

Abstract
Immunotherapy has emerged as a promising strategy for glioblastoma (GBM), a disease that remains universally fatal despite currently available standard-of-care. Adoptive T cell therapy has been shown to produce potent antitumor immunity while obviating the need for traditional antigen presentation and primary immune responses. Chimeric antigen receptors (CARs) are specialized molecules that can be expressed on the surface of T cells allowing for redirected cytotoxicity against tumor antigens of interest. To date, the application of CAR T cells for GBM has been relatively limited, in large part due to a dearth of well-described tumor specific antigens that are both homogenously and frequently expressed. A mutated version of the epidermal growth factor receptor, EGFRvIII, is a constitutively activated tyrosine kinase that is expressed on the surface of GBM and other common neoplasms, but completely absent from all normal tissues. We have recently generated CAR T cells directed against EGFRvIII and reported results from a Phase I clinical trial investigating this platform in patients with EGFRvIII-expressing GBM. Our study showed that despite conventional notions of central nervous system “immune-privilege,” EGFRvIII CAR T cells trafficked to intracerebral tumors, leading to successful targeting and eradication of this antigen in the brain. Here, we review our experience with EGFRvIII CAR T cells and highlight important considerations for the clinical translation of this therapy in patients with GBM.
Glioblastoma (GBM) is the most common and most deadly primary malignant brain tumor. Despite aggressive multimodal therapy including surgical resection, radiation therapy, anti-angiogenic treatments, and temozolomide chemotherapy, a diagnosis of GBM remains uniformly lethal with an expected survival of less than 15 months from the time of diagnosis.1 In addition to providing only incremental benefit in overall survival, these conventional therapies are also nonspecific and can result in incapacitating damage to systemic tissues as well as adjacent eloquent brain. Conversely, the immune system, with its inherent biological specificity, has the theoretical capacity to eliminate tumors while leaving healthy cells intact.
One immunotherapeutic platform that has shown great potential is adoptive cellular therapy (ACT), or the infusion of antitumor cytotoxic T lymphocytes (CTLs) for patients with cancer. Tumor-reactive CTLs can either be derived from the patient’s tumor itself (e.g., tumor infiltrating lymphocytes, TILs) or otherwise engineered to express T-cell receptors (TCRs) with antitumor specificity.2 A major drawback of this approach however is that TCRs are by nature reliant on interaction with a tumor-associated peptide presented by human leukocyte antigen molecules (HLA), down-regulation of which is a common mechanism of tumor escape.3,4 By contrast, chimeric antigen receptors (CARs) are highly specialized, recombinant surface molecules that combine the antigen-recognizing variable region of an antibody in tandem with intracellular T-cell signaling moieties (e.g., CD3ζ, costimulatory signal domains) (Fig. 1).5 As such, CARs have the capacity to bind tumor antigen more avidly than endogenous TCR and do so without the need for classically defined peptide-HLA complex or co-stimulation. In addition, they may incorporate several downstream pathways that make them less susceptible to host mechanisms of immune suppression, including intracellular sequences that have been shown to enhance T-cell survival and proliferation.
Figure 1. Chimeric antigen receptor and T-cell receptor structure.
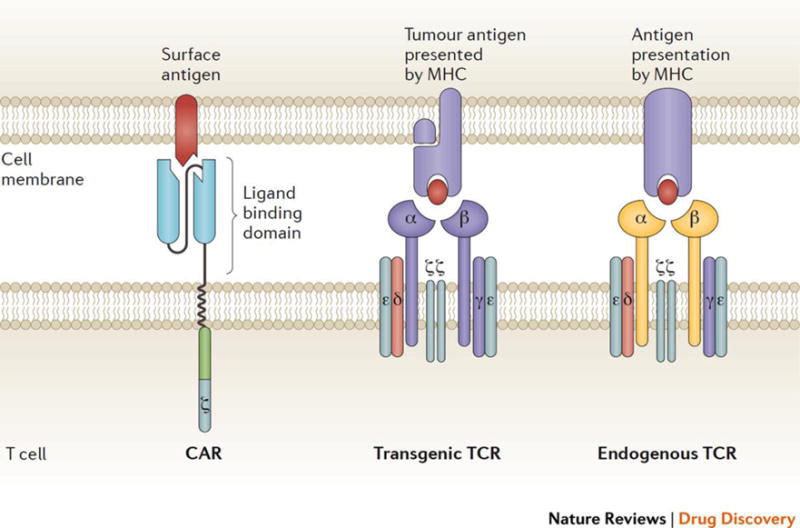
A chimeric antigen receptor (CAR; left) has an extracellular antigen-binding domain made of antibody variable fragments, fused via a linker to an intracellular domain composed of the CD3ζ signaling domain of the T cell receptor (TCR) and a co-stimulatory domain. Transgenic TCRs (middle) are similar to endogenous TCRs (right) but have engineered antigen-binding domains specific for a tumor-associated target. TCRs require co-stimulation from separate receptors for full T cell activation. CARs bind to cell surface antigen, whereas TCRs recognize intracellular antigens that are presented on human leukocyte antigen molecules (HLA). Adapted from Nat. Rev. Drug Discov. 16, 301–304 (2017).5
To date perhaps the most dramatic responses in clinical trials have been for CARs targeting CD19, a cell surface antigen expressed exclusively on the surface of B cells and many B-cell malignancies. In patients with relapsed or refractory B-cell acute lymphoblastic leukemia (ALL), treatment with CD19 CAR T cells demonstrated complete response rates of approximately 90%, with sustained remission rates at 6 months of nearly 70%.6 Despite successful targeting of CD19 in hematologic malignancies, the application of CAR immune therapy to solid tumors has been relatively limited, in large part due to the dearth of tumor antigens that are homogenously expressed in the setting of malignancy yet completely absent in healthy tissues. Indeed, given the potent cytotoxicity of CAR T cells, cross-expression of tumor surface antigens in healthy cells can lead to severe “off-tumor, on-target” autoimmune effects that can vary widely depending on the affected organ tissue.7
Towards this end, a tumor-specific mutation of the epidermal growth factor receptor, EGFRvIII, has emerged as an ideal surface target for CAR T cell technology. EGFRvIII is most frequently seen in patients with GBM, but it is also found in a broad array of other human cancers.8 It is derived from an in-frame deletion of 801 base pairs from the extracellular domain of the wild-type receptor, which results in the translation of an otherwise absent glycine residue at the fusion junction. This novel domain that stems from the union of normally distant parts of the protein creates an antigenic epitope that is not present in any normal tissues.9 Functionally, EGFRvIII has constitutive activity that enhances neoplasticity and confers radiation and chemotherapeutic resistance to tumor cells, which in turn become “addicted” to its downstream signaling pathways and are unable to survive when this stimulus is removed. The exquisite tumor-specificity of EGFRvIII and its importance in the pathobiology of tumors make the mutation an ideal target for CAR T-cell therapy.
We recently described a systematic approach for the creation and development of a new humanized CAR directed against EGFRvIII.10 Using vector backbones based on murine CAR T cells that have already been tested in xenograft GBM models, anti-EGFRvIII antibodies were screened and assayed extensively prior to constructing an EGFRvIII CAR with desired functionality and reactivity, followed by testing for antitumor effects in vitro and in preclinical murine studies. These data ultimately led to our opening of a Phase I clinical trial of EGFRvIII CAR T cells in patients with residual or recurrent GBM (NCT02209376). In this first-in-human clinical trial, we have found that infusion of CART-EGFRvIII cells was feasible through standard technique (Fig. 2) and safe, without evidence of off-tumor toxicity such as cross-reactivity to wild-type EGFR. No clinical or laboratory signs of systemic cytokine release syndrome was observed, and no off-tumor toxicity occurred. Several patients had post-CART-EGFRvIII surgical intervention, which allowed for tissue-specific analysis of certain endpoints. Specifically, CART-EGFRvIII T cells were demonstrated to traffic to tumor by immunohistochemistry, resulting in subsequent eradication of EGFRvIII in residual lesion, implying successful immunological targeting in the brain. In our study, we used a single intravenous dose of CAR T cells. Other investigators have used multiple doses, and administered CAR T cells intratumorally or intrathecally through a catheter in the ventricles. In one case, intracranial administration of multiple CAR T cell doses directed to the IL-13Rα2 antigen were able to induce a transient but complete regression of metastatic GBM.11 Ahmed and colleagues induced at least one partial response out of 17 patients with progressive GBM treated with virus-specific T cells transduced with a CAR directed against the HER2 antigen.12
Figure 2. Overview of CAR T-cell therapy in the clinic.
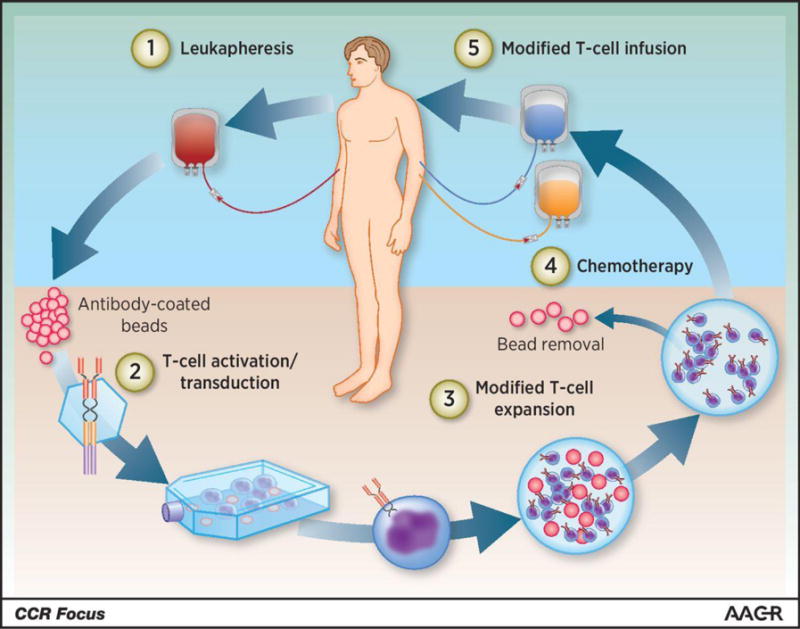
A patient’s T cells are harvested through leukapheresis, followed by T-cell activation on antibody-coated beads serving as artificial dendritic cells. The activated T cells are then genetically reprogrammed ex vivo by transduction with a construct encoding the CAR, and the CAR T cells are further expanded ex vivo. When the CAR T-cell product has been prepared and has passed all quality control testing, the patient receives lymphodepleting chemotherapy and CAR T-cell infusion. Adapted from Clin. Cancer Res. 22 (8), 1875–1884 (2016).18
Despite these promising findings, several hurdles to successful translation of CAR T cells for patients with GBM remain. While it is still too soon to conclude on objective clinical benefits in survival, early data suggest that further investigation will be needed for optimal efficacy and durable antitumor responses. First, despite eradication of EGFRvIII antigen expression following CAR therapy, EGFRvIII-negative tumors persisted in several patients, ultimately leading to disease progression. Analogous observations were also noted in clinical studies of an EGFRvIII peptide vaccine, in which patients achieved significantly prolonged survival but eventually had tumor recurrence owing to outgrowth of EGFRvIII-negative tumor.13 On the other hand, early evidence in immune competent murine models suggests that EGFRvIII CARs, under certain circumstances, may elicit host immunity against additional tumor antigens, thereby protecting against EGFRvIII antigen loss.14 Further research will be needed to determine whether this effect is recapitulated in clinical studies.
Another critical factor to consider in the successful clinical translation of an EGFRvIII CAR is the growing consensus that, due to the harsh immune suppressive environment known to be associated with GBM and other solid tumors, combination therapies may be required to achieve durable antitumor responses. In a HER2-transgenic mouse model of breast cancer, mice treated with anti-HER2 CAR T cells and repeated injections of well-described antibodies for checkpoint blockade showed greater CD8+ CAR T-cell function, less T-cell exhaustion and enhanced tumor cell killing.15 Recently, in models of renal cell carcinoma, CAR T cells have been designed to secrete immune-modulating antibodies in the tumor milieu to achieve superior antitumor immune responses.16 Although this strategy has been shown to have potentially important implications on CAR technology, its efficacy has yet to be explored for patients with brain tumors. Of particular note, if shown to be effective, the ability for CAR T cells to secrete checkpoint inhibitors at the site of tumor antigen expression may have particular relevance to the treatment of GBM, where immune checkpoint molecules have not only been shown to be expressed at high levels,17 but also where the blood-brain barrier may otherwise inhibit the localization and activity of systemically administered antibody-mediated blockade.
Overall, our work with the EGFRvIII CAR contributes to the growing body of research committed to discovering a novel therapy for GBM. To date, the success of immune therapy has been modest for the treatment of brain tumors due to several factors, the most prominent of which include the relative dearth of tumor-associated and tumor-specific antigens, the associated immunosuppressive microenvironment, the presence of the blood-brain barrier, and toxicities associated with non-specific activity. EGFRvIII-specific CARs promise to overcome many of these critical barriers, though further work will be needed to successfully translate this platform for patients with GBM, where the need for effective treatment is great.
Take home points.
Immune therapy with CAR technology shows great promise in the treatment of GBM and other solid tumors.
The EGFRvIII antigen represents an ideal target for CAR T cells since it is expressed on the surface of tumor cells, is not found in any healthy tissues, and plays a central role in the pathobiology of GBM.
EGFRvIII CARs have demonstrated clinical safety and feasibility in early phase clinical trials.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005 Mar 10;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer–what clinicians need to know. Nature reviews. Clinical oncology. 2011 Oct;8(10):577–585. doi: 10.1038/nrclinonc.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nature reviews. Cancer. 2005 Apr;5(4):263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 4.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nature reviews. Immunology. 2006 Oct;6(10):715–727. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 5.Kingwell K. CAR T therapies drive into new terrain. Nature reviews. Drug discovery. 2017 Apr 28;16(5):301–304. doi: 10.1038/nrd.2017.84. [DOI] [PubMed] [Google Scholar]
- 6.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014 Oct 16;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular therapy: the journal of the American Society of Gene Therapy. 2010 Apr;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995 Jul 15;55(14):3140–3148. [PubMed] [Google Scholar]
- 9.Choi BD, Archer GE, Mitchell DA, et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain pathology. 2009 Oct;19(4):713–723. doi: 10.1111/j.1750-3639.2009.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Science translational medicine. 2015 Feb 18;7(275):275ra222. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown CE, Alizadeh D, Starr R, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. The New England journal of medicine. 2016 Dec 29;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed N, Brawley V, Hegde M, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA oncology. 2017 Apr 20; doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010 Nov 01;28(31):4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampson JH, Choi BD, Sanchez-Perez L, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014 Feb 15;20(4):972–984. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013 Oct 15;19(20):5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 16.Suarez ER, Chang de K, Sun J, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016 Jun 7;7(23):34341–34355. doi: 10.18632/oncotarget.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nature reviews. Neurology. 2015 Sep;11(9):504–514. doi: 10.1038/nrneurol.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maus MV, June CH. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016 Apr 15;22(8):1875–1884. doi: 10.1158/1078-0432.CCR-15-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]