

Abstract
Core fucosylation of N-glycoproteins plays a crucial role in modulating the biological functions of glycoproteins. Yet, the synthesis of structurally well-defined, core-fucosylated glycoproteins remains a challenging task due to the complexity in multi-step chemical synthesis or the inability of the biosynthetic α1,6-fucosyltransferase (FUT8) to directly fucosylate full-size mature N-glycans in a chemoenzymatic approach. We report in this paper the design and generation of potential α1,6-fucosynthase and fucoligase for direct core-fucosylation of intact N-glycoproteins. We found that mutation at the nucleophilic residue (D200) did not provide a typical glycosynthase from this bacterial enzyme, but several mutants with mutation at the general acid/base residue E274 of the Lactobacillus casei α1,6-fucosidase, including E274A, E274S, and E274G, acted as efficient glycoligases that could fucosylate a wide variety of complex N-glycopeptides and intact glycoproteins by using α-fucosyl fluoride as a simple donor substrate. Studies on the substrate specificity revealed that the α1,6-fucosidase mutants could introduce an α1,6-fucose moiety specifically at the Asn-linked GlcNAc moiety not only to GlcNAc-peptide, but also to high-mannose and complex type N-glycans in the context of N-glycopeptides, N-glycoproteins, and intact antibodies. This discovery opens a new avenue to a wide variety of homogeneous, core-fucosylated N-glycopeptides and N-glycoproteins that are hitherto difficult to obtain for structural and functional studies.
Graphical Abstract
INTRODUCTION
Asparagine-linked glycosylation, namely the N-glycosylation, is one of the most prevalent posttranslational modifications of proteins in mammals, which plays important roles in modulating the intrinsic properties and biological functions of the underlying proteins 1,2. For example, the N-glycans attached can have a profound effect on protein’s folding, stability, antigenicity, and immunogenicity 2,3. On the other hand, the N-glycans can directly participate in a variety of biological recognition processes, including cell adhesion, host-pathogen interaction, cancer metastasis, and immune response 1,4–9. While all mammalian N-glycans share a common oligosaccharide core structure, further decoration of the core, such as sialylation and fucosylation, adds another level of structural diversity in modulating biological functions.
Core-fucosylation, the attachment of an α1,6-linked fucose to the innermost, asparagine-linked N-acetylglucosamine (GlcNAc) moiety in the N-glycans, is an important modification of N-glycoproteins. Compelling data have shown that core-fucosylation of glycoproteins regulates a wide range of cellular functions. For example, many studies have revealed that increased core fucosylation is often associated with cancer progression 10–12. AFP-L3, the core-fucosylated α-fetoprotein has been approved by regulatory agency as a biomarker for hepatocellular carcinoma, a major form of liver cancer 13. The roles of core-fucosylation in development has been demonstrated by the experiments that knockout of FUT8 gene in mouse models induces severe growth retardation and death during postnatal development 14. Core fucosylation also directly modulates the biological activities of glycoproteins, such as the antibody dependent cellular cytotoxicity (ADCC) of therapeutic monoclonal antibodies 15, the signaling functions of growth factor receptors and adhesion molecules 14,16–20, and the antigen recognition of IgG B cell receptors 21. On the other hand, structural studies have suggested that core-fucosylation could affect the conformations of N-glycans 22,23.
Given the difficulties to isolate homogeneous glycoforms from natural sources, synthesis of structurally well-defined, core-fucosylated glycopeptides and glycoproteins is essential for various glycomics studies aiming to further decipher the structural and functional impact of core-fucosylation 24–26. In animals and humans, core-fucosylation is catalyzed solely by the mammalian α1,6-fucosyltransferase, FUT8 27,28. However, FUT8 has a very strict substrate specificity, requires the presence of a free GlcNAc at the α1,3-linked mannose arm in the N-glycan as the substrate and usually is unable to fucosylate full-size mature N-glycans 29–32. Only until recently we have provided the first examples showing that FUT8 could catalyze in vitro fucosylation of some high-mannose N-glycans lacking a free GlcNAc at the α1,3-linked mannose arm when the glycan is present in an appropriate protein or other context 33. This strict substrate specificity makes the α1,6-fucosyltransferase of limited usefulness for chemoenzymatic synthesis. On the other hand, chemical synthesis of core-fucosylated N-glycopeptides and N-glycoproteins is more complex than the synthesis of those non-fucosylated glycoconjugates, due to the difficulty in control of the α-stereo-selectivity in glycosylation and the acid-labile nature of the α1,6-fucosidic linkage 34,35. A method for direct fucosylation of intact glycopeptides and glycoproteins is highly desirable. We report in this paper the discovery of novel mutants derived from Lactobacillus casei α-fucosidase, which are able to use α-fucosyl fluoride as the substrate for direct core-fucosylation of intact N-glycopeptides and N-glycoproteins without product hydrolysis. We found that the rationally designed mutants, E274A/S/G derived from Lactobacillus casei α-fucosidase carrying a single mutation at the general acid/base residue (E274) acted as an efficient fucoligase and were able to fucosylate a wide variety of substrates including large synthetic N-glycopeptides, natural N-glycoproteins, and intact monoclonal antibodies, paving a way to obtain core-fucosylated glycopeptides and glycoproteins for structural and functional studies.
RESULTS AND DISCUSSION
Design
The goal of this study is to generate glycosidase mutants capable of using simple glycosyl donors for direct core-fucosylation of intact N-glycopeptides and N-glycoproteins, which could not be achieved by the catalysis of the biosynthetic pathway α1,6-fucosyltransferase (FUT8) because of its strict substrate specificity. There are two general glycosidase engineering strategies that may convert a glycosidase into a synthetically useful mutant. One is the glycosynthase concept through site-directed mutagenesis at the critical nucleophilic residue of a retaining glycosidase to generate a mutant that is devoid of hydrolysis activity but can take an activated glycosyl donor (usually a glycosyl fluoride) with an opposite anomeric configuration for transglycosylation 36–38. Glycosynthases derived from several GH family glycosidases have been successfully created using this strategy 39–43. The other is the glycoligase approach, first developed by Withers and co-workers, in which the general acid/base residue of a retaining glycosidase is mutated to eliminate the hydrolysis activity, and the enzymatic transglycosylation is enabled by using an activated glycosyl donor with the same anomeric configuration 44–47. For β-glycosynthases derived from the corresponding retaining β-glycosidases, the readily synthesized and relatively stable α-glycosyl fluorides have become the common glycosyl donor substrates 39,40. However, the evaluation of the transglycosylation activity of potential α-fucosynthases usually requires a β-fucosyl fluoride, which is quite unstable in aqueous solution and will be hydrolyzed spontaneously with a half-life of ca. 20 min 48. Previously several α1,2- and α1,3–1,4-fucosynthases have been generated from the Bifidobacterium bifidum α-L-fucosidases and examined for enzymatic transfucosylation of glycoconjuagtes, but the dependence on the use of the highly unstable β-glycosyl fluoride renders these mutants less attractive for synthetic purpose 48–51. As an alternative approach, Moracci and co-workers have shown that the stable β-fucosyl azide could serve as a glycosyl donor for the α-fucosynthases derived from the hyperthermophilic archaeon Sulfolobus solfataricus α-L-fucosidase 52. The β-galactosyl azide was also successfully applied as a substrate for an α-galactosynthase 53,54.
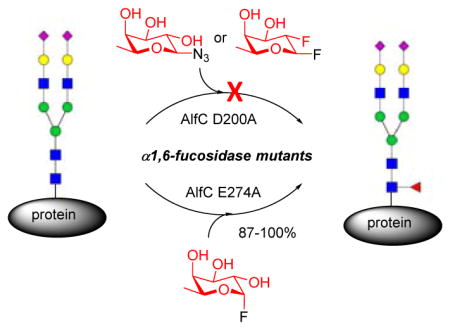
Despite these successes, no α1,6-fucosynthase or any α-fucoligase has been reported so far. To explore this possibility, we started our pursuit by choosing the α1,6-fucosidase from Lactobacillus casei (AlfC) as the model enzyme, which was shown to hydrolyze specifically α1,6-fucosidic linkage 55 and was recently reported to have transglycosylation activity, capable of making Fucα1,6GlcNAc disaccharide, using p-nitrophenyl α-fucopyranoside as the donor substrate and GlcNAc as the acceptor 56,57. Nevertheless, the wild type enzyme also hydrolyzes the disaccharide product rapidly leading to low synthetic efficiency. Moreover, it has been unclear if this enzyme could act on more complex substrates for transglycosylation other than a simple GlcNAc substrate. Thus, our design is to generate potential glycosynthase and glycoligase mutants from the Lactobacillus casei α1,6-fucosidase, and to test their ability to core-fucosylate various acceptor substrates (Figure 1). For evaluating the potential glycosynthase mutants, we would use the stable β-fucosyl azide (1) 52 as the substrate. In addition, since β-fucosyl fluoride is highly unstable with a half-life of ca. 20 min in a neutral aqueous solution 48, we sought to synthesize the 2-deoxy-2-fluoro-β-fucosyl fluoride (2) as a more stable substrate to test the potential α-fucosynthase mutants. For evaluating the potential glycoligase mutants, the α-fucosyl fluoride (3) would be used as a donor substrate.
Figure 1.
Evaluation of fucosidase mutants for direct core fucosylation of N-glycans
Alignment of Amino acid sequence with GH29 α-fucosidases
GH29 retaining α-L-fucosidases catalyze the removal of non-reducing terminal L-fucose residues in the α1,2, α1,3, α1,4, or α1,6-glycosidic linkages in oligosaccharides and glycoconjugates. In order to determine the key catalytic residues of AlfC α-fucosidase, several α-fucosidases belonging to the GH29 family were chosen in alignment of amino acid sequences, due to their distinct substrate specificities and positions of the two catalytic residues clarified in previous reports 58–61. The results were shown in Figure 2. All the positions of catalytic nucleophile, an aspartate residue, turned out to be completely aligned up among the five α-fucosidases including AlfC (Indicated by blue arrowhead and box in Figure 2). It has been previously shown that the nucleophile residue is fully conserved across the GH29 family 62. Thus residue D200 in AlfC which is aligned with the nucleophilic residues of the other GH29 family enzymes was very likely the nucleophile residue of the AlfC enzyme. The positions of general acid/base seemed more complicated, as these residues scattered around in these enzyme sequences (Indicated by purple box in Figure 2). The glutamate residue E274 of AlfC aligned with the E289 of Homo sapiens A1 (Hfuc), while no polar amino acid residue of AlfC showed up at the positions of the general acid/base residues in other α-fucosidases. Interestingly, both Hfuc and AlfC turned out to be α1,6-fucosidases. Considering the alignment results and the substrate specificity, we assumed that the E274 residue could be the catalytic general acid/base residue for the AlfC enzyme.
Figure 2.

Sequence alignment of AlfC and selected GH29 fucosidases including human α-(1,6)-fucosidase (Hfuc). Blue arrowhead and blue box indicate the fully conserved nucleophile site across GH29 family. Purple arrowhead shows the experimentally confirmed general acid/base site E289 for Hfuc. Purple box indicates that the E274 of AlfC aligns with the E289 of Hfuc.
Generation of potential α-fucosidase-based glycosynthases and glycoligases
Following the glycosynthase concept proposed by Withers and co-workers 36, we performed site-directed mutagenesis at the identified nucleophile in the AlfC α1,6-fucosidase, D200, to generate selected mutants, including D200G, D200S, D200A, and D200T. Similarly, selected mutants at the putative general acid/base residue, E274, were generated to provide potential glycoligases, including E274A, E274S, E274G, and E274D (Table S1). These mutants were expressed as a fusion protein with a C-terminal Vibrio cholera MARTX toxin cysteine protease domain (CPD) carrying a 10× His tag, as previous experiments have demonstrated that the CPD tag can enhance the solubility and stability of recombinant proteins 63–66. All these mutants were efficiently expressed in E. coli with a relatively high yield (more than 40 mg/L) and were readily purified by Nickel-NTA affinity chromatography 63. We then tested the hydrolysis activity of these mutants together with wild type AlfC using p-nitro α-fucoside (pNPFuc) as the substrate. As expected, all these mutants, except E274D, showed only trace residual hydrolysis activity due to the mutations at the critical residues, while (Figure S1). This study confirms that the D200 residue is the nucleophile and that the E274 residue is most likely the general acid/base residue. In the case of E294D, the similarity between the Glu and Asp suggests that the Asp residue in place of the E274 could still play the role of the Glu residue as the general acid/base for promoting hydrolysis of the substrate.
Assessment of the mutants as potential glycosynthases or glycoligases
First, we examined the D200 mutants as potential glycosynthases using two types of activated fucosyl donors with opposite anomeric configuration as the donor substrates. As β-glycosyl fluorides are quite unstable and are subjected to spontaneous hydrolysis in aq. solutions 48, we synthesized 2-deoxy-2-fluoro-β-fucosyl fluoride (2) as a more stable substrate (Scheme S1) 67. We tested its stability in an aqueous buffer and found that the half-life of 2 was about 6 h at 37 °C in a phosphate buffer (100 mM, pH 7.4) (Figure S2), which was significantly more stable than the corresponding β-fucosyl fluoride (t1/2 ca. 20 min) 48,67. β-Fucosyl azide (1) was used before as substrate for testing α1,2- and α1,3-fucosidase mutants 52. Thus, the potential glycosylation activity of the nucleophilic mutants, including D200G, D200S, D200A, and D200T, were tested using both the β-glycosyl azide (1) and the β-glycosyl fluoride (2) as the donor substrates and the Fmoc-Asn(GlcNAc)-OH (4) 68 as the acceptor substrate (Scheme 1). The reactions were run with different amounts of enzymes and were monitored by reverse-phase HPLC analysis. However, no glycosylation products were observed with either the β-fucosyl azide (1) or the 2-dexoy-2-fluoro-β-fucosyl fluoride (2) donors by any of the nucleophilic residue mutants. This result suggested that those nucleophilic mutants so far tested did act as a glycosynthase.
Scheme 1.

Transglycosylation with potential α-fucosynthase and α-fucoligase
Next, we tested the potential glycoligase activity of the general acid/base residue mutants using the α-fucosyl fluoride (3) as the donor substrate (Scheme 1). Interestingly, the E274A mutant exhibited excellent catalytic activity to transfer a fucose residue to the GlcNAc moiety of acceptor 4 to give the disaccharide derivative (5) with regio- and stereo-specificity (Scheme 1). Using a moderate excess of the fucosyl fluoride (donor/acceptor, 2:1 molar equivalent), an almost quantitative yield of product 5 was achieved by incubation of E274A (0.1 mg/ml, pH 7.5) with 3 and 4 at 42 °C within 1 h. The other two mutants, E274G and E274S, were also excellent catalysts for the α1,6-fucosylation and gave similar results.
The structure of 5 was confirmed by ESI-MS (calculated, M = 703.3 Da; found, 704.5 [M + H]+) and 1H-1H COSY NMR (Figure S3). A doublet at δ 4.62 with a relatively small coupling constant (J = 3.2 Hz) for the H-1 of the fucose moiety suggested that the attached fucose was in an α-glycosidic linkage. On the other hand, an obvious shift for the H-6 in the GlcNAc moiety of 5 toward the low field (from δ3.6 in 4 to δ3.8 in 5), while other protons on the GlcNAc moieties in 4 and 5 did not show obvious change, suggested that the fucosylation occurred at the 6-hydroxy group of the GlcNAc moiety to form an α1,6-fucosyl glycosidic linkage.
Interestingly, we observed that in the absence of the GlcNAc acceptor, the AlfC mutants (E274A, E274G, and E274S) only hydrolyzed α-fucosyl fluoride slowly but the wild type AlfC could quickly hydrolyze the donor substrate. Taken together, these results indicated that the AlfC mutants represented a class of unique O-fucoligase for core-fucosylation, which could use synthetic α-fucosyl fluoride as the simple donor substrate instead of the expensive GDP-fucose as the substrate as required by the α1,6-fucosyltransferase (FUT8). To examine whether the fused CPD domain in the enzyme played a role in catalyzing fucosylation, we removed the CPD tag in the AlfC E274A mutant by using inositol hexaphosphate (Figure S4). The purified mutant without CPD tag showed comparable catalytic activity as its CPD tagged counterpart, indicating that CPD tag did not affect the catalytic activity of the mutant (Data not shown). However, we found that the mutants fused with the CPD were more thermostable than the free enzyme. While incubation of the free enzyme at 55 °C for 5 h led to partial precipitation probably due to aggregation, the CPD fused mutant was still active, indicating that the CPD domain could stabilize the enzyme. A detailed study on the fucosylation activity of E274A indicated that the optimal pH of its catalysis was 7–8.5 and the optimal temperature was 40–50 °C (Figure S5).
We also performed kinetic studies on the mutants and the results were summarized in Table S2 and Figure S6. The E274A mutant exhibited the highest fucosyl transfer activity with the catalytic efficiency (kcat/KM) of up to 1.6 × 103 min−1mM−1. The E274S and E274G mutants also showed high activities (Table S2). In particular, kinetic analysis revealed that these mutants had high turnover numbers (kcat > 200 min−1) implying that these mutants, with mutation at the general acid/base residue, could efficiently use the α-fucosyl fluoride for transglycosylation without hydrolysis of the product.
Direct core-fucosylation of various GlcNAc-peptides
The discovery that the E274A and related mutants were able to efficiently catalyze the trans-fucosylation on the GlcNAc-Asn derivative prompted us to test if the mutants could also fucosylate GlcNAc moiety in the context of polypeptides. We chose three distinct GlcNAc-containing peptides for the test: the hexapeptide (6) derived from the sialylglycopeptide (SGP) isolated from egg yolks, a 19-mer GlcNAc-peptide (8) that consisting of the CD52 antigen and a sortase A signal peptide sequence, and a potent HIV inhibitor GlcNAc-C34 (10) derived from the HIV-1 gp41 envelope glycoprotein 69.
We found that the E274A could transfer a fucose moiety to all the three GlcNAc-peptides to form the corresponding fucosylated peptides (7, 9, and 11), respectively (Scheme 2). While the small GlcNAc-peptide (6) was transformed faster than the large ones (8 and 10), all the enzymatic fucosylation could be achieved essentially quantitatively within a relatively short time of incubation (1–2 h) when a sufficient amount of enzyme was used. The glycopeptide products were readily purified and its identity was confirmed by LC-MS analysis (Figure S7). The Fucα1,6GlcNAc-peptides could serve as excellent acceptor substrates for the Endo-F3 D165A glycosynthase to synthesize core-fucosylated complex N-glycopeptides 66.
Scheme 2. Core fucosylation of GlcNAc-peptides by the fucoligasea).

a)Reagents and conditions: i) 0.001–0.0015 mol. equiv. AlfC E274A, PBS (pH7.5), 42°C, 0.5–2 h
Direct core fucosylation of intact N-glycopeptides
Next, we tested the feasibility of E274A-catalyzed directed core-fucosylation of intact N-glycopeptides carrying a full-size N-glycan. Several complex N-glycopeptides (12, 14, 16, and 18) carrying a sialylated biantennary complex type N-glycan were used as potential acceptor substrates, including a large cyclic HIV-1 V3 glycopeptide (18). Surprisingly, the E274 mutant could efficiently add a core fucose to the intact N-glycopeptides to give the corresponding core-fucosylated N-glycopeptides (13, 15, 17, and 19), respectively in excellent yields (Scheme 3). The glycopeptide products were separated by RP-HPLC and their identities were first characterized by ESI-MS analysis (Figures S8).
Scheme 3. Fucoligase catalyzed direct core fucosylation of intact N-glycopeptidesa.

aReagents and conditions: a) 0.0015 mol equiv. of AlfC E274A, PBS (pH7.5), 42°C, 1–3 h.
To verify that the fucose was specifically added to the innermost GlcNAc moiety in the glycopeptide, the isolated glycopeptide product (15) was treated with two specific hydrolytic enzymes PNGase F and Endo-F3 that hydrolyze N-glycopeptides and N-glycoproteins. PNGase F completely removes the N-glycans from N-glycopeptides by hydrolyzing the amide linkage between asparagine and the N-glycans, while Endo-F3 specifically cleaves at the β1,4-glycosidic bond between the two GlcNAc moieties in N-glycopeptides/proteins. ESI-MS analysis indicated that treatment of 15 with PNGase F efficiently converted 15 into a single peptide species that was corresponding to the free polypeptide without any sugar attached (Calcd for the plain polypeptide, M = 1848.75 Da; found, M= 1848.83 Da) (Figure S9A), suggesting that the fucose must be transferred to the N-glycan portion instead of any residues on the peptide portion. It should be pointed out that enzyme PNGase F is highly sensitive to the site of fucosylation in the N,N-diacetylchitobiose core, which could tolerate core fucosylation at the 6-OH of the innermost GlcNAc moiety, but could not release N-glycans with an α1,3-fucose being attached to the core, as usually found in plant N-glycoproteins 70. The efficient release of the N-glycans from the N-glycopeptide by PNGase F treatment further confirmed that the fucose moiety was α1,6 attached to the innermost GlcNAc moiety. On the other hand, treatment of 15 with Endo-F3 gave a species that corresponded to Fucα1,6GlcNAc-peptide (9) (ESI-MS, calculated, M= 2195.38 Da; found, M = 2196.64 Da, deconvolution data) (Figure S9B). Taken together, all these experimental data suggested that indeed the fucose moiety was added specifically to the innermost, Asn-linked GlcNAc moiety of the glycopeptide in the glycoligase-catalyzed fucosylation. The ability to directly core-fucosylated intact N-glycopeptides opens a new avenue to access core-fucosylated N-glycopeptides directly from natural and synthetic N-glycopeptides.
Direct core fucosylation of intact glycoproteins
The success in direct core-fucosylation intact glycopeptides by the glycoligase mutants encouraged us to also test the direct core-fucosylation of intact N-glycoproteins. Thus, we chose bovine ribonuclease B (RNase B, 20), a natural glycoprotein with heterogeneous high-mannose type N-glycan, as a model system to investigate whether this engineered enzyme was capable of fucosylating the natural glycoprotein. RNase B is a good model for the study of protein glyco-remodeling, as it has only a single N-glycosylation site, N34, located around the cleft of the spheroidal protein. The N-glycans on natural RNase B are a mixture of high mannose type from five to nine mannose residues. Firstly, we examined whether the E274A mutant could perform direct core-fucosylation on the glycoform mixtures and, if yes, whether the mutant showed any selectivity on the N-glycans of different size. Thus, RNase B (20) was incubated with 3 and the E274A mutant at 37 °C (a lower temperature to prevent protein denaturation). The reaction was monitored with LC-MS. To calculate the reaction yields, glycans were cleaved from the protein with PNGase F and then detected and quantitated with HPLC after Fmoc-labelling (See experimental section) 71. We found that RNase B (20) could serve as an acceptor substrate of AlfC E274A for core-fucosylation (Scheme 4), but the reaction was relatively slow, giving partial transformations to the fucosylated RNase B glycoforms 21 under the conditions after 5 h (Figure S10). Interestingly, the E274A mutant exhibited some substrate specificity on the different glycoforms. The yields of core fucosylation of respective glycoforms, as judged by the HPLC analysis of the released core-fucosylated N-glycans were 72%, 54%, 51%, 55% and 43%, for the Man5 to Man9 glycoforms, respectively (Figure S11). These results suggested that for high-mannose type N-glycoforms, the larger size N-glycans were less favorable substrate for E274A, probably due to steric hindrance. This study also demonstrated that high-mannose type glycoproteins could serve as substrates for core-fucosylation by the fucoligase mutant.
Scheme 4. Fucoligase mediated glyco-remodeling of natural glycoprotein RNase Ba).

a)Reagents and conditions: i) AlfC E274A (0.042 mol. equiv. of 20), PBS (pH 7.5), 42 °C, 5 h; ii) Endo-H (0.01 mol. equiv. of 20), PBS (pH6.0), 37 °C, 1.5 h; iii) AlfC E274A (0.021 mol. equiv. of 22), PBS (pH 7.5), 42 °C, 6 h; iv) Endo-F3 D126A (0.015 mol. equiv. of 23), PBS (pH6.5), 37°C, 0.5 h.
Since the direct core-fucosylation of RNase B was less efficient, we tested an alternative approach that consists of deglycosylation, glycoligase-catalyzed core-fucosylation, and Endo-F3 catalyzed transglycosylation to constitute a core-fucosylated complex type RNase glycoform (Ribonuclease C) (Scheme 4). Thus, The high mannose N-glycans in RNase B (20) were removed by Endo-H to afford the deglycosylated protein GlcNAc-RNase (22) in quantitative yield 72. We found that, in contrast to the slow fucosylation of the intact RNase B (20), the core fucosylation of GlcNAc-RNase (22) was much more efficient to give the Fucα1,6GlcNAc-RNase (23) in essentially quantitative yield within 5 h under the same conditions. Deconvolution of the ESI-MS spectrum gave a single species confirming the identity of the fucosylated product (Calculated for 23, M = 14031 Da; found, M = 14034 Da) (Figure S12A). Subsequently, Endo-F3 D165A catalyzed glycosylation of the Fucα1,6GlcNAc-RNase (23) with a complex glycan oxazoline (24), following our previously reported procedure 66, gave the core fucosylated complex glycoform (25) in 97% yield (Scheme 4). ESI-MS analysis verified the glycosylation product (Calculated for 25, M = 16033 Da; found, M = 16036 Da) (Figure S12B). This was the first synthesis of core-fucosylated complex type glycoform of RNase (RNase C). The synthesis was more efficient than the preparation of the non-fucosylated RNase C that we have previously reported using Endo-M mutant for transglycosylation 73. The ability of the fucoligase mutant to directly fucosylate intact glycopeptides and glycoproteins significantly expands the repertoire of toolkits for making homogeneous core fucosylated glycoforms for various glycomic applications.
Direct enzymatic core-fucosylation of intact monoclonal therapeutic antibody
The potent transfucosylation activity of AlfC α-fucoligase towards glycopeptides and glycoproteins prompted us to explore its application for glycoengineering of therapeutic monoclonal antibodies. Core fucosylation of the Fc domain N-glycans plays a pivotal role in modulating the functions of antibodies and their therapeutic efficacy 8,74. We have previously developed a chemoenzymatic Fc glycan remodeling method that consists of deglycosylation and glycosynthase-catalyzed glycan transfer to produce homogeneous Fc glycoforms 65,66,75. If the fucoligase mutant could act on intact antibody, then it would further expand the repertoire of the toolbox for antibody Fc glycan engineering. To test this possibility, we selected rituximab (26) as a model system. First, rituximab was deglycosylated with Endo-S followed by defucosylation with the wild type AlfC α-fucosidase to give the GlcNAc-rituximab (27) (Scheme 5). Interestingly, Wild-type AlfC α-fucosidase showed good hydrolytic activity, to afford the deglycosylated product (27) with essentially complete transformation within 3 h. However, we found that Wild-type AlfC α-fucosidase was unable to remove the fucose moiety in the intact rituximab before Endo-S treatment, even with significantly large amount of the enzyme and prolonged incubation time (data not shown). The non-fucosylated sialyl complex type glycoform of rituximab (S2G2-rituximab, 28) was synthesized by the EndoS-D233Q catalyzed glycosylation with glycan oxazoline (24) as the donor, following our previously published procedure 75. These two glycoforms (27 and 28) were used as potential substrates for examining the AlfC fucoligase mutants. The GlcNAc-rituximab (27) was incubated with α-fucosyl fluoride (3) and AlfC E274A mutant at 37 °C and the reaction was monitored by LC-MS analysis. We found that GlcNAc-rituximab acted as an excellent substrate of the fucoligase AlfC E274A and the enzymatic reaction resulted in complete core-fucosylation of 27 to give the Fucα1,6GlcNAc-rituximab (29) as a single product within 6 h under the conditions. The product was readily isolated by protein A affinity chromatography. To confirm the identity of the Fucα1,6GlcNAc-rituximab (29), the product was treated with protease IdeS which specifically hydrolyzes IgG antibody to release the monomeric Fc domain, and the released Fc was analyzed by ESI-MS. The deconvolution of the ESI-MS of core fucosylated Fc domain exhibited a single species with the expected molecular mass (Calculated for Fucα1,6GlcNAc-Fc, M = 24112 Da; found, M = 24110 Da) (Figure S13). The 146 Da increase from 27 to 29 suggested an addition of a single fucose moiety in the monomeric Fc domain. As described in our previous study, Fucα1,6GlcNAc-rituximab (29) was an excellent substrate for EndoS-D233Q 75. Indeed, incubation of 29 with EndoS-D233Q and glycan oxazoline (24) gave the expected core-fucosylated sialyl complex type glycoform of rituximab (S2G2F-rituximab, 30) in excellent yield, further confirming that the 29 was the expected Fucα1,6GlcNAc-rituximab.
Scheme 5. Chemoenzymatic glycoengneering of human IgG combined with fucoligase promoted core fucosylationa.

a)Reagents and conditions: i) (a) Endo-S (0.005 mol. equiv. of 26), Tris-HCl (pH 8.0), 37 °C, 1 h; (b) AlfC (0.01 mol. equiv. of antibody), PBS (pH7.0), 37 °C, 3 h. ii) Endo-S D233Q (0.025 mol. equiv. of 27), Tris-HCl (pH7.4), 30 °C, 0.5 h; iii) AlfC 274A (0.225 mol. equiv. of 27), PBS (pH 7.5), 42 °C, 6 h; iv) Endo-F3 D126A (0.1 mol. equiv. of 29), PBS (pH6.5), 37 °C, 0.5 h; v) AlfC E274A (0.45 mol. equiv. of 28), PBS (pH 7.5), 42 °C, 8 h (Note: the LC-MS analysis showed almost complete converstion for the enzymatic reactions; the yields indicated isolated yields after a single protein A affinity chrmatogarphic purification and quantified by nanodrop analysis.
Finally, we tested the direct fucosylation of S2G2-rituximab (28) carrying the full size complex type Fc N-glycans. Interestingly, we found that the AlfC E274 mutant could also smoothly transfer the fucose moiety to the intact rituximab, albeit at a slow rate in comparison with the GlcNAc-rituximab substrate. Nevertheless, an increased amount of the enzyme could lead to complete transformation of the S2G2-rituximab (28) to the core-fucosylated glycoform (S2G2F-rituximab, 30) within a few hours at 37 °C (Scheme 5). This result is remarkable given the fact that the Fc domain N-glycans are buried between two Fc domains, and that wild type AlfC α-fucosidase actually shows almost no activity to remove the core fucose in the intact Fc N-glycans. It is puzzling why the wild type α-fucosidase was unable to hydrolyze the fucose in the intact antibody but the E274A mutant could manage to access the site to efficiently perform the trans-fucosylation on the intact antibody.
To confirm that the fucose was specifically attached at the core position and that there was no non-enzymatic attachment of the fucose to any residues in the protein portions, we performed ESI-MS analysis of the intact antibody product, coupled with specific enzymatic transformations (Figure 3). First, the observed molecular mass of 30 from the ESI-MS deconvolution data matched well the expected molecular mass (Calcd, M = 148888 Da, found, M = 148886 Da) (Figure 3A). The data suggested an addition of two fucose moieties on top of the S2G2-rituximab (28) that was a dimer carrying two N-glycans (each heavy chain has an N-glycan at the Asn-297 site); second, PNGase F treatment of 30 gave a single species of the antibody, the ESI-MS deconvolution data (144182 Da) of which agreed with the polypeptide backbone of rituximab (calcd, M = 144181 Da) (Figure 3B). This result suggested that there was no any additional modification of the protein portions of rituximab during the AlfC E274A catalyzed reactions; Third, Endo-S treatment of 30 converted it back to the Fucα1,6GlcNAc-rituximab (29), which appeared as a single species at 144882 Da (Figure 3C).
Figure 3.

Characterization of the fucosylated rituximab (30). A) ESI-MS spectrum and its deconvolution of 30; B) ESI-MS spectrum and its deconvolution of the product from the PNGase F treatment of 30; C) ESI-MS spectrum and its deconvolution of the product from the Endo-S treatment of 30.
Lectin binding studies with core-fucosylated RNase B and rituximab
The interactions of glycoproteins and core fucose-specific lectins play important roles in biological recognition processes. To demonstrate the specific recognition, we performed the binding analysis of the interactions between the Aleuria aurantia lectin (AAL) and a few core-fucosylated glycoproteins synthesized via the AlfC fucoligase catalyzed reactions. AAL is a commonly used lectin, which has high affinity for α1,6-fucosylated glycoproteins, including core fucosylated antibody and haptoglobin 76,77. First we tested the binding of 21 carrying core fucosylated high-mannose glycans and the natural glycoprotein (20) with AAL. The AAL showed specific binding to 21, while it didn’t show any binding to the non-fucosylated glycoprotein (20) (Figure 4A). AAL also demonstrated specific recognition of the S2G2F-rituximab (30) but did not show any detectable affinity to the S2G2-rituximab (28) (Figure 4B). Interestingly, a significant difference in the affinities of 21 and 30 for lectin AAL was observed (EC50: 275 μM for 21 vs. 15 μM for 30), with the affinity of the core-fucosylated antibody being about 17 fold higher than that of core-fucosylated RNase B. To verify whether the observed difference majorly came from the difference in the nature of the N-glycans (core fucosylated high-mannose type in 21 vs. complex type in 30) or in the nature of proteins (the context of RNase vs. antibody), we released the N-glycans from the proteins by PNGase F treatment with in situ Fmoc labeling 71. ELISA analysis of the AAL binding indicated that the affinity of the core-fucosylated complex type N-glycans (EC50, 1.8 μM) released from antibody (30) was about 8 fold higher than that of the core-fucosylated high-mannose type N-glycans (EC50, 16 μM) released from (21). Thus, the difference in AAL affinities between core-fucosylated RNase B (21) and core-fucosylated antibody (30) came mainly from the difference in the nature of the N-glycans (high mannose vs. complex type), and the protein part contributed to a less extent (ca. 2-fold).
Figure 4.

ELISA analysis of the binding between lectin AAL and glycoproteins or released N-glycans. The glycoproteins or released N-glycans (tagged with Fmoc) were immobilized on the plates and probed by serial dilutions of the lectin. A) AAL binding with RNase B (20) and fucosylated RNase B (21); B) AAL binding with non-fucosylated glycoform of rituximab (28) and fucosylated rituximab glycoform (30); C) AAL binding with the PNGase F released high-mannose N-glycans released from 20 and 21; D) AAL binding with the PNGase F released complex type N-glycans from 28 and 30.
Comparative SPR binding studies with core-fucosylated and non-fucosylated glycoforms of antibody rituximab
Previous studies have demonstrated that core fucosylation adversely impacts the binding of IgG antibodies to the FcγIIIA receptor (FcγRIIIA) on natural killer cells and thus down-regulates the antibody-dependent cellular cytotoxicity (ADCC) 74,78,79. To verify the effects of fucosylation on Fc receptor binding of the core-fucosylated rituximab glycoforms obtained through the fucoligase technology, we performed the surface plasmon resonance (SPR) analysis on the binding of the FcγRIIIA-V158 (a major high-affinity FcγIIIA receptor allele) to the non-fucosylated glycoforms (27 and 29) vs. the corresponding core-fucosylated glycoforms (28 and 30) (Figure 5). The rituximab glycoforms were site-specifically immobilized on protein A chips and the FcγRIIIA-V158 at various concentrations were injected as analytes, following our previously reported procedures 74. As expected, the non-fucosylated sialyl complex type glycoform (S2G2-rituximab, 28) showed much higher affinity than that of the corresponding core-fucosylated glycoform (S2G2F-rituximab, 30). As estimated by the dissociation constant (KD), the affinity of S2G2-rituximab (28) (KD = 2.2 nM) for FcγRIIIA-V158 was about 56-fold higher than that of the corresponding fucosylated S2G2F glycoform (30) (KD = 131 nM) (Figure 5A and 5B). This result was consistent with our previously reported binding data 74. On the other hand, the deglycosylated glycoforms (27 and 29) showed much lower affinity than that of the glycosylated glycoforms (Figure 5C and 5D). Even in this case, the effects of core-fucosylation was apparent. While the GlcNAc-rituximab (27) carrying only the innermost GlcNAc moiety at the Fc domain showed a moderate affinity for the FcγIIIA receptor with a KD of 784 nM, the corresponding core-fucosylated glycoform, Fucα1,6GlcNAc-rituximab (29), demonstrated only marginal binding to the receptor under the measurement condition, for which the KD was not determined. These data confirm that core fucosylation could remarkably reduce the affinity of IgG antibodies for the activation receptor (FcγIIIA), which would significantly decrease antibody-dependent cellular cytotoxicity, as demonstrated in clinical trials 79. On the other hand, a decreased affinity for the activation Fc receptor could be beneficial if the antibody would be used for the treatment of autoimmune diseases, as demonstrated by the Fc sialylation-enriched intravenous immunoglobulin (IVIG) that showed significantly enhanced anti-inflammatory activity in a mouse model of rheumatoid arthritis (RA) 80,81. Thus, the fucoligase-catalyzed direct core-fucosylation of intact antibodies provides an efficiency avenue to modulating antibody’s immunological properties.
Figure 5.

SPR analysis of the binding between FcγRIIIA-V158 and various glycoforms of rituximab. The antibodies were immobilized on a protein A chip and and the Fc receptor was run as analytes at 2X serial dilutions starting at 2 μM. A) binding with non-fucosylated sialyl complex type rituximab (28); B) binding with core-fucosylated sialyl complex type rituximab (30); C) binding with GlcNAc-rituximab (27); D) binding with Fucα1,6GlcNAc-rituximab (29).
CONCLUSION
A highly efficient chemoenzymatic method for direct core-fucosylation of intact N-glycopeptides, N-glycoproteins, and therapeutic antibodies is established. This method was enabled by the discovery of an array of AlfC α-fucosidase mutants that act as novel glycoligases for transglycosylation using α-fucosyl fluoride as the simple donor substrates. The AlfC α-fucosidase mutants represent the first examples of glycoligases capable of specifically attaching an α1,6-fucose moiety to intact N-glycans of glycoproteins. The discovery of the novel α-fucoligases opens a new avenue to quickly constructing library of core-fucosylated N-glycopeptides and N-glycoproteins directly from the corresponding non-fucosylated counterparts, which have been hitherto difficult to obtain for various glycomics studies.
EXPERIMENTAL SECTION
Materials and Methods
All chemicals, reagents and solvents were purchased from Sigma–Aldrich and TCI, and unless specially noted, applied in the reaction without further purification. Monoclonal antibody rituximab was purchased from Premium Health Services Inc. (Columbia, MD). Silica gel (200–425 mesh) for flash chromatography was purchased from Sigma–Aldrich. Analytical reverse-phase chromatography was performed on a Waters 626 HPLC instrument equipped with an XBridge BEH130 C18 column (3.5 μm, 4.6 × 250 mm) for reversed phase or YMC-Pack NH2 column (5 μm, 4.6 × 250 mm) for normal phase. The XBridge column was eluted with a linear gradient of acetonitrile (0–30%, v/v) with water containing TFA (0.1%) over 35 min at a flowrate of 0.5 mL/min under UV 214nm. The YMC-Pack NH2 column was eluted with a linear gradient of ammonium formate (100mM, pH4.5, 10–60%, v/v) with acetonitrile containing TFA (0.1%) over 80 min at a flowrate of 0.5 mL/min under UV 266nm. Preparative HPLC was performed on a Waters 600 HPLC instrument equipped with a SymmetryPrepTM C18 column (7 μm, 19 × 300 mm). High-Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD) was performed on a Dionex ICS-5000 chromatography system (Fischer Scientific) equipped with an electrochemical detector (ED50) and an anion exchange column (CarboPac PA10 4 × 250 mm). The PA10 column was eluted with a constant buffer composed of 80% 100 mM NaOH and 20% water at a flowrate 1.0 mL/min. Liquid Chromatography Electrospray Mass spectrometry (LC-ESI-MS) was used to analyze transfer products including core fucosylated glycopeptides and glycoproteins. The LC-ESI-MS was performed on an Exactive™ Plus Orbitrap Mass Spectrometer (Thermo Scientific) equipped with a C18 column (proZap Expedite MS C18, 2.1 × 10 mm, 1.5 μm, P.J. Cobert Associates, Inc.) for glycopeptides, C8 column (Poroshell 300SB-C8, 1.0 × 75 mm, 5 μm, Agilent) for glycoproteins and C-4 column (XBridgeTM BEH300 C4, 2.1 × 50 mm, 3.5 μm, Waters) for antibody analysis. 1H, 13C and 1H-1H COSY spectra were recorded on a 400 or 600 MHz spectrometer (Bruker, Tokyo, Japan) with D2O or DMSO-d6 as the solvent. Amino acid sequence alignment for different fucosidases was performed using MAFFT, a multiple sequence alignment tool. Plasmid pCPD vector (CPD-Lasso) was created by engineering the CPD protein as a C-terminal tag into pET31(b) (Novagen) vector. PfuUltra II fusion HS DNA polymerase was purchased from Agilent. Restriction enzymes and Escherichia coli competent cells including 5-alpha and BL21 (DE3), used for DNA manipulation and protein production, were purchased from New England BioLabs Inc. Enzyme Kinetics was performed by GraFit (Erithacus Software, UK).
Construction, expression and purification of AlfC fucosidase
The DNA sequence encoded AlfC α1,6-fucosidase from Lactobacillus casei was synthesized (GenScript) and inserted into CPD-Lasso plasmid at NdeI and BamHI sites. The plasmid was transformed to BL21 (DE3) E. coli competent cell and AlfC α-fucosidases were overexpressed as CPD and 10× His tagged proteins in Luria–Bertani (1 L) broth with carbenicillin (0.1 mg/ml) at 37 °C for 4 h until the OD600 up to 0.5. After induced by isopropyl β-D-1-thiogalactoside (0.2 mM) at 20 °C overnight, the cells were harvested by centrifugation at 8000 rpm and then disrupted by sonication. The supernatant of crude overexpressed enzyme was subjected to a Nickel-affinity chromatography in an AKTA prime plus system (GE Healthcare) equipped with a His-Trap column as previously described. The eluted target protein (69 kDa) was dialyzed against sodium phosphate (100 mM, pH7.4) and its concentration was determined by NanoDrop 2000c (Thermo Scientific).
Site-directed mutagenesis of AlfC α-fucosidase
The site-directed mutagenesis of AlfC α1,6-fucosidase was performed using Stratagene protocol. Two complementary primers (0.02 nmol) were employed in the polymerase chain reaction (PCR). After 14 cycles for PCR reaction, the reaction was mixed with DpnI (2,000 U/ml) and CutSmart buffer (1X), and incubated at 37 °C for 1 h. Then the plasmids in reaction mixture were transformed to NEB 5-alpha high efficiency competent cell for miniprep. After gene sequencing, the plasmid was transformed to BL21 (DE3) E. coli competent cell, following the same procedure of expression and purification described above. The purified AlfC mutants were characterized by SDS-PAGE.
To remove the cysteine protease domain (CPD) tag, inositol hexaphosphate (10 mM) was mixed with AlfC mutants (1 mg) in PBS buffer (0.1 M, pH 7.0) at 4 °C overnight. The enzyme without CPD tag was then purified by Ni-NTA spin kit (QIAGEN). Purified AlfC mutants (44 kDa) were characterized by SDS-PAGE, compared with its counterpart with CPD tag.
Enzymatic kinetics of hydrolysis and transfucosylation of AlfC and its mutants
Kinetic studies on the hydrolysis were carried out at 42 °C in sodium phosphate (0.1 M, pH 7.0). The amount of the released free fucose was detected by using Dionex chromatography. The concentration of the substrate 4-nitrophenyl α-fucoside (pNPFuc) was varied from 0.2 to 2.0 mM. KM and kcat values were determined by fitting the initial velocity curves to the Michaelis–Menten equation by nonlinear regression in GraFit (Erithacus Software).
The kinetic studies on transglycosylation were carried out at 42 °C in sodium phosphate (0.1 M, pH 7.5). The transglycosylation product was detected and quantitated by HPLC analysis. The concentration of the donor substrates (1–3) was varied from 0.2 to 2.0 mM, while that of the acceptor (4) was fixed (2 mM). KM and kcat values (strictly, apparent KM and kcat, as they were determined at a fixed, nonsaturating, co-substrate concentration) were determined by fitting the initial velocity curves to the Michaelis–Menten equation by nonlinear regression in GraFit.
Fucosylation of Fmoc-Asn(GlcNAc)-OH (4)
To a mixture of 3 (0.249 mg, 1.5 μmol) and acceptor 4 (0.557 mg, 1 μmol) in a buffer (PBS, 100 mM, pH 7.5, 500 μL) containing 20% DMSO was added the E274A mutant (0.05 mg, 0.1 mg/mL), and the solution was incubated at 42 °C. The reaction was monitored by LC-MS analysis on a C-18 column. After the reaction completed (30 min), the reaction mixture was quenched by 0.1% TFA, and then centrifuged and filtered through 0.45 μm syringe filter. The filtrate was concentrated and the residue was purified by reverse-phase (C18) HPLC to obtain the product (5) (0.63 mg, 99%) as a white powder after lyophilizaion. 1H NMR (DMSO-d6 + 1% D2O, 400 MHz): δ = 7.88 (d, J = 7.6 Hz, 2H, Ph-H), 7.71 (d, J = 7.6 Hz, 2H, Ph-H), 7.41 (dd, J = 7.6, 7.2 Hz, 2H, Ph-H), 7.32 (dd, J = 7.2, 7.6 Hz, 2H, Ph-H), 4.80 (d, J1,2 = 6.4 Hz, 1H, H-1), 4.62 (d, J1,2 = 3.2 Hz, 1H, H-1′), 4.22 (m, 4H, Asn-H-2, Fmoc-H-2, Fmoc-H-1), 3.85 (m 1H, H-6), 3.77 (d, J = 10 Hz, 1H, H-6), 3.50 (m, 3H, H-2′, H-3′, H-2), 3.46 (m, 1H, H-4′), 3.33 (m, 3H, H-3, H-4, H-5′), 3.09 (m, 1H, H-5), 2.54 (m, 2H, Asn-H-1), 1. 78 (s, 3H, -COCH3), 1.06 (d, J = 6.8 Hz, 3H, -CH3). 13C NMR (DMSO-d6 + 1% D2O, 100 MHz): δ = 174.52 (C=O), 172.64 (C=O), 171.37 (C=O), 158.20 (C=O), 145.59 (Ph-C, 2C), 145.26 (Ph-C, 2C), 131.44 (Ph-C, 2C), 129.36 (Ph-C, 2C), 126.48 (Ph-C, 2C), 121.91 (Ph-C, 2C), 102.10 (C-1′), 88.62 (C-1), 77.03, 75.28, 72.77, 71.31, 70.85, 69.93, 68.48, 67.73, 67.02, 55.63, 51.85, 47.09, 36.52, 23.03 (COCH3), 17.85 (C-6′). ESI-MS: calc. for 5, M = 703.3 Da; found (m/z), 704.5 [M + H]+, 726.6 [M + Na]+. Analytical RP-HPLC, tR = 28.5 min.
Fucosylation of GlcNAc-peptides. The fucosylation of 6 as a representative procedure
A mixture of 3 (0.249 mg, 1.5 μmol) and acceptor 6 (0.863 mg, 1 μmol) in a buffer (PBS, 100 mM, pH 7.5, 500 μL) containing the glycoligase mutant E274A (0.1 mg, 0.2 mg/mL) was incubated at 42 °C. The reaction was monitored by LC-MS analysis. After the reaction was complete, the reaction mixture was quenched by 0.1% TFA, and then centrifuged and filtered through 0.45 μm syringe filter. The filtrate was dried, and the product was purified by RP-HPLC to give the fucosylated product (7) (1.00 mg, 99%). ESI-MS: Calcd. for 7, M = 1008.6 Da; found (m/z), 505.43 [M + 2H]2+, 1009.68 [M + H]+. Analytical RP-HPLC, tR = 6.5 min. the fucosylation of 8 and 10 followed the same procedure to give the fucosylated products 9 and 11 in almost quantitative yield.
Fucosylated glycopeptide 9
ESI-MS: Calcd. for 9, M = 2197.0 Da; found (m/z), 550.79 [M + 4H]4+, 733.63 [M + 3H]3+, 1099.43 [M + 2H]2+. Analytical RP-HPLC, tR = 22.4 min.
Fucosylated glycopeptide 9
ESI-MS: calcd. for 11, M = 4647.3 Da; found (m/z), 929.36 [M + 5H]5+, 1161.67 [M + 4H]4+, 1547.66 [M + 3H]3+. Analytical RP-HPLC, tR = 26.7 min.
Direct core fucosylation of intact glycopeptides. Fucosylation of 12 as a representative procedure
To a mixture of 3 (0.249 mg, 1.5 μmol) and the acceptor (12) (2.86 mg, 1 μmol) in a buffer (PBS, 100mM, pH 7.5, 500 μL) was added mutant E274A (0.14 mg, 0.28 mg/mL). The solution was incubated at 42 °C. The reaction was monitored by HPLC and LC-MS analysis. HPLC indicated the completion of the reaction after 45 min. Then the reaction was quenched wiht 0.1% TFA, and the mixture was centrifuged and filtered through 0.45 μm syringe filter. The filtrate was dried and the product was purified by RP-HPLC to give the fucosylated product (13) (2.81 mg, 93.4 %). ESI-MS: calcd. for 13, M = 3010.2 Da; found (m/z), 1004.73 [M + 3H]3+, 1506.68 [M + 2H]2+. Analytical RP-HPLC, tR = 4.2 min. The fucosylation of 14, 16, and 18 was performed in a similar manner and the reaction took 1–5 h to completion as monitored by HPLC analysis. The products were purified by RP-HPLC.
Fucosylated glycopeptide 15 (92%)
ESI-MS: calcd. for 15, M = 4198.2 Da; found (m/z), 840.60 [M + 5H]5+, 1050.48 [M + 4H]4+, 1400.72 [M + 3H]3+. Analytical RP-HPLC, tR = 16.0 min.
Fucosylated glycopeptide 17 (91%)
ESI-MS: calcd. for 17, M = 6641.7 Da; found (m/z), 1107.29 [M + 6H]6+, 1329.27 [M + 5H]5+, 1661.48 [M + 4H]4+. Analytical RP-HPLC, tR = 19.6 min.
Fucosylated glycopeptide 19 (90%)
ESI-MS: calcd. for 19, M = 6479.3 Da; found (m/z), 926.76 [M + 7H]7+, 1080.81 [M + 6H]6+, 1296.40 [M + 5H]5+, 1620.62 [M + 4H]4+. Analytical RP-HPLC, tR = 22.5 min.
Core fucosylation of ribonuclease B (20)
To a mixture of the α-fucosyl fluoride (3) (0.224 mg, 1.34 μmol) and the RNase B (20) (1.0 mg, 0.067 μmol) win the buffer (PBS, 100 mM, pH 7.5, 100 μL) was added mutant E274A (0.2 mg, 2 mg/mL). The solution was incubated at 37 °C. The reaction was monitored by LC-MS analysis. After 5 h, the reaction was quenched by adding 0.1% TFA and the mixture was filtrated through a 0.45 μm syringe filter. The filtrate was subjected to RP-HPLC purification. The fractions containing the fucosylated (21) and afucosylated RNase B (20) were pooled and lyophilized. The residue was then dialyzed against sodium phosphate (100 mM, pH 7.4) at 4 °C. Concentration of the RNase B mixture was determined by NanoDrop quantitation. ESI-MS: calcd. for 21, M = 15042 Da (M5F), 15205 Da (M6F), 15368 Da (M7F), 15528 Da (M8F) and 15692 Da (M9F) Da; found (deconvolution data) (m/z): 15044 (M5F), 15207 (M6F), 15369 (M7F), 15530 (M8F) and 15693 (M9F). Analytical RP-HPLC, tR = 18.4 min.
To quantitate the M5-M9 glycan forms of RNase B, normal phase HPLC equipped with NH2 column was used to separate and quantitate the fucosylated and afucosylated N-glycans after PNGase F releasing and labeling with Fmoc tag, as described below. The RNase B mixture (100 μg) was first denatured by treatment with 1 mL of guanidine hydrochloride (8 M) containing 1,4-dithiothreitol (180 mM). The mixture was incubated at 37 °C for 1 h. Then iodoacetamide (0.7 M) was added and the mixture was incubated at 37 °C for 1 h. The denatured RNase B mixture was dialyzed against sodium phosphate (100 mM, pH 8.5) at ambient temperature. To release and tag the N-glycans, the mixture was treated with PNGase F (10 U) (37 °C for 2 h). After that, 200 μL of Fmoc-Cl in acetone (50 mg/mL) was added and the mixture was incubated at 37 °C for 1h. After centrifugation, the reaction mixture was washed with chloroform (3 × 200 μL) and the aqueous layer was passed through a Sep-Pak® C-18 cartridge to remove deglycosylated proteins. The purified Fmoc-labeling N-glycans was eluted and analyzed by normal HPLC. The ratios of each N-glycan form were calculated based on the peak integration. (M5F: 72%, M6F: 54%, M7F: 51%, M8F: 55% and M9F: 43%). NP-HPLC, tR = 39.8, 49.1, 56.9, 63.8 and 70.3 min, respectively.
Fucosylation of GlcNAc-rituximab (27)
To a mixture of the α-fucosyl fluoride (3) (96 μg, 0.56 μmol) and the GlcNAc-rituximab (27) (2.0 mg, 0.014 μmol) in a buffer (PBS, 100 mM, pH 7.5, 100 μL) was added mutant E274A (200 μg, 2.0 mg/mL). The solution was incubated at 37 °C for 7 h, when LC-MS indicated complete conversion of 27 to the fully fucosylated product (29). The mixture was then loaded on a protein A affinity column (HiTrap Protein A HP, GE Healthcare). After washing, the desired product was eluted with citrate buffer (50 mM, pH 3.5) and promptly dialyzed against sodium phosphate (100 mM, pH 7.4) at 4 °C. The solution was concentrated and the amount of fucosylated rituximab (29) was quantitated by NanoDrop analysis (1.80 mg, 90%). To verify the complete fucosylation at the Fc domain, the fucosylated rituximab (29) was treated with the IdeS protease (0.2 mg/mL) to release the monomeric Fc domain, which was then subjected to LC ESI-MS analysis. The ESI-MS revealed a single Fc species confirming the complete fucosylation of the Fc domain. ESI-MS: calcd. for the IdeS released Fc domain of 29, M = 24108 Da; found (m/z), 965.53 [M + 25H]25+, 1005.36 [M + 24H]24+, 1049.23 [M + 23H]23+, 1096.91 [M + 22H]22+, 1149.06 [M + 21H]21+, 1206.02 [M + 20H]20+, 1269.72 [M + 19H]19+, 1340.18 [M + 18H]18+, and 1419.02 [M + 17H]17+; Deconvolution of the ESI-MS, M = 24110 Da.
Fucosylation of intact rituximab (28) and ESI-MS analysis of the fucosylated intact antibody (30)
To a mixture of the α-fucosyl fluoride (3) (46.3 μg, 0.27 μmol) and the intact rituximab (28) (1.0 mg, 0.0067 μmol) in a buffer (PBS, 100 mM, pH 7.5, 100 μL) was added mutant E274A (200 μg, 2.0 mg/mL). The fucosylation of 28 was performed in a similar manner with that of 27 and the reaction took 8 h to completion as monitored by LC-MS analysis. The products were purified by protein A affinity column and was quantitated by NanoDrop analysis (0.87 mg, 87%). The ESI-MS revealed a single species confirming the complete fucosylation in the intact rituximab (30). ESI-MS: calcd. for 30, M = 148888 Da; found (m/z), 2708.01 [M + 55H]55+, 2758.17 [M + 54H]54+, 2810.16 [M + 53H]53+, 2864.21 [M + 52H]52+, 2920.29 [M + 51H]51+, 2978.64 [M + 50H]50+, 3039.40 [M + 49H]49+, 3102.70 [M + 48H]48+, 3168.70 [M + 47H]47+; Deconvolution of the ESI-MS, M = 148886 Da.
Lectin binding studies of the core-fucosylated RNase B and rituximab
The glycoprotein (20, 21, 28 or 30) (10 μg/ml) in a phosphate buffer (pH 7.5) was coated onto a 96-well plate (UltraCruz®) at 4 °C for overnight. After wash twice, 2% bovine serum albumin in PBS containing Tween® 20 (PBST buffer) was added to block the plate for 2 h. Subsequently, after washed twice again, a serial dilution of AAL-biotin ranging from 0.1 μM to 1000 μM in PBST buffer was added and the plate was incubated for 1h. After washing, peroxidase streptavidin (2 μg/mL) (Jackson ImmunoResearch Inc.) was added and incubated for 1 h. Finally, 100 μL of substrate, 3, 3′, 5, 5′-tetramethylbenzidine was added for signal development. The reaction was stopped by the adding 100 μL of 20% sulfuric acid (v/v). The absorbance at 450 nm was measured using SpectraMax M5 microplate reader (Molecular Devices). For the binding of the N-glycans, the Fmoc-labeled N-glycans released from 20, 21, 28 and 30 (1 μg/ml) were coated on the plate and the ELISA analysis was performed in the same manner as that of the glycoproteins.
SPR binding analysis on the interactions between the FcγIIIA receptor and different glycoforms of antibody rituximab
The experiment was carried out by capturing each antibody glycoform onto the protein A chip and flowing serial dilutions of FcγRIIIA V158 as the analyte. After each cycle, the surface was regenerated by injecting a glycine HCl buffer (10 mM, pH 2.0). The antibodies were captured at 200 RU. The receptor in 2X serial dilutions (7.8 nM – 2 μM) was injected at 30 μL/min for 180 s, followed by a 300-s dissociation. The experimental data were fit to a 1:1 Langmuir binding model using the BIA Evaluation software (GE Healthcare) to obtain the kinetic data.
Supplementary Material
Acknowledgments
We thank Tiezheng Li, John Giddens, Joseph Lomino and other members of the Wang lab for technical assistance and helpful discussions. This work was supported by the National Institutes of Health (NIH grants R01GM080374 and R01GM096973 to LXW).
Footnotes
NOTES
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website. Scheme S1; Tables S1 and S2; Figures S1–S13.
References
- 1.Dwek RA. Chem Rev. 1996;96:683. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 2.Helenius A, Aebi M. Science. 2001;291:2364. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 3.Petrescu AJ, Wormald MR, Dwek RA. Curr Opin Struct Biol. 2006;16:600. doi: 10.1016/j.sbi.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Varki A. Glycobiology. 1993;3:97. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haltiwanger RS, Lowe JB. Annu Rev Biochem. 2004;73:491. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 6.Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 7.Nimmerjahn F, Ravetch JV. Nat Rev Immunol. 2008;8:34. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 8.Jefferis R. Nat Rev Drug Discov. 2009;8:226. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 9.Hart GW, Copeland RJ. Cell. 2010;143:672. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taniguchi N, Kizuka Y. Adv Cancer Res. 2015;126:11. doi: 10.1016/bs.acr.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Pinho SS, Reis CA. Nat Rev Cancer. 2015;15:540. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 12.Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, Yang PC, Hsiao M, Hsu TL, Wong CH. Proc Natl Acad Sci USA. 2013;110:630. doi: 10.1073/pnas.1220425110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato Y, Nakata K, Kato Y, Shima M, Ishii N, Koji T, Taketa K, Endo Y, Nagataki S. N Engl J Med. 1993;328:1802. doi: 10.1056/NEJM199306243282502. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Inoue S, Gu J, Miyoshi E, Noda K, Li W, Mizuno-Horikawa Y, Nakano M, Asahi M, Takahashi M, Uozumi N, Ihara S, Lee SH, Ikeda Y, Yamaguchi Y, Aze Y, Tomiyama Y, Fujii J, Suzuki K, Kondo A, Shapiro SD, Lopez-Otin C, Kuwaki T, Okabe M, Honke K, Taniguchi N. Proc Natl Acad Sci USA. 2005;102:15791. doi: 10.1073/pnas.0507375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mimura Y, Katoh T, Saldova R, O’Flaherty R, Izumi T, Mimura-Kimura Y, Utsunomiya T, Mizukami Y, Yamamoto K, Matsumoto T, Rudd PM. Protein Cell. 2017 Jun 8; doi: 10.1007/s13238-017-0433-3. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SH, Takahashi M, Honke K, Miyoshi E, Osumi D, Sakiyama H, Ekuni A, Wang X, Inoue S, Gu J, Kadomatsu K, Taniguchi N. J Biochem. 2006;139:391. doi: 10.1093/jb/mvj039. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Gu J, Ihara H, Miyoshi E, Honke K, Taniguchi N. J Biol Chem. 2006;281:2572. doi: 10.1074/jbc.M510893200. [DOI] [PubMed] [Google Scholar]
- 18.Pinho SS, Seruca R, Gartner F, Yamaguchi Y, Gu J, Taniguchi N, Reis CA. Cell Mol Life Sci. 2011;68:1011. doi: 10.1007/s00018-010-0595-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, Wang D, Wu T, Dong C, Shen N, Sun Y, Xie H, Wang N, Shan L. Am J Physiol Renal Physiol. 2011;300:F1017. doi: 10.1152/ajprenal.00426.2010. [DOI] [PubMed] [Google Scholar]
- 20.Venkatachalam MA, Weinberg JM. Kidney Int. 2013;84:11. doi: 10.1038/ki.2013.95. [DOI] [PubMed] [Google Scholar]
- 21.Li W, Yu R, Ma B, Yang Y, Jiao X, Liu Y, Cao H, Dong W, Liu L, Ma K, Fukuda T, Liu Q, Ma T, Wang Z, Gu J, Zhang J, Taniguchi N. J Immunol. 2015;194:2596. doi: 10.4049/jimmunol.1402678. [DOI] [PubMed] [Google Scholar]
- 22.Andre S, Kozar T, Kojima S, Unverzagt C, Gabius HJ. Biol Chem. 2009;390:557. doi: 10.1515/BC.2009.072. [DOI] [PubMed] [Google Scholar]
- 23.Andre S, Kozar T, Schuberth R, Unverzagt C, Kojima S, Gabius HJ. Biochemistry. 2007;46:6984. doi: 10.1021/bi7000467. [DOI] [PubMed] [Google Scholar]
- 24.Schmaltz RM, Hanson SR, Wong CH. Chem Rev. 2011;111:4259. doi: 10.1021/cr200113w. [DOI] [PubMed] [Google Scholar]
- 25.Rillahan CD, Paulson JC. Annu Rev Biochem. 2011;80:797. doi: 10.1146/annurev-biochem-061809-152236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiessling LL, Splain RA. Annu Rev Biochem. 2010;79:619. doi: 10.1146/annurev.biochem.77.070606.100917. [DOI] [PubMed] [Google Scholar]
- 27.Voynow JA, Kaiser RS, Scanlin TF, Glick MC. J Biol Chem. 1991;266:21572. [PubMed] [Google Scholar]
- 28.Yang Q, Wang LX. J Biol Chem. 2016;291:11064. doi: 10.1074/jbc.M116.720789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Liu Y, Ma C, Qu J, Calderon AD, Wu B, Wei N, Wang X, Guo Y, Xiao Z, Song J, Sugiarto G, Li Y, Yu H, Chen X, Wang PG. Chem Sci. 2015;6:5652. doi: 10.1039/c5sc02025e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brzezicka K, Echeverria B, Serna S, van Diepen A, Hokke CH, Reichardt NC. ACS Chem Biol. 2015;10:1290. doi: 10.1021/cb501023u. [DOI] [PubMed] [Google Scholar]
- 31.Calderon AD, Liu Y, Li X, Wang X, Chen X, Li L, Wang PG. Org Biomol Chem. 2016;14:4027. doi: 10.1039/c6ob00586a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tseng TH, Lin TW, Chen CY, Chen CH, Lin JL, Hsu TL, Wong CH. J Am Chem Soc. 2017;139:9431. doi: 10.1021/jacs.7b03729. [DOI] [PubMed] [Google Scholar]
- 33.Yang Q, Zhang R, Cai H, Wang LX. J Biol Chem. 2017;292:14796. doi: 10.1074/jbc.M117.804070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J Am Chem Soc. 2009;131:5792. doi: 10.1021/ja809554x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun B, Srinivasan B, Huang X. Chem Eur J. 2008;14:7072. doi: 10.1002/chem.200800757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacKenzie LF, Wang Q, Warren RAJ, Withers SG. J Am Chem Soc. 1998;120:5583. [Google Scholar]
- 37.Moracci M, Trincone A, Perugino G, Ciaramella M, Rossi M. Biochemistry. 1998;37:17262. doi: 10.1021/bi981855f. [DOI] [PubMed] [Google Scholar]
- 38.Malet C, Planas A. FEBS Lett. 1998;440:208. doi: 10.1016/s0014-5793(98)01448-3. [DOI] [PubMed] [Google Scholar]
- 39.Perugino G, Trincone A, Rossi M, Moracci M. Trends Biotechnol. 2004;22:31. doi: 10.1016/j.tibtech.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 40.Hancock SM, Vaughan MD, Withers SG. Curr Opin Chem Biol. 2006;10:509. doi: 10.1016/j.cbpa.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 41.Shaikh FA, Withers SG. Biochem Cell Biol. 2008;86:169. doi: 10.1139/O07-149. [DOI] [PubMed] [Google Scholar]
- 42.Cobucci-Ponzano B, Moracci M. Nat Prod Rep. 2012;29:697. doi: 10.1039/c2np20032e. [DOI] [PubMed] [Google Scholar]
- 43.Wang LX, Amin MN. Chem Biol. 2014;21:51. doi: 10.1016/j.chembiol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jahn M, Marles J, Warren RA, Withers SG. Angew Chem Int Ed. 2003;42:352. doi: 10.1002/anie.200390114. [DOI] [PubMed] [Google Scholar]
- 45.Kim YW, Zhang R, Chen H, Withers SG. Chem Commun (Camb) 2010;46:8725. doi: 10.1039/c0cc03168b. [DOI] [PubMed] [Google Scholar]
- 46.Li C, Ahn HJ, Kim JH, Kim YW. Carbohydr Polym. 2014;99:39. doi: 10.1016/j.carbpol.2013.08.056. [DOI] [PubMed] [Google Scholar]
- 47.Danby PM, Withers SG. ACS Chem Biol. 2016;11:1784. doi: 10.1021/acschembio.6b00340. [DOI] [PubMed] [Google Scholar]
- 48.Sakurama H, Fushinobu S, Hidaka M, Yoshida E, Honda Y, Ashida H, Kitaoka M, Kumagai H, Yamamoto K, Katayama T. J Biol Chem. 2012;287:16709. doi: 10.1074/jbc.M111.333781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wada J, Honda Y, Nagae M, Kato R, Wakatsuki S, Katayama T, Taniguchi H, Kumagai H, Kitaoka M, Yamamoto K. FEBS Lett. 2008;582:3739. doi: 10.1016/j.febslet.2008.09.054. [DOI] [PubMed] [Google Scholar]
- 50.Sugiyama Y, Gotoh A, Katoh T, Honda Y, Yoshida E, Kurihara S, Ashida H, Kumagai H, Yamamoto K, Kitaoka M, Katayama T. Glycobiology. 2016;26:1235. doi: 10.1093/glycob/cww085. [DOI] [PubMed] [Google Scholar]
- 51.Sugiyama Y, Katoh T, Honda Y, Gotoh A, Ashida H, Kurihara S, Yamamoto K, Katayama T. Biosci Biotechnol Biochem. 2017;81:283. doi: 10.1080/09168451.2016.1254532. [DOI] [PubMed] [Google Scholar]
- 52.Cobucci-Ponzano B, Conte F, Bedini E, Corsaro MM, Parrilli M, Sulzenbacher G, Lipski A, Dal Piaz F, Lepore L, Rossi M, Moracci M. Chem Biol. 2009;16:1097. doi: 10.1016/j.chembiol.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Cobucci-Ponzano B, Zorzetti C, Strazzulli A, Bedini E, Corsaro MM, Sulzenbacher G, Rossi M, Moracci M. Biocatalysis and Biotransformation. 2012;30:288. [Google Scholar]
- 54.Cobucci-Ponzano B, Zorzetti C, Strazzulli A, Carillo S, Bedini E, Corsaro MM, Comfort DA, Kelly RM, Rossi M, Moracci M. Glycobiology. 2011;21:448. doi: 10.1093/glycob/cwq177. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez-Diaz J, Monedero V, Yebra MJ. Appl Environ Microbiol. 2011;77:703. doi: 10.1128/AEM.01906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodriguez-Diaz J, Carbajo RJ, Pineda-Lucena A, Monedero V, Yebra MJ. Appl Environ Microbiol. 2013;79:3847. doi: 10.1128/AEM.00229-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Becerra JE, Coll-Marques JM, Rodriguez-Diaz J, Monedero V, Yebra MJ. Appl Microbiol Biotechnol. 2015;99:7165. doi: 10.1007/s00253-015-6666-2. [DOI] [PubMed] [Google Scholar]
- 58.Ashida H, Miyake A, Kiyohara M, Wada J, Yoshida E, Kumagai H, Katayama T, Yamamoto K. Glycobiology. 2009;19:1010. doi: 10.1093/glycob/cwp082. [DOI] [PubMed] [Google Scholar]
- 59.Sakurama H, Tsutsumi E, Ashida H, Katayama T, Yamamoto K, Kumagai H. Biosci Biotechnol Biochem. 2012;76:1022. doi: 10.1271/bbb.111004. [DOI] [PubMed] [Google Scholar]
- 60.Sulzenbacher G, Bignon C, Nishimura T, Tarling CA, Withers SG, Henrissat B, Bourne Y. J Biol Chem. 2004;279:13119. doi: 10.1074/jbc.M313783200. [DOI] [PubMed] [Google Scholar]
- 61.Liu SW, Chen CS, Chang SS, Mong KK, Lin CH, Chang CW, Tang CY, Li YK. Biochemistry. 2009;48:110. doi: 10.1021/bi801529t. [DOI] [PubMed] [Google Scholar]
- 62.Shaikh FA, Lammerts van Bueren A, Davies GJ, Withers SG. Biochemistry. 2013;52:5857. doi: 10.1021/bi400183q. [DOI] [PubMed] [Google Scholar]
- 63.Shen A, Lupardus PJ, Morell M, Ponder EL, Sadaghiani AM, Garcia KC, Bogyo M. PloS One. 2009;4:e8119. doi: 10.1371/journal.pone.0008119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lomino JV, Tripathy A, Redinbo MR. J Bacteriol. 2011;193:2089. doi: 10.1128/JB.01231-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li T, Tong X, Yang Q, Giddens JP, Wang LX. J Biol Chem. 2016;291:16508. doi: 10.1074/jbc.M116.738765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giddens JP, Lomino JV, Amin MN, Wang LX. J Biol Chem. 2016;291:9356. doi: 10.1074/jbc.M116.721597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams SJ, Withers SG. Carbohydr Res. 2000;327:27. doi: 10.1016/s0008-6215(00)00041-0. [DOI] [PubMed] [Google Scholar]
- 68.Huang W, Li J, Wang LX. ChemBioChem. 2011;12:932. doi: 10.1002/cbic.201000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang LX, Song H, Liu S, Lu H, Jiang S, Ni J, Li H. ChemBioChem. 2005;6:1068. doi: 10.1002/cbic.200400440. [DOI] [PubMed] [Google Scholar]
- 70.Tretter V, Altmann F, Marz L. Eur J Biochem. 1991;199:647. doi: 10.1111/j.1432-1033.1991.tb16166.x. [DOI] [PubMed] [Google Scholar]
- 71.Kamoda S, Nakano M, Ishikawa R, Suzuki S, Kakehi K. J Proteome Res. 2005;4:146. doi: 10.1021/pr049825o. [DOI] [PubMed] [Google Scholar]
- 72.Amin MN, Huang W, Mizanur RM, Wang LX. J Am Chem Soc. 2011;133:14404. doi: 10.1021/ja204831z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang W, Yang Q, Umekawa M, Yamamoto K, Wang LX. ChemBioChem. 2010;11:1350. doi: 10.1002/cbic.201000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Proc Natl Acad Sci USA. 2017;114:3485. doi: 10.1073/pnas.1702173114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang W, Giddens J, Fan SQ, Toonstra C, Wang LX. J Am Chem Soc. 2012;134:12308. doi: 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakano M, Nakagawa T, Ito T, Kitada T, Hijioka T, Kasahara A, Tajiri M, Wada Y, Taniguchi N, Miyoshi E. Int J Cancer. 2008;122:2301. doi: 10.1002/ijc.23364. [DOI] [PubMed] [Google Scholar]
- 77.Lai JI, Licht AF, Dugast AS, Suscovich T, Choi I, Bailey-Kellogg C, Alter G, Ackerman ME. J Virol. 2014;88:2799. doi: 10.1128/JVI.03130-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. J Biol Chem. 2002;277:26733. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 79.Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, Matsushima K, Ueda R, Hanai N, Shitara K. Cancer Res. 2004;64:2127. doi: 10.1158/0008-5472.can-03-2068. [DOI] [PubMed] [Google Scholar]
- 80.Kaneko Y, Nimmerjahn F, Ravetch JV. Science. 2006;313:670. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 81.Washburn N, Schwab I, Ortiz D, Bhatnagar N, Lansing JC, Medeiros A, Tyler S, Mekala D, Cochran E, Sarvaiya H, Garofalo K, Meccariello R, Meador JW, 3rd, Rutitzky L, Schultes BC, Ling L, Avery W, Nimmerjahn F, Manning AM, Kaundinya GV, Bosques CJ. Proc Natl Acad Sci USA. 2015;112:E1297. doi: 10.1073/pnas.1422481112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.