

Abstract
Hypoxia in the embryo is a frequent cause of intra-uterine growth retardation, low birth weight and multiple organ defects. In the kidney this can lead to low nephron endowment predisposing to chronic kidney disease and arterial hypertension. A key component in cellular adaptation to hypoxia is the hypoxia-inducible factor pathway, which is regulated by prolyl-4-hydroxylase domain (PHD) dioxygenases PHD1, PHD2 and PHD3. In the adult kidney PHD oxygen-sensors are differentially expressed in a cell type-dependent manner and control the production of erythropoietin in interstitial cells. However, the role of interstitial cell PHDs in renal development has not been examined. Here we used a genetic approach in mice to interrogate PHD function in FOXD1-expressing stroma during nephrogenesis. We demonstrate that PHD2 and PHD3 are essential for normal kidney development as the combined inactivation of stromal PHD2 and PHD3 resulted in renal failure that was associated with reduced kidney size, decreased numbers of glomeruli and abnormal postnatal nephron formation. In contrast, nephrogenesis was normal in animals with individual PHD inactivation. We furthermore demonstrate that the defect in nephron formation in PHD2/PHD3 double mutants required intact hypoxia-inducible factor-2 signaling and was dependent on the extent of stromal hypoxia-inducible factor activation. Thus, hypoxia-inducible factor prolyl-4-hydroxylation in renal interstitial cells is critical for normal nephron formation.
Keywords: Hypoxia, renal development, chronic kidney disease, hypoxia-inducible factor, prolyl-4-hydroxylase, pericytes
INTRODUCTION
Hypoxia not only occurs under pathologic conditions, but also physiologically during normal development regulating stem cell behavior, cellular differentiation, proliferation and migration, as well as the reciprocal interactions between different cell types on multiple levels, thus affecting morphogenesis of the embryo and placenta. Molecular mechanisms that permit cells to adequately respond to discrepancies between oxygen demand and supply are therefore critically important for normal embryonic development. A disruption of these responses may lead to developmental abnormalities in multiple organ systems and in the worst-case scenario to intra-embryonic demise.1
A major and critical component of cellular hypoxia responses is the prolyl-4-hydroxylase domain (PHD) / hypoxia-inducible factor (HIF) axis, which enables cells to respond to changes in tissue oxygen levels in a rapid and controlled fashion. HIF-1 and HIF-2 are pleiotropic basic helix-loop-helix transcription factors that consist of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit, also known as the aryl hydrocarbon receptor nuclear translocator ARNT, and regulate a multitude of hypoxia responses, thus allowing cells to adapt to and survive low oxygen environments.2 Under normoxic conditions, oxygen-, iron- and 2-oxoglutarate (2OG)-dependent prolyl-4-hydroxylase domain proteins, PHD1, PHD2 and PHD3, also known as egl nine homolog (EGLN) 2, EGLN1 and EGLN3 respectively, function as oxygen sensors of this pathway. PHD enzymes initiate rapid proteasomal degradation of constitutively synthesized HIF-α subunits through hydroxylation of specific proline residues.3 A reduction in PHD catalytic activity, for example, under hypoxic conditions or due to pharmacologic inhibition, results in HIF-α stabilization and activation of HIF transcriptional programs.3
Sustained discrepancies between oxygen demand and supply can result from maternal disease, utero-placental insufficiency or life at high altitude. This frequently leads to intra-uterine growth retardation (IUGR), low birth weight and increases the risk of developing diabetes, cardiopulmonary disease, stroke, arterial hypertension or chronic kidney disease (CKD) in adults.4–6 In the developing kidney hypoxia reduces ureteric bud (UB) branching and nephron formation7 and results in low nephron endowment, which by itself associates with increased risk of developing CKD and/or arterial hypertension.8, 9
Normal kidney development is driven by multiple reciprocal and cyclical interactions between the UB and the metanephric mesenchyme (MM), which result in repeated UB branching and nephron formation.10, 11 However, little detail is known about the role of stromal cells in this process. Renal stroma is identified by the expression of the forkhead box D1 (FOXD1) transcription factor and surrounds the cap mesenchyme (CM). FOXD1-expressing stroma plays a critical role in renal capsule development, renal progenitor differentiation and nephron formation, is important for normal vascular patterning, and ultimately gives rise to cortical and medullary interstitial fibroblast-like cells, pericytes, mesangial cells and vascular smooth muscle cells (VSMC).12–17 As hypoxia occurs physiologically during kidney development both HIF-1α and HIF-2α have been detected in the developing kidney in a cell-type dependent manner.18–20 Despite clear evidence of HIF pathway activation, the functional roles of cell-specific HIF signaling during renal development, however, are poorly understood and information from genetic models is limited.
In order to examine the role of interstitial HIF oxygen sensing in renal development and homeostasis, we used the Cre-loxP system to target all 3 HIF-PHDs in conjunction with HIF-1α or HIF-2α in FOXD1-expressing stromal cells. We found that mice with individual Phd1, Phd2 or Phd3 deletion or Phd1/Phd2 and Phd1/Phd3 double deletion were born with normal kidneys, whereas the combined inactivation of Phd2 and Phd3 resulted in abnormal kidney development, renal failure and premature death. Kidney defects in Phd2/Phd3 double knockout mice became apparent after postnatal day (P) 7, correlated with the degree of interstitial HIF activation and were characterized by a HIF-2-dependent reduction in the number of mature nephrons and glomeruli as well as abnormal renal vasculature. Taken together, our data establish that the ability to regulate HIF prolyl-4-hydroxylation in FOXD1 stroma-derived cells is essential for normal nephron formation. Our data have implications for the therapeutic use of HIF prolyl-4-hydroxylase inhibitors, which are currently in phase 3 clinical development for renal anemia.21
RESULTS
Combined inactivation of Phd2 and Phd3 in FOXD1 stroma is associated with renal failure and juvenile lethality
Interstitial cells play an important role in the regulation of renal hypoxia responses. A classic example is the hypoxic induction of EPO. In order to examine the role of individual PHDs in these responses, we utilized Foxd1cre/+ transgenic mice. In this transgenic line, the Cre transgene, which consists of an enhanced green fluorescent protein/Cre-recombinase (EGFP/Cre) fusion protein, is under transcriptional control of the Foxd1 promoter (Figure 1A).22, 23 FOXD1-expressing cells surround the CM in the nephrogenic zone, give rise to all stromal components of the developing kidney and express Phd1, 2 and 3 (Supplemental Figure S1).
Figure 1. Combined inactivation of Phd2 and Phd3 in FOXD1 stroma results in renal failure.

(A) Schematic illustrating the experimental approach and location of targeted sequences within the Phd2 and Phd3 floxed alleles. (B) Survival curve of Cre− littermate controls (Foxd1+/+ Phd2fl/fl and Foxd1+/+ Phd2fl/fl Phd3fl/fl mice), Foxd1-Phd2−/− and Foxd1-Phd2−/−-Phd3−/− mice. Kaplan-Meier curves were plotted and compared using the log-rank test (n>10), ***p<0.001. (C) Blood urea nitrogen (BUN) from Cre− littermate control and Foxd1-Phd2−/−-Phd3−/− mice at P14–P17 (n=3 each). (D) Shown are representative H&E images and immunostaining for ACTA2 and CD31 of kidney sections from Cre− control and Foxd1-Phd2−/−Phd3−/− mice at P14 and P30. Asterisks depict cysts, # depicts dilated vessels, yellow arrows indicate vascular walls, and blue arrows indicate glomeruli. Scale bars represent 1 mm for whole kidney cross-sections, 100 μm for high-power H&E images, and 50 μm for ACTA2/CD31 immunostaining. (E and F) Epo, collagen type 1 alpha 1 (Col1a1), and F4/80 mRNA levels in Cre− littermate control and Foxd1-Phd2−/−-Phd3−/− mutant kidneys at P14 (n=8–10). Vascular lumina were quantified and are represented as total lumen area per tissue area (n=3 each). Data are represented as mean ± SEM; 2-tailed Student’s t-test; *p<0.05, **p<0.01 and ***p<0.001.
Utilizing Foxd1cre/+ transgenic mice, we developed animals with individual or combined inactivation of PHD1, PHD2 and PHD3. Whereas Foxd1cre/+ Phd1fl/fl, Foxd1cre/+ Phd2fl/fl and Foxd1cre/+ Phd3fl/fl mutant mice, from hereon referred to as Foxd1-Phd1−/−, Foxd1-Phd2−/− and Foxd1-Phd3−/− mutants, developed normally into adulthood, Foxd1cre/+ Phd2fl/fl Phd3fl/fl double knockout mice, from hereon referred to as Foxd1-Phd2−/−-Phd3−/− mutants, were small and died prematurely (Figure 1B). Differences in whole body weight between mutants and Cre− littermate controls (Foxd1+/+ Phd2fl/fl Phd3fl/fl) became statistically significant by postnatal day (P) 14 (5.0 ± 0.4 g for mutants vs. 7.3 ± 0.5 g for controls; n=8 and 12 respectively; p=0.003; Supplemental Table S1). Juvenile lethality in the mutant cohort was associated with renal failure and severe pathological changes in the kidney (Figures 1C and 1D). Kidney weight was significantly reduced in mutants compared to controls (28.2 ± 2.1 mg vs. 47.9 ± 2.5 mg in controls; n=16 and 24 respectively; p<0.0001; Supplemental Table 1), and renal defects in Foxd1-Phd2−/−-Phd3−/− mutants at weaning age were characterized by tubular and vascular dilatations, tubular cyst formation, accumulation of α-smooth muscle actin (α-SMA/ACTA2)-positive interstitial and glomerular cells, glomerular sclerosis, increase in collagen matrix and collagen type 1 alpha 1 (Col1a1) mRNA production and increased F4/80 mRNA levels (Figure 1E, 1F and Supplemental Figure S2). Since PHD inhibition results in normoxic HIF-α stabilization and activation of HIF signaling, we assessed the mRNA expression levels of HIF target gene Epo. Despite the presence of severe morphologic defects Epo mRNA levels in the kidney were significantly increased in 2 week-old Foxd1-Phd2−/−-Phd3−/− mice, indicating that the combined deletion of Phd2 and Phd3 resulted in robust activation of the HIF system in FOXD1 stroma-derived interstitial cells (~80-fold increase; n=9 and 10 respectively; p<0.0001; Figure 1E). Increased Epo was accompanied by a small but significant increase in hematocrit (38.8% ± 1.1% vs. 35.5% ± 0.8% in controls; n=14 and 14, respectively; p<0.05).
Since HIF is known to promote angiogenesis,24, 25 we examined the renal vasculature in Foxd1-Phd2−/−-Phd3−/− kidneys. For this, we used real time PCR in conjunction with IHC to characterize the tissue expression patterns of endothelial cell marker CD31 and ACTA2. At P14 we observed significant increases in microvessel density, which correlated with elevated Cd31 mRNA expression in whole kidney homogenates (~1.4-fold increase) (Figure 1F). Furthermore, small and medium-sized arterial vessels in Foxd1-Phd2−/−-Phd3−/− kidneys were dilated and were characterized by relatively thin vessel walls (Figure 1D). Vascular changes in Foxd1-Phd2−/−-Phd3−/− kidneys were associated with increased transcription of vascular endothelial growth factor (Vegf) in renal interstitium as demonstrated by fluorescent RNA in situ hybridization (RNA-FISH, data not shown), which is consistent with findings in adult Foxd1-Phd2−/− mice.26 Differences in Acta2 mRNA expression were not observed. Taken together our data demonstrate that inactivation of both PHD2 and PHD3 in stromal cells resulted in interstitial HIF activation, which led to renal failure and was associated with juvenile lethality.
Combined loss of Phd2 and Phd3 in FOXD1 stroma results in reduced nephron formation
In the mouse metanephric kidney development begins at ~ embryonic day (E) 10.5 with the formation of the UB, which induces the CM.10, 15 Since FOXD1 stromal cells, which surround the CM, are an important source of metanephric regulatory signals and have been shown to play a critical role in the maintenance and differentiation of epithelial progenitors,14, 16, 17 we sought to determine at what time point during development renal defects in Foxd1-Phd2−/−-Phd3−/− mice became apparent. For this we harvested kidneys from Cre− control, Foxd1-Phd2−/− and Foxd1-Phd2−/−-Phd3−/− mice at P0, P7 and P14 and used routine histological methods for morphologic assessment.
Whereas Foxd1-Phd2−/− kidneys were morphologically normal and could not be distinguished from controls at either time point, kidneys from Foxd1-Phd2−/−-Phd3−/− mutants were characterized by the presence of undifferentiated epithelial cells in the subcapsular cortex and a reduction in the number of differentiated proximal tubules (PT) (Figure 2). Periodic acid-Schiff (PAS) staining, which identifies polysaccharides of tubular basement and brush border membranes, demonstrated a lack of PT brush border staining in Foxd1-Phd2−/−-Phd3−/− kidneys at P7 and P14 (Figure 2). In contrast, normal cortical PAS staining patterns were found in Cre− control and Foxd1-Phd2−/− kidneys at P7 indicating that nephron formation was not affected by Phd2 inactivation.
Figure 2. Defective nephron formation in Foxd1-Phd2−/−-Phd3−/− mice.
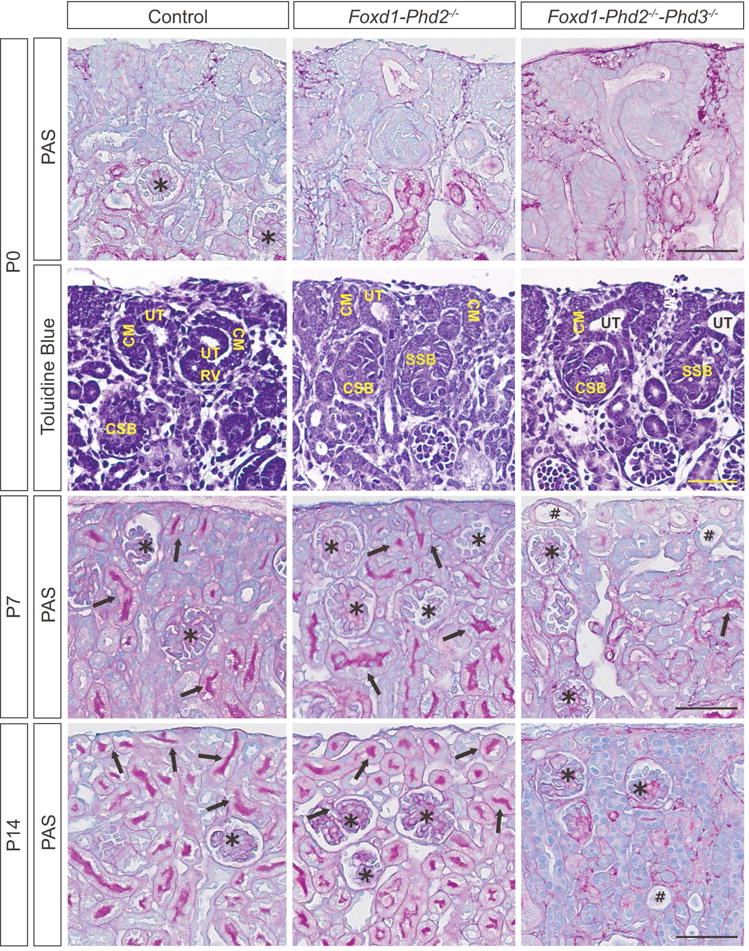
Shown are representative images of Periodic acid-Schiff (PAS)-stained kidney sections from Cre− control (Foxd1+/+ Phd2fl/fl Phd3fl/fl for P0, and Foxd1+/+ Phd2fl/fl for P7 and P14), Foxd1-Phd2−/− and Foxd1-Phd2−/−-Phd3−/− mice at P0, P7 and P14, and toluidine blue-stained kidney sections from P0 animals. Arrows indicate PAS-positive mature tubules, asterisks depict glomeruli, and # marks dilated tubules. Scale bar represents 50 μm.
Toluidine blue staining demonstrated that the nephrogenic zone, which consists of CM, ureteric tips (UT) and early nephron structures such as renal vesicles (RV), comma-shaped bodies (CSB) and S-shaped bodies (SSB) was morphologically similar between control, Foxd1-Phd2−/−, and Foxd1-Phd2−/−-Phd3−/− kidneys at P0. Differences in the size of the stromal cell compartment were not observed between control and Foxd1-Phd2−/−-Phd3−/− kidneys (Supplemental Figure S3). Taken together our findings suggest that the combined inactivation of Phd2 and Phd3 primarily impacted the later stages of tubulogenesis, which became morphologically evident by P7 (Figure 2).
To further assess the Foxd1-Phd2−/−-Phd3−/− phenotype, we performed immunohistochemistry (IHC) and analyzed nephron segment-specific gene expression in whole kidney homogenates by real time PCR. The pattern of Lotus tetragonolobus lectin (LTL) staining was consistent with a reduction in the number of mature PT (Figure 3A), which was statistically significant at P7 (156.8 ± 12.4 tubules/mm2 for mutants vs. 300.7 ± 20.8 tubules/mm2 for controls; n=3 each; p=0.004). Statistically significant differences were not observed at P0 (152.3 ± 15.0 tubules/mm2 for mutants vs. 174.3 ± 14.8 tubules/mm2 for Cre− controls; n=3 each), supporting the notion that the combined inactivation of PHD2 and PHD3 impacted postnatal nephron development, which in mice terminates within the first week after birth.27, 28 The expression of two additional PT maturation markers, megalin and cubilin, was also significantly decreased in Foxd1-Phd2−/−-Phd3−/− mutants (Supplemental Figure S4). In contrast, the staining patterns and density of Tamm-Horsfall protein (THP) / uromodulin expression, a marker for the thick ascending limb of the loop of Henle (mTAL), and lectin Dolichos biflorus agglutinin (DBA) staining, which identifies the collecting duct (CD), were comparable between control and Foxd1-Phd2−/−-Phd3−/− kidneys (Figure 3B). Morphologic findings were consistent with a significant decrease in aquaporin 1 (Aqp1) and sodium-phosphate co-transporter-2a (NaPi2a) mRNA levels, whereas the mRNA expression levels of mTAL gene Nkcc2, which encodes the Na-K-2Cl co-transporter 2, transient receptor potential cation channel subfamily V member 5 (Trpv5), which is associated with the distal tubule, and CD-specific aquaporin 2 (Aqp2) were not reduced. In contrast mRNA levels of uromodulin, NaCl co-transporter (Ncc) and sodium channel epithelial 1 alpha subunit (Scnn1a) were significantly reduced compared to control (Figure 3C). The reduction in the number of mature nephrons in Foxd1-Phd2−/−-Phd3−/− mutants was associated with a decrease in the number of glomeruli at P7 (45.0 ± 2.5 glomuruli / mm2 in mutant vs. 58.0 ± 4.1 glomeruli / mm2 in control; n=5 and 4, respectively; p<0.05; Figure 3D). In summary, our data suggest that the combined loss of PHD2 and PHD3 catalytic activity in FOXD1 stromal progenitors suppressed nephron formation, which in turn led to renal failure in juvenile mice.
Figure 3. Combined inactivation of Phd2 and Phd3 in FOXD1 stroma reduces nephron formation.
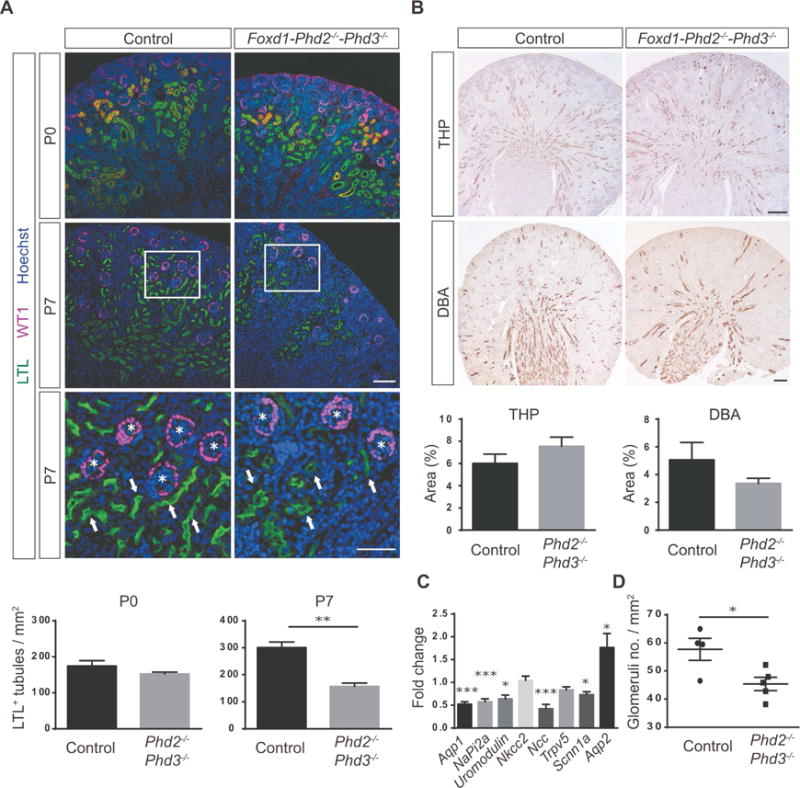
(A) Shown are representative images of Lotus tetragonolobus lectin (LTL) staining (green) and IHC staining for Wilms tumor 1 (WT1) (pink); kidney sections were obtained from Cre− littermate control or Foxd1-Phd2−/−-Phd3−/− mutants at P0 or P7. Nuclei were stained with Hoechst dye. Arrows indicate proximal tubules and asterisks depict glomeruli. Scale bars: 100 μm (top and middle panels) and 50 μm (bottom panel). LTL+ tubules were quantified (n=3 each) for P0 and P7. (B) Shown are representative images of IHC staining for Tamm-Horsfall protein (THP) and Dolichos biflorus agglutinin (DBA) lectin staining of kidney sections from Cre− littermate controls and Foxd1-Phd2−/−-Phd3−/− mice at P7. THP+ and DBA+ tubules were quantified (n=3–5). Scale bar: 200 μm. (C) Fold changes in mRNA expression levels of nephron segment-specific genes in total kidney homogenates fromFoxd1-Phd2−/−-Phd3−/− mutants compared to Cre− littermate controls (n=8 each). (D) Quantification of glomerular numbers. Shown are number of glomeruli per mm2 (n=4–5). Data are represented as mean ± SEM; 2-tailed Student’s t-test, *p<0.05, **p<0.01 and ***p<0.001. Acta 2, α-smooth muscle actin; Aqp1, aquaporin 1; Aqp2, aquaporin 2; NaPi2a, sodium-phosphate co-transporter-2a; Ncc NaCl co-transporter; Nkcc2, Na-K-2Cl co-transporter, Scnn1a, sodium channel epithelial 1 alpha subunit; Trpv5, transient receptor potential cation channel subfamily V member 5.
SIX2-expressing progenitors are not reduced in Foxd1-Phd2−/−-Phd3−/− kidneys
FOXD1 stroma-derived interstitial cells play a critical role in the maintenance and differentiation of renal epithelial progenitor cells, which are contained within the CM. The CM represents induced mesenchyme and contains several layers of cells that express multiple transcription factors, including paired box 2 (PAX2), sine oculis-related homeobox 2 (SIX2) and spalt-like transcription factor 1 (SALL1), as well as several secreted molecules.10, 11 In order to better understand the pathogenesis of defective nephron formation in Foxd1-Phd2−/−-Phd3−/− mice, we used morphologic analysis and gene expression studies to assess for potential abnormalities in the nephrogenic zones of mutant kidneys. IHC for SIX2, cytokeratin, SALL1, PAX2, neural cell adhesion molecule 1 (NCAM), E-cadherin (ECAD) and LIM homeobox protein 1 (LHX1) at E15.5 and at P0 indicated that CM volume was not reduced and that renal vesicle formation was not defective in Foxd1-Phd2−/−-Phd3−/− mutants (Figure 4A and 4B). Real time PCR analysis at P0, however, demonstrated that Six2, Sall1, and Pax2 mRNA levels in whole-kidney homogenates from Foxd1-Phd2−/−-Phd3−/− mice were significantly increased compared to Cre− control (~1.8-, ~1.5- and ~1.4-fold, respectively), suggesting a moderate expansion in CM volume (Figure 4C). By P7, SIX2 expression, which is highly associated with the epithelial progenitor compartment, was no longer detectable in both control and Foxd1-Phd2−/−-Phd3−/− kidneys (Figure 4A), indicating that reduced nephron formation in Foxd1-Phd2−/−-Phd3−/− mutants was not associated with abnormal persistence of epithelial progenitor cells. Taken together our data suggest that Foxd1-Phd2−/−-Phd3−/− mutant mice were not characterized by major defects in prenatal nephrogenesis or the diminished presence of renal progenitors.
Figure 4. Nephron progenitors are not decreased in Foxd1-Phd2−/−-Phd3−/− kidneys.
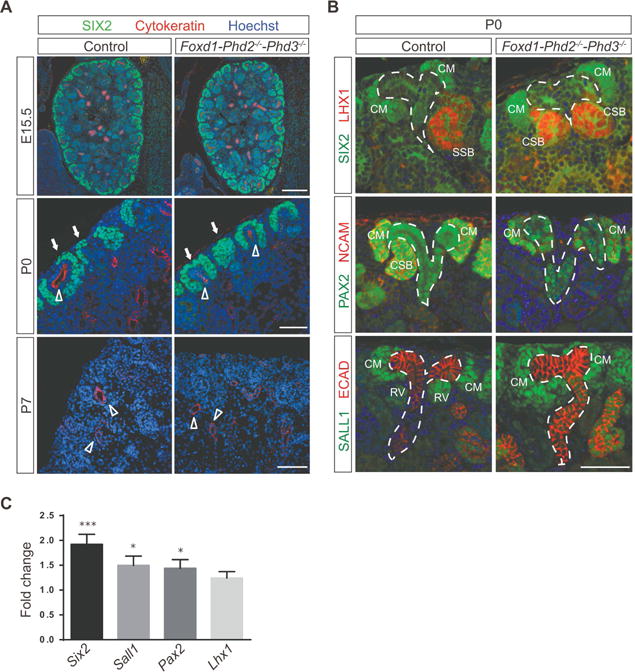
(A) Representative images of IHC staining of formalin-fixed paraffin-embedded kidney sections for nephron progenitor marker SIX2 (green) and UB marker pan-cytokeratin (red); shown are results for Cre− littermate control and Foxd1-Phd2−/−-Phd3−/− mutants at embryonic day (E) 15.5, P0 or P7. Nuclei were stained with Hoechst dye. Scale bar: 200 μm (E15.5) and 50 μm (P0 and P7). Arrows indicate cap mesenchyme and open arrowheads depict ureteric bud. (B) Shown are representative images of SIX2 (green) and LXH1 (red) (top panels), PAX2 (green) and NCAM (red) (middle panels), and SALL1 (green) and ECAD (red) (bottom panels) IHC staining of formalin-fixed paraffin-embedded kidney sections from Cre− littermate control and Foxd1-Phd2−/−-Phd3−/− mice at P0. Nuclei were stained with Hoechst dye. Ureteric trees are outlined by dashed white lines and cap mesenchyme (CM), renal vesicle (RV), comma shaped body (CSB), and S-shaped body (CSB) are annotated. Scale bar: 50 μm. (C) Fold changes in Six2, Sall1, Pax2 and Lhx1 mRNA expression levels in total kidney homogenates from Foxd1-Phd2−/−-Phd3−/− at P0 compared to Cre− littermate controls (n=12 for control and n=10 for mutants). Data are represented as mean ± SEM; 2-tailed Student’s t-test; ***p<0.001 and *p<0.05.
Reduced nephron formation in Foxd1-Phd2−/−-Phd3−/− kidneys is dependent on the extent of stromal HIF activation
We have previously shown in adult mice that Foxd1-Cre-mediated inactivation of PHD2 resulted in HIF-2 activation in a subset of renal interstitial cells. This was associated with increased Epo and Vegf transcription, and suggested that renal interstitial cells were heterogeneous with regard to their responsiveness to PHD2 inactivation.26 Although HIF prolyl-4-hydroxylases regulate the activity of both HIF-1α and HIF-2α, HIF-1α was not detectable in adult Foxd1-Phd2−/− kidneys. To examine whether HIF-1α was expressed in renal stroma during development, we used IHC to investigate the cellular and spatial distribution of HIF-1α and HIF-2α in kidneys from newborn Cre− control, Foxd1-Phd2−/− and Foxd1-Phd2−/−-Phd3−/− mice. In contrast to adults, nuclear HIF-1α staining was detectable in the cortex of both Foxd1-Phd2−/− and Foxd1-Phd2−/−-Phd3−/− kidneys at P0 and localized predominantly to the nephrogenic zone, whereas the majority of cortical HIF-2α-expressing cells were found in the sub-nephrogenic zone, suggesting a homolog-dependent spatial distribution pattern for HIF-α subunits in the developing cortical interstitium (Figure 5A). In the renal medulla, however, both HIF-1α- and HIF-2α-expressing interstitial cells were detected (data not shown). Consistent with the spatial distribution of HIF-2α was the distribution of Epo transcripts, which were not detected in the nephrogenic cortex by RNA-FISH (Figure 5B).
Figure 5. Phd3 inactivation in Phd2−/− FOXD1 stroma increases the number of HIF-α expressing cells.

(A) Shown are representative images of HIF-1α and HIF-2α IHC staining of formalin-fixed, paraffin-embedded kidney sections from Cre− control (Foxd1+/+ Phd2fl/fl and Foxd1+/+ Phd2fl/fl Phd3fl/fl), Foxd1-Phd2−/− and Foxd1-Phd2−/−-Phd3−/− mice at P0 (n=3–4). Arrows depict HIF-α+ cells. To facilitate visualization and to provide an overview of HIF-α+ cell distribution at low magnification, HIF-α+ cells were annotated with red circles (bottom right panels). The extent of stromal HIF activation is expressed as HIF-α+ cell number per 0.01 mm2. The Cre− control group consisted of one Foxd1+/+ Phd2fl/fl and three Foxd1+/+ Phd2fl/fl Phd3fl/fl mice. Data represent mean ± SEM and were analyzed by 1-way ANOVA; **p<0.01. Scale bar: 50 μm (top panels), 10 μm (high-magnification images, bottom left panels), and 100 μm (low-magnification images, bottom, right panels). (B) Shown are images illustrating the distribution of Epo+ cells in Foxd1-mJ/mG-Phd2−/−-Phd3−/− kidneys by RNA-FISH. To facilitate visualization and to provide an overview of Epo+ cell distribution at low magnification, Epo+ cells were annotated with red circles (right panel). Epo transcripts were detected by red fluorescence and EGFP transcripts (cells with a history of Foxd1-cre expression) were detected by green fluorescence (left panel). Scale bars: 10 μm (left) and 100 μm (right). Labeling of low-magnification images in (A) and (B): (a), superficial cortex with nephrogenic zone; (b), deeper cortex and(c), medulla.
To determine whether abnormal nephron formation in Foxd1-Phd2−/−-Phd3−/− mice correlated with the degree of interstitial HIF activation, we determined the number of HIF-1α- and HIF-2α-expressing cells in Cre− control, Foxd1-Phd2−/− and Foxd1-Phd2−/−-Phd3−/− kidneys. We found that compared to Foxd1-Phd2−/− mice, the number of both HIF-1α- and HIF-2α-expressing stromal cells was significantly increased in Foxd1-Phd2−/−-Phd3−/− kidneys (16.5 ± 3.3 vs. 6.5 ± 1.2 cells/0.01mm2 for HIF-1α, and 3.32 ± 1.25 vs. 0.77 ± 0.14 cells/0.01mm2 for HIF-2α; n = 3 and 4 and p<0.01 and p<0.05, respectively; Figure 5A). Increased presence of HIF-α expressing cells was also found in Foxd1-von Hippel-Lindau (Vhl)−/− mice, which are characterized by renal maturation defects similar to those observed in Foxd1-Phd2−/−-Phd3−/− mutants (Supplemental Figure S5).
Taken together our data suggest that a) HIF-1α and HIF-2α are differentially expressed in FOXD1 stroma-derived cells during kidney development, b) that FOXD1 stroma-derived cells respond differentially to PHD2 inactivation, and c) that the development of renal maturation defects is dependent on the extent of HIF activation in renal stroma, i.e. the number of cells that express HIF-α.
Nephron formation defect in Foxd1-Phd2−/−-Phd3−/− mice is dependent on HIF-2 activation
To examine to what degree HIF-1 or HIF-2 signaling contributed to defective nephron formation in Foxd1-Phd2−/−-Phd3−/− mice we generated Foxd1-Phd2−/−-Phd3−/−-Hif1a−/− and Foxd1-Phd2−/−-Phd3−/−-Hif2a−/− triple mutant mice in which PHD2 and PHD3 were inactivated together with either HIF-1α or HIF-2α {recombination for all targeted 2-lox alleles was equally efficient in Foxd1-Phd2−/−-Phd3−/−-Hif1a−/− and Foxd1-Phd2−/−-Phd3−/−-Hif2a−/− mutants (Supplemental Figure S6)}. Foxd1-Phd2−/−-Phd3−/−-Hif1a−/− mutants developed nephron formation defects similar to those seen in Foxd1-Phd2−/−-Phd3−/− mice and were characterized by small kidneys and abnormal PAS, LTL and THP staining patterns (Figure 6A and Supplemental Figure S7). Morphologic abnormalities were consistent with the reduced expression of mRNAs encoding segment-specific nephron markers (Figure 6B). Nephron formation defects in Foxd1-Phd2−/−-Phd3−/−-Hif1a−/− mutants were furthermore associated with enhanced ACTA2 staining in glomeruli and renal interstitium (Figure 6A). In contrast, Foxd1-Phd2−/−-Phd3−/−-Hif2a−/− triple mutant mice survived to adulthood, had normal-sized kidneys and did not display any developmental defects or differences in the expression of mRNAs encoding epithelial or vascular markers compared to Cre− littermate controls. However, Epo mRNA levels were severely reduced in Foxd1-Phd2−/−-Phd3−/−-Hif2a−/− kidneys, which is an expected finding as renal Epo transcription is HIF-2-dependent (Figure 6B).29 Foxd1-Cre-mediated deletion of Hif1a or Hif2a alone did not result in renal maturation defects although smaller body and kidney weight were observed in Foxd1-Hif1a−/− mice at P14 (Supplemental Figure S8). Our data demonstrate that inactivation of HIF-2α but not HIF-1α in FOXD1 stroma was sufficient to restore normal nephron formation in Foxd1-Phd2−/−-Phd3−/− mutant mice.
Figure 6. Nephron formation defects in Foxd1-Phd2−/−-Phd3−/− mice require HIF-2α.

(A) Shown are representative images of Periodic acid-Schiff (PAS)-stained kidney sections (top) and CD31-and ACTA2 IHC staining (middle and lower panels) for Cre− control (Foxd1+/+ Phd2fl/fl Phd3fl/fl Hif2afl/fl), Foxd1-Phd2−/−-Phd3−/−-Hif1a−/− and Foxd1-Phd2−/−-Phd3−/−-Hif2a−/− mutant kidneys at P14. Asterisks depict glomeruli and arrows indicate renal tubules with PAS-positive brush borders. Scale bars represent 50μm. (B) mRNA expression levels of Epo, nephron-segment-specific and vascular markers in whole kidney homogenates from Foxd1-Phd2−/−-Phd3−/−-Hif1a−/− and Foxd1-Phd2−/−-Phd3−/−-Hif2a−/− compared to Cre− controls (n=4 for Foxd1+/+ Phd2fl/fl Phd3fl/fl Hif2afl/fl control mice; n=3 for Foxd1-Phd2−/−-Phd3−/−-Hif1a−/− mutants; n=5 each for Foxd1-Phd2−/−-Phd3−/−-Hif2a−/− mice). Data are represented as mean ± SEM; 2-tailed Student’s t-test; *p<0.05, **p<0.01 and ***p<0.001. Acta 2, α-smooth muscle actin; Aqp1, aquaporin 1; Aqp2, aquaporin 2; NaPi2a, sodium-phosphate co-transporter-2a; Ncc NaCl co-transporter; Nkcc2, Na-K-2Cl co-transporter Scnn1a, sodium channel epithelial 1 alpha subunit; Trpv5, transient receptor potential cation channel subfamily V member 5.
DISCUSSION
Here we have developed several genetic models to investigate the role of individual HIF prolyl-4-hydroxylases in renal homeostasis. We demonstrate that the HIF oxygen-sensing pathway in FOXD1 stroma has a crucial function during nephrogenesis and that the ability to hydroxylate HIF-2α via PHD2 and PHD3 is required for normal nephron formation. Our data suggest that PHD2 is indispensable for normal physiologic control of interstitial HIF activity during renal development, as it functions as the main HIF prolyl-4-hydroxylase in FOXD1 stroma-derived interstitial cells, whereas PHD3 is dispensable in the presence of PHD2, but becomes rate-limiting when PHD2 catalytic activity is inhibited or absent (Figure 7). We furthermore provide evidence for a distinct and non-overlapping spatial distribution of interstitial HIF-1α and HIF-2α during development and propose that physiologic control of HIF-2 signaling in renal stroma is critically important for normal kidney development.
Figure 7. Schematic depicting the role of PHD dioxygenases in renal development.
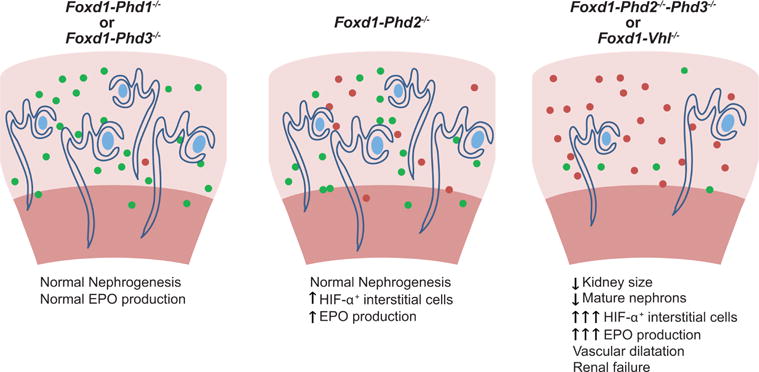
HIF-α+ stromal cells are shown as filled red circles, filled green circles depict targeted stromal cells that fail to stabilize HIF-α. Normal EPO production in Foxd1-Phd1−/− or Foxd1-Phd3−/− mutant mice is indicated by a single filled red circle.
Experimental intrauterine hypoxia has been shown to affect kidney development and postnatal renal function at multiple levels and is associated with low kidney weight at birth, low nephron endowment, reduced glomerular number and abnormal intrarenal vascular regulation.7, 30–33 Our genetic studies establish that the stromal PHD/HIF axis is a critical component of the renal hypoxia response system that regulates late nephrogenesis in a HIF-2-dependent manner. However, the developmental impact of HIF activation and the role of individual HIF transcription factors in other cell types is less clear. Whereas mice with germline HIF-1α deletion die in utero between E9.5 and E11 precluding studies of renal development,34, 35 hypoxia and wide-spread induction of HIF-1 signaling has been shown to restrict branching morphogenesis in vitro, possibly through the release of anti-branching factors.33 Genetic HIF activation in UB cells specifically increased kidney size and number of glomeruli and very modestly enhanced UB branching, whereas UB-directed HIF-1α inactivation had the opposite effect, suggesting cell type-dependent functions of renal HIF signaling during development.33 In contrast renal development was reported to be normal in a mixed-background strain of mice with germ line HIF-2α inactivation.20 Normal kidney development was also reported in a genetic model of renal epithelial HIF activation generated by Vhl gene deletion in SIX2+ epithelial progenitor cells.36 Six2-Cre-Vhl mutant mice, however, developed renal failure as adults. Whether nephron endowment was affected in this model is unclear. Furthermore, defects in renal development were not reported in endothelial cell-specific HIF knockout models.37 These studies, together with our data, support the concept of cell type-dependent roles of individual HIF transcription factors in renal organogenesis.
Our studies have implications for human biology that reach beyond gestational hypoxia as PHD inhibitors are currently clinical development for renal anemia.38 Some of the inhibiting compounds display preferential activity for certain PHDs, to what degree individual compounds have negative impact on nephrogenesis will have to be examined.21
Although the Foxd1-Phd2−/−-Phd3−/− mutant phenotype is complex and involves multiple cell types, our studies indicate that it is dependent on the extent on stromal HIF activation, i.e. the number/density of HIF-α-expressing cells in the kidney (Figure 7). Although PHD2 is the main regulator of HIF in many cell types, PHD1 and PHD3 dioxygenases play a significant role in controlling HIF activity in a context and cell type-dependent manner.3, 39–41 Our genetic data establish that PHD2 catalysis alone is sufficient for HIF-α degradation in FOXD1 stroma-derived interstitial cells, as the individual or combined inactivation of Phd1 and Phd3 alone does not result in detectable HIF-α stabilization. We also establish that PHD3, but not PHD1 (data not shown), is capable of suppressing HIF-α stabilization in a large subpopulation of Phd2−/− interstitial cells, as Foxd1-Phd2−/−-Phd3−/− mutants were characterized by a significant increase in the number of stromal cells with stabilized HIF-α compared to Foxd1-Phd2−/− mutants. Differential sensitivity of renal stroma to genetic Phd2 deletion may be due to differences in PHD3 expression levels as previously suggested,26 which may indicate that renal interstitial cells display differential responsiveness to hypoxia.39, 41
In summary, our genetic studies establish a critical role for stromal HIF oxygen sensing in nephrogenesis. Our findings provide strong rationale for further investigations into oxygen-regulated signals that control intercellular crosstalk during renal development and have implications for clinical studies that investigate the therapeutic potential of PHD inhibitors in humans.
METHODS AND MATERIALS
Generation and genotyping of mice and animal procedures
The generation and genotyping of mice with floxed alleles for Phd1 (Egln2), Phd2 (Egln1), and Phd3 (Egln3), Hif1a, and Hif2a (Epas1) has been described elsewhere.42–44 Foxd1-Phd2−/−-Phd3−/− were generated by breeding Foxd1cre/+ Phd2fl/+ Phd3fl/fl mice with Cre-negative Foxd1+/+ Phd2fl/fl Phd3fl/fl homozygous mice. Only Cre− littermates served as controls. Details regarding animal work can be found in Supplemental Methods and Materials.
RNA analysis and immunohistochemistry
Technical details can be found in Supplemental Methods and Materials.
Statistics
Data are reported as mean ± SEM. Statistical analysis were performed with Prism 6 software (GraphPad Software Inc.) using Student’s t test or 1-way ANOVA with Tukey’s post hoc analysis. The survival curve was analyzed using the Kaplan-Meier method and groups were compared by log-rank test. P-values of less than 0.05 were considered statistically significant. Study approval and ethical permits. All procedures involving mice were performed in accordance with NIH guidelines for the use and for care of live animals and were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Supplementary Material
Acknowledgments
VHH is supported by the Krick-Brooks chair in Nephrology, NIH grants R01-DK101791 and R01-DK081646, and a Department of Veterans Affairs Merit Award (1I01BX002348). Additional support was provided by Vanderbilt’s Diabetes Research and Training Center (P30-DK20593). The authors wish to thank Peter Ratcliffe and Tammie Bishop, University of Oxford, for generously providing the PM8 HIF2α antibody. Whole slide imaging was performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHORSHIP CONTRIBUTIONS
HK and VHH conceived and designed the research studies, analyzed and interpreted data, wrote the manuscript and made the figures. HK, JL, AAU, MB, KI, MR and OL performed experiments, acquired and analyzed data. SZ provided conceptual input and helped with the interpretation of data.
DISCLOSURE
The authors declare that no conflict of interest exists. Volker H. Haase serves on the Scientific Advisory Board of Akebia Therapeutics, a company that develops prolyl-4-hydroxylase inhibitors for the treatment of anemia.
References
- 1.Dunwoodie SL. The role of hypoxia in development of the Mammalian embryo. Dev Cell. 2009;17:755–773. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 3.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 4.Marsal K. Intrauterine growth restriction. Curr Opin Obstet Gynecol. 2002;14:127–135. doi: 10.1097/00001703-200204000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Rueda-Clausen CF, Morton JS, Davidge ST. Effects of hypoxia-induced intrauterine growth restriction on cardiopulmonary structure and function during adulthood. Cardiovasc Res. 2009;81:713–722. doi: 10.1093/cvr/cvn341. [DOI] [PubMed] [Google Scholar]
- 6.Moore LG, Charles SM, Julian CG. Humans at high altitude: hypoxia and fetal growth. Respir Physiol Neurobiol. 2011;178:181–190. doi: 10.1016/j.resp.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkinson LJ, Neal CS, Singh RR, et al. Renal developmental defects resulting from in utero hypoxia are associated with suppression of ureteric beta-catenin signaling. Kidney Int. 2015;87:975–983. doi: 10.1038/ki.2014.394. [DOI] [PubMed] [Google Scholar]
- 8.Keller G, Zimmer G, Mall G, et al. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–108. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- 9.Hoy WE, Hughson MD, Bertram JF, et al. Nephron number, hypertension, renal disease, and renal failure. J Am Soc Nephrol. 2005;16:2557–2564. doi: 10.1681/ASN.2005020172. [DOI] [PubMed] [Google Scholar]
- 10.Little MH, McMahon AP. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause M, Rak-Raszewska A, Pietila I, et al. Signaling during Kidney Development. Cells. 2015;4:112–132. doi: 10.3390/cells4020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatini V, Huh SO, Herzlinger D, et al. Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev. 1996;10:1467–1478. doi: 10.1101/gad.10.12.1467. [DOI] [PubMed] [Google Scholar]
- 13.Levinson RS, Batourina E, Choi C, et al. Foxd1-dependent signals control cellularity in the renal capsule, a structure required for normal renal development. Development. 2005;132:529–539. doi: 10.1242/dev.01604. [DOI] [PubMed] [Google Scholar]
- 14.Das A, Tanigawa S, Karner CM, et al. Stromal-epithelial crosstalk regulates kidney progenitor cell differentiation. Nat Cell Biol. 2013;15:1035–1044. doi: 10.1038/ncb2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez IG, Duffield JS. The FOXD1 lineage of kidney perivascular cells and myofibroblasts: functions and responses to injury. Kidney Int Suppl (2011) 2014;4:26–33. doi: 10.1038/kisup.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fetting JL, Guay JA, Karolak MJ, et al. FOXD1 promotes nephron progenitor differentiation by repressing decorin in the embryonic kidney. Development. 2014;141:17–27. doi: 10.1242/dev.089078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakagawa N, Xin C, Roach AM, et al. Dicer1 activity in the stromal compartment regulates nephron differentiation and vascular patterning during mammalian kidney organogenesis. Kidney Int. 2015;87:1125–1140. doi: 10.1038/ki.2014.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeburg PB, Robert B, St John PL, et al. Podocyte expression of hypoxia-inducible factor (HIF)-1 and HIF-2 during glomerular development. J Am Soc Nephrol. 2003;14:927–938. [Google Scholar]
- 19.Bernhardt WM, Schmitt R, Rosenberger C, et al. Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int. 2006;69:114–122. doi: 10.1038/sj.ki.5000062. [DOI] [PubMed] [Google Scholar]
- 20.Steenhard BM, Freeburg PB, Isom K, et al. Kidney development and gene expression in the HIF2alpha knockout mouse. Dev Dyn. 2007;236:1115–1125. doi: 10.1002/dvdy.21106. [DOI] [PubMed] [Google Scholar]
- 21.Haase VH. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial Int. 2017;21(Suppl 1):S110–S124. doi: 10.1111/hdi.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi A, Mugford JW, Krautzberger AM, et al. Identification of a multipotent self-renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Reports. 2014;3:650–662. doi: 10.1016/j.stemcr.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi H, Liu Q, Binns TC, et al. Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J Clin Invest. 2016;126:1926–1938. doi: 10.1172/JCI83551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartman HA, Lai HL, Patterson LT. Cessation of renal morphogenesis in mice. Dev Biol. 2007;310:379–387. doi: 10.1016/j.ydbio.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rumballe BA, Georgas KM, Combes AN, et al. Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev Biol. 2011;360:110–122. doi: 10.1016/j.ydbio.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapitsinou PP, Liu Q, Unger TL, et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. 2010;116:3039–3048. doi: 10.1182/blood-2010-02-270322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Figueroa H, Lozano M, Suazo C, et al. Intrauterine growth restriction modifies the normal gene expression in kidney from rabbit fetuses. Early Hum Dev. 2012;88:899–904. doi: 10.1016/j.earlhumdev.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Tang J, Zhu Z, Xia S, et al. Chronic hypoxia in pregnancy affected vascular tone of renal interlobar arteries in the offspring. Sci Rep. 2015;5:9723. doi: 10.1038/srep09723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez-Rodriguez P, Jr, Tong W, Xue Q, et al. Fetal hypoxia results in programming of aberrant angiotensin ii receptor expression patterns and kidney development. Int J Med Sci. 2013;10:532–538. doi: 10.7150/ijms.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schley G, Scholz H, Kraus A, et al. Hypoxia inhibits nephrogenesis through paracrine Vegfa despite the ability to enhance tubulogenesis. Kidney Int. 2015;88:1283–1292. doi: 10.1038/ki.2015.214. [DOI] [PubMed] [Google Scholar]
- 34.Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang SS, Gu YF, Wolff N, et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc Natl Acad Sci U S A. 2014;111:16538–16543. doi: 10.1073/pnas.1414789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapitsinou PP, Sano H, Michael M, et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest. 2014;124:2396–2409. doi: 10.1172/JCI69073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koury MJ, Haase VH. Anaemia in kidney disease: harnessing hypoxia responses for therapy. Nat Rev Nephrol. 2015;11:394–410. doi: 10.1038/nrneph.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 40.Berra E, Benizri E, Ginouves A, et al. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minamishima YA, Moslehi J, Padera RF, et al. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol Cell Biol. 2009;29:5729–5741. doi: 10.1128/MCB.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeda K, Ho VC, Takeda H, et al. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gruber M, Hu CJ, Johnson RS, et al. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci U S A. 2007;104:2301–2306. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryan HE, Poloni M, McNulty W, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.