

Abstract
Over the last half-century, transition metal-mediated cross-coupling reactions have changed the way in which complex organic molecules are synthesized. Indeed, the predictable and chemoselective nature of these transformations has led to their widespread adoption across a vast array of chemical research areas1. However, the construction of sp3–sp3 bonds, a fundamental unit of organic chemistry, remains an important yet elusive objective for cross-coupling reaction engineering2. In comparison to related procedures with sp2-hybridized species, the development of methods for sp3–sp3 bond formation via transition metal catalysis has been historically hampered by deleterious side-reactions, such as β-hydride elimination with Pd-catalysis, and the reluctance of alkyl halides to undergo oxidative addition3,4. To address this issue, a number of research groups have demonstrated the feasibility of nickel-catalyzed cross-coupling processes to form sp3–sp3 bonds that utilize organometallic nucleophiles and alkyl electrophiles5–7. In particular, the coupling of alkyl halides with pregenerated organozinc8–10, Grignard11,12, and organoborane13 species has been used to furnish diverse molecular structures. However, the poor step and atom economies along with the operational difficulties associated with making, carrying, and using these sensitive coupling partners has hindered their widespread adoption. The prospect of establishing a generically useful sp3–sp3 coupling technology that employs bench-stable, native organic functional groups, without the need for pre-functionalization or substrate derivatization, would therefore be a valuable addition to fields of research that rely on organic molecule construction. Here, we demonstrate that the synergistic merger of photoredox and nickel catalysis enables the direct formation of sp3–sp3 bonds using only simple carboxylic acids and alkyl halides as the nucleophilic and electrophilic coupling partners, respectively. The outlined protocol is suitable for a wide array of primary and secondary carboxylic acids and does not require the presence of radical stabilizing groups. The merit of this coupling strategy is illustrated by the expedient synthesis of the pharmaceutical tirofiban in four steps from commercially available starting materials.
Within the field of drug discovery, there is a demonstrated statistical correlation between clinical success and the molecular complexity of medicinal candidates with respect to the inherent ratio of sp2–sp3 to sp3–sp3 bond content14. Not surprisingly, these findings have created an emerging demand within medicinal chemistry for new reaction technologies that enable rapid access to drug-like molecules via the coupling of fragments that incorporate or build novel sp3–sp3 bonds. However, a major hurdle associated with achieving sp3–sp3 bond formation via transition metal catalysis is the limited availability of a diverse suite of nucleophilic coupling partners that are bench-stable, inexpensive, and easily procured. An attractive option would be to employ simple carboxylic acids, an abundant native functional group that is chemically robust yet can be readily exploited as a latent leaving group after multistep synthetic sequences (Fig. 1).
Figure 1. Carboxylic acids as coupling partners in a metallaphotoredox-mediated process to form sp3–sp3 bonds.
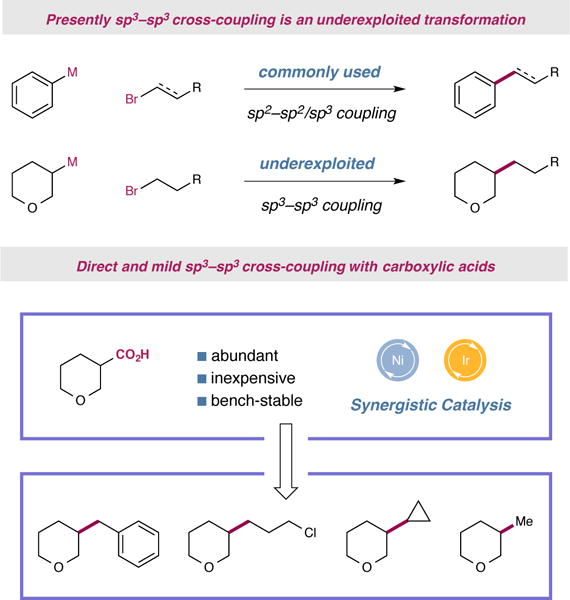
The majority of transition metal-catalyzed cross-couplings generally employ at least one sp2-hydridized coupling partner. The direct utilization of robust, widely available, native functional groups such as carboxylic acids should encourage greater adoption of sp3–sp3 bond forming methods.
The emergence of visible light-mediated photoredox catalysis within the field of synthetic organic chemistry has enabled the discovery and invention of numerous unique and valuable transformations15,16. Indeed, the electronic duality of photocatalyst excited states (which are simultaneously strong oxidants and reductants) has prompted the exploitation of these polypyridyl transition metal complexes in unconventional bond disconnections17 and facilitated the manipulation of oxidation states in organometallic complexes to enable previously elusive reactions18–22. For example, the synergistic merger of photon-driven single-electron transfer (SET) processes with nickel-activated electrophiles has promoted the formation of valuable sp2–sp3 bonds whilst broadening the field of cross-coupling chemistry via the use of non-conventional reaction substrates23,24. On this basis, we hypothesized that a straightforward and generic procedure might be developed to enable analogous sp3–sp3 bond formations via the application of ubiquitous carboxylic acids in a decarboxylative cross-coupling with alkyl halides using photoredox catalysis. Moreover, we hoped to introduce a paradigm for carbon–carbon bond construction that would (i) provide rapid access to complex molecular fragments via sp3–sp3 coupling, (ii) systematically streamline synthetic routes towards drug candidates, and (iii) enable alkyl–alkyl cross-coupling broadly in medicinal, process, and natural product chemistry programs.
A detailed mechanism for the proposed decarboxylative sp3–sp3 coupling is delineated in Fig. 2. Initial visible-light excitation of the iridium(III) photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine] (1) would generate the long-lived (τ = 2.3 μs)25 excited-state *IrIII complex 2. This species is a strong single-electron oxidant (E1/2red [*IrIII/IrII] = +1.21 V vs. SCE in CH3CN)25 and should undergo reduction by a carboxylate anion derived from deprotonation of the acid 3. The resultant carboxyl radical is expected to rapidly extrude CO2 to produce alkyl radical 4 and the reduced IrII catalyst 5. Concurrently, the ligated nickel(0) complex 6 is generated in situ via two discrete SET reductions of (dtbbpy)Ni(II)Cl2 by the iridium(II) state of the photocatalyst through sacrificial carboxylic acid consumption (E1/2red [IrIII/IrII] =−1.37 V vs. SCE in CH3CN, (E1/2red [NiII/Ni0] = −1.2 V vs. SCE in DMF)25,26. The Ni0 complex 6 can intercept radical 4 to produce alkyl-NiI intermediate 727. Subsequent oxidative addition with alkyl halide 8 forms the putative organometallic NiIII species 9, which after reductive elimination forges the desired sp3–sp3 bond to furnish the coupled product 10 and NiI adduct 1128–30. At this stage the two catalytic cycles would converge by reduction of nickel(I) intermediate 11 by the reduced form of the iridium photocatalyst to reestablish both the IrIII complex 1 and the Ni0 catalyst 625. Presently, we cannot rule the possibility of an alternative mechanism that involves Ni0-mediated oxidative addition and trapping of the alkyl radical 4 by a NiII-species23,27.
Figure 2. Proposed mechanism for the metallaphotoredox-mediated cross-coupling of carboxylic acids to generate sp3–sp3 bonds.
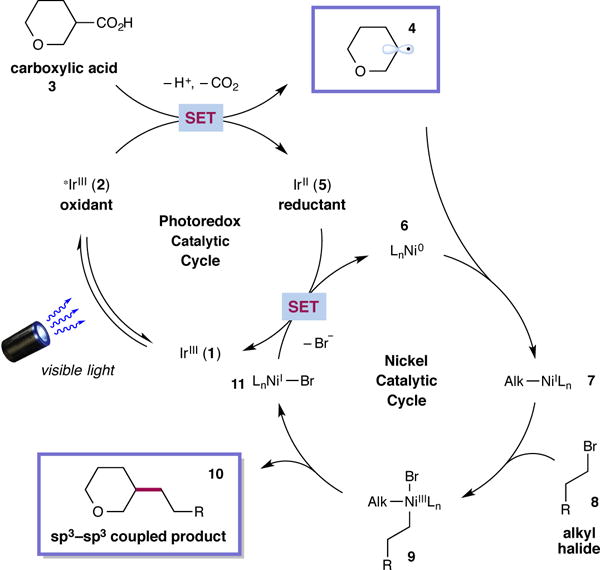
The photoredox catalytic cycle commences with excitation of IrIII 1 to give the excited state 2. Single electron oxidation of the carboxylate anion derived from acid 3 produces the alkyl radical 4 after CO2-extrusion along with IrII 5. Ni0 catalyst 6 captures the alkyl radical 4 to form the NiI species 7. Ensuing oxidative addition with alkyl halide 8 leads to nickel(III) intermediate 9. Reductive elimination would then liberate the desired product 10 and NiI 11. Both catalytic cycles converge to complete a single turnover via a SET event that regenerates the photoredox and nickel catalysts
Predicated on our envisioned decarboxylative sp3–sp3 coupling mechanism, we began our primary investigations with consideration of the metallaphotoredox conditions previously developed within our group23. In these studies we employed N-Boc proline and 1-bromo-3-phenylpropane as coupling partners. Unfortunately, owing to the basic reaction conditions in combination with the polar aprotic solvent dimethylformamide, exclusive ester formation was observed. Therefore, we recognized that judicious selection of solvent and base would be necessary to suppress this unwanted byproduct formation without obstructing the desired photocatalytic oxidation reaction pathway. To accomplish this goal a survey of solvents and inorganic bases was conducted in the presence of Ir[dF(CF3)ppy]2(dtbbpy)PF6, NiCl2×glyme, and dtbbpy with visible light irradiation from blue light-emitting diodes (LEDs). Pleasingly, this revealed that the combination of acetonitrile and K2CO3 greatly reduced the rate of carboxylate alkylation and furnished the desired product in 68% yield. Further optimization improved conversion to product through switching to the more electron-rich ligand 4,4′-dOMe-bpy (4,4′-dimethoxy-2,2′-bipyridine). Finally, the addition of water, which decelerated ester formation, provided an additional enhancement and an isolated yield of 85%. A series of control experiments omitting each individual reaction component highlighted the importance of photocatalyst, nickel, and light for promoting this decarboxylative sp3–sp3 bond-forming reaction (see Supplementary Information).
With optimal metallaphotoredox conditions in hand, we probed the generality of this process with respect to the aliphatic halide electrophile. As representative nucleophilic coupling partners, N-Boc- and N-Cbz proline were utilized interchangeably for this purpose (Fig. 3). Simple unfunctionalized primary alkyl halides such as 1-bromo-3-phenylpropane and 1-bromobutane were examined, and these underwent efficient cross-coupling (12 and 13, 85% and 76% yield, respectively). A substrate bearing a terminal olefin performed favorably under the reaction conditions to deliver the desired product (14, 84% yield). As this protocol is conducted at near ambient temperature, hydrolysis of an ethyl ester does not occur under the optimized basic reaction conditions, and the corresponding carbamate-protected amine was isolated in good yield (15, 64% yield). In addition, perfect chemoselective functionalization of an alkyl bromide in the presence of a primary chloroalkane was observed (16, 96% yield). A deprotection-cyclization sequence with this material would provide rapid access to the bicyclic tertiary amine core of the naturally occurring pyrrolizidine alkaloids31. The presence of free hydroxyl groups is fully compatible with the iridium photocatalyst and the nickel complex (17, 86% yield). Moreover, reactive Lewis basic functionalities, such as epoxides and aldehydes, are well tolerated in this cross-coupling procedure and provide numerous opportunities for further derivatization (18 and 19, 83% and 62% yield, respectively). The influence of substitution on the alkyl halide was also investigated to probe the steric limits of the electrophilic coupling partner. No detrimental effects to the efficiency of this process were observed when a β,β-disubstituted bromoalkane was employed, and pleasingly, neopentyl bromide coupled in modest yield (20 and 21, 75% and 52% yield, respectively). The higher propensity for activated electrophiles to promote esterification led to the utilization of benzyl chloride, as opposed to benzyl bromide, which generated a homobenzylic amine in good yield (22, 84% yield). Notably, bromomethane was a competent coupling partner in this protocol which formally affords the product of a fully reduced carboxylic acid moiety in a single step (23, 62% yield).
Figure 3. Carboxylic acid and alkyl halide scope in the dual nickel-photoredox catalyzed sp3–sp3 coupling reaction.
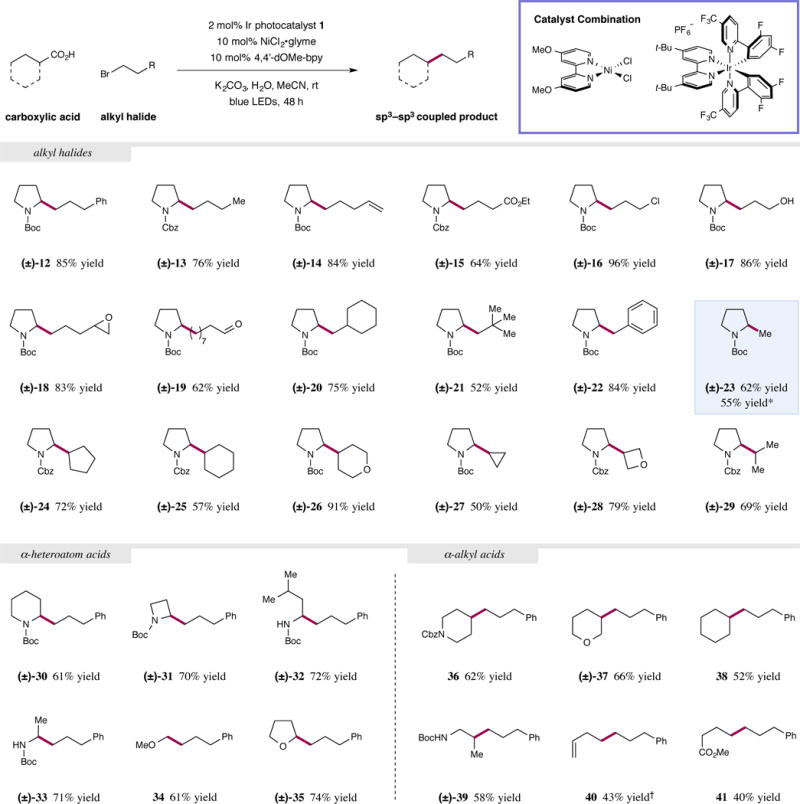
A broad array of alkyl halides and carboxylic acids are amenable coupling partners in this transformation. Top, generalized reaction; bottom, substrate scope. Primary and secondary electrophiles were coupled efficiently with proline derivatives. Alternative α-heteroatom substituted carboxylic acids could also be employed to form functionalized amines and ethers. Challenging substrates lacking apparent radical stabilization could also be employed successfully. Isolated yields are reported below each entry. See Supplementary Information for full experimental details. *Reaction run in flow (GC yield), see S.I. †Cyclopropylacetic acid was used.
Expanding the substrate scope to encompass secondary alkyl halides permitted us to forge sp3–sp3 bonds with adjacent tertiary carbon centers. For example, five- and six-membered cyclic bromoalkanes smoothly reacted to form the desired alkylated products in good to excellent yields (24–26, 57–91% yield). Smaller ring systems, including cyclopropane and oxetane, were also introduced via this metallaphotoredox procedure (27 and 28, 50% and 79% yield, respectively). These motifs have found application in drug discovery programs as chemically and metabolically stable bioisosteres32. Lastly, an acyclic secondary alkyl bromide was also successfully cross-coupled to generate the desired Cbz-protected amine (29, 69% yield).
We subsequently examined the scope of the nucleophilic component and we were pleased to discover that an assortment of readily available carboxylic acids were viable for this transformation. For instance, Boc-protected pipecolic acid and an azetidine derivative both underwent decarboxylative coupling to furnish alkylated products in good yields (30 and 31, 61% and 70% yield, respectively). Naturally occurring amino acids, which are inexpensive and obtainable from ample biomass feedstocks, can also be exploited to form α-functionalized amines with excellent efficiency (32 and 33, 72% and 71% yield, respectively). Similarly, O-methylated glycolic acid functions well to provide access to linear ethers in a straightforward manner (34, 61% yield). The cyclic substrate tetrahydrofuran-2-carboxylic acid was also coupled with 1-bromo-3-phenylpropane under these dual nickel-photoredox conditions to afford the ethereal product in very good yield (35, 74% yield). Although beneficial, the inclusion of an α-heteroatom on the acid fragment is not a prerequisite for this sp3–sp3 bond forming process. For example, Cbz-protected isonipecotic acid and a tetrahydro-2H-pyran derivative were cleanly converted to the corresponding coupled products in an effective fashion (36 and 37, 62% and 66% yield, respectively). Simple alkyl precursors lacking heteroatoms can also be utilized, with cyclohexanecarboxylic acid reacting in reasonable yield (38, 52% yield). An acyclic β-amino acid that would generate a secondary radical upon decarboxylation exhibited respectable efficiency and offers the opportunity to synthesize β-functionalized amines with ease (39, 58% yield).
Finally, two primary substrates were evaluated in this system to fully exemplify the power of this new sp3–sp3 coupling paradigm. The rearrangement of a cyclopropyl system under the reaction conditions presents the opportunity to produce alkylated homoallylic products in a single step (40, 43% yield). Moreover, the monomethyl ester of glutaric acid was subjected to the optimized metallaphotoredox procedure and provided an encouraging quantity of the desired product (41, 40% yield). This substrate highlights the potential for downstream modification of latent carboxylates since hydrolysis of the methyl ester would unlock the potential for further sp3–sp3 bond formation. Thus, molecules that contain multiple carboxylic acids can function as linchpin reagents for the rapid assembly of complex molecular architectures. It should be noted that when tertiary acids were applied, the coupled products could only be obtained limited efficiencies (~5–10%). Attempts to improve the yields to synthetically useful levels are currently ongoing.
To further demonstrate the synthetic utility of this decarboxylative coupling protocol, we applied it to the synthesis of the antiplatelet drug tirofiban33. As shown in Fig. 4, Boc-isonipecotic acid 42 and alkyl bromide 43 (protected to avoid cyclization to THF) were exposed to the optimized metallaphotoredox conditions to afford alcohol 44 in good yield, following deprotection of the silyl ether. Thereafter, utilization of the previously established dual photoredox-nickel catalytic etherification reaction enabled direct formation of the desired C–O bond18. Following acidic deprotection, tirofiban 45 was synthesized in 59% yield over the final two steps.
Figure 4. Application of two metallaphotoredox strategies to the synthesis of tirofiban.
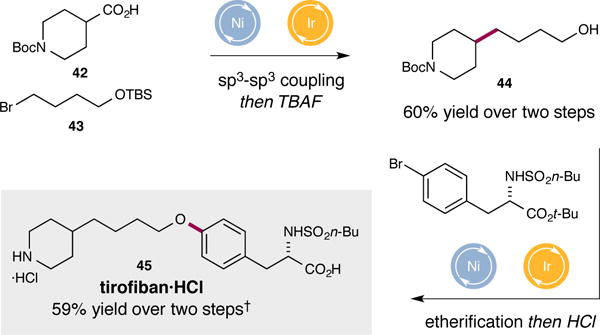
The cross-coupling of acid 42 and alkyl halide 43 generates a new sp3–sp3 bond and subsequent TBAF deprotection provides alcohol 44. Tirofiban 45 is then synthesized in two further steps in good yield via a Ni-photoredox-mediated etherification reaction and acidic deprotection. †34% of bromide starting material recovered.
In summary, we have established a robust strategy for the direct formation of sp3–sp3 bonds from abundant carboxylic acids and alkyl halides. This new platform for carbon–carbon bond construction is enabled by the catalytic activation of both coupling partners through the synergistic merger of photoredox and nickel catalysis. The benign nature of the reaction conditions has been exemplified by the breadth of functional groups tolerated in this transformation. We believe that the generality of this methodology and the ready availability of the starting materials used will aid the uptake of sp3–sp3 cross-coupling across several fields of synthetic organic chemistry.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 GM093213-01) and kind gifts from Merck, AbbVie, and Bristol-Myers Squibb. C.P.J. thanks Marie Curie Actions for an international outgoing fellowship (PIOF-GA-2013-627695). S.A. thanks the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral fellowship (AL 1860/2-1).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions CPJ, RTS, and SA performed and analyzed experiments. CPJ, RTS, SA, and D.W.C.M. designed experiments to develop this reaction and probe its utility, and also prepared this manuscript.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.de Meijere A, Diederich F. Metal-catalyzed cross-coupling reactions. Wiley-VCH; 2004. [Google Scholar]
- 2.Geist E, Kirschning A, Schmidt T. sp3-sp3 Coupling reactions in the synthesis of natural products and biologically active molecules. Nat Prod Rep. 2014;31:441. doi: 10.1039/c3np70108e. [DOI] [PubMed] [Google Scholar]
- 3.Haas D, Hammann JM, Greiner R, Knochel P. Recent developments in Negishi cross-coupling reactions. ACS Catal. 2016;6:1540–1552. [Google Scholar]
- 4.Phapale VB, Cárdenas DJ. Nickel-catalysed Negishi cross-coupling reactions: scope and mechanisms. Chem Soc Rev. 2009;38:1598. doi: 10.1039/b805648j. [DOI] [PubMed] [Google Scholar]
- 5.Jana R, Pathak TP, Sigman MS. Advances in transition metal (Pd, Ni, Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tasker SZ, Standley EA, Jamison TF. Recent advances in homogeneous nickel catalysis. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For an elegant extension of our concepts for decarboxylative coupling with Nickel see:; Qin T, et al. A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science. 2016 doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giovannini R, Stüdemann T, Dussin G, Knochel P. An efficient nickel-catalyzed cross-coupling between sp3 carbon centers. Angew Chem Int Ed. 1998;37:2387–2390. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2387::AID-ANIE2387>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Fu GC. Cross-couplings of unactivated secondary alkyl halides: room-temperature nickel-catalyzed Negishi reactions of alkyl bromides and Iodides. J Am Chem Soc. 2003;125:14726–14727. doi: 10.1021/ja0389366. [DOI] [PubMed] [Google Scholar]
- 10.Cordier CJ, Lundgren RJ, Fu GC. Enantioconvergent cross-couplings of racemic alkylmetal reagents with unactivated secondary alkyl electrophiles: catalytic asymmetric negishi α-alkylations of N-Boc-pyrrolidine. J Am Chem Soc. 2013;135:10946–10949. doi: 10.1021/ja4054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terao J, Watanabe H, Ikumi A, Kuniyasu H, Kambe N. Nickel-catalyzed cross-coupling reaction of Grignard reagents with alkyl halides and tosylates: remarkable effect of 1,3-butadienes. J Am Chem Soc. 2002;124:4222–4223. doi: 10.1021/ja025828v. [DOI] [PubMed] [Google Scholar]
- 12.Vechorkin O, Hu X. Nickel-catalyzed cross-coupling of non-activated and functionalized alkyl halides with alkyl grignard reagents. Angew Chem Int Ed. 2009;48:2937–2940. doi: 10.1002/anie.200806138. [DOI] [PubMed] [Google Scholar]
- 13.Saito B, Fu GC. Alkyl–alkyl Suzuki cross-couplings of unactivated secondary alkyl halides at room temperature. J Am Chem Soc. 2007;129:9602–9603. doi: 10.1021/ja074008l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lovering F, Bikker J, Humblet C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 15.Narayanam JMR, Stephenson CRJ. Visible light photoredox catalysis: applications in organic synthesis. Chem Soc Rev. 2010;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 16.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeffrey JL, Terrett JA, MacMillan DWC. O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols. Science. 2015;349:1532–1536. doi: 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC. Switching on elusive organometallic mechanisms with photoredox catalysis. Nature. 2015;524:330–334. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. Room-temperature C–H arylation: merger of Pd-catalyzed C–H functionalization and visible-light photocatalysis. J Am Chem Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rueping M, et al. Dual Catalysis: Combination of photocatalytic aerobic oxidation and metal catalyzed alkynylation reactions—C–C bond formation using visible light. Chem - Eur J. 2012;18:5170–5174. doi: 10.1002/chem.201200050. [DOI] [PubMed] [Google Scholar]
- 21.Sahoo B, Hopkinson MN, Glorius F. Combining gold and photoredox catalysis: Visible light-mediated oxy- and aminoarylation of alkenes. J Am Chem Soc. 2013;135:5505–5508. doi: 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]
- 22.Fabry DC, Zoller J, Raja S, Rueping M. Combining rhodium and photoredox catalysis for C-H functionalizations of arenes: Oxidative heck reactions with visible light. Angew Chem Int Ed. 2014;53:10228–10231. doi: 10.1002/anie.201400560. [DOI] [PubMed] [Google Scholar]
- 23.Zuo Z, et al. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science. 2014;345:437–440. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tellis JC, Primer DN, Molander GA. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science. 2014;345:433–436. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowry MS, et al. Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex. Chem Mater. 2005;17:5712–5719. [Google Scholar]
- 26.Durandetti M, Devaud M, Perichon J. Investigation of the reductive coupling of aryl halides and/or ethylchloroacetate electrocatalyzed by the precursor NiX2(bpy) with X- = Cl-, Br- or MeSO3- and bpy = 2,2′-dipyridyl. New J Chem. 1996;20:659. [Google Scholar]
- 27.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. Nickel-catalyzed cross-coupling of photoredox-generated radicals: Uncovering a general manifold for stereoconvergence in nickel-catalyzed cross-couplings. J Am Chem Soc. 2015;137:4896–4899. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin X, Phillips DL. Density functional theory studies of negishi alkyl–alkyl cross-coupling reactions catalyzed by a methylterpyridyl-Ni(I) complex. J Org Chem. 2008;73:3680–3688. doi: 10.1021/jo702497p. [DOI] [PubMed] [Google Scholar]
- 29.Ren P, Vechorkin O, Allmen K, von Scopelliti R, Hu X. A Structure–activity study of Ni-catalyzed alkyl–alkyl Kumada coupling. Improved catalysts for coupling of secondary alkyl halides. J Am Chem Soc. 2011;133:7084–7095. doi: 10.1021/ja200270k. [DOI] [PubMed] [Google Scholar]
- 30.Schley ND, Fu GC. Nickel-catalyzed Negishi arylations of propargylic bromides: A mechanistic investigation. J Am Chem Soc. 2014;136:16588–16593. doi: 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robertson J, Stevens K. Pyrrolizidine alkaloids. Nat Prod Rep. 2014;31:1721–1788. doi: 10.1039/c4np00055b. [DOI] [PubMed] [Google Scholar]
- 32.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 33.Hartman GD, et al. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J Med Chem. 1992;35:4640–4642. doi: 10.1021/jm00102a020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.