

Abstract
Biodegradable synthetic polymers have been widely used as tissue engineering scaffold materials. Even though they have shown excellent biocompatibility, they have failed to resemble the low stiffness and high elasticity of soft tissues because of the presence of massive rigid ester bonds. Herein, we synthesized a new thermoplastic polyurethane elastomer (CTC-PU(BET)) using poly ester ether triblock copolymer (polycaprolactone-block-polytetrahydrofuran-block-polycaprolactone triblock copolymer, PCTC) as the soft segment, aliphatic diisocyanate (hexamethylene diisocyanate, HDI) as the hard segment, and degradable diol (bis(2-hydroxyethyl) terephthalate, BET) as the chain extender. PCTC inhibited crystallization and reduced the melting temperature of CTC-PU(BET), and BET dramatically enhanced the thermal decomposition and hydrolytic degradation rate when compared with conventional polyester-based biodegradable TPUs. The CTC-PU(BET) synthesized in this study possessed a low tensile modulus and tensile strength of 2.2 MPa and 1.3 MPa, respectively, and an elongation-at-break over 700%. Meanwhile, it maintained a 95.3% recovery rate and 90% resilience over ten cycles of loading and unloading. In addition, the TPU could be electrospun into both random and aligned fibrous scaffolds consisting of major microfibers and nanobranches. 3T3 fibroblast cell culture confirmed that these scaffolds outperformed the conventional biodegradable TPU scaffolds in terms of substrate–cellular interactions and cell proliferation. Considering the advantages of this TPU, such as ease of synthesis, low cost, low stiffness, high elasticity, controllable degradation rate, ease of processability, and excellent biocompatibility, it has great prospects to be used as a tissue engineering scaffold material for soft tissue regeneration.
Keywords: Thermoplastic polyurethane, biodegradable elastomer, soft tissue engineering, electro-spinning scaffolds
1 Introduction
In recent years, significant interest has been paid to the investigation and development of biodegradable and bioresorbable tissue engineering scaffolds. Efforts have been made in developing both new materials and new fabrication methods.1, 2 Electrospinning has been recognized as a versatile technique capable of producing continuous fibers with diameters ranging from tens of nanometers to a few micrometers. It has been highly praised because the fabricated fibrous scaffolds can resemble the microstructure of native extracellular matrix (ECM).3 One of the current challenges is developing new biomaterials that can better mimic the properties of native tissue. To date, synthetic biodegradable polymers—especially polyesters such as poly(lactic acid) (PLA), poly(glycolic acid) (PGA), poly (lactic-co-glycolic acid) (PLGA), poly(caprolactone) (PCL), and polyhydroxybutyrate (PHB)—and their blends or block copolymers have been widely used to prepare scaffolds for bone, cartilage, muscle, tendon, nerve, and myocardial tissue engineering applications.4-7 However, typical polyesters are relatively stiff and have very limited flexibility, which makes them not ideal for engineering soft and flexible tissues that can undergo dynamic loading, such as skin, tendons, ligaments, cartilage, blood vessels, and muscles, since they may mechanically fail in long term implantation applications, although they could support cell growth in short term.8, 9 Therefore, there is an urgent demand for biodegradable elastomeric materials that could mimic soft tissue properties.
Having been extensively used in the medical field for almost half a century, segmented polyurethanes (PUs) continue to gain the interest of researchers owing to their tunable mechanical properties and flexibility.10, 11 PUs are a category of segmented polymers that are generally synthesized via the step-growth polymerization of polyols (a relatively long and flexible component contributing to the soft segment) with diisocyanates and low molecular weight diol or diamine chain-extenders (contributing to the hard segment).12 Thermoplastic polyurethane (TPU) is a kind of PU that possesses linear molecular chains that can be easily processed via solution or melt methods.13 In recent years, various biodegradable TPUs have been synthesized as tissue engineering materials for cardiovascular tissues,14 soft tissues,15 bone tissues,16 blood vessels,17 and wound dressings applications.18 These TPUs are recognized as biodegradable because they are normally synthesized using either polyester diols as soft segments or degradable chain extenders.11, 19-21 The degradation rate can be tuned by varying the components and compositions of the reactants. PCL has been the most commonly used soft segment in biodegradable TPU synthesis although its degradation rate is relatively low.22-24 To improve the biodegradability of PUs, Blakney et al. synthesized a new class of polyurethanes using PLGA as the soft segment. These TPUs degraded much faster than PCL-based TPUs and could be used in drug release applications.25 To regulate cell behavior during tissue regeneration, PLLA has been employed as the soft segment and amine-capped aniline trimer has served as an electroactive hard segment to synthesize conductive polyurethane–urea copolymers to combine the advantages of polyurethane and conducting polymers.26
However, the presence of abundant ester bonds leads to the material’s high stiffness and low elasticity.21, 27 These materials tend to undergo permanent deformation under dynamic loading. Therefore, it is still questionable as to whether or not these biodegradable PUs can have potential long-term success as scaffold materials for soft tissues subjected to dynamic loading.28 To address these problems, researchers have employed block copolymers as the soft segments to adjust the elasticity and biodegradability of PUs. Trinca et al. synthesized biodegradable soft TPU using PEG, PLLA, and poly(trimethylene carbonate) (PTMC) blocks as the soft segments and fabricated electrospun fibrous scaffolds;29 however, the mechanical properties and degradation behaviors were not studied. Henry et al. synthesized a soft TPU combining different poly diols as the soft segments (PHB for the crystalline segment and poly(caprolactone-co-glycolide) for the amorphous segment). The material had a tensile modulus of about 8 MPa and a tensile strength of over 20 MPa, and porous scaffolds showed good in vivo biocompatibility and angiogenic responses, but the material did not show significant weight loss after incubating in PBS for 346 days and no obvious signs of degradation were found after implantation for 63 days.30, 31 John et al. synthesized a series of poly(ether ester) thermoplastic elastomers using various poly diols as the soft segment. The elastomer synthesized using the polycaprolactone-block-polytetrahydrofuran-block-polycaprolactone (PCTC) soft segment showed the lowest tensile strength of 7.8 MPa and a 350% elongation-at-break; however, the degradation behavior was not studied.32 Tataia et al. made an effort to develop degradable chain extender (DCE) PUs. They found that using DCEs increased the degradation rate of TPUs.20 The poly(caprolactone ethylene glycol) triblock copolymer (PCL-PEG-PCL) has been used to synthesize soft TPU in recent years. Although PCL-PEG-PCL-based TPU can achieve a low modulus of 5 MPa, it showed a slow degradation rate.33
It is well recognized that cells receive mechanical feedback from the substrate to which they adhere, which is influential for cell growth, differentiation, and tissue development and regeneration. Thus it has been widely accepted that an ideal engineered scaffold must reproduce the properties of the intended target tissue.9, 34, 35 Human soft tissues like the cerebral vein, ureter, vascular elastin, smooth muscle, knee articular cartilage, and skin9, 36, 37 have tensile moduli ranging from 0.01 to 12 MPa, which is far below commonly used polyesters. Other polyol/dicarboxylic acid based-elastomers that possess low moduli (less than 1 MPa) have been developed especially for soft tissue engineering applications, such as poly(polyol sebacate) (PPS) and poly(glycerol sebacate) (PGS).37, 38 While they are commonly fabricated as gel scaffolds, they require a polymer carrier in electrospinning. Moreover, crosslinking is also required to maintain the produced architecture. All of these attributes limit the ability of post processing and scaffold production on a large scale. Therefore, there is still a great need to develop biodegradable thermoplastic elastomers with low moduli for soft tissue engineering applications.
The aim of the present work is to design and synthesize novel, biocompatible, and biodegradable TPU elastomers with a low modulus but high elasticity for application as soft tissue engineering scaffold materials. To achieve this, PCTC triblock copolymer diol, which consists of a PCL degradable moiety and a PTHF amorphous moiety, was used as the soft segment, and an aliphatic diisocyanate and a DCE containing aromatic ester bonds were used as the hard segment and chain extender, respectively, in the synthesis of novel TPU. This design combines the advantage of using block copolymers, which provide flexibility to molecular chains, as well as the advantage of using DCE, which increases the degradation rate of PU. Commonly used PCL-based biodegradable TPU and PCTC-based TPU with a non-degradable chain extender were also synthesized for comparison. In this study, the chemical structure, thermal properties, mechanical properties, and degradability of different TPUs were investigated in detail. The synthesized soft TPU achieved a low tensile modulus and strength of 2.2 MPa and 1.3 MPa, respectively, while it retained high recoverability and resilience. The TPUs synthesized have high enough molecular weights to be electrospun into both random and aligned fibrous scaffolds, with the fibers consisting of major microfibers and nanobranches. 3T3 fibroblast cell culture tests on these scaffolds confirmed their biocompatibility, with the soft TPU scaffold being more favorable for cell proliferation as compared with hard TPUs.
2 Experimental
2.1 Materials
Reactant materials, including polycaprolactone diol (PCL, Mw = 2000), was purchased from Polysciences, Inc. Polycaprolactone-block-polytetrahydrofuran-block-polycaprolactone (PCTC, Mw = 2000, equal polymerization degree for each block), hexamethylene diisocynate (HDI), 1,4-butanediol (BD), bis(2-hydroxyethyl) terephthalate (BET), and dibutyltin dilaurate (DBTDL) were purchased from Sigma–Aldrich. PCL diol, PCTC diol, BD, and BET were vacuum dried at 75 °C for 2 h prior to use. HDI was dried using a 4Å molecular sieve and then vacuum distilled. Chemical reagents such as dimethyl formamide (DMF), chloroform (CF), anhydrous dimethylsulfoxide (DMSO, ≥99.9%), and isopropanol were purchased from Sigma–Aldrich and used as received. Deionized (DI) water was used throughout the experiment.
2.2 Synthesis of TPU Elastomers
TPU elastomers were synthesized though a two-step polymerization process according to the following procedure. (1) Pre-polymer synthesis: PCL diol or PCTC diol was dissolved in anhydrous DMSO and slowly added into a dried three-necked reaction flask equipped with a mechanical stirrer and containing HDI under nitrogen protection followed by drop-wise addition of DBTDL catalyst (0.05 wt.% with respect to the monomer). The reaction was carried out at 70 °C for 3 h to yield an isocynate-ended, viscous pre-polymer. (2) Chain extender (BD or BET) was dissolved in anhydrous DMSO and then added drop-wise to the pre-polymer within 10 min. The reaction was maintained at 70 °C for 3 h to yield the TPU elastomers. After the reaction, the synthesized TPUs were precipitated in excess DI water and then quenched in isopropanol for 3 days to remove any impurities in the TPUs. Finally, the TPUs were dried in a vacuum oven at 60 °C for 2 days and kept in a desiccator until use. The yields of the TPUs ranged from 84% to 89%. Schematic 1 illustrates the synthesis procedures of the three TPUs. The molar ratio of soft segment to hard segment to chain extender was kept constant at 1:2:1 for all TPUs to yield a minimum proportion of hard segment, which should lead to the softest form of TPU. The three TPUs were named according to their soft segment and chain extender; for example, CL-PU(BD) indicates that PCL was the soft segment and BD was the chain extender; CTC-PU(BD) indicates that PCTC was the soft segment and BD was the chain extender; and CTC-PU(BET) indicates that PCTC was the soft segment and BET was the chain extender.
Schematic 1.
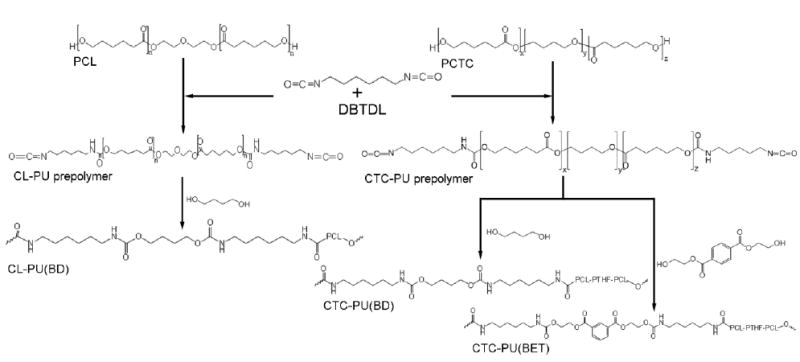
Synthesis flowchart of CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) elastomers.
2.3 Preparation of Test Samples and Electrospun Random and Aligned Fibers
Synthesized TPUs were dissolved in DMF to yield 10 wt% solutions at 70 °C for 5 h under magnetic stirring. The solutions were then cast into polypropylene petri dishes, degassed using a bath sonicator, dried at 50 °C under vacuum for 3 days, and air dried for 14 days to ensure complete evaporation of the solvent. After drying, samples were soaked in DI-water for 1 h, then detached and dried in a desiccator until use. Films with thicknesses around 1 mm, and sheets with thicknesses around 3 mm, were prepared for mechanical tests and degradation tests, respectively.
Electrospinning was carried out on a lab-built electrospinning setup. The TPUs were dissolved in a chloroform/DMF (v/v = 6:4) solvent mixture for 8 h under magnetic stirring at room temperature. The solution concentrations were optimized in our preliminary studies to yield continuous bead-free fibers for the three TPUs. The concentrations were 30 wt/vol% for CL-PU(BD), 25% wt/vol% for CTC-PU(BD), and 33 wt/vol% for CTC-PU(BET). The prepared solution was loaded into a plastic syringe connected to an 18 gauge blunt-end needle mounted on a digital syringe pump. Electrospinning was performed with a 15 cm needle-to-collector distance, a 0.5 mL/h flow rate, and an 18 kV applied voltage. Non-woven random fibers were obtained using an aluminum foil collector, and aligned fibers were prepared using a rotating collector (diameter of 10 cm) with a rotational speed of 8000 RPM.
2.4 Polymer Characterization
The chemical structure of synthesized TPU elastomers was characterized by 1H NMR (300 MHz, Bruker Biospin Co., Billerica, MA) using DMSO-d6 (TMS, 99.9+%, NMR grade, Sigma–Aldrich) as a solvent with tetramethylsilane as an internal reference. Fourier transform infrared (FTIR) spectra were recorded in transmittance mode using a Bruker Tensor 27 spectrometer in the range of 4000–600 cm-1, with a resolution of 4 cm-1. Molecular weights (number average molecular weight (Mn) and weight average molecular weight (Mw)) and polydispersity indices (PDI) were measured by a gel permeation chromatographer (GPC, Viscotek, USA) equipped with a D6000M column and calibrated using PMMA-65K standards. Samples with concentrations of 1mg/mL in DMF were tested. The thermal properties of thin film samples were measured via differential scanning calorimetry (DSC Q20, TA) and thermogravimetric analysis (TGA Q50, TA). DSC measurements were performed at a rate of 10 °C/min across a temperature range of −100 °C to 150 °C. During testing, each sample was first heated to 150 °C, and then kept at 150 °C for 3 min to eliminate any prior thermal history, then cooled down to −100 °C and reheated to 150 °C. The TGA tests were initiated at room temperature and heated to 600 °C at a heating rate of 10 °C/min. The change in weight as the temperature increased was recorded. X-ray diffraction (XRD) patterns were measured on a D8-Discovery diffractometer (Bruker, USA) in a θ–2θ mode at a scanning rate of 5°/min. The wettability of the materials was measured by a video contact angle instrument (Dataphysics, OCA 15) using 4 μL droplets. Scanning electron microscopy (SEM) was used to characterize the morphological properties. Samples were first coated with a thin layer of gold and then imaged using a fully digital LEO GEMINI 1530 SEM (Zeiss, Germany) at a voltage of 3 kV. The fiber diameter and fiber orientation angle of the electrospun scaffolds were measured from SEM images using Image Pro-Plus software. At least 50 fibers were measured for each sample.
2.5 Mechanical Testing
Rectangular strips 8 mm × 30 mm in dimension were cut from the casted thin films and electrospun scaffolds for mechanical testing. Tensile tests were performed on a universal mechanical testing machine (Instron 5967) at room temperature. Samples were stretched at a crosshead speed of 5 mm/min for solid films and 1mm/min for electrospun scaffolds until fracture. Five samples were tested for each group and the tensile modulus, tensile strength, and strain-at-break were recorded.
Cyclical tensile tests of casted thin films were performed using the same instrument to investigate the sustainability of these TPU elastomers. Samples were stretched to 50% strain and released at a crosshead speed of 5 mm/min for 10 cycles. The change of tensile stress versus tensile strain was recorded and the recovery ratio in each cycle was determined by Equation (1),
| (1) |
where ε was the strain when the stress decreased to zero in the unloading step. Resilience, which is the property of the material to absorb energy after elastic deformation, was calculated using Equation (2),39
| (2) |
where Wload and Wunload were represented by the area beneath the stress–strain curves in the stretch-and-release steps, respectively.
2.6 Polymer Degradation
The hydrolytic degradation behavior of synthesized TPU elastomers was simulated by PBS soaking experiments. Briefly, polymer sheets were cut into 5 mm × 8 mm pieces, then vacuum dried and weighed. Each sample was individually sealed in a plastic container filled with a 10× PBS solution, and then incubated at 37 °C on a parallel shaker (MaxQ 4000, Thermo Scientific) at a shaking rate of 225 rpm. PBS was changed every 3 days to ensure a constant pH. The degradation test was performed for up to 10 weeks. At each time point, the samples were rinsed with DI water three times, followed by sufficient drying and weighing. The weight loss of the materials was calculated using Equation (3). The samples, which degraded for 10 weeks, were also characterized by FTIR and SEM.
| (3) |
where W0 is the original sample weight and Wn is the weight of the same sample after degradation for a time period n.
2.7 3T3 Fibroblast Cell Culture
Swiss mouse NIH 3T3 fibroblasts were cultured on the electrospun fibers to evaluate the cytotoxicity and biocompatibility of various TPU scaffolds. Briefly, electrospun fibers were wrapped on coverslips and UV sterilized for 30 min prior to cell seeding. 3T3 cells were treated with ethylenediaminetetraacetic acid (EDTA) for 5 min and washed with phosphate-buffered saline (PBS) solution, and then seeded on the samples at a cell density of 1.25×105 cells/cm2. Cells were fed high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 20% fetal bovine serum (FBS) (WiCell), 1% penicillin–streptomycin (10,000 U/ml, Life Technologies), and 1% L-glutamine (200 mM, Life Technologies). Spent medium was aspirated and replaced with 1 mL of fresh medium daily for screening samples. Samples were characterized at day 5 and day 10 time points.
2.8 Biological Characterization
Cell viability was determined after culturing on the scaffolds for 5 days and 10 days. Viability was assessed via a Live/Dead viability/cytotoxicity kit (Life Technologies). The staining protocol followed the manufacturer’s instructions. The green fluorescent calcein-AM was used to target the esterase activity within the cytoplasm of living cells, while the red fluorescence ethidium homodimer-1(EthD-1) was used to indicate cell death. Stained cells were imaged with a Nikon Eclipse Ti Microscope with an attached Photometrics CoolSNAP HQ2 camera, and Nis-D Elements Advanced Research v3.22 software was used for image analysis.
The number of collected cells that fluoresced red and green were counted with an Accuri C6 (BD Biosciences) flow cytometer to obtain viability data. Briefly, the stained cells of the live/dead assay were detached from the scaffolds by incubation in 250 μL of Trypsin (Life Technologies) per well at 37 °C for 5 min. Then the cells were collected and centrifuged at 1000 rpm for 5 min. The supernatant was then aspirated and the cells were resuspended in 600 μL of PBS and filtered prior to analysis.
The viable cell number of cells after culturing for 5 days and 10 days was determined by (3-(4,5-dimethylthiazol-z-yl)-5-(3-carboxylmethoxyphenyl)-2-(4-sulfo-phenyl)-2H-tetrazolium (MTS) assays using the CellTiter 96 Aqueous One Solution kit from Promega (Promega Life Sciences). The whole procedure followed the manufacturer’s instructions. Upon testing, cells were treated with media containing 20% MTS solution and allowed to incubate for exactly 1 h. After incubation, 100 μL of spent media was transferred into a clear 96-well plate. The absorbance of the plates at the 450 nm wavelength was read with a Glomax-Multi+Multiplate Reader (Promega). The subsequent number of cells was determined relative to the negative control.
The shape and cytoskeleton organization of the cells were determined by phalloidin–tetramethylrhodamine B isothiocyanate (phalloidin–TMRho, Sigma) staining. For this assay, cells were first fixed in 4% paraformaldehyde and then diluted in PBS for 15 minutes at room temperature. Next, they were washed with PBS and permeabilized with 0.1% Triton-X in PBS for 5 min. The cells were then washed once again and treated with 0.3 μM phalloidin–TMRho with 4’, 6-diamidino-2-phenylindole (DAPI) for 1 hour at room temperature. Samples were then washed with PBS and imaged using a Nikon A1RSi inverted confocal microscope.
2.9 Statistical Analysis
All biological results are presented as mean ± standard deviation. All of the values were averaged at least in triplicate and expressed as mean ± standard deviation (SD). The data were analyzed using the one-way analysis of variance method (ANOVA). The Turkey’s test was then used to evaluate the specific differences of the data, and these differences were considered statistically significant at p < 0.05.
3 Results and Discussion
3.1 TPU Elastomers Characterization
Synthesized PCL-PU(BD), CTC-PU(BD), and CTC-PU(BET) elastomers were characterized using 1H NMR and FTIR to verify their molecular structures. The 1H NMR spectrums (Fig. 1) for the three TPUs showed many peaks at the same ppm due to similarities in their chemical structures. Generally, the peaks between 1–2 ppm were assigned to the CH2 group in the PCL, HDI, and BD. The peak at 2.92 ppm corresponded to the NH–CH2 group in the hard segment.40 The peaks at 3.98 and 2.25 ppm corresponded to the protons adjacent to the carbon–oxygen single and double bonds in the ester group of PCL, respectively. All of the bonds belonging to PCL and HDI were observed in all three materials. The distinct characteristic peaks for CL-PU(BD) located at 3.59 and 4.11 ppm correspond to the O–CH2–CH2–O linkage from ether and ester bonds that connected the two PCL moieties.41 The absence of these peaks was the major difference among the TPUs synthesized using PCL and PCTC as the soft segments. In CTC-PU(BET), the change of chain extender was indicated by the peaks’ shift to 4.32 and 8.13 ppm, which was ascribed to the CH–O bond adjacent to the ester bonds and the CH from the benzene ring, respectively.19 These results suggested the successful synthesis of the three kinds of TPU elastomers.
Fig. 1.
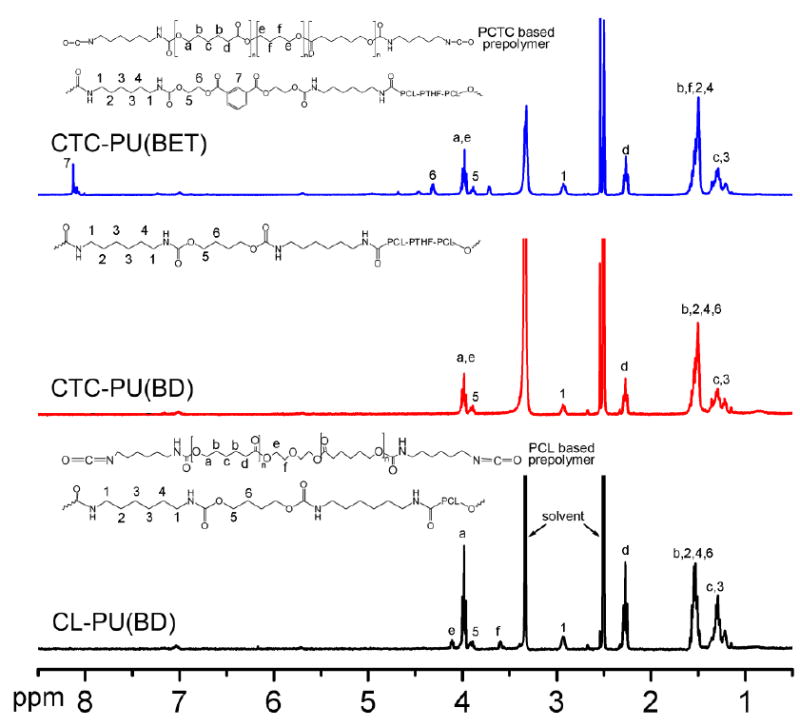
1H NMR spectra of PCL-PU(BD), CTC-PU(BD), and CTC-PU(BET) using DMSO-d6 as the solvent.
FTIR measurements (Fig. 2) were performed to further confirm the chemical structure of the TPUs. The absorption bands at 3320 cm-1 (N–H stretching), 1724 cm-1 (C=O stretching), and 1580 cm-1 (C–N stretching) indicated urethane linkage formation in the TPUs.42 In addition, the absorption band at 1532 cm-1 corresponded to the amide II in the urethane, which is a strong evidence of successfully synthesis of PU.43 The characteristic peaks located at 2935 cm-1 and 2863 cm-1 corresponded to the asymmetric and symmetric stretching vibrations of the −CH2 groups, respectively. Specifically, there was an obvious absorption peak at 1105 cm-1 for CTC-PU(BD) and CTC-PU(BET), corresponding to the C–O–C linkages in the PTHF moiety of the PCTC soft segment. Compared with PCL-PU(BD) and CTC-PU(BD), there was a peak at 697 cm-1 assigned to the stretching vibrations for C–H in the benzene ring of the BET chain extender. Therefore, the FTIR results further confirmed the successful synthesis of the three kinds of TPUs. Even though the molecular structure is the major factor that determines the chemical and physical properties of polymers, the length of the molecules (molecular weights) also plays an important role in determining properties, especially the elasticity. Table 1 lists the molecular weights of the three TPU elastomers. It is difficult to synthesize three TPUs with the same molecular weight in the lab; however, the synthesized TPUs achieved a high molecular weight—over 60,000 g/mol—with PDI values of less than 2.
Fig. 2.
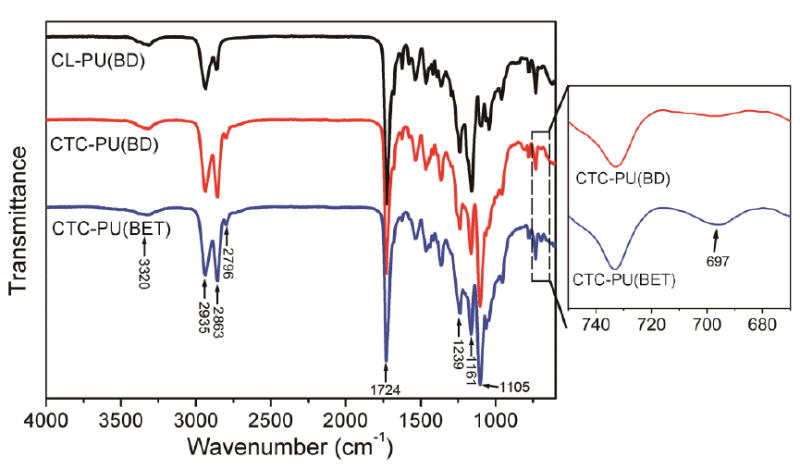
FTIR spectra of synthesized CL-PU(BD), CTC-PU(BD), and CTC-PU(BET).
Table 1.
Mw, Mn, and PDI results from GPC measurements using DMF as the solvent. Unit for molecular weight is g/mol.
| CL-PU(BD) | CTC-PU(BD) | CTC-PU(BET) | |
|---|---|---|---|
| Mw | 62,000 | 80,000 | 67,000 |
| Mn | 42,000 | 48,000 | 38,000 |
| PDI | 1.5 | 1.7 | 1.8 |
3.2 Thermal Properties
To understand the thermal properties of the synthesized polymers in this study, DSC and TGA were carried out, and the results are shown in Fig. 3. The first heating scan was performed to remove any prior thermal history of the casted films. In the second heating scan (Fig. 3 (a)), CL-PU(BD) showed a distinct sharp melting peak at 43.7 °C corresponding to the melting of the PCL crystalline phase. Accordingly, a sharp crystallization peak was observed at −1.2 °C in the cooling scan (Fig. 3 (b)). Crystallization in the polymer would significantly enhance the stiffness and reduce the flexibility of the bulk material; hence, low crystallinity is preferred in this study for soft tissue applications. Comparing all three materials, their glass transition temperatures were all lower than −50 °C, which means that they were in the rubbery state at room temperature. The TPUs synthesized using PCTC as a soft segment showed a very different thermogram pattern. They had two wide melting peaks and two weak crystallization peaks. This dramatic difference with CL-PU(BD) was due to the incorporation of the PTHF moiety in the PCTC copolymer which reduced the crystallization ability of the PCL molecules by interrupting the arrangement of the PCL chains. Interestingly, it was found that the first melting peaks of CTC-PU(BD) and CTC-PU(BET) were lower than room temperature while the material still maintained their elastomeric properties. The first melting peak was attributed to the low melting temperature of PTHF, and it has been reported that TPU synthesized using PTHF as the soft segment sometimes behaves like wax.19 The PCL blocks and amide bonds might have formed hard network points that prevented free deformation of the material as indicated by another melting peak at a higher temperature. Compared to PCL-diol and PCTC-diol (Fig. S1), both the Tm and Tc of the TPUs were lower than the soft segments, thus indicating that crystallization of the soft segments was greatly hindered after they were synthesized into macromolecules. The effect of the chain extender on the thermal properties can be seen by comparing CTC-PU(BD) and CTC-PU(BET). It is clear that the two melting peaks were closer together and the crystallization peaks were almost merged into a single wide peak for CTC-PU(BET), thus indicating that the hard segment composed by HDI and BET had better miscibility with the PCTC soft segment than that comprised with HDI and BD. Both CTC-PU(BD) and CTC-PU(BET) showed weak crystallization peaks in the cooling scan; therefore, XRD was utilized to further analyze the crystalline domains of the TPUs (Fig. S2). XRD results showed distinct crystalline peaks of PCL (110) and (200) lattice planes in the CL-PU(BD) sample,44 while there was no identifiable peaks for CTC-PU(BD) and CTC-PU(BET) besides a big arc representing the amorphous phase, which indicated their low crystallinity.
Fig. 3.
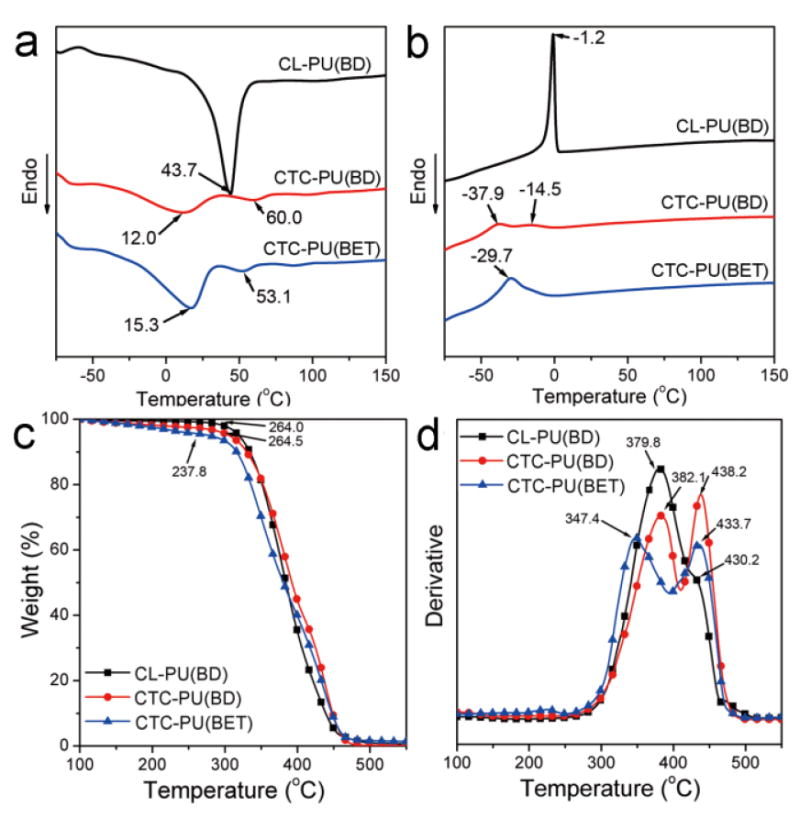
Thermal properties of synthesized CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) elastomers: (a) DSC second heating scan, (b) DSC cooling scan, (c) TGA weight loss results, and (d) derivative TG results.
The TGA results in Fig. 3 (c) reflected the thermal decomposition behavior of the materials and the composition of blended or block polymers. All three materials showed continuous weight loss as the temperature increased, while the weight loss rates and starting temperatures were different. CL-PU(BD) and CTC-PU(BD) showed close onset decomposition temperatures, as well as a maximum weight loss temperature, which were both higher than that of CTC-PU(BET), thus suggesting that the ester bonds in BET were cleaved at lower temperatures than the ester bonds in PCL. Another distinct difference in the derivative TG (D-TG) curves in Fig. 3 (d) was that CL-PU(BD) had one peak at 379.8 °C, with a shoulder peak at 430.2 °C, while CTC-PU(BD) and CTC-PU(BET) had two separate peaks with wider temperature differences. Furthermore, the difference for CTC-PU(BET) was wider than that for CTC-PU(BD). These results suggest the stepwise decomposition of these TPUs.
It is common for PUs to show two-step decomposition behavior, with the first peak usually indicating the decomposition of the amide bond in the hard segment and the second peak at a higher temperature representing the decomposition of the ether bonds in the soft segment.45 However, it has been found that the ester bonds in PCL cleaved at a lower temperature than the amide bonds in the hard segment for PCL-based biodegradable PUs,41 which was also the case for the CL-PU(BD) we synthesized. For CTC-PU(BD), the first peak was assigned to the PCL proportion suggested by the same temperature with CL-PU(BD), while the second peak was attributed to the PTHF and the hard segment. The first peak shifted much lower while the second peak remained at about the same temperature for CTC-PU(BET) as compared with CTC-PU(BD), thus suggesting that the use of BET as chain extender enhanced the decomposition of PCL. In addition, the areas under each peak indicated the proportion of the segments that decomposed within that temperature range. The D-TG curves were Gaussian peak fitted and the area of each peak was integrated (Fig. S3). It was found that the peak area indicating decomposition at high temperatures was 8.1% for CL-PU(BD), which represented a proportion of the hard segment. Meanwhile, the area of this peak increased to 28.2% for CTC-PU(BD) and 18.7% for CTC-PU(BET), thus suggesting the decomposition of not only the hard segment, but also the PTHF block. Noticeably, an additional peak (49.7%) appeared at a much lower temperature than the peak from PCL decomposition attributed to the cleavage of the BET chain extender, thus suggesting that BET caused significant acceleration of thermal decomposition. Similar accelerated decomposition with the use of degradable chain extenders was also reported elsewhere.19, 46
3.3 Mechanical Properties
Mechanical properties are one of the most important factors for tissue engineering scaffolds, not only because the scaffolds need to support the surrounding tissue and cells, but also proper stiffness of the substrate has been found to be beneficial for stimulating cell growth and differentiation towards the target tissue.47, 48 The properties of bulk scaffolds can be controlled by varying the scaffold’s porous structure, while the cellular response from the substrate’s stiffness is mainly determined by the intrinsic properties of the material itself. Most commonly used synthetic biodegradable scaffold materials, such as PCL, PLA, PLGA, PHBV, and polyester-based PU, are rigid polymers that lack elasticity. However, most human tissues are soft and highly flexible, including most organs, skin, cartilage, and blood vessels. Therefore, there is an urgent demand for soft biodegradable materials in tissue engineering. Figure 4 shows the mechanical property results of the synthesized TPUs. It is obvious that CL-PU(BD) has a much higher tensile strength and tensile modulus but a lower strain-at-break than CTC-PU(BD) and CTC-PU(BET) (Figs. 4 (a)–(d)). The tensile modulus and strength for CTC-PU(BD) were 4.6 MPa and 3.0 MPa, respectively, and 2.2 MPa and 1.3 MPa for CTC-PU(BET), respectively. It is rarely reported that synthetic polymers have as low of mechanical properties as the TPUs we synthesized in this study.49 Most commonly used ester-based synthetic materials, such as PDLLA,4, 50 PLLA,50, 51 PCL,52 PGA,53 PLGA,54 and PHBV,55 have moduli of at least a few hundred MPa and strength in the tens of MPa. Researchers have been developing new biodegradable PUs by varying their soft and hard segments, as well as by changing chain extenders. To date, the moduli of most of these TPUs have been in the range of 8 to a few hundred MPa.49, 56, 57 For example, the CL-PU(BD) we synthesized in this study showed a modulus of 102.2 MPa and a strength of 7.4 MPa. In addition, the PCTC-based soft TPUs showed high elongation-at-break values of over 700%, thus indicating excellent flexibility. The soft mechanical properties of these TPUs can mainly be attributed to increased molecular chain flexibility and diminished crystallinity. Table 2 compares the mechanical properties and degradation properties of biodegradable PUs that have been reported with the CTC-PU(BET) synthesized in this study. It was found that CTC-PU(BET) possesses the lowest tensile modulus and strength while maintaining a high strain-at-break.
Fig. 4.
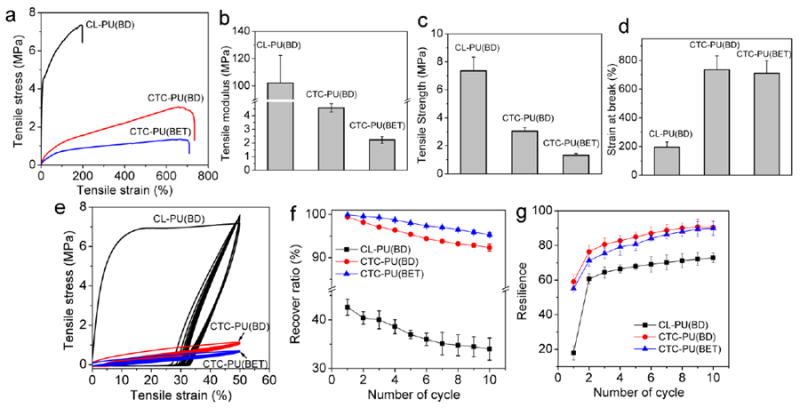
Tensile tests on CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) TPU films. (a) Representative stress–strain curves in tensile tests. Statistical results of (b) tensile modulus, (c) tensile strength, (d) strain-at-break, (e) representative cyclical tensile test curves, (f) film recover ratio, and (g) resilience in ten cycles of loading and unloading.
Table 2.
Mechanical properties and degradation rates of degradable PUs from the literature compared with the CTC-PU(BET) synthesized in this study.
| Formula of PU | Modulus (MPa) | Strength (MPa) | Elongation at Break (%) | Degradation Properties | Ref. |
|---|---|---|---|---|---|
| S=PCL; H=MDI; E=BD or MIDE | n/a | 41–64 | 756–780 | 65% Mw reduction after in PBS after 30 days at 77 °C | 21 |
| S=Sulfobetaine-diol; H=HDI; E=DB | 22–265 | 21–48 | 85–1001 | 2–90% weight loss in lipase PBS after 35 days at 37 °C | 66 |
| S= PCL-PEG-PCL; H=HDI; E=DB | 5–75 | 8–20 | 325–560% | 10–20% weight loss in PBS after 56 days | 33 |
| S=PCL; H=HDI or LDI; E=diester-diphenol | 5–56 | 1–7 | 7–1006 | n/a | 49 |
| S=PCL+ N-Boc-serinol; H=BDI; E=BD | 13–140 | 25–38 | 246–823 | 1–20% weight loss in PBS after 16 weeks at 37 °C | 24 |
| S=PCL+DTT;H=LDI or BDI; E=BD | 2.9–18.4 | 38–45 | 680–830 | 3–8% weight loss in PBS after 16 weeks at 37 °C | 67 |
| S= PCL; H= HDI; E= CaM | 16 | 4.3 | 500 | 24% weight loss in PBS after 180 days at 37 °C | 40 |
| S=PGS+AP;C=HDI | 1.5–75.5 | 1–5.3 | 17–104 | 5–86% weight loss in lipase PBS after 50 hours at 37 °C | 11 |
| S=PHB+PCL-PEG; H=TMDI | >20 | ~8 | 400% | 50% Mw reduction in PBS after 346 days at 37 °C | 30 |
| S=star-shaped PCL polyols; C=HDI | 62–1876 | 4.5–39.1 | 2.1–10 | 1.5–2.5% weight loss in sea water after 60 days | 68 |
| S=PCTC; H=HDI; E=BET | 2.2 | 1.30 | 710 | 2.3% weight loss in PBS after 10 weeks at 37 °C | This work |
S=soft segment; H=hard segment; E=chain extender; C=crosslinker
MIDE= 2,2-(methylimino)diethanol; MDI= Methylene diphenyl diisocyanate; DB= butanediamine; AP= aniline pentamer; PHB= poly(hydroxybutyrate); TMDI=2,2,4-trimethyl hexamethylene diisocyanate; LDI= L-Lysine ethyl ester diisocyanate; BDI=1,4-diisocyanatobutane; DTT= DL-dithiothreitol; calcium salt of p-aminobenzoic acid (CaM)
Cyclical tensile tests were performed on the TPUs to investigate their properties in dynamic loading circumstances. The recoverability under cyclical loading is considered to be a pivotal characteristic required to mimic biological tissues such as skin, tendons, blood vessels, and cartilage. This requirement has limited the application of various ester-based synthetic polymers and rigid nature materials. Herein, we found that PCTC-based soft TPUs showed excellent recoverability within ten cycles of loading and unloading (Figs. 4 (e)–(g)). The recovery ratio for CL-PU(BD) decreased from 42% to 34%, thus indicating poor recoverability, while CTC-PU(BD) showed a high recovery ratio of 92.3% after ten cycles. CTC-PU(BET) even outperformed CTC-PU(BD), showing an initial recovery ratio of 99.9%, and a ratio of 95.3% after the tenth cycle. Resilience, which is a measure of a material’s ability to deform reversibly without energy loss,39 was evaluated on the TPU films (Fig. 4 (g)). As can be seen, all samples lost the most energy in the first cycle of the test, with the energy loss decreasing in subsequent cycles as indicated by the increasing resilience. It should be noted that the resilience of CTC-PU(BD) and CTC-PU(BET) was about 90% after 8 cycles of testing, thus indicating that the molecular chains were able to dissipate energy by unfolding and folding effectively. A resilience of 90% is believed to be able to mimic the biomechanical properties of muscles.58 A small reduction in tensile moduli was seen in the cyclical tensile tests, which further lowered the stiffness of the materials in dynamic circumstances. Overall, these PCTC-based TPU elastomers possessed low stiffness, high flexibility, and high recoverability, thereby extending the lower boundary of the mechanical properties of biodegradable TPUs and enabling their potential use in various tissue engineering and biomedical device applications.
3.4 Polymer Degradation in PBS
Although the synthesized TPUs contain degradable ester bonds and amide bonds, it is worth investigating the degradation behavior of them since there has been a wide variation in degradation times for ester-based synthetic polymers.25, 41 Moreover, PCTC copolymers have been considered biodegradable in many publications and patents due to the presence of ester bonds from PCL.59, 60 However, the biodegradability of PCTC-based materials has rarely been reported. In this study, the hydrolytic degradation of TPU sheets was performed by immersion in 10× PBS for up to 10 weeks on a parallel shaker at 225 rpm at 37 °C. PBS was changed every 3 days to maintain a constant pH to resemble in vivo environmental conditions. The degradation results are summarized in Fig. 5. Overall, all three TPUs showed relatively slow degradation rates compared with some aliphatic polyesters like PLA, PLGA, and PGA.54, 61 It was found that CTC-PU(BET) showed the fastest initial degradation rate in the first 5 weeks, although it slowed down in later weeks. The degradation trends of CL-PU(BD) and CTC-PU(BET) were closer to a linear pattern. This difference was mainly caused by the ease of hydrolytic cleavage of chemical bonds in the different TPUs. The results indicate that the ester bonds in the BET chain extender accelerated the initial degradation. The fast degradation of BET chain extenders has been demonstrated elsewhere.19, 46 It is obvious that the PCL soft segment degrades faster than the PCTC soft segment, which is reasonable because the PTHF moiety in the PCTC could not be hydrolyzed. Figure 5 (c) shows the FTIR results of the TPUs before and after degradation for 10 weeks. It was found that the intensity of the peaks at 1157 cm-1 and 1098 cm-1 obviously decreased for CL-PU(BD), thus indicating the cleavage of ester bonds. The intensity of these bonds decreased slightly for CTC-PU(BET), while it remained almost the same for CTC-PU(BD), thus suggesting slower degradation of PCL segments in PCTC, which supports the weight loss results. Moreover, the shoulder peak at 1680.4 cm-1 assigned to the C=O bonds in the urethane group decreased for CTC-PU(BET) after degradation, thus indicating the degradation of urethane bonds in the hard segment. The change in surface morphology of the TPUs before and after degradation is shown in Fig. 5 (b). The sample before degradation had all smooth surfaces, and no significant changes were seen at low magnification for the degraded samples. However, micro- and nanovoids were observed at high magnification. The CL-PU(BD) sample showed the highest number of voids, followed by CTC-PU(BET) and CTC-PU(BD). The CTC-PU(BET) surface also showed some cracks, which verified the enhanced degradation rate when BET was used as a chain extender. Considering the chemical structure of these TPUs, their degradation products are supposed to be metabolized in vivo; therefore, they have the potential to be used as tissue engineering scaffolds with slow but controllable degradation rates, which would be suitable for long term implantation applications.46, 59
Fig. 5.
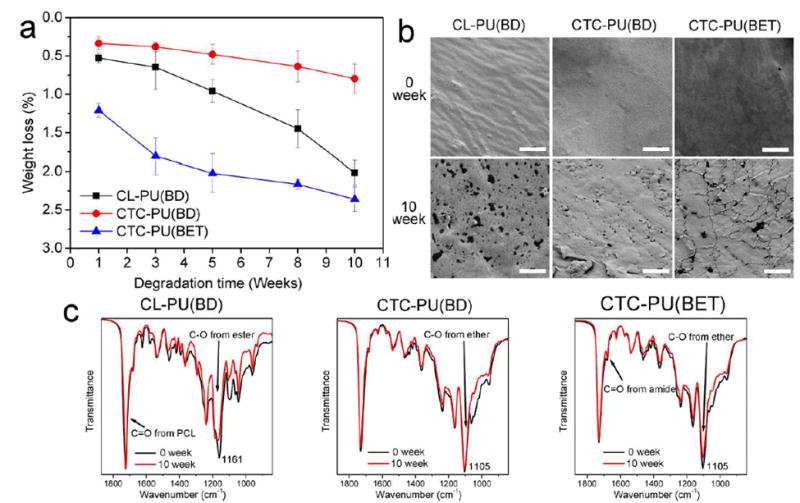
Degradation test on CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) TPU films: (a) weight loss over 10 weeks of degradation, (b) surface morphology of TPUs before and after degradation, and (c) FTIR measurements on TPUs before and after degradation. Scale bars in SEM images are 5 μm.
3.5 Scaffold Morphology and Mechanical Properties
Since these TPU elastomers were intended for soft tissue engineering scaffolding, it is important to investigate their ability to be electrospun into fibrous scaffolds. A number of biodegradable TPUs have been discussed in the literature, but for many of them, only the biological properties of their casted films were studied.24, 26, 33 Polymers with highly branched chains and low molecular weights were found to be difficult to be electrospun because it is hard for them to form a viscose solution with enough chain entanglements. Hence, aside from creating a new molecular structure, developing a biomaterial that can be easily processed is also a challenge. In this study, both random and aligned fibrous scaffolds were prepared for all three TPUs. By optimizing the solution concentration and electrospinning parameters, uniform bead-free fibers were obtained for three kinds of TPUs (Fig. 6). We found in our preliminary study that polymers with lower molecular weights required higher concentrations to create bead-free fibers, and it was difficult to electrospin TPUs with molecular weights below 50 kDa. The relevant fiber diameter and fiber orientation angle results suggest that CL-PU(BD) formed fibers with a more uniform diameter distribution, while CTC-PU(BD) and CTC-PU(BET) showed a wider fiber distribution. From the SEM images, the aligned and random fibers of CTC-PU(BD) and CTC-PU(BET) had branched nanofibers that extended from their major micron or submicron fibers forming a bimodal fiber size morphology. In addition, there were more branched fibers on the CTC-PU(BET) scaffolds. (It should be noted that the fiber diameter and orientation angle were both measured on the major fibers.) This phenomenon might have been caused by the difference in molecular structure.62 The molecular chains of CTC-PU(BD) and CTC-PU(BET) are less homogenous than that of CL-PU(BD) due to the existence of PTHF flexible segments. This might cause different stiffness regions in the molecular coil and further lead to some degree of separation during fiber deposition, yielding the formation of a bimodal fibrous structure. The benzene ring in the BET chain extender might further increase the intermolecular space and possibly cause more separation, thus resulting in more branched fibers. These branched fibers should be favorable for cell attachment since our previous study verified that micro- and nanosized hybrid fibrous scaffolds have a higher cell affinity.63 In addition, aligned fibers showed smaller fiber diameters than random fibers for CL-PU(BD) and CTC-PU(BD) due to the elongation effect. However, aligned CTC-PU(BD) scaffolds showed slightly larger fiber diameters than random fibers because some branched fibers had merged with the major fibers. The orientation angles of the major fibers in the aligned scaffolds were within ±35° of the defined orientation direction, as indicated by the arrows on the images, suggesting a high orientation rate.
Fig. 6.
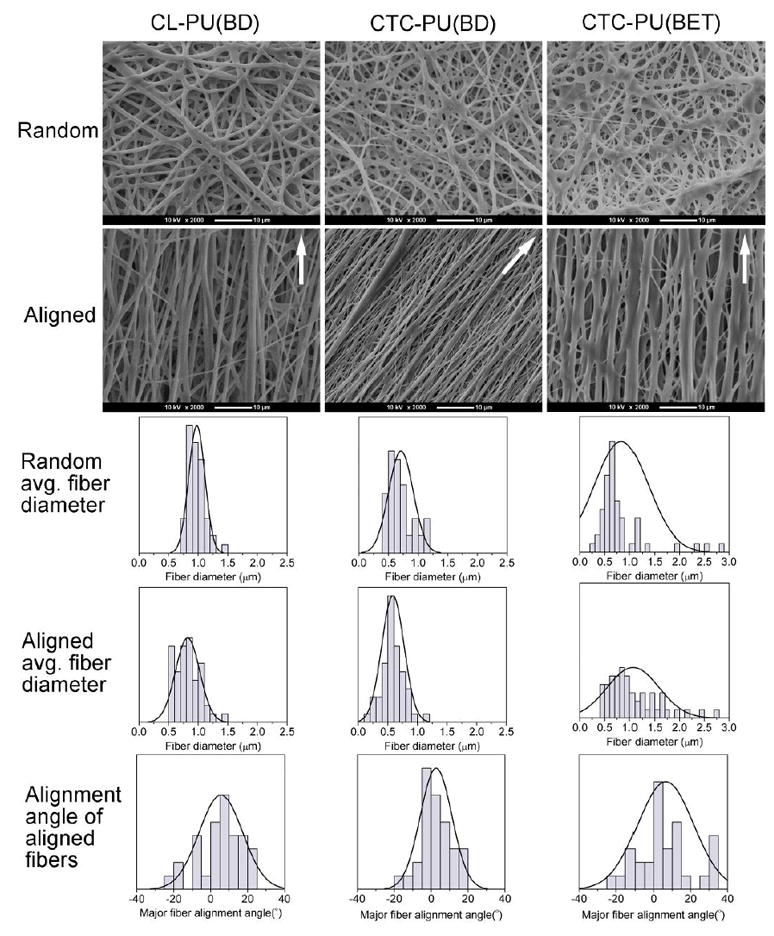
Electrospun random and aligned scaffolds of CL-PU(BD), CTC-PU(BD), and CTC-PU(BET). The first and second rows are SEM images of electrospun random and aligned fibers, scale bar = 10 μm; the third and fourth rows are the fiber diameter distribution histograms of electrospun fibers; and last row is the fiber alignment angle of electrospun aligned fibers.
Considering that the first melting peak of CTC-PU(BD) and CTC-PU(BET) were lower than 37 °C, it is reasonable to investigate the stability of the fiber structure in an aqueous environment at body temperature. Electrospun random fibers of the three TPUs were incubated in PBS at 37 °C for 1 day and imaged via SEM. No obvious structural changes were observed after incubation (Fig. S4). As discussed in the thermal and mechanical properties, the PTHF block provided viscidity and resulted in the first low melting temperature, while the PCL block and hard segment provided elasticity and prohibited the free movement of the PTHF chains. Therefore, the morphology of the electrospun fibers would not change unless the temperature reached the second melting point.
The tensile properties of electrospun scaffolds are shown in Fig. 7. Along with the solid film, CL-PU(BD) showed a much higher modulus and strength, but a lower strain-at-break, as compared to CTC-PU(BD) and CTC-PU(BET). The aligned scaffolds showed a higher modulus and strength but a lower strain-at-break than the random scaffolds since they were stretched in the fiber alignment direction. Notably, the modulus and strength of CTC-PU(BET) random scaffolds were 0.2 MPa and 1.1 MPa, respectively, while they were 0.8 MPa and 1.8 MPa for CTC-PU(BET) aligned scaffolds, respectively. Interestingly, we found that electrospun scaffolds showed a much lower initial modulus and strain-at-break than their solid counterparts, which was attributable to the pores among the fibers.
Fig. 7.
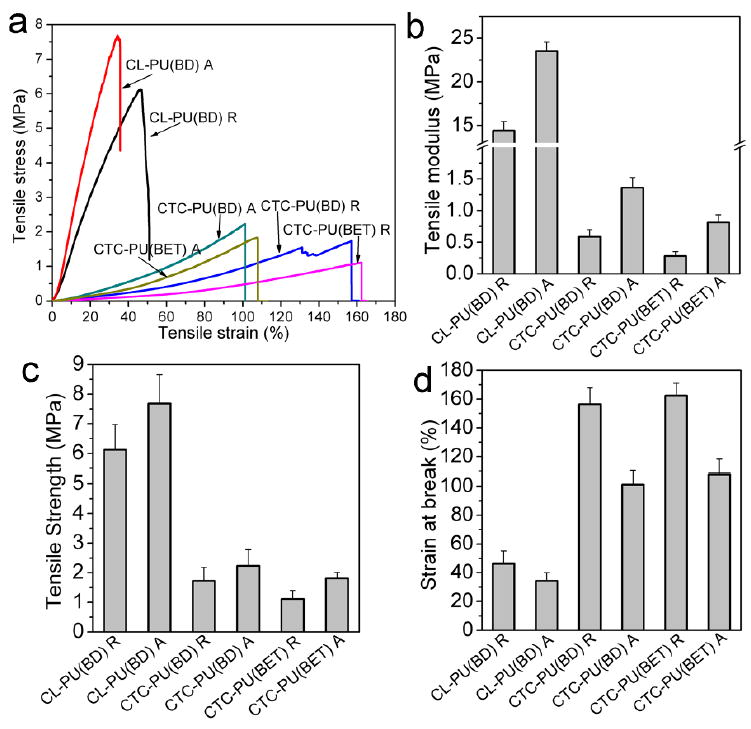
Tensile properties of CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) electrospun random and aligned scaffolds. (a) Representative stress strain curves in tensile tests, and statistical results of (b) tensile modulus, (c) tensile strength, and (d) strain-at-break.
3.6 Biocompatibility Evaluations
The biocompatibility of the scaffolds was studied by culturing 3T3 fibroblasts on UV-sterilized scaffolds for up to 10 days. Cell viability, proliferation, and morphology were investigated. From the cytoskeleton images (Fig. 8 (a)), it was found that the cells cultured on random fibers showed a round or polygonal shape, while on all three aligned TPU fiber scaffolds, the cells were mostly stretched, showing a spindle-like shape with the cells orientated along the fibers. In addition, the cells were more rounded on the CL-PU(BD) scaffolds, while they tended to extend and spread out on the CTC-PU(BD) and CTC-PU (BET) scaffolds, thus demonstrating a flourishing growth state. Moreover, the cells were also denser on the PCTC-based soft TPUs. The live/dead assay verified that all scaffolds were non-cytotoxic since the cell viability was over 90% on all scaffolds at both time points (Fig. 8 (b) and Fig. S5). There was no significant difference among different scaffolds in terms of cell viability. The MTS assay results indicated that the cell population on aligned fibers was significantly higher than that of random fibers for the three TPUs at both day 5 and day 10. This was because the cells were able to present as their naturally aligned shape on the aligned fibers, which might have induced stronger proliferation. In comparing the different materials for the same type of fiber (random and aligned fibers), the difference wasn’t significant at day 5. However, there were significantly more cells on CTC-PU(BD) and CTC-PU(BET) aligned fibers than on CL-PU(BD) aligned fibers at day 10. The statistical results (Fig. 8 (c)) were in agreement with the observations of cell areas from fluorescence images (Fig. 8 (a) and Fig. S5) and SEM images (Fig. 9).
Fig. 8.
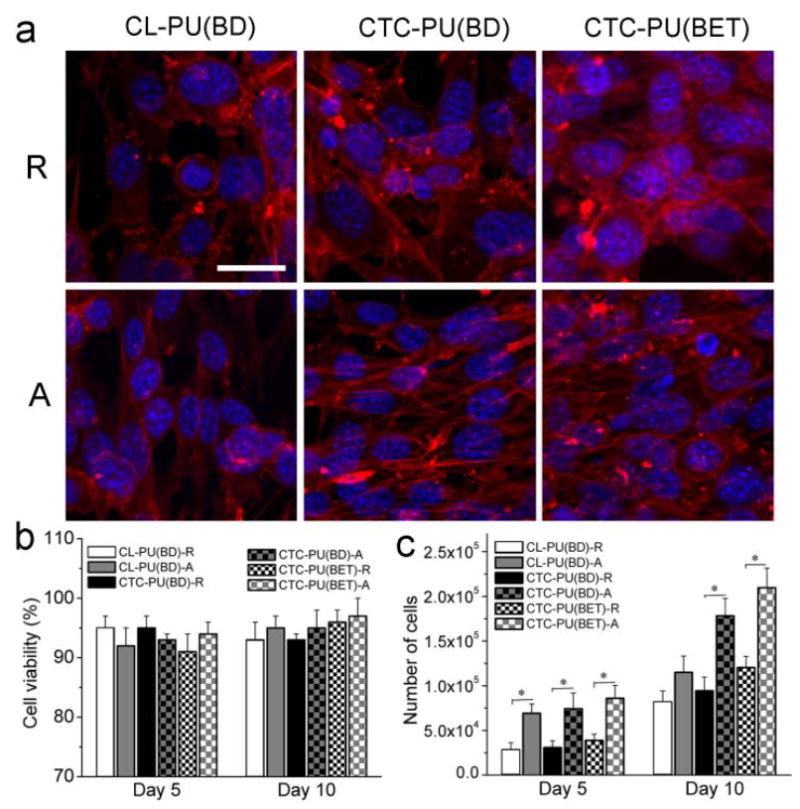
3T3 fibroblast cell culture results on electrospun random and aligned scaffolds of CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) scaffolds. (a) Cytoskeleton images of cells cultured on scaffolds for 5 days; scale bar = 25 μm. (b) Cell viability. (c) Cell proliferation statistical results on different scaffolds at day 5 and day 10 (p < 0.05).
Fig. 9.
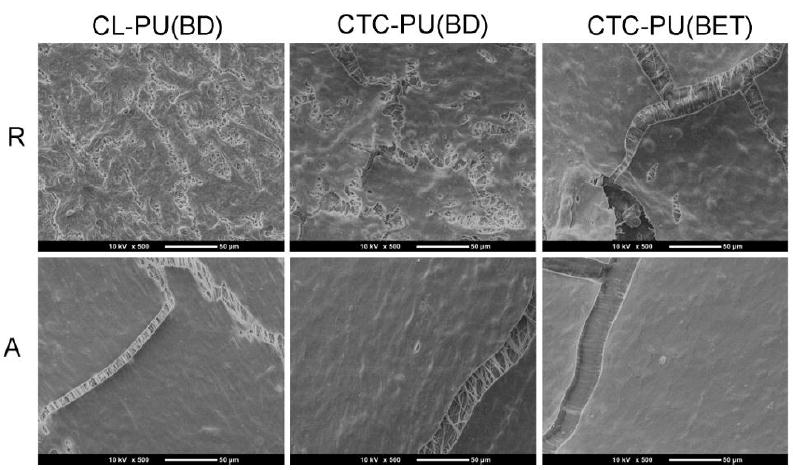
SEM images of cells cultured on random and aligned scaffolds of CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) scaffolds for 10 days. Scale bar = 50 μm.
From the SEM images (Fig. 9), it was found that after 10 days of culture, cells were able to form continuous membranes on the scaffolds, especially on the aligned fibers. The cracking on the cell membranes was caused by dehydration during sample preparation for SEM imaging. From the cracks, we were able to see the structure of the fibers beneath the cell membrane. Individual fibers could be observed clearly for CL-PU(BD) and CTC-PU(BD) scaffolds, thus indicating that the fibers were able to maintain their shape under cell culture conditions and they did not significantly degrade in 10 days of culture. However, the CTC-PU(BET) fibers showed a flat structure and it was already difficult to distinguish individual fibers, which is a good indication of biodegradation since we have proven that the fibers would not melt when incubated at 37 °C in PBS (Fig. S4). Moreover, the fast degradation of CTC-PU(BET) scaffolds is in good agreement with in vitro degradation results (Fig. 5).
Overall, we concluded that PCTC copolymer-based TPU scaffolds showed better fibroblast cell affinity than traditional PCL-based TPU scaffolds in terms of cell morphology and proliferation. Generally, structural, mechanical, and chemical cues affect substrate–cellular interaction. For these specific fibrous scaffolds with different TPUs, there might have been a synergetic effect of these three cues. First, CL-PU(BD) scaffolds possess a uniform and smooth fibrous structure, while CTC-PU(BD) and CTC-PU(BET) scaffolds exhibit a wider major diameter distribution and have nanobranches adhered to the major fibers (Fig. 6). This special micro- and nano-hybrid fibrous structure might have assisted with initial cell attachment and cell spreading.63 Second, the substrate stiffness has been found to be influential on cell behavior, and substrates with stiffness close to native tissues are highly preferred in supporting cell growth.47, 48 Fibroblasts are a major component cell type in various soft tissues; therefore, scaffolds with low moduli (e.g., CTC-PU(BD) and CTC-PU(BET)) should be more favorable for cells than relatively hard substrates (e.g., CL-PU(BD)). Third, surface chemistry is determined by the functional groups on the substrate surface. Most synthetic polymers do not have functional groups that directly attach to cells. Instead, synthetic polymers attract cells by immobilizing proteins from culture media.64 For materials with a similar molecular structure, the water contact angle (WCA) would affect the ability of cell to attach to a given substrate. A low contact angle of 40° has been reported to be the best for attachment for 3T3 fibroblasts.65 The average WCAs for the CL-PU(BD), CTC-PU(BD), and CTC-PU(BET) scaffolds were 91.3°, 93.5°, and 88.6°, respectively (Fig. S6). Hence, CTC-PU(BET) should be the easiest substrate for cells to attach to, which corresponds to the highest cell population on electrospun CTC-PU(BET) scaffolds.
Conclusions
In conclusion, a novel biodegradable soft TPU based on the ester ether triblock copolymer (PCTC) and the degradable chain extender (BET) has been successfully synthesized in this study in order to fulfill the mechanical requirements of soft tissue regeneration and meet the urgent demand for soft biodegradable thermoplastic elastomers. Conventional polyester (PCL)-based TPU and TPU synthesized based on PCTC and a non-degradable chain extender (BD) were used for comparison. 1H NMR and FTIR results confirmed the successful synthesis of all three TPUs, and they all had a uniform molecular weight distribution with molecular weights over 60 kDa. DSC results showed that the use of PCTC soft segments reduced the melting temperature and inhibited the crystallization of the elastomers. Furthermore, the BET chain extender increased the miscibility of the soft and hard segments. TGA results showed that the weight loss of CTC-PU(BET) was significantly faster than the other TPUs because the ester bonds in the BET cleaved at a much lower temperature than ester bonds in PCL, while the PTHF segment decomposed last. The tensile modulus and tensile strength of CTC-PU(BET) were as low as 2.2 MPa and 1.3 MPa, respectively, while they were 102.2 MPa and 7.4 MPa for conventional PCL-based TPU. Meanwhile, CTC-PU(BET) showed an elongation-at-break of over 700%, and maintained a 95.3% recovery rate and a 90% resilience over ten cycles of loading and unloading. The in vitro degradation test proved that the BET chain extender dramatically accelerated the initial degradation rate, and the PCTC soft segment degraded slower than the PCL soft segment. These results were in agreement with the thermal decomposition results. In addition, these TPUs were able to be electrospun into both random and aligned fibers. Notably, TPUs synthesized using PCTC as the soft segment formed fibers containing major microfibers and nano-branches which were better able to mimic the structure of ECM. 3T3 cell culture confirmed that these novel biodegradable soft TPU scaffolds outperformed conventional PCL-based TPU scaffolds in terms of cell proliferation and substrate–cellular interactions. All of these results suggest that the TPU synthesized in this study has great potential for use as tissue engineering scaffold materials for soft tissue regeneration.
Supplementary Material
Acknowledgments
Research reported in this paper was partially supported by the NHLBI of the National Institutes of Health under award number U01HL134655. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would also like to acknowledge the financial support of the National Natural Science Foundation of China (51603075; 21604026), the Fundamental Research Funds for the Central Universities (2015ZM093), the Kuo K. and Cindy F. Wang Professorship, and the Wisconsin Institute for Discovery at the University of Wisconsin–Madison.
Footnotes
Electronic Supplementary Information (ESI) available: DSC of diols, XRD, Gaussian fitted DTG, SEM of PBS incubated electrospun fibers, fluorescence images of live/dead stained cells, and water contact angle results.
Notes and References
- 1.Diez-Pascual AM, Diez-Vicente AL. J Mater Chem B. 2016;4:600–612. doi: 10.1039/c5tb01861g. [DOI] [PubMed] [Google Scholar]
- 2.Wu W, Allen RA, Wang YD. Nat Med. 2012;18:1148–+. doi: 10.1038/nm.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jing X, Mi HY, Wang XC, Peng XF, Turng LS. Acs Appl Mater Inter. 2015;7:6955–6965. doi: 10.1021/acsami.5b00900. [DOI] [PubMed] [Google Scholar]
- 4.Place ES, George JH, Williams CK, Stevens MM. Chem Soc Rev. 2009;38:1139–1151. doi: 10.1039/b811392k. [DOI] [PubMed] [Google Scholar]
- 5.Kim IG, Hwang MP, Du P, Ko J, Ha CW, Do SH, Park K. Biomaterials. 2015;50:75–86. doi: 10.1016/j.biomaterials.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 6.Lee P, Tran K, Zhou G, Bedi A, Shelke NB, Yu XJ, Kumbar SG. Soft Matter. 2015;11:7648–7655. doi: 10.1039/c5sm01909e. [DOI] [PubMed] [Google Scholar]
- 7.Chiono V, Tonda-Turo C. Prog Neurobiol. 2015;131:87–104. doi: 10.1016/j.pneurobio.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Folliguet TA, Rucker-Martin C, Pavoine C, Deroubaix E, Henaff M, Mercadier JJ, Hatem SN. J Thorac Cardiov Sur. 2001;121:510–519. doi: 10.1067/mtc.2001.112528. [DOI] [PubMed] [Google Scholar]
- 9.Serrano MC, Chung EJ, Ameer GA. Adv Funct Mater. 2010;20:192–208. [Google Scholar]
- 10.Lin HH, Hsieh FY, Tseng CS, Hsu SH. J Mater Chem B. 2016;4:6694–6705. doi: 10.1039/c6tb01501h. [DOI] [PubMed] [Google Scholar]
- 11.Wu YB, Wang L, Guo BL, Shao YP, Ma PX. Biomaterials. 2016;87:18–31. doi: 10.1016/j.biomaterials.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Mi HY, Salick MR, Jing X, Crone WC, Peng XF, Turng LS. J Biomed Mater Res A. 2015;103:593–603. doi: 10.1002/jbm.a.35208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Woodruff TM, Clark RJ, Martin DJ, Minchin RF. Acta Biomater. 2016;41:264–272. doi: 10.1016/j.actbio.2016.05.034. [DOI] [PubMed] [Google Scholar]
- 14.Guan JJ, Fujimoto KL, Sacks MS, Wagner WR. Biomaterials. 2005;26:3961–3971. doi: 10.1016/j.biomaterials.2004.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karchin A, Simonovsky FI, Ratner BD, Sanders JE. Acta Biomater. 2011;7:3277–3284. doi: 10.1016/j.actbio.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kavlock KD, Pechar TW, Hollinger JO, Guelcher SA, Goldstein AS. Acta Biomater. 2007;3:475–484. doi: 10.1016/j.actbio.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Punnakitikashem P, Truong D, Menon JU, Nguyen KT, Hong Y. Acta Biomater. 2014;10:4618–4628. doi: 10.1016/j.actbio.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong Y, Ye SH, Pelinescu AL, Wagner WR. Biomacromolecules. 2012;13:3686–3694. doi: 10.1021/bm301158j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baudis S, Ligon SC, Seidler K, Weigel G, Grasl C, Bergmeister H, Schima H, Liska R. J Polym Sci Pol Chem. 2012;50:1272–1280. [Google Scholar]
- 20.Tatai L, Moore TG, Adhikari R, Malherbe F, Jayasekara R, Griffiths I, Gunatillake PA. Biomaterials. 2007;28:5407–5417. doi: 10.1016/j.biomaterials.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 21.Zhang CH, Zhang N, Wen XJ. J Biomed Mater Res B. 2006;79B:335–344. doi: 10.1002/jbm.b.30547. [DOI] [PubMed] [Google Scholar]
- 22.Heijkants RGJC, van Calck RV, van Tienen TG, de Groot JH, Buma P, Pennings AJ, Veth RPH, Schouten AJ. Biomaterials. 2005;26:4219–4228. doi: 10.1016/j.biomaterials.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Guan J, Sacks MS, Beckman EJ, Wagner WR. Journal of biomedical materials research. 2002;61:493–503. doi: 10.1002/jbm.10204. [DOI] [PubMed] [Google Scholar]
- 24.Fang J, Ye SH, Shankarraman V, Huang YX, Mo, Wagner WR. Acta Biomater. 2014;10:4639–4649. doi: 10.1016/j.actbio.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blakney AK, Simonovsky FI, Suydam IT, Ratner BD, Woodrow KA. Acs Biomater Sci Eng. 2016;2:1595–1607. doi: 10.1021/acsbiomaterials.6b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Dong RN, Ge J, Guo BL, Ma PX. Acs Appl Mater Inter. 2015;7:28273–28285. doi: 10.1021/acsami.5b10829. [DOI] [PubMed] [Google Scholar]
- 27.Ou BL, Chen ML, Huang R, Zhou H. Rsc Adv. 2016;6:47138–47144. [Google Scholar]
- 28.Dey J, Xu H, Shen JH, Thevenot P, Gondi SR, Nguyen KT, Sumerlin BS, Tang LP, Yang J. Biomaterials. 2008;29:4637–4649. doi: 10.1016/j.biomaterials.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trinca RB, Abraham GA, Felisberti MI. Mat Sci Eng C-Mater. 2015;56:511–517. doi: 10.1016/j.msec.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 30.Henry JA, Simonet M, Pandit A, Neuenschwander P. J Biomed Mater Res A. 2007;82A:669–679. doi: 10.1002/jbm.a.31094. [DOI] [PubMed] [Google Scholar]
- 31.Henry JA, Burugapalli K, Neuenschwander P, Pandit A. Acta Biomater. 2009;5:29–42. doi: 10.1016/j.actbio.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 32.John JV, Moon BK, Kim I. React Funct Polym. 2013;73:1213–1222. [Google Scholar]
- 33.Guan JJ, Sacks MS, Beckman EJ, Wagner WR. Biomaterials. 2004;25:85–96. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- 34.Discher DE, Janmey P, Wang YL. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 35.Breuls RG, Jiya TU, Smit TH. The open orthopaedics journal. 2008;2:103–109. doi: 10.2174/1874325000802010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agache PG, Monneur C, Leveque JL, De Rigal J. Archives of dermatological research. 1980;269:221–232. doi: 10.1007/BF00406415. [DOI] [PubMed] [Google Scholar]
- 37.Liu QY, Jiang L, Shi R, Zhang LQ. Prog Polym Sci. 2012;37:715–765. [Google Scholar]
- 38.Loh XJ, Karim AA, Owh C. J Mater Chem B. 2015;3:7641–7652. doi: 10.1039/c5tb01048a. [DOI] [PubMed] [Google Scholar]
- 39.Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K. Philos T Roy Soc B. 2002;357:121–132. doi: 10.1098/rstb.2001.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nair PA, Ramesh P. J Biomed Mater Res A. 2013;101:1876–1887. doi: 10.1002/jbm.a.34490. [DOI] [PubMed] [Google Scholar]
- 41.Chan-Chan LH, Solis-Correa R, Vargas-Coronado RF, Cervantes-Uc JM, Cauich-Rodriguez JV, Quintana P, Bartolo-Perez P. Acta Biomater. 2010;6:2035–2044. doi: 10.1016/j.actbio.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 42.Guelcher SA, Gallagher KM, Didier JE, Klinedinst DB, Doctor JS, Goldstein AS, Wilkes GL, Beckman EJ, Hollinger JO. Acta Biomater. 2005;1:471–484. doi: 10.1016/j.actbio.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 43.Zhang LS, Huang MM, Yu RL, Huang JC, Dong X, Zhang RY, Zhu J. J Mater Chem A. 2014;2:11490–11498. [Google Scholar]
- 44.Li XY, Liu HC, Wang JN, Li CJ. Polymer. 2012;53:248–253. [Google Scholar]
- 45.Mi HY, Jing X, Jacques BR, Turng LS, Peng XF. J Mater Res. 2013;28:2339–2350. [Google Scholar]
- 46.Bergmeister H, Seyidova N, Schreiber C, Strobl M, Grasl C, Walter I, Messner B, Baudis S, Frohlich S, Marchetti-Deschmann M, Griesser M, di Franco M, Krssak M, Liska R, Schima H. Acta Biomater. 2015;11:104–113. doi: 10.1016/j.actbio.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Kim TH, An DB, Oh SH, Kang MK, Song HH, Lee JH. Biomaterials. 2015;40:51–60. doi: 10.1016/j.biomaterials.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 48.Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King CA. Acta Biomater. 2013;9:4635–4644. doi: 10.1016/j.actbio.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caracciolo PC, Buffa F, Abraham GA. J Mater Sci-Mater M. 2009;20:145–155. doi: 10.1007/s10856-008-3561-8. [DOI] [PubMed] [Google Scholar]
- 50.Middleton JC, Tipton AJ. Biomaterials. 2000;21:2335–2346. doi: 10.1016/s0142-9612(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 51.Yang SF, Leong KF, Du ZH, Chua CK. Tissue Eng. 2001;7:679–689. doi: 10.1089/107632701753337645. [DOI] [PubMed] [Google Scholar]
- 52.Webb AR, Yang J, Ameer GA. Expert Opin Biol Th. 2004;4:801–812. doi: 10.1517/14712598.4.6.801. [DOI] [PubMed] [Google Scholar]
- 53.Ramakrishna S, Huang ZM, Kumar GV, Batchelor AW, Mayer J. An introduction to biocomposites. Imperial College Press; London: 2004. [Google Scholar]
- 54.Seal BL, Otero TC, Panitch A. Mat Sci Eng R. 2001;34:147–230. [Google Scholar]
- 55.Engelberg I, Kohn J. Biomaterials. 1991;12:292–304. doi: 10.1016/0142-9612(91)90037-b. [DOI] [PubMed] [Google Scholar]
- 56.Cohn D, Salomon AF. Biomaterials. 2005;26:2297–2305. doi: 10.1016/j.biomaterials.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 57.Lee SH, Kim BS, Kim SH, Choi SW, Jeong SI, Kwon IK, Kang SW, Nikolovski J, Mooney DJ, Han YK, Kim YH. J Biomed Mater Res A. 2003;66A:29–37. doi: 10.1002/jbm.a.10497. [DOI] [PubMed] [Google Scholar]
- 58.Lv S, Dudek DM, Cao Y, Balamurali MM, Gosline J, Li HB. Nature. 2010;465:69–73. doi: 10.1038/nature09024. [DOI] [PubMed] [Google Scholar]
- 59.Guo L, Ma MM, Zhang N, Langer R, Anderson DG. Adv Mater. 2014;26:1427–1433. doi: 10.1002/adma.201304140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labhasetwar V, Vasir JK, Reddy MK. The Cleveland Clinic Foundation; US: 2016. [Google Scholar]
- 61.Nair LS, Laurencin CT. Prog Polym Sci. 2007;32:762–798. [Google Scholar]
- 62.McKee MG, Park T, Unal S, Yilgor I, Long TE. Polymer. 2005;46:2011–2015. [Google Scholar]
- 63.Jing X, Mi HY, Peng J, Peng XF, Turng LS. Carbohyd Polym. 2015;117:941–949. doi: 10.1016/j.carbpol.2014.10.025. [DOI] [PubMed] [Google Scholar]
- 64.Arima Y, Iwata H. Biomaterials. 2007;28:3074–3082. doi: 10.1016/j.biomaterials.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 65.Webb K, Hlady V, Tresco PA. Journal of biomedical materials research. 1998;41:422–430. doi: 10.1002/(sici)1097-4636(19980905)41:3<422::aid-jbm12>3.0.co;2-k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye SH, Hong Y, Sakaguchi H, Shankarraman V, Luketich SK, D’Amore A, Wagner WR. Acs Appl Mater Inter. 2014;6:22796–22806. doi: 10.1021/am506998s. [DOI] [PubMed] [Google Scholar]
- 67.Fang J, Ye SH, Wang J, Zhao T, Mo XM, Wagner WR. Biomacromolecules. 2015;16:1622–1633. doi: 10.1021/acs.biomac.5b00192. [DOI] [PubMed] [Google Scholar]
- 68.Yi J, Huang CS, Zhuang HY, Gong H, Zhang CY, Ren RT, Ma YP. Prog Org Coat. 2015;87:161–170. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.