cells to effectively maintain epigenetic information due to the fact that nucleosomes must be disassembled and stripped from DNA to allow for progression of the replication fork (McKnight and Miller, 1977, Sogo et al., 1986). The assembly of newly synthesized DNA into chromatin, termed replication-coupled nucleosome assembly, is defined by the coordination of two processes: recycling of old histones and incorporation of new histones (Burgess and Zhang, 2013, Bannister and Kouzarides, 2011) (Figure 7). Although significant progress has been made in understanding the process by which new histones are incorporated, much less is known regarding the molecular mechanisms by which old histones are recycled. As old histones contain posttranslational modifications (PTMs) capable of regulating gene expression, a better understanding of histone recycling should provide invaluable insights into understanding how the process of DNA replication impacts epigenetic inheritance. Several models have been proposed to explain the how old histones are efficiently recycled after passage of the replication fork.
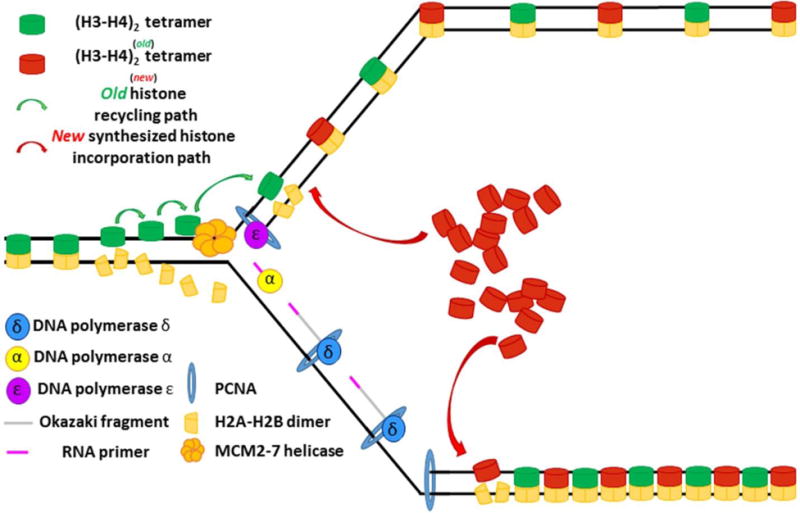
Figure 7. Overview of replication-coupled nucleosome assembly.
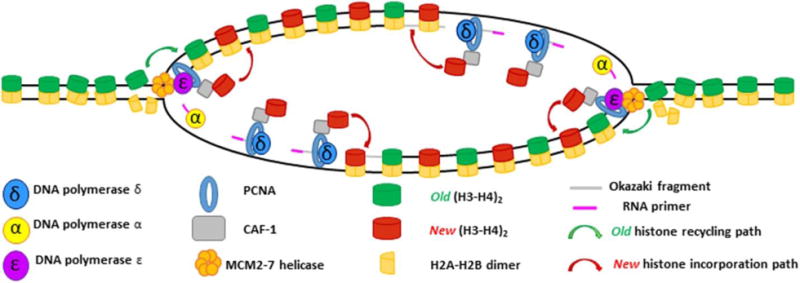
The MCM2-7 helicase sits at the foremost edge of the replication fork where its primary function is to unwind non-replicated DNA. Torsional strain ahead of the fork begins to break apart nucleosomes, which must then be recycled to newly synthesized DNA on the other side of the fork. The MCM2 protein can bind (H3–H4)2 tetramers dissociated in the wake of the advancing fork to allow for their subsequent deposition on nascent DNA. Due to its ability to bind (H3–H4)2 tetramers, CAF-1 may help coordinate the deposition of preexisting histones after fork passage. CAF-1 is recruited to the edge of the advancing fork by PCNA, which also serves as a processivity factor for the replicative polymerases ε and δ. CAF-1 then coordinates new H3–H4 deposition onto nascent DNA. The subsequent incorporation of two (H2A-H2B) dimers reforms the nucleosome structure and signifies the end of the process of replication coupled histone deposition.
3.3.1. Three models for histone recycling: Semi-conservative model
The histone octamer is composed of one (H3–H4)2 tetramer and two (H2A-H2B) dimers. As a majority of the PTMs known to regulate gene expression have been found on the N-terminal tails of H3 and H4, many studies of histone inheritance have focused primarily on understanding the behavior of the (H3–H4)2 tetramer as it is displaced and recycled during passage of the replication fork (Hammond et al., 2017). The quasi-symmetry of the histone octamer initially prompted researchers to propose the semiconservative model of histone inheritance, in which the (H3–H4)2 tetramer would be split into two dimers, each of which would be inherited by newly synthesized sister chromatids. The model further postulated that each of the (H3–H4) dimers would contain the relevant epigenetic information necessary to propagate the parental chromatin state to each of the newly synthesized sister chromatids (Zhu and Reinberg, 2011) (Figure 8A). However, recent findings suggest that semiconservative histone inheritance is not the primary means by which epigenetic information is passed from the mother strand to each of the newly synthesized daughter strands. Mass spectrometry analysis of pre-existing and newly synthesized histones has revealed several key features of the (H3–H4)2 tetramer and its behavior during DNA replication which argue against a semiconservative mode of histone inheritance: First and foremost, (H3–H4)2 tetramers infrequently split, meaning that mixed tetramers [comprising old and new (H3–H4) dimers] rarely ever form. Second, the (H3–H4) dimers which comprise the (H3–H4)2 tetramers are not symmetrically modified (van Rossum et al., 2012, Voigt et al., Chen et al., 2011). This means that even in the case of a rare (H3–H4)2 splitting event, the likelihood of both (H3–H4) dimers containing the same PTMs is extremely low (Xu et al., 2010). Interestingly, tetramers containing the histone H3 variant H3.3 show a higher frequency of splitting than do H3-containing tetramers (~23% for H3.3 versus ~3% for H3). Furthermore, these splitting events appear to be associated with cell type–specific enhancer elements (Huang et al., 2013), suggesting that, although semi-conservative histone inheritance is unlikely to regulate epigenetic inheritance on a genome-wide scale, semi-conservative histone inheritance could play an important role in propagating epigenetic information at particular genomic loci.
Figure 8. Different models for histone inheritance.
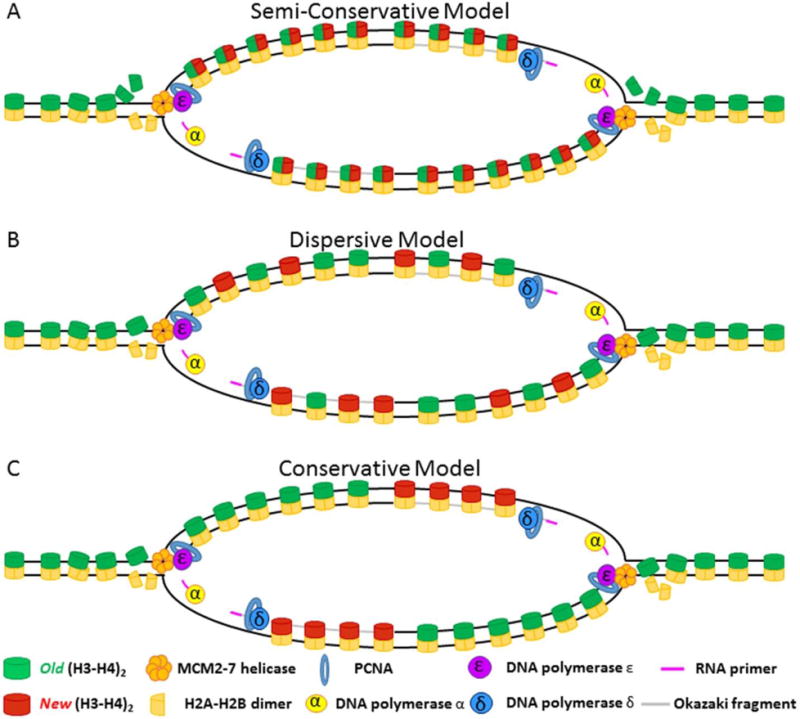
(A) Semi-conservative model: Old (H3–H4)2 tetramers are split at the replication fork, allowing (H3–H4) dimers to be inherited equally on both the leading and the lagging strand. New (H3–H4) dimers pair with old (H3–H4) dimers to recreate the tetramer structure. (B) Dispersive model: Old (H3–H4)2 tetramers remain together at the fork and are randomly segregated to leading or lagging strand in roughly equal numbers. New histones fill in gaps left by old histones to reconstitute nucleosome density. (C) Conservation model: Old (H3–H4)2 tetramers remain un-split at the fork and are inherited as a tetramer. In this model, tetramers are biased in their inheritance such that either the leading strand (shown) or the lagging strand (not shown) inherits a majority of the old (H3–H4)2 tetramers. New histones fill in gaps left by old histones to reconstitute nucleosome density.
3.3.2. Three models for histone recycling: Dispersive model
If semi-conservative histone inheritance is confined to very limited regions of genome replication, how then are the vast majority of histones recycled during DNA replication? A majority of research both past and present supports the dispersive model of histone inheritance (Figure 8B) (Jackson and Chalkley, 1981, Jackson and Chalkley, 1985, Herz et al., 2014, Hammond et al., 2017, Alabert and Groth, 2012, Alabert et al., 2015). According to this model, (H3–H4)2 tetramers dislodged by the advancing helicase remain intact during the process of nucleosome breakdown and histone recycling. Old (H3–H4)2 tetramers are segregated randomly at the fork, such that approximately half of the (H3–-H4)2 displaced from the parental strand are recycled to the leading strand and the other half are incorporated onto the lagging strand (Figure 8B). New histones are incorporated as necessary to maintain nucleosome density. As new histones are largely devoid of PTMs (Lin et al., 2016), old histones must play an instructive role by assisting new histones in the process of acquiring the appropriate epigenetic signatures needed to recapitulate the parental chromatin state. In support of this hypothesis, chromatin maturation studies have demonstrated that for a variety of PTMs, old histones are able to recruit the appropriate histone modifying enzymes needed to established the correct PTMs on newly synthesized histones (Alabert et al., 2015, Alabert and Groth, 2012, Ayyanathan et al., 2003, Hansen et al., 2008, Margueron et al., 2009, Ragunathan et al., 2015, Audergon et al., 2015). In summation, the dispersive model argues that by randomly segregating old histones to both newly synthesized sister chromatids, the epigenetic identity of each genomic region can be maintained and propagating to each daughter cell during the process of cell division.
3.3.3. Three models for histone recycling: Conservative model
Intriguingly, evidence from several studies suggests that histone inheritance may not always proceed in a dispersive fashion. Early studies in chromatin inheritance found that old histones are incorporated in a non-random manner, displaying a strand preference during recycling events (Weintraub, 1976, Seale, 1976, Riley and Weintraub, 1979, Roufa and Marchionni, 1982, Leffak et al., 1977). This model, termed the conservative model of histone segregation, hypothesizes that (H3–H4)2 tetramers are biased in their incorporation at the fork such that either the leading strand (Figure 8C) or the lagging strand preferentially inherits more of old (H3–H4)2 tetramers recycled from the parental strand. Subsequent experiments further specified the nature of the bias in tetramer deposition, indicating that old histones are most often preferentially reincorporated onto the leading strand (Seidman et al., 1979).
How could similar studies into the mechanisms of histone inheritance yield such contrasting results? A closer look at some of the data supporting both models reveals that the story of replication-coupled nucleosome assembly may be more complicated than depicted in our simplified models (Figure 8). In 1979, the team of Riley and Weintraub utilized electron microscopy (EM) to test whether old histones undergo dispersive or conservative segregation following passage of the replication fork (Riley and Weintraub, 1979). With the aid of cyclohexamide to block new histone synthesis, Riley and Weintraub were able to observe the distribution of old histones on newly synthesized sister chromatids in the absence of new histone incorporation. Strikingly, the authors observed that instead of both strands being populated equally by recycled old histones (as predicted by the dispersive model), one strand was densely labeled with recycled histones whereas the other stand consisted of entirely of naked DNA. The asymmetry observed in old histone recycling patterns clearly supports a conservative model of histone segregation. In contrast to these initial findings, a later publication by the team of Jackson and Chalkley found that using alternate fixation conditions, histones could be detected on both nascent sister chromatids following passage of the replication fork. Based on these findings, Jackson and Chalkley propose that non-physiological fixation conditions (low ionic strength, pH 9) destabilize nucleosomes in newly replicated regions of the genome, resulting in a loss of histone proteins in EM images of sister chromatids (Jackson and Chalkley, 1981).
While these observations offer compelling evidence that non-physiological fixation can destabilize newly deposited histones, they do not fully explain the initial asymmetries in histone localization observed by Riley and Weintraub. Indeed, taken together, these observations argue that non-physiological fixation conditions destabilize histones in a strand-specific manner, possibly indicating transient asymmetries in the stability of nucleosomes in the leading strand vs. the lagging strand. While it remains unclear whether recycled histones associate more strongly with the leading strand or the lagging strand, we speculate that if such an asymmetry exists, it could serve as means to bias histone recycling in a more gradient-like manner (Figure 9), rather than the all-or-none outcome that has been suggested previously by conservative DNA replication models (Figure 8C) Furthermore, a more subtle, gradient-like distribution of old histones between the leading and the lagging strand may make asymmetries in present in histone segregation more challenging to detect.
Figure 9. Old histone distribution in the absence of new histone synthesis.
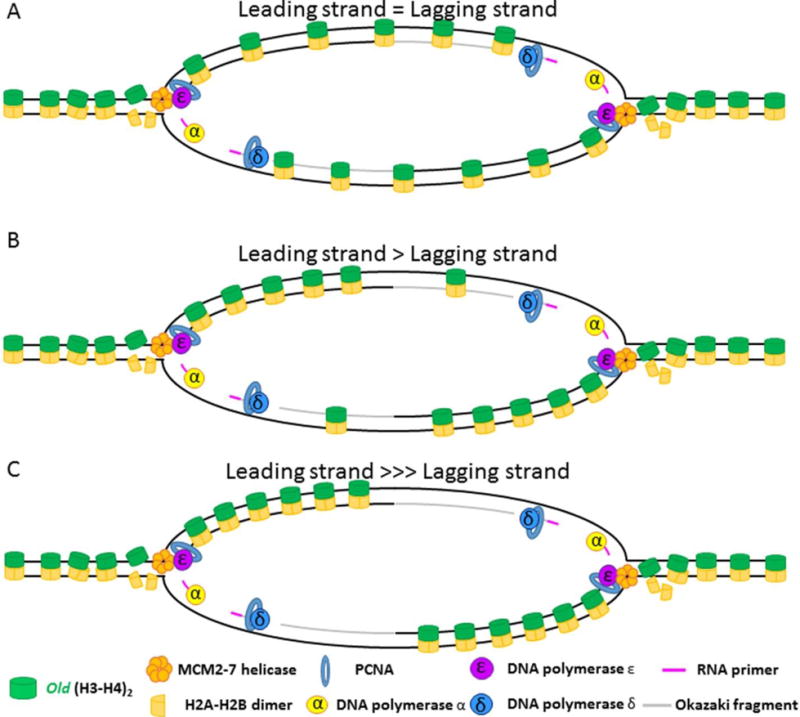
(A) In the event that old histones have an equal likelihood of binding the leading or lagging strand, old histones should distribute themselves on the leading and lagging strands in roughly equal numbers. (B) If the likelihood of stable binding the leading strand is higher than on the lagging strand, then more old histones should be bound to the leading strand in a manner that is proportional to increased favorability of leading strand association. (C) If the lagging strand is incapable of binding old histones in the immediate aftermath of replication fork passage, then all old histones should be recycled onto the leading strand.
Observations of asymmetry in replication-coupled nucleosome assembly (such as those described above) hint at the possibility that asymmetries in the synthesis of the leading strand vs. the lagging strand have the potential to bias histone incorporation during DNA replication (Annunziato, 2013). However, the wealth of published data demonstrating dispersive histone segregation demonstrates that in most conditions, the asymmetries inherent to the process of DNA replication are insufficient to bias histone inheritance. Rather, the fact that evidence can be found supporting both conservative and dispersive models suggests that histone segregation may be a more plastic process than has been previously appreciated. Much in the same way that semi-conservative histone segregation can be detected at specific genomic regions enriched with histone H3.3 (Huang et al., 2013), it is conceivable that at certain genomic loci, conservative DNA segregation may be utilized to generate localized asymmetries in epigenetic information. We speculate that for cells undergoing asymmetric cell division, localized asymmetries at key developmental loci could serve as a valuable intrinsic mechanism to ensure that divergent cell fates are reliably specified during the process of cell division. In the following section, we will discuss several lines of evidence suggesting that DNA replication may be playing a causal role in shaping the epigenetic landscape in preparation for ACD.
3.4. Replication Asymmetries Pattern Cell Fate Decisions in Caenorhabditis elegans
Recent research efforts have revealed an ACD in C. elegans that may utilize molecular asymmetries associated with replication-coupled nucleosome assembly to generate distinct cell fates. A mutagenesis screen targeting factors affecting cell fate decisions during C. elegans development has described a gain-of-function mutation in an H3-encoding gene (his-9) that is sufficient to disrupt neuronal bilateral asymmetry, affecting the formation of the MI motor neuron and the e3D pharyngeal epithelial cell (Sulston et al., 1983). The his-9 (Q125ochre) mutation eliminates two residues, leucine 126 and isoleucine 130, which are thought to engage in the H3-H3 interaction. This mutation disrupts (H3–H4)2 tetramer formation, leading to the inhibition of CAF-1-mediated nucleosome assembly at the replication fork (Figure 7). A similar loss of neuronal asymmetry phenotype could be recapitulated upon knocking down either components of the worm CAF-1 complex PCNA, a finding that further implicates the process of replication-coupled nucleosome assembly in specifying divergent cell fates. These results suggest that replication-coupled nucleosome assembly has an essential role in regulating asymmetric cell fate decision (for example: neuron versus muscle) during C. elegans development (Nakano et al., 2011).
Although the precise mechanisms behind the loss of asymmetric cell fate are yet to be elucidated, the authors of Nakano et al. hypothesize that asymmetries present during DNA replication, specifically nucleosomal density differences hypothesized to exist between the leading and lagging strands, may underlie the divergent cell fate choices made by MI and e3D cells. The elevated density of PCNA molecules present on the lagging strand has led some to propose that the lagging strand may contain a higher nucleosomal density relative to the leading strand (Shibahara and Stillman, 1999, Waga and Stillman, 1998b, Waga and Stillman, 1998a, Yu et al., 2014). The authors speculate that if nucleosomal density differences are present at critical developmental genes, such density differences could drive differential gene expression and distinct cell fate decisions during development (Figure 10). To further explore DNA replication’s impact on histone inheritance, we now discuss interesting recent observations concerning asymmetric segregation of histone proteins during ACD of D. melanogaster germline stem cells (GSCs).
Figure 10. Hypothetical asymmetric nucleosome density in leading vs. lagging strand.
As PCNA has been shown to play a central role in recruiting new histones for deposition on newly synthesized DNA, the increased density of PCNA on the lagging strand could serve to increase histone density on the lagging strand when compared to the leading strand. Increased histone density would chance the chromatin state of these regions, possibly as a way to bias the transcriptional output of the two sister chromatids.
3.5. Asymmetric Histone Inheritance in the Germline of Drosophila melanogaster
In addition to changes in nucleosomal density, many other aspects of chromatin structure germane to cell fate specification could be affected via the process of replication-coupled nucleosome assembly. Recent work has demonstrated that sister chromatids enriched with distinct histone populations are asymmetrically segregated during GSC division. Using a dual-color strategy to label preexisting versus newly synthesized histones, researchers observed that the preexisting H3 is segregated to the male GSCs, whereas newly synthesized H3 is enriched toward the differentiating daughter cell. Subsequent studies have further demonstrated that disruption of asymmetric histone inheritance can lead to adverse phenotypes ranging from cell death to tumorigenesis. Interestingly, asymmetric histone inheritance is specific to the canonical H3, but not to histone variant H3.3 (Figure 11). Because H3 is incorporated during DNA replication whereas H3.3 is incorporated in a replication-independent manner, these findings suggest a role for DNA replication in establishing epigenetic information differentially in preparation for ACD (Tran et al., 2012).
Figure 11. Asymmetric vs. Symmetric histone distribution in Drosophila melanogaster germline stem cells (GSC) sister chromatids.
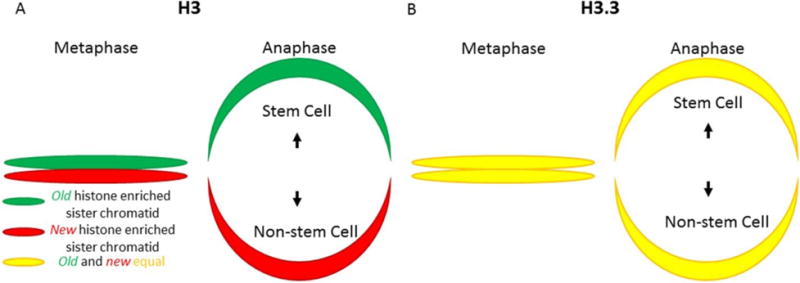
(A) Old H3 are enriched in the sister chromatids inherited by the cell fated to remain a GSC whereas newly-synthesized H3 are enriched in the sister chromatids inherited by the daughter cell destined to differentiate. (B) Old and new H3.3 are present in equal quantities on all sister chromatids and are inherited equally by GSC and non-GSC daughter cells.
These findings suggest that, similar to the mechanisms concerning neuronal cell fate specification described above, asymmetries present in the synthesis of the leading strand versus the lagging strand could be an initial mechanism to bias histone inheritance during DNA replication. As has been hypothesized and previously described for transcription factor binding, the temporal and molecular asymmetries that define continuous (leading-strand) vs. discontinuous (lagging-strand) synthesis could serve as a mechanism to influence the binding of histone proteins following DNA replication. As discussed above, synthesis and processing of the lagging strand require the combined action of many more proteins than for the leading strand. Consequently, the lagging strand (as its name implies) takes longer than the leading strand to generate double-stranded DNA that is capable of being folded around the histone octamer to form the nucleosome structure. The leading strand, in contrast, emerges from the replicative polymerase fully capable of serving as a landing site from histone proteins recently dislodged by the advancing helicase. Whether by the aid of histone chaperones or by hopping from un-replicated strand to replicated strand, if lagging-strand processing were sufficiently delayed, old histones would be biased in their re-association with DNA to preferentially bind the leading strand (Annunziato, 2013, Brennan et al., 2016, Huang et al., 2015). An important consequence of this binding would be that newly synthesized histones would, by default, preferentially incorporate onto the lagging strand. As histone proteins represent a key component of the epigenome, such a mechanism would mean that intrinsic asymmetries present in DNA replication may play an important role in helping to coordinate cellular processes and to regulate cell fate decisions during metazoan development.
3.6. Selective Sister Chromatid Segregation
The asymmetric segregation of sister chromatids enriched with distinct histone species represents a unique example of a previously described phenomenon known as selective sister chromatid segregation. To begin to understand the biological relevance of selective sister chromatid segregation, several different models have been proposed: the immortal strand hypothesis (Cairns, 1975), the silent sister chromatid hypothesis (Lansdorp, 2007), and the strand-specific imprinting and selective chromatid segregation model (Klar, 1994, Klar, 2008, Klar, 2014). All of these models are similar in principle, in that they all involve non-random segregation of sister chromatids during mitosis. The primary difference between these models lies in the molecular features responsible for distinguishing sister chromatids during the process of selective segregation.
The immortal strand hypothesis states that the age of the template serves as the primary feature responsible for differentiating sister chromatids during the process of selective segregation. The theory goes on to posit that by segregating by age of the template strand, continuously dividing populations of cells (stem cells) are able to minimize the accumulation of deleterious mutations which could compromise cellular function (Cairns, 1975, Cairns, 2006). In the context of the immortal strand hypothesis, selective sister chromatid segregation serves a protective role; safeguarding the integrity of the stem cell genome by segregating mutation-prone newly synthesized DNA to differentiating daughter cells.
The silent sister chromatid hypothesis argues that rather than a protective role, selective sister chromatid segregation serves a more instructive role during the processes of development and tissue homeostasis. In this model, sister chromatids carrying distinct epigenetic signatures at specific genomic loci are segregated asymmetrically as a mechanism to bias cell fate outcomes, specifically in the case of ACD events (Lansdorp, 2012, Lansdorp, 2007) (Figure 12, 13). Recent studies in both the Drosophila male GSC system and the mouse satellite cell system have shown biased sister chromatid segregation during stem cell ACDs (Yadlapalli and Yamashita, 2013, Rocheteau et al., 2012). However, this bias applies to either particular chromosomes or all chromosomes, suggesting potential chromosome-specific regulation(s).
Figure 12. Asymmetric vs. symmetric epigenetic regions on metaphase sister chromatids.
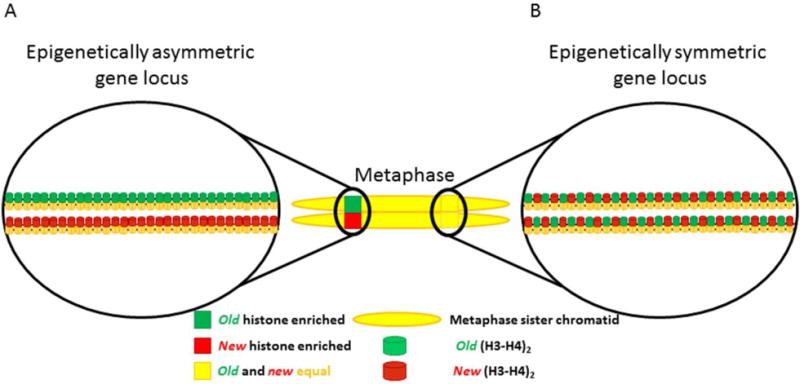
(A) Epigenetically asymmetric gene locus. Epigenetic information is asymmetrically partitioned between the two sister chromatids. Age of histone is shown as a representative epigenetic asymmetry. Other epigenetic modifications such as histone post-translational modifications, nucleosome density, and histone variant incorporation could also establish epigenetically distinct sister chromatid regions. (B) Epigenetically symmetric gene locus. Epigenetic modifications are equally distributed between sister chromatids.
Figure 13. Segregation of asymmetrically modified sister chromatids vs. symmetrically modified sister chromatids during asymmetric cell division.
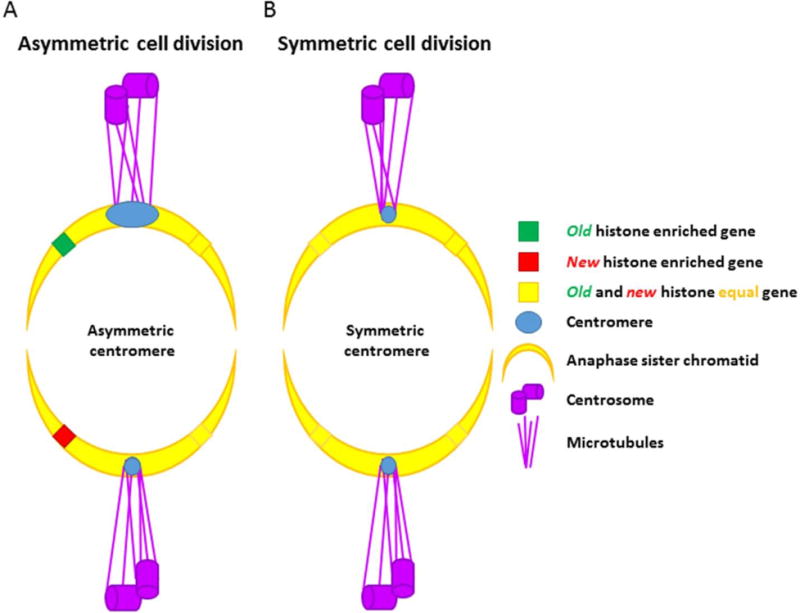
(A) Asymmetric cell division. Sister chromatids bearing asymmetric epigenetic information at key developmental loci are selectively recognized and segregated by asymmetric modifications in the centromeric region. These asymmetries in the centromere could allow the microtubules to bind and recognize these molecularly-distinct sisters in order to segregate them to the appropriate daughter cell. (B) Symmetric cell division. Sister chromatids containing identical epigenetic information have identical centromere structures, and are segregated randomly during the process of cell division.
The strand-specific imprinting and selective chromatid segregation model also argues that epigenetic differences are the primary means by which sister chromatids are selectively segregated (Figure 12, 13). Using a genetic manipulation specifically introduced to mouse chromosome 7, Armakolas and Klar demonstrated that biased sister chromatid segregation patterns appear in a cell type–specific manner (Armakolas and Klar, 2006). The authors of this study further suggest that these sister chromatids contain differential epigenetic information and that these unique segregation patterns may serve as a mechanism of pattern formation during development. Furthermore, these researchers demonstrated that these unique segregation patterns are dependent on the left-right dynein gene, a finding that the authors conclude implicates mitotic machinery in playing a key role in biased sister chromatid segregation (Armakolas and Klar, 2007). However, it is unclear in these disparate systems how distinct sister chromatids are differentially marked and recognized in a manner that would ensure their proper segregation during mitosis. One possible mechanism involves asymmetric DNA replication at the centromeric regions of asymmetrically segregating chromosomes.
In metazoans, the centromere is an epigenetically defined structure marked by the deposition of the histone H3 variant CENP-A, which regulates sister chromatid segregation during mitosis (McKinley and Cheeseman, 2016). Asymmetries inherent in the process of DNA replication have been invoked as a possible mechanism by which sister chromatid centromeres could be asymmetrically marked to allow for selective segregation during mitosis. The temporal and molecular differences present in the continuous synthesis of the leading strand versus the discontinuous synthesis of the lagging strand could provide asymmetrically dividing cells with the necessary tools to mark sister chromatids in a molecularly distinct fashion, likely through the asymmetric segregation of key chromatin factors. It has been further proposed that, to ensure uniform asymmetry across the centromeric region, replication forks could be biased in their progression to ensure that one of the two sisters is replicated in a predominantly leading-strand mode of synthesis, whereas the other sister chromatid is replicated largely via discontinuous lagging-strand synthesis (Lew et al., 2008). Although whether centromeric regions are replicated in such a coordinated fashion has yet to be demonstrated, investigations have revealed that epigenetic asymmetries underlie the proper recognition and segregation of sister chromatids bearing distinct asymmetric signatures during ACD of the Drosophila GSC (Figure 13). The team of Xie et al. was recently able to demonstrate that a peri-centromere-enriched, mitosis-specific phosphorylation of threonine three in H3 (H3T3P) selectively labels old histones during the transition from prophase to metaphase (Xie et al., 2015, Lin et al., 2016). Furthermore, the authors found that this asymmetry serves as a mechanism to allow the mitotic spindle to recognize sister chromatids enriched with different populations of histones as a means to assure their proper segregation during GSC ACD.
4. CONCLUSIONS: ASYMMETRY OF DNA REPLICATION AS A TOOLKIT
Life accomplishes a huge diversity of tasks using simple building blocks, organized and manipulated through millennia of selective pressures to ever more precisely match form and function. The structure of DNA represents one of these building blocks. In the past, we gained a great appreciation for how the structure of the double-stranded DNA molecule allows it to serve the essential function of genome duplication. Recent work has given us an appreciation for how the structure of DNA may be functioning to assist in other processes necessary for growth and development. By virtue of the structure of DNA, the process of DNA replication is able to serve as a fundamental tool for allowing life to create heterogeneity. In unicellular organisms, the asymmetry of DNA replication is used to drive gene evolution, maximizing potentially beneficial mutations and limiting deleterious mutations, and can even be used to drive processes like mating type switches in yeast. In metazoans, DNA replication contributes to the crucial generation of asymmetric cell fates which have been demonstrated to play a critical role in asymmetric stem cell division and development. Going forward, it will be essential to test these hypothesis more rigorously in the context of asymmetric cell divisions to determine the extent to which asymmetries present during DNA replication impact asymmetric cell fate outcomes essential to the process of growth and development.
Footnotes
The authors declare no conflicts of interest.
References
- AHMAD K, HENIKOFF S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9:1191–200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- ALABERT C, BARTH TK, REVERON-GOMEZ N, SIDOLI S, SCHMIDT A, JENSEN ON, IMHOF A, GROTH A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015;29:585–90. doi: 10.1101/gad.256354.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALABERT C, GROTH A. Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol. 2012;13:153–67. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- ALBERTSON TM, OGAWA M, BUGNI JM, HAYS LE, CHEN Y, WANG Y, TREUTING PM, HEDDLE JA, GOLDSBY RE, PRESTON BD. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106:17101–4. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALLIS CD, JENUWEIN T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- ANNUNZIATO AT. Assembling chromatin: the long and winding road. Biochim Biophys Acta. 2013;1819:196–210. doi: 10.1016/j.bbagrm.2011.07.005. [DOI] [PubMed] [Google Scholar]
- ARCANGIOLI B, DE LAHONDES R. Fission yeast switches mating type by a replication-recombination coupled process. EMBO J. 2000;19:1389–96. doi: 10.1093/emboj/19.6.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARMAKOLAS A, KLAR AJ. Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis. Science. 2006;311:1146–9. doi: 10.1126/science.1120519. [DOI] [PubMed] [Google Scholar]
- ARMAKOLAS A, KLAR AJ. Left-right dynein motor implicated in selective chromatid segregation in mouse cells. Science. 2007;315:100–1. doi: 10.1126/science.1129429. [DOI] [PubMed] [Google Scholar]
- AUDERGON PN, CATANIA S, KAGANSKY A, TONG P, SHUKLA M, PIDOUX AL, ALLSHIRE RC. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science. 2015;348:132–5. doi: 10.1126/science.1260638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AVERY OT, MACLEOD CM, MCCARTY M. STUDIES ON THE CHEMICAL NATURE OF THE SUBSTANCE INDUCING TRANSFORMATION OF PNEUMOCOCCAL TYPES: INDUCTION OF TRANSFORMATION BY A DESOXYRIBONUCLEIC ACID FRACTION ISOLATED FROM PNEUMOCOCCUS TYPE III. J Exp Med. 1944;79:137–58. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AYYANATHAN K, LECHNER MS, BELL P, MAUL GG, SCHULTZ DC, YAMADA Y, TANAKA K, TORIGOE K, RAUSCHER FJ., 3RD Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 2003;17:1855–69. doi: 10.1101/gad.1102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALAKRISHNAN L, BAMBARA RA. Okazaki Fragment Metabolism. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAMBARA RA, MURANTE RS, HENRICKSEN LA. Enzymes and reactions at the eukaryotic DNA replication fork. J Biol Chem. 1997;272:4647–50. doi: 10.1074/jbc.272.8.4647. [DOI] [PubMed] [Google Scholar]
- BANNISTER AJ, KOUZARIDES T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELL SP, STILLMAN B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357:128–34. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- BESSMAN MJ, KORNBERG A, LEHMAN IR, SIMMS ES. Enzymic synthesis of deoxyribonucleic acid. Biochim Biophys Acta. 1956;21:197–8. doi: 10.1016/0006-3002(56)90127-5. [DOI] [PubMed] [Google Scholar]
- BESSMAN MJ, LEHMAN IR, SIMMS ES, KORNBERG A. Enzymatic synthesis of deoxyribonucleic acid. II. General properties of the reaction. J Biol Chem. 1958;233:171–7. [PubMed] [Google Scholar]
- BIRD A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- BRENNAN LD, FORTIES RA, PATEL SS, WANG MD. DNA looping mediates nucleosome transfer. Nat Commun. 2016;7:13337. doi: 10.1038/ncomms13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURATOWSKI S, KIM T. The role of cotranscriptional histone methylations. Cold Spring Harb Symp Quant Biol. 2010;75:95–102. doi: 10.1101/sqb.2010.75.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURGESS RJ, ZHANG Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAIRNS J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- CAIRNS J. Cancer and the Immortal Strand Hypothesis. Genetics. 2006;174:1069–72. doi: 10.1534/genetics.104.66886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAYROU C, COULOMBE P, VIGNERON A, STANOJCIC S, GANIER O, PEIFFER I, RIVALS E, PUY A, LAURENT-CHABALIER S, DESPRAT R, MECHALI M. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011;21:1438–49. doi: 10.1101/gr.121830.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CERRITELLI SM, CROUCH RJ. Ribonuclease H: the enzymes in Eukaryotes. Febs j. 2009;276:1494–505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN P, ZHAO J, WANG Y, WANG M, LONG H, LIANG D, HUANG L, WEN Z, LI W, LI X, FENG H, ZHAO H, ZHU P, LI M, WANG QF, LI G. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013;27:2109–24. doi: 10.1101/gad.222174.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN X, XIONG J, XU M, CHEN S, ZHU B. Symmetrical modification within a nucleosome is not required globally for histone lysine methylation. EMBO Rep. 2011;12:244–51. doi: 10.1038/embor.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALGAARD JZ, KLAR AJ. Orientation of DNA replication establishes mating-type switching pattern in S. pombe. Nature. 1999;400:181–4. doi: 10.1038/22139. [DOI] [PubMed] [Google Scholar]
- DALGAARD JZ, KLAR AJS. Does S. pombe exploit the intrinsic asymmetry of DNA synthesis to imprint daughter cells for mating-type switching? Trends in Genetics. 2001;17:153–157. doi: 10.1016/s0168-9525(00)02203-4. [DOI] [PubMed] [Google Scholar]
- EGEL R. DNA replication: stalling a fork for imprinting and switching. Curr Biol. 2004;14:R915–7. doi: 10.1016/j.cub.2004.10.012. [DOI] [PubMed] [Google Scholar]
- FERRARO T, ESPOSITO E, MANCINI L, NG S, LUCAS T, COPPEY M, DOSTATNI N, WALCZAK AM, LEVINE M, LAGHA M. Transcriptional Memory in the Drosophila Embryo. Curr Biol. 2016;26:212–8. doi: 10.1016/j.cub.2015.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIJALKOWSKA IJ, JONCZYK P, TKACZYK MM, BIALOSKORSKA M, SCHAAPER RM. Unequal fidelity of leading strand and lagging strand DNA replication on the Escherichia coli chromosome. Proceedings of the National Academy of Sciences. 1998;95:10020–10025. doi: 10.1073/pnas.95.17.10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANCINO MP, OCHMAN H. Strand asymmetries in DNA evolution. Trends Genet. 1997;13:240–5. doi: 10.1016/S0168-9525(97)01118-9. [DOI] [PubMed] [Google Scholar]
- FRANKLIN RE, GOSLING RG. Evidence for 2-chain helix in crystalline structure of sodium deoxyribonucleate. Nature. 1953a;172:156–7. doi: 10.1038/172156a0. [DOI] [PubMed] [Google Scholar]
- FRANKLIN RE, GOSLING RG. Molecular configuration in sodium thymonucleate. Nature. 1953b;171:740–1. doi: 10.1038/171740a0. [DOI] [PubMed] [Google Scholar]
- FUJIHARA I, FURUSAWA M. Disparity mutagenesis model possesses the ability to realize both stable and rapid evolution in response to changing environments without altering mutation rates. Heliyon. 2016;2 doi: 10.1016/j.heliyon.2016.e00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FULLER RS, FUNNELL BE, KORNBERG A. The dnaA protein complex with the E. coli chromosomal replication origin (oriC) and other DNA sites. Cell. 1984;38:889–900. doi: 10.1016/0092-8674(84)90284-8. [DOI] [PubMed] [Google Scholar]
- FURUSAWA M. Implications of fidelity difference between the leading and the lagging strand of DNA for the acceleration of evolution. Front Oncol. 2012;2:144. doi: 10.3389/fonc.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURUSAWA M. The disparity mutagenesis model predicts rescue of living things from catastrophic errors. Front Genet. 2014;5:421. doi: 10.3389/fgene.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURUSAWA M, DOI H. Promotion of evolution: disparity in the frequency of strand-specific misreading between the lagging and leading DNA strands enhances disproportionate accumulation of mutations. J Theor Biol. 1992;157:127–33. doi: 10.1016/s0022-5193(05)80761-1. [DOI] [PubMed] [Google Scholar]
- FURUSAWA M, DOI H. Asymmetrical DNA replication promotes evolution: disparity theory of evolution. Genetica. 1998;102–103:333–47. [PubMed] [Google Scholar]
- GAMBUS A, JONES RC, SANCHEZ-DIAZ A, KANEMAKI M, VAN DEURSEN F, EDMONDSON RD, LABIB K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 2006;8:358–366. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- GREER EL, BLANCO MA, GU L, SENDINC E, LIU J, ARISTIZABAL-CORRALES D, HSU CH, ARAVIND L, HE C, SHI Y. DNA Methylation on N6-Adenine in C. elegans. Cell. 2015;161:868–78. doi: 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HABER JE. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics. 2012;191:33–64. doi: 10.1534/genetics.111.134577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMMOND CM, STROMME CB, HUANG H, PATEL DJ, GROTH A. Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol. 2017;18:141–158. doi: 10.1038/nrm.2016.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANSEN KH, BRACKEN AP, PASINI D, DIETRICH N, GEHANI SS, MONRAD A, RAPPSILBER J, LERDRUP M, HELIN K. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- HANSON SJ, WOLFE KH. An Evolutionary Perspective on Yeast Mating-Type Switching. Genetics. 2017;206:9–32. doi: 10.1534/genetics.117.202036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HATEBOER G, WOBST A, PETERSEN BO, LE CAM L, VIGO E, SARDET C, HELIN K. Cell Cycle-Regulated Expression of Mammalian CDC6 Is Dependent on E2F. Mol Cell Biol. 1998;18:6679–97. doi: 10.1128/mcb.18.11.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HELMRICH A, BALLARINO M, NUDLER E, TORA L. Transcription-replication encounters, consequences and genomic instability. Nat Struct Mol Biol. 2013;20:412–8. doi: 10.1038/nsmb.2543. [DOI] [PubMed] [Google Scholar]
- HELMRICH A, BALLARINO M, TORA L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011;44:966–77. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- HERZ HM, MORGAN M, GAO X, JACKSON J, RICKELS R, SWANSON SK, FLORENS L, WASHBURN MP, EISSENBERG JC, SHILATIFARD A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. 2014;345:1065–70. doi: 10.1126/science.1255104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLMES AM, KAYKOV A, ARCANGIOLI B. Molecular and Cellular Dissection of Mating-Type Switching Steps in Schizosaccharomyces pombe. Mol Cell Biol. 2005;25:303–11. doi: 10.1128/MCB.25.1.303-311.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG C, ZHANG Z, XU M, LI Y, LI Z, MA Y, CAI T, ZHU B. H3.3-H4 tetramer splitting events feature cell-type specific enhancers. PLoS Genet. 2013;9:e1003558. doi: 10.1371/journal.pgen.1003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG H, STROMME CB, SAREDI G, HODL M, STRANDSBY A, GONZALEZ-AGUILERA C, CHEN S, GROTH A, PATEL DJ. A unique binding mode enables MCM2 to chaperone histones H3–H4 at replication forks. Nat Struct Mol Biol. 2015;22:618–26. doi: 10.1038/nsmb.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUBSCHER U, MAGA G, SPADARI S. Eukaryotic DNA polymerases. Annu Rev Biochem. 2002;71:133–63. doi: 10.1146/annurev.biochem.71.090501.150041. [DOI] [PubMed] [Google Scholar]
- HUBSCHER U, SEO YS. Replication of the lagging strand: a concert of at least 23 polypeptides. Mol Cells. 2001;12:149–57. [PubMed] [Google Scholar]
- JACKSON V, CHALKLEY R. A new method for the isolation of replicative chromatin: selective deposition of histone on both new and old DNA. Cell. 1981;23:121–34. doi: 10.1016/0092-8674(81)90277-4. [DOI] [PubMed] [Google Scholar]
- JACKSON V, CHALKLEY R. Histone segregation on replicating chromatin. Biochemistry. 1985;24:6930–8. doi: 10.1021/bi00345a027. [DOI] [PubMed] [Google Scholar]
- JOHNSON RE, KLASSEN R, PRAKASH L, PRAKASH S. A Major Role of DNA Polymerase delta in Replication of Both the Leading and Lagging DNA Strands. Mol Cell. 2015;59:163–75. doi: 10.1016/j.molcel.2015.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAR AJ. Differentiated parental DNA strands confer developmental asymmetry on daughter cells in fission yeast. Nature. 1987;326:466–70. doi: 10.1038/326466a0. [DOI] [PubMed] [Google Scholar]
- KLAR AJ. The developmental fate of fission yeast cells is determined by the pattern of inheritance of parental and grandparental DNA strands. EMBO J. 1990;9:1407–15. doi: 10.1002/j.1460-2075.1990.tb08256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAR AJ. A model for specification of the left-right axis in vertebrates. Trends Genet. 1994;10:392–6. doi: 10.1016/0168-9525(94)90055-8. [DOI] [PubMed] [Google Scholar]
- KLAR AJ. Support for the selective chromatid segregation hypothesis advanced for the mechanism of left-right body axis development in mice. Breast Dis. 2008;29:47–56. doi: 10.3233/bd-2008-29106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAR AJ. The yeast mating-type switching mechanism: a memoir. Genetics. 2010;186:443–9. doi: 10.1534/genetics.110.122531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAR AJ, BONADUCE MJ, CAFFERKEY R. The mechanism of fission yeast mating type interconversion: seal/replicate/cleave model of replication across the double-stranded break site at mat1. Genetics. 1991;127:489–96. doi: 10.1093/genetics/127.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAR AJS. Selective Chromatid Segregation Mechanism Invoked For the Human Congenital Mirror Hand Movement Disorder Development by RAD51 Mutations: A Hypothesis. Int J Biol Sci. 2014;10:1018–23. doi: 10.7150/ijbs.9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KORNBERG A, LEHMAN IR, BESSMAN MJ, SIMMS ES. Enzymic synthesis of deoxyribonucleic acid. 1956. Biochim Biophys Acta. 1989;1000:57–8. [PubMed] [Google Scholar]
- KORONA DA, LECOMPTE KG, PURSELL ZF. The high fidelity and unique error signature of human DNA polymerase ε. Nucleic Acids Research. 2011;39:1763–1773. doi: 10.1093/nar/gkq1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KULIS M, ESTELLER M. 2 - DNA Methylation and Cancer. In: ZDENKO H, TOSHIKAZU U, editors. Advances in Genetics. Academic Press; 2010. [DOI] [PubMed] [Google Scholar]
- KUNKEL TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–92. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LABIB K, TERCERO JA, DIFFLEY JFX. Uninterrupted MCM2-7 Function Required for DNA Replication Fork Progression. Science. 2000;288:1643. doi: 10.1126/science.288.5471.1643. [DOI] [PubMed] [Google Scholar]
- LANSDORP PM. Immortal strands? Give me a break. Cell. 2007;129:1244–7. doi: 10.1016/j.cell.2007.06.017. [DOI] [PubMed] [Google Scholar]
- LANSDORP PM. Epigenetic differences between sister chromatids? 2012;1266:1–6. doi: 10.1111/j.1749-6632.2012.06505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LARK KG. Evidence for the direct involvement of RNA in the initiation of DNA replication in Escherichia coli 15T. J Mol Biol. 1972a;64:47–60. doi: 10.1016/0022-2836(72)90320-8. [DOI] [PubMed] [Google Scholar]
- LARK KG. Genetic control over the initiation of the synthesis of the short deoxynucleotide chains in E. coli. Nat New Biol. 1972b;240:237–40. doi: 10.1038/newbio240237a0. [DOI] [PubMed] [Google Scholar]
- LEFFAK IM, GRAINGER R, WEINTRAUB H. Conservative assembly and segregation of nucleosomal histones. Cell. 1977;12:837–45. doi: 10.1016/0092-8674(77)90282-3. [DOI] [PubMed] [Google Scholar]
- LEHMAN IR, BESSMAN MJ, SIMMS ES, KORNBERG A. Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J Biol Chem. 1958;233:163–70. [PubMed] [Google Scholar]
- LEONHARDT H, RAHN HP, WEINZIERL P, SPORBERT A, CREMER T, ZINK D, CARDOSO MC. Dynamics of DNA replication factories in living cells. J Cell Biol. 2000;149:271–80. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEW DJ, BURKE DJ, DUTTA A. The immortal strand hypothesis: how could it work? Cell. 2008;133:21–3. doi: 10.1016/j.cell.2008.03.016. [DOI] [PubMed] [Google Scholar]
- LIN S, YUAN ZF, HAN Y, MARCHIONE DM, GARCIA BA. Preferential Phosphorylation on Old Histones during Early Mitosis in Human Cells. J Biol Chem. 2016;291:15342–57. doi: 10.1074/jbc.M116.726067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU B, ALBERTS B. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995;267:1131–1137. doi: 10.1126/science.7855590. [DOI] [PubMed] [Google Scholar]
- LOPEZ-VERNAZA MA, LEACH DR. Symmetries and asymmetries associated with non-random segregation of sister DNA strands in Escherichia coli. Semin Cell Dev Biol. 2013;24:610–7. doi: 10.1016/j.semcdb.2013.05.010. [DOI] [PubMed] [Google Scholar]
- LOW DA, WEYAND NJ, MAHAN MJ. Roles of DNA Adenine Methylation in Regulating Bacterial Gene Expression and Virulence. Infect Immun. 2001;69:7197–204. doi: 10.1128/IAI.69.12.7197-7204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUEBBEN WR, SHARMA N, NYBORG JK. Nucleosome eviction and activated transcription require p300 acetylation of histone H3 lysine 14. Proc Natl Acad Sci U S A. 2010;107:19254–9. doi: 10.1073/pnas.1009650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARGUERON R, JUSTIN N, OHNO K, SHARPE ML, SON J, DRURY WJ, 3RD, VOIGT P, MARTIN SR, TAYLOR WR, DE MARCO V, PIRROTTA V, REINBERG D, GAMBLIN SJ. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–7. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASAI H, MATSUMOTO S, YOU Z, YOSHIZAWA-SUGATA N, ODA M. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- MCKINLEY KL, CHEESEMAN IM. The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol. 2016;17:16–29. doi: 10.1038/nrm.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKNIGHT SL, MILLER OL., JR Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell. 1977;12:795–804. doi: 10.1016/0092-8674(77)90278-1. [DOI] [PubMed] [Google Scholar]
- MESELSON M, STAHL FW. The replication of DNA in Escherichia coli. Proceedings of the National Academy of Sciences. 1958;44:671–682. doi: 10.1073/pnas.44.7.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLDOVAN GL, PFANDER B, JENTSCH S. PCNA controls establishment of sister chromatid cohesion during S phase. Mol Cell. 2006;23:723–32. doi: 10.1016/j.molcel.2006.07.007. [DOI] [PubMed] [Google Scholar]
- MOYER SE, LEWIS PW, BOTCHAN MR. Isolation of the Cdc45/Mcm2– 7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proceedings of the National Academy of Sciences. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAMATSU S, HIRAI K, TAK YS, KAMIMURA Y, ARAKI H. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ε, and GINS in budding yeast. Genes Dev. 2010;24:602–12. doi: 10.1101/gad.1883410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAMOTO T, MÜLLER I, THOMAS G, MELVIN A, CHUBB JR. Methylation of H3K4 Is Required for Inheritance of Active Transcriptional States. Current Biology. 2010;20:397–406. doi: 10.1016/j.cub.2010.01.017. [DOI] [PubMed] [Google Scholar]
- NAKANO S, STILLMAN B, HORVITZ HR. Replication-coupled chromatin assembly generates a neuronal bilateral asymmetry in C. elegans. Cell. 2011;147:1525–36. doi: 10.1016/j.cell.2011.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKAYAMA J, ALLSHIRE RC, KLAR AJ, GREWAL SI. A role for DNA polymerase alpha in epigenetic control of transcriptional silencing in fission yeast. EMBO J. 2001;20:2857–66. doi: 10.1093/emboj/20.11.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISHITANI H, LYGEROU Z, NISHIMOTO T, NURSE P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature. 2000;404:625–628. doi: 10.1038/35007110. [DOI] [PubMed] [Google Scholar]
- NOUGAREDE R, DELLA SETA F, ZARZOV P, SCHWOB E. Hierarchy of S-phase-promoting factors: yeast Dbf4-Cdc7 kinase requires prior S-phase cyclin-dependent kinase activation. Mol Cell Biol. 2000;20:3795–806. doi: 10.1128/mcb.20.11.3795-3806.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’DONNELL M, LANGSTON L, STILLMAN B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGAWA T, OKAZAKI T. Discontinuous DNA replication. Annu Rev Biochem. 1980;49:421–57. doi: 10.1146/annurev.bi.49.070180.002225. [DOI] [PubMed] [Google Scholar]
- OKAZAKI R, OKAZAKI T, SAKABE K, SUGIMOTO K, SUGINO A. Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains. Proc Natl Acad Sci U S A. 1968;59:598–605. doi: 10.1073/pnas.59.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAUL S, MILLION-WEAVER S, CHATTOPADHYAY S, SOKURENKO E, MERRIKH H. Accelerated gene evolution through replication-transcription conflicts. Nature. 2013;495:512–5. doi: 10.1038/nature11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRINDLE MJ, LOEB LA. DNA polymerase delta in DNA replication and genome maintenance. Environ Mol Mutagen. 2012;53:666–82. doi: 10.1002/em.21745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROBST AV, DUNLEAVY E, ALMOUZNI G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- PURSELL ZF, ISOZ I, LUNDSTROM EB, JOHANSSON E, KUNKEL TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAGUNATHAN K, JIH G, MOAZED D. Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science. 2015;348:1258699. doi: 10.1126/science.1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REIJNS MAM. Lagging strand replication shapes the mutational landscape of the genome. 2015;518:502–6. doi: 10.1038/nature14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RHIND N. DNA replication timing: random thoughts about origin firing. Nat Cell Biol. 2006;8:1313–6. doi: 10.1038/ncb1206-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RILEY D, WEINTRAUB H. Conservative segregation of parental histones during replication in the presence of cycloheximide. Proc Natl Acad Sci U S A. 1979;76:328–32. doi: 10.1073/pnas.76.1.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROCHA EP, DANCHIN A. Gene essentiality determines chromosome organisation in bacteria. Nucleic Acids Res. 2003;31:6570–7. doi: 10.1093/nar/gkg859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROCHETEAU P, GAYRAUD-MOREL B, SIEGL-CACHEDENIER I, BLASCO MA, TAJBAKHSH S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148:112–25. doi: 10.1016/j.cell.2011.11.049. [DOI] [PubMed] [Google Scholar]
- ROSSI ML, PIKE JE, WANG W, BURGERS PM, CAMPBELL JL, BAMBARA RA. Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. J Biol Chem. 2008;283:27483–93. doi: 10.1074/jbc.M804550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROUFA DJ, MARCHIONNI MA. Nucleosome segregation at a defined mammalian chromosomal site. Proc Natl Acad Sci U S A. 1982;79:1810–4. doi: 10.1073/pnas.79.6.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RYBA T, HIRATANI I, LU J, ITOH M, KULIK M, ZHANG J, SCHULZ TC, ROBINS AJ, DALTON S, GILBERT DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–70. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKABE K, OKAZAKI R. A unique property of the replicating region of chromosomal DNA. Biochim Biophys Acta. 1966;129:651–4. doi: 10.1016/0005-2787(66)90088-8. [DOI] [PubMed] [Google Scholar]
- SANKAR TS, WASTUWIDYANINGTYAS BD, DONG Y, LEWIS SA, WANG JD. The nature of mutations induced by replication–transcription collisions. Nature. 2016;535:178–181. doi: 10.1038/nature18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEALE RL. Studies on the mode of segregation of histone nu bodies during replication in HeLa cells. Cell. 1976;9:423–9. doi: 10.1016/0092-8674(76)90087-8. [DOI] [PubMed] [Google Scholar]
- SEIDMAN MM, LEVINE AJ, WEINTRAUB H. The asymmetric segregation of parental nucleosomes during chrosome replication. Cell. 1979;18:439–49. doi: 10.1016/0092-8674(79)90063-1. [DOI] [PubMed] [Google Scholar]
- SEQUEIRA-MENDES J, DIAZ-URIARTE R, APEDAILE A, HUNTLEY D, BROCKDORFF N, GOMEZ M. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 2009;5:e1000446. doi: 10.1371/journal.pgen.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBAHARA K, STILLMAN B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell. 1999;96:575–85. doi: 10.1016/s0092-8674(00)80661-3. [DOI] [PubMed] [Google Scholar]
- SHIMODA C, ITADANI A, SUGINO A, FURUSAWA M. Isolation of thermotolerant mutants by using proofreading-deficient DNA polymerase delta as an effective mutator in Saccharomyces cerevisiae. Genes Genet Syst. 2006;81:391–7. doi: 10.1266/ggs.81.391. [DOI] [PubMed] [Google Scholar]
- SMITH DJ, WHITEHOUSE I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–8. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH ZD, MEISSNER A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- SOGO JM, STAHL H, KOLLER T, KNIPPERS R. Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J Mol Biol. 1986;189:189–204. doi: 10.1016/0022-2836(86)90390-6. [DOI] [PubMed] [Google Scholar]
- SPITZ F, FURLONG EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- STANCHEVA I, KOLLER T, SOGO JM. Asymmetry of Dam remethylation on the leading and lagging arms of plasmid replicative intermediates. Embo j. 1999;18:6542–51. doi: 10.1093/emboj/18.22.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SULSTON JE, SCHIERENBERG E, WHITE JG, THOMSON JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- TAJBAKHSH S, GONZALEZ C. Biased segregation of DNA and centrosomes [mdash] moving together or drifting apart? Nat Rev Mol Cell Biol. 2009;10:804–810. doi: 10.1038/nrm2784. [DOI] [PubMed] [Google Scholar]
- TANABE K, KONDO T, ONODERA Y, FURUSAWA M. A conspicuous adaptability to antibiotics in the Escherichia coli mutator strain, dnaQ49. FEMS Microbiol Lett. 1999;176:191–6. doi: 10.1111/j.1574-6968.1999.tb13661.x. [DOI] [PubMed] [Google Scholar]
- THURMAN RE, RYNES E, HUMBERT R, VIERSTRA J, MAURANO MT, HAUGEN E, SHEFFIELD NC, STERGACHIS AB, WANG H, VERNOT B, GARG K, JOHN S, SANDSTROM R, BATES D, BOATMAN L, CANFIELD TK, DIEGEL M, DUNN D, EBERSOL AK, FRUM T, GISTE E, JOHNSON AK, JOHNSON EM, KUTYAVIN T, LAJOIE B, LEE BK, LEE K, LONDON D, LOTAKIS D, NEPH S, NERI F, NGUYEN ED, QU H, REYNOLDS AP, ROACH V, SAFI A, SANCHEZ ME, SANYAL A, SHAFER A, SIMON JM, SONG L, VONG S, WEAVER M, YAN Y, ZHANG Z, ZHANG Z, LENHARD B, TEWARI M, DORSCHNER MO, HANSEN RS, NAVAS PA, STAMATOYANNOPOULOS G, IYER VR, LIEB JD, SUNYAEV SR, AKEY JM, SABO PJ, KAUL R, FUREY TS, DEKKER J, CRAWFORD GE, STAMATOYANNOPOULOS JA. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRAN V, LIM C, XIE J, CHEN X. Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science. 2012;338:679–82. doi: 10.1126/science.1226028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSAKRAKLIDES V, BELL SP. Dynamics of pre-replicative complex assembly. J Biol Chem. 2010;285:9437–43. doi: 10.1074/jbc.M109.072504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN ROSSUM B, FISCHLE W, SELENKO P. Asymmetrically modified nucleosomes expand the histone code. Nat Struct Mol Biol. 2012;19:1064–1066. doi: 10.1038/nsmb.2433. [DOI] [PubMed] [Google Scholar]
- VASHEE S, CVETIC C, LU W, SIMANCEK P, KELLY TJ, WALTER JC. Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev. 2003;17:1894–908. doi: 10.1101/gad.1084203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VASSEUR P, TONAZZINI S, ZIANE R, CAMASSES A, RANDO OJ, RADMAN-LIVAJA M. Dynamics of Nucleosome Positioning Maturation following Genomic Replication. Cell Rep. 2016;16:2651–65. doi: 10.1016/j.celrep.2016.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOIGT P, LEROY G, DRURY WILLIAM J, III, ZEE BARRY M, SON J, BECK DAVID B, YOUNG NI, GARCIA COLASL, BENJAMIN A, REINBERG D. Asymmetrically Modified Nucleosomes. Cell. 151:181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAGA S, STILLMAN B. Cyclin-dependent kinase inhibitor p21 modulates the DNA primer-template recognition complex. Mol Cell Biol. 1998a;18:4177–87. doi: 10.1128/mcb.18.7.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAGA S, STILLMAN B. The DNA replication fork in eukaryotic cells. Annu Rev Biochem. 1998b;67:721–51. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- WATSON JD, CRICK FHC. Genetical Implications of the Structure of Deoxyribonucleic Acid. Nature. 1953a;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- WATSON JD, CRICK FHC. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature. 1953b;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- WEINTRAUB H. Cooperative alignment of nu bodies during chromosome replication in the presence of cycloheximide. Cell. 1976;9:419–22. doi: 10.1016/0092-8674(76)90086-6. [DOI] [PubMed] [Google Scholar]
- WILKINS MHF, STOKES AR, WILSON HR. Molecular Structure of Nucleic Acids: Molecular Structure of Deoxypentose Nucleic Acids. Nature. 1953;171:738–740. doi: 10.1038/171738a0. [DOI] [PubMed] [Google Scholar]
- WOHLSCHLEGEL JA, DHAR SK, PROKHOROVA TA, DUTTA A, WALTER JC. Xenopus Mcm10 Binds to Origins of DNA Replication after Mcm2-7 and Stimulates Origin Binding of Cdc45. Molecular Cell. 2002;9:233–240. doi: 10.1016/s1097-2765(02)00456-2. [DOI] [PubMed] [Google Scholar]
- WU TP, WANG T, SEETIN MG, LAI Y, ZHU S, LIN K, LIU Y, BYRUM SD, MACKINTOSH SG, ZHONG M, TACKETT A, WANG G, HON LS, FANG G, SWENBERG JA, XIAO AZ. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature. 2016;532:329–333. doi: 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE J, WOOTEN M, TRAN V, CHEN BC, POZMANTER C, SIMBOLON C, BETZIG E, CHEN X. Histone H3 Threonine Phosphorylation Regulates Asymmetric Histone Inheritance in the Drosophila Male Germline. Cell. 2015;163:920–33. doi: 10.1016/j.cell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU M, LONG C, CHEN X, HUANG C, CHEN S, ZHU B. Partitioning of histone H3–H4 tetramers during DNA replication-dependent chromatin assembly. Science. 2010;328:94–8. doi: 10.1126/science.1178994. [DOI] [PubMed] [Google Scholar]
- YADLAPALLI S, YAMASHITA YM. Chromosome-specific nonrandom sister chromatid segregation during stem-cell division. Nature. 2013;498:251–4. doi: 10.1038/nature12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMADA-INAGAWA T, KLAR AJS, DALGAARD JZ. Schizosaccharomyces pombe Switches Mating Type by the Synthesis-Dependent Strand-Annealing Mechanism. Genetics. 2007;177:255–65. doi: 10.1534/genetics.107.076315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMASHITA YM. Nonrandom template segregation: A way to break the symmetry of stem cells. The Journal of Cell Biology. 2013;203:7–9. doi: 10.1083/jcb.201308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAN J, ENGE M, WHITINGTON T, DAVE K, LIU J, SUR I, SCHMIERER B, JOLMA A, KIVIOJA T, TAIPALE M, TAIPALE J. Transcription Factor Binding in Human Cells Occurs in Dense Clusters Formed around Cohesin Anchor Sites. Cell. 2013;154:801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
- YU C, GAN H, HAN J, ZHOU ZX, JIA S, CHABES A, FARRUGIA G, ORDOG T, ZHANG Z. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol Cell. 2014;56:551–63. doi: 10.1016/j.molcel.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG G, HUANG H, LIU D, CHENG Y, LIU X, ZHANG W, YIN R, ZHANG D, ZHANG P, LIU J, LI C, LIU B, LUO Y, ZHU Y, ZHANG N, HE S, HE C, WANG H, CHEN D. N6-methyladenine DNA modification in Drosophila. Cell. 2015;161:893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- ZHU B, REINBERG D. Epigenetic inheritance: Uncontested? Cell Research. 2011;21:435–441. doi: 10.1038/cr.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZOU L, MITCHELL J, STILLMAN B. CDC45, a novel yeast gene that functions with the origin recognition complex and Mcm proteins in initiation of DNA replication. Mol Cell Biol. 1997;17:553–63. doi: 10.1128/mcb.17.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]