

Abstract
Background & Aims
Intrahepatic cholangiocarcinoma (ICC) is a lethal malignancy without effective treatment options. MLN0128, a second-generation pan-mTOR inhibitor, shows efficacy for multiple tumor types.
Methods
We established a novel ICC mouse model via hydrodynamic transfection of activated forms of AKT (myr-AKT) and Yap (YapS127A) protooncogenes (that will be referred to as AKT/YapS127A). Genetic approaches were applied to study the requirement of mTORC1 and mTORC2 in mediating AKT/YapS127A driven tumorigenesis. Gemcitabine/Oxaliplatin and MLN0128 were administered in AKT/YapS127A tumor-bearing mice to study their antitumor efficacy in vivo. Multiple human ICC cell lines were used for in vitro experiments. Hematoxylin and eosin staining, immunohistochemistry and immunoblotting were applied for characterization and mechanistic study.
Results
Co-expression of myr-AKT and YapS127A promoted ICC development in mice. Both mTORC1 and mTORC2 complexes were required for AKT/YapS127A ICC development. Gemcitabine/Oxaliplatin had limited efficacy in treating late stage AKT/YapS127A ICC. In contrast, partial tumor regression was achieved when MLN0128 was applied in the late stage of AKT/YapS127A cholangiocarcinogenesis. Furthermore, when MLN0128 was administered in the early stage of AKT/YapS127A carcinogenesis, it led to disease stabilization. Mechanistically, MLN0128 efficiently inhibited AKT/mTOR signaling both in vivo and in vitro, inducing strong ICC cell apoptosis and only marginally affecting proliferation.
Conclusions
Altogether, our study suggests that mTOR kinase inhibitors may be beneficial for the treatment of ICC, even in tumors that are resistant to standard of care chemotherapeutics such as gemcitabine/Oxaliplatin based regimen, especially in the subset exhibiting activated AKT/mTOR cascade.
Lay Summary
We established a novel mouse model of intrahepatic cholangiocarcinoma (ICC). Using this new preclinical model, we evaluated the therapeutic potential of mTOR inhibitor MLN0128 versus Gemcitabine/Oxaliplatin (the standard chemotherapy for ICC treatment). Our study shows the anti-neoplastic potential of MLN0128, suggesting that it may be superior to Gemcitabine/Oxaliplatin based chemotherapy for the treatment of ICC, especially in the tumors exhibiting activated AKT/mTOR cascade.
Keywords: Intrahepatic cholangiocarcinoma, Dual mTOR inhibitor, Targeted therapy, Gemcitabine, Translational medicine, Mouse model, MLN0128
Graphical abstract
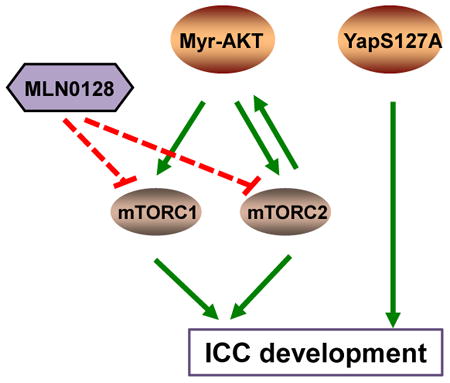
Introduction
Intrahepatic cholangiocarcinoma (ICC) is the second most common liver tumor, whose incidence is rapidly rising in the Western countries [1]. Patients with ICC are commonly diagnosed at advanced stages. This excludes them from surgical resection, the only possible curative treatment [2]. Gemcitabine/Oxaliplatin based regimen is used as a standard chemotherapy for advanced ICC, but its efficacy is rather limited [3]. Therefore, there is an urgent need to develop novel therapeutic strategies for the treatment of this lethal malignancy.
Aberrant activation of v-akt murine thymoma viral oncogene homolog (AKT)/mammalian target of rapamycin (mTOR) cascade has been found in diverse cancer types [4,5]. Evidence shows frequent activation of this pathway in both intrahepatic and extrahepatic CCA, >60% and >80% respectively [6,7]. AKT is activated by phosphoinositide (PI) 3-kinase (PI3K) through phosphorylation. Once phosphorylated, AKT activates mTOR, a key downstream effector. As the catalytic subunit, mTOR together with regulatory-associated protein of mTOR (RAPTOR) forms mTOR complex 1 (mTORC1), and with rapamycin-insensitive companion of mTOR (RICTOR) forms mTORC2 [8]. mTORC1 phosphorylates p70 ribosomal protein S6 kinase (p70 S6K) and eukaryotic initiation factor 4E binding protein 1 (4EBP1), which activates the ribosomal protein S6 (RPS6) and relieves eukaryotic initiation factor 4E (eIF4E), respectively. This leads to increased mRNA translation and protein synthesis, thus resulting in cell proliferation and growth. mTORC2 exerts its cell proliferation and survival-promoting effects through activating several kinases including AKT [9-11].
MLN0128 is a selective, highly potent and orally active mTOR inhibitor with Ki for mTOR at 1.4nM, and Ki for PI3K greater than 200nM [12]. It belongs to the second generation of mTOR inhibitors, which are ATP analogues and exert their inhibitory effects on the catalytic activity of both mTORC1 and mTORC2 by binding to the kinase domain of mTOR [4, 13]. Because of its promising pharmacological effects, MLN0128 is under intensive investigation. MLN0128 has already entered many phase I and II clinical trials as a single agent or in combined therapy, mostly for solid tumors [14]. In preclinical settings, it shows significant therapeutic effects in various tumor types including leukemia, sarcoma, breast cancer, renal cell carcinoma, and colorectal cancer [14-18]. Compared to allosteric inhibitors such as rapamycin, MLN0128 suppresses mTORC1 activity more completely, as indicated by robust and lasting inhibition of 4EBP1, and it also targets mTORC2 to inhibit AKT signaling, thus leading to superior therapeutic effect [14,15,18,19]. However, to date, no study has been conducted investigating the therapeutic effect of second generation mTOR inhibitors in ICC cell lines and animal models.
Here, we established a novel ICC mouse model via hydrodynamic transfection of activated forms of AKT and Yes-associated protein (Yap) oncogenes. Using genetic approaches, we demonstrated that both mTORC1 and mTORC2 are required for ICC development in this model. Noticeably, we found that while Gemcitabine/Oxaliplatin had limited efficacy in this model, especially in late stage ICC, MLN0128 showed significant therapeutic effects. Our study therefore suggests a role for mTOR inhibitors in treating ICC.
Materials and Methods
Human Tissue Samples
A collection of formalin-fixed, paraffin-embedded ICC (n=94) samples was used in the present study. The clinicopathological features of liver cancer patients are summarized in Supplementary Table 1. ICC specimens were collected at the Medical Universities of Greifswald (Greifswald, Germany) and Sassari (Sassari, Italy). Institutional Review Board approval was obtained at the local Ethical Committee of the Medical Universities of Greifswald and Sassari. Informed consent was obtained from all subjects.
Hydrodynamic Injection and Mouse Treatment
Wild-type (WT) FVB/N and Rictorflox/flox mice were obtained from Jackson Laboratory (Sacramento, CA). Hydrodynamic injection was performed as described [20]. To generate the ICC model, we injected 20μg pT3-EF1α-HA-myr-AKT and 30μg pT3-EF1α-YapS127A in FVB/N mice. To inhibit mTORC1 signaling, 20μg pT3-EF1 α-HAmyr-AKT-shRaptor or 20μg pT3-EF1 α-HA-myr-AKT-shLuc was injected together with 30μg pT3-EF1 α-YapS127A in FVB/N mice. To inhibit mTORC2 signaling, we injected 60μg pT3-EF1 α-Cre or 60μg pT3-EF1α (empty vector control) together with 20μg pT3-EF1α-HA-myr-AKT and 30μg pT3-EF1α-YapS127A to delete Rictor while co-expressing AKT and YapS127A in Rictorflox/flox mice. Gemcitabine (50mg/kg) plus Oxaliplatin (4mg/kg) or vehicle was intraperitoneally injected weekly for 3 weeks starting 3.5 weeks or 5.5 weeks after plasmid injection for early and late stage AKT/YapS127A ICC treatment, respectively. MLN0128 (1mg/kg/day) or vehicle was orally administered via gavage. For early stage MLN0128 therapy model, we started therapy administration 3.5 weeks post injection for 3 consecutive weeks, and mice were sacrificed 6.5 weeks after hydrodynamic injection. For late stage therapeutic model, MLN0128 or vehicle was administered in mice 5.5 weeks post injection, and mice were sacrificed after 3 days, 1, 2 or 3 weeks of therapy. Everolimus (1mg/kg/day) or vehicle was orally administered via gavage. Mice were housed, fed, and monitored according to protocols approved by the committee for animal research at the University of California San Francisco (San Francisco, CA).
Statistical analysis
Data analysis was performed with Prism 6 (GraphPad, San Diego, CA). Data are presented as Means ± SE. Comparisons between two groups were performed with two-tailed unpaired t test. P values < 0.05 were considered statistically significant.
Additional information can be found in Supplementary Methods.
Results
Concomitant upregulation of Yap and AKT in human ICC samples
First, to evaluate the importance of the Yap and AKT pathways in human cholangiocarcinogenesis, we determined the protein levels of activated/phosphorylated AKT in a collection of human ICC samples by immunohistochemistry. As a surrogate marker of Yap activation, its nuclear accumulation was investigated. We found that activated/phosphorylated levels of AKT are upregulated, when compared with nontumorous surrounding counterparts, in 68 of 94 (72.34%) ICC specimens (Fig. 1). Equivalent levels of AKT immunoreactivity in ICC and corresponding non-neoplastic livers were detected in the remaining samples. In accordance with previous findings [21,22], nuclear accumulation of Yap was almost ubiquitously detected in ICC (90/94; 95.74%), whereas only faint nuclear and/or cytoplasmic immunoreactivity of Yap was observed in surrounding non-neoplastic counterparts. Noticeably, 63 of 94 (67.02%) ICC displayed simultaneous activation of phosphorylated AKT and Yap, thus suggesting the relevance of the two concomitant oncogenic events along cholangiocarcinogenesis. No association between the status of AKT or Yap and clinicopathologic features of the patients, including age, gender, etiology, presence of cirrhosis, and tumor size was detected (Supplementary Table 2 and 3).
Figure 1. Frequent concomitant activation of AKT and Yap pathways along human cholangiocarcinogenesis.
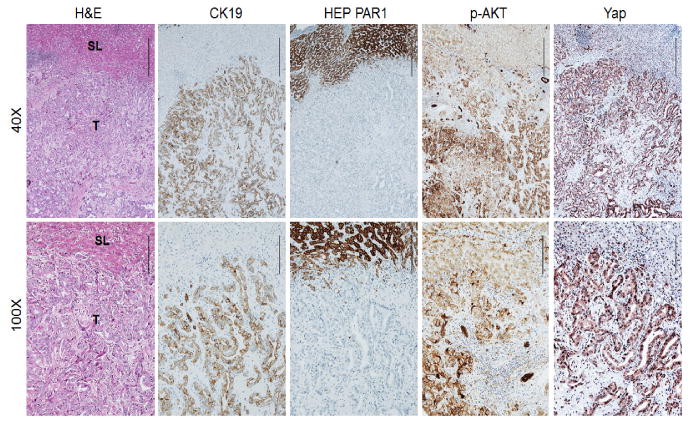
Immunohistochemical pattern of cytokeratin 19 (CK19), HEP PAR1, phosphorylated/activated (p-) AKT, and Yap in a human intrahepatic cholangiocellular carcinoma (ICC). The ICC case depicted in two magnifications (40× and 100×) shows upregulation of p-AKT in the tumor part (T) and activation of the Yap cascade (as indicated by Yap nuclear accumulation) when compared with non-tumorous surrounding liver (SL). (Scale bar: 500μm for 40×; 200μm for 100×). H&E, haematoxylin and eosin staining. CK19 and HEP PAR1 were used as biliary and hepatocellular markers, respectively.
Coordinated activation of AKT and Yap induces ICC in mice
Due to the high frequency of simultaneous activation of AKT and Yap in human ICC, we stably expressed activated forms of human AKT1 (myristoylated AKT1 or myr-AKT1, with C-terminal HA tag) and an unphosphorylatable form of Yap that cannot be targeted for proteasomal degradation by the Hippo kinase pathway (YapS127A) in the mouse liver via hydrodynamic injection (which will be referred to as AKT/YapS127A). As we previously reported, overexpression of myr-AKT alone led to hepatic steatosis and HCC formation ∼30 weeks post injection [23]. Overexpression of YapS127A did not result in any liver abnormality [21]. In contrast, when mice were injected with AKT/YapS127A, as early as 3 weeks post injection, tumor lesions could be observed macroscopically and histologically. Tumors grew rapidly and all mice became moribund, requiring to be euthanized by 5.5 to 7.5 weeks post injection (Supplementary Fig. 1A). Extra-hepatic metastases were not observed in these mice, presumably due to the rapid progression of liver tumors leading to mouse mortality. Histological analysis showed that the tumors displayed a ductular phenotype, resembling human ICC. Consistently, all tumor cells were positive for CK19, a bile duct marker (Fig. 2A). Furthermore, AKT/YapS127A tumors exhibited positive staining for markers of tissue desmoplasia, such as vimentin and smooth muscle actin (α -SMA) in the stromal cells, a prominent feature of human ICC, which was further proven by Sirius Red staining (Supplementary Fig. 1B). Western blotting was performed to determine the activation of AKT and Yap signaling in wild-type livers and AKT/YapS127A ICC lesions. As expected, total AKT and phosphorylated AKT at S473 and T308 sites as well as key downstream components of mTORC1 and mTORC2 signaling (p-mTOR, p-PRS6, p-4E-BP1, p-PRAS40 and p-NDRG1) were elevated in AKT/YapS127A mouse cancer tissues. Levels of Yap and its downstream target Jag1 were also increased in AKT/YapS127A tissues. Ras/MAPK pathway was activated, as shown by increased level of p-ERK. Furthermore, we detected high levels of Hexokinase 1/2 (HK1/2) and pyruvate kinase M1/M2 (PKM1/2), thus indicating increased glycolysis, while increased survivin suggested a stronger tendency to survive of AKT/YapS127A tumors (Fig. 2B). In summary, co-expression of activated AKT and Yap induces ICC development in mice.
Figure 2. Coordinated activation of AKT and Yap induces ICC in mice.
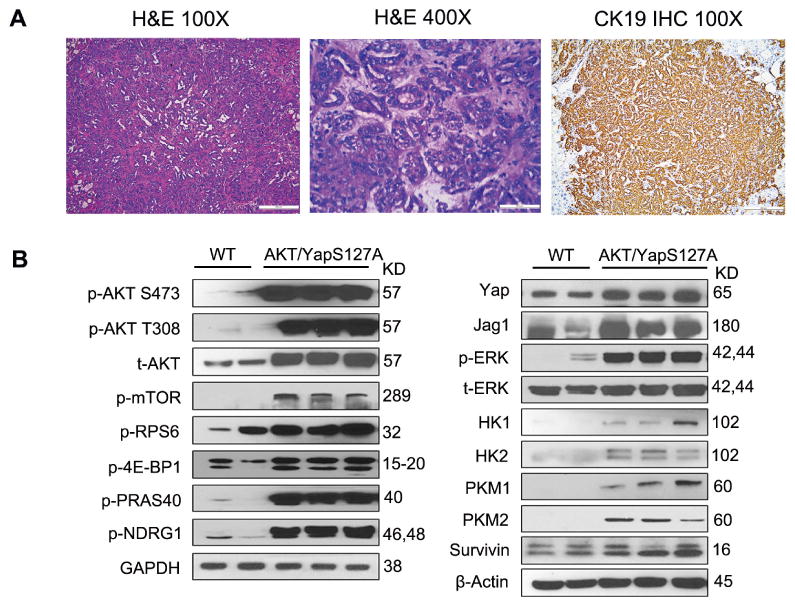
(A) HE and CK19 staining in mouse liver with lethal burden of ICC after hydrodynamic injection of AKT and YapS127A. (Scale bar: 200μm for 100×; 50μm for 400×). (B) Representative immunoblotting in wild-type (WT) and AKT/YapS127A liver tissues. GAPDH and β-Actin were used as loading controls. IHC, immunohistochemistry; t, total; p, phosphorylated.
mTORC1 signaling is required for AKT/YapS127A-induced cholangiocarcinogenesis
To investigate whether mTORC1 signaling is necessary for ICC development induced by AKT/YapS127A, we constructed AKT-shRaptor plasmid and control AKT-shLuc (shRNA against Renilla Luciferase) plasmid [24], which were hydrodynamically co-injected with YapS127A into mice (AKT-shRaptor/YapS127A or AKTshLuc/YapS127A) (Fig. 3A). Silencing of Raptor by shRNA substantially delayed the development of ICC by AKT/YapS127A expression in mice. Indeed, while control AKTshLuc/YapS127A mice developed lethal tumor burden by 6.5 weeks post injection, only few ICC lesions occurred in AKT-shRaptor/YapS127A mice at this time point (Fig. 3B). Even as late as 13 weeks post injection, the tumor burden as indicated by liver weight in AKT-shRaptor/YapS127A mice was still significantly lagging behind, and the survival was greatly improved (Fig. 3C). This suggests that mTORC1 is required for AKT/YapS127A-induced cholangiocarcinogenesis. Consistently, clonogenic capacity of the HuCCT1 and KMCH human ICC cell lines was significantly suppressed after inhibiting Raptor via lentivirus transfection of shRNA (Supplementary Fig. 2). Altogether, our data indicate that mTORC1 signaling plays a key role in cholangiocarcinogenesis.
Figure 3. mTORC1 signaling is required for AKT/YapS127A-induced cholangiocarcinogenesis.
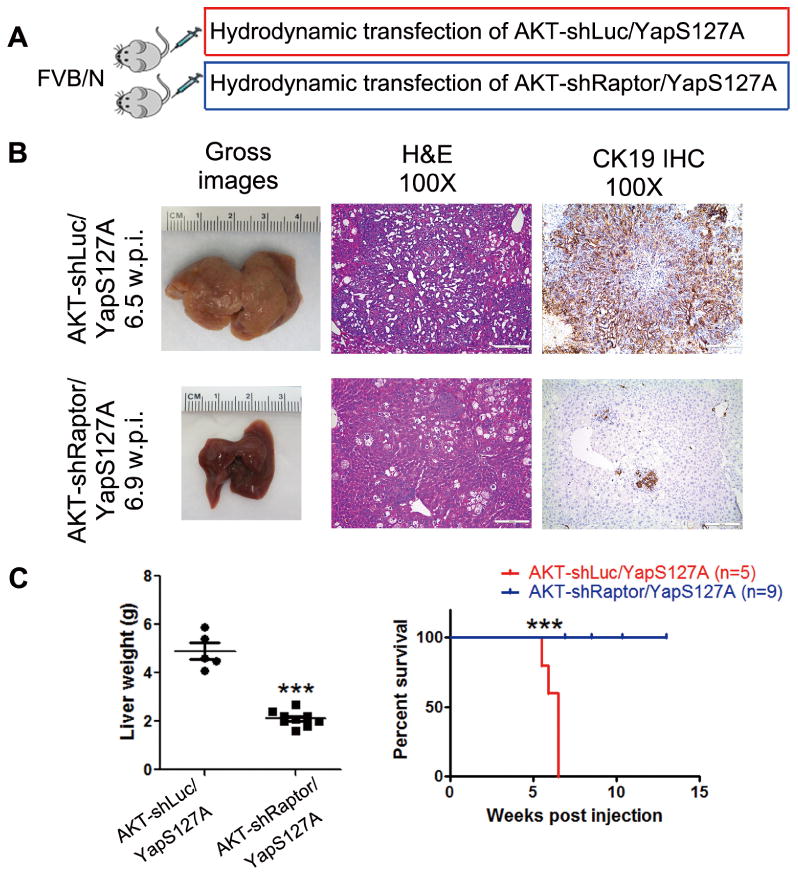
(A) Study design. (B) Gross images, H&E and CK19 staining of AKT-shLuc/YapS127A and AKT-shRaptor/YapS127A mouse livers at 6.5 weeks and 6.9 weeks post hydrodynamic injection respectively. (Scale bar: 200μm). (C) Liver weight and survival curve of AKT-shLuc/YapS127A and AKT-shRaptor/YapS127A mice. *** P < 0.001; w.p.i., weeks post injection.
mTORC2 is required for AKT/YapS127A-induced cholangiocarcinogenesis
Subsequently, we investigated whether mTORC2 signaling is required for ICC development induced by AKT/YapS127A as well. Thus, we hydrodynamically injected pT3-EF1α (empty vector) or pT3-EF1α-Cre together with AKT/YapS127A in Rictorflox/flox mice (which will be referred to as AKT/YapS127A/pT3 and AKT/YapS127A/Cre mice, respectively) (Fig. 4A). Co-injection of Cre with AKT/YapS127A plasmids into Rictorflox/flox mice allowed the expression of AKT/YapS127A oncogenes in Rictor deleted cells (Fig. 4A). Starting from 4.9 weeks post injection, AKT/YapS127A/pT3 mice developed lethal burden of ICC. In striking contrast, no tumor lesions occurred in AKT/YapS127A/Cre mice at this time point, and bulky tumors only developed after 14 weeks post injection. Importantly, histological evidence indicated that AKT/YapS127A/Cre mice developed hepatocellular lesions instead of ICC (Fig. 4B) and the survival was significantly improved (Fig. 4C). Consistently, CK19 staining demonstrated that few ICC lesions could be observed in AKT/YapS127A/Cre Rictorflox/flox mouse cancer tissues (Fig. 4B). These findings indicate that, similar to mTORC1, mTORC2 is also required for AKT/YapS127A-induced cholangiocarcinogenesis. Western blotting showed that, compared with AKT/YapS127A/pT3 mice, Rictor was effectively knocked down and p-AKT expression was suppressed in AKT/YapS127A/Cre mice (Fig. 4D). Consistently, clonogenic capacity of HuCCT1 and KMCH human ICC cells was impaired after inhibiting Rictor via lentivirus transfection of shRictor (Supplementary Fig. 3). Overall, our results suggest that mTORC2 signaling also plays an important role in AKT/YapS127A-driven cholangiocarcinogenesis.
Figure 4. mTORC2 signaling is required for AKT/YapS127A-induced cholangiocarcinogenesis.
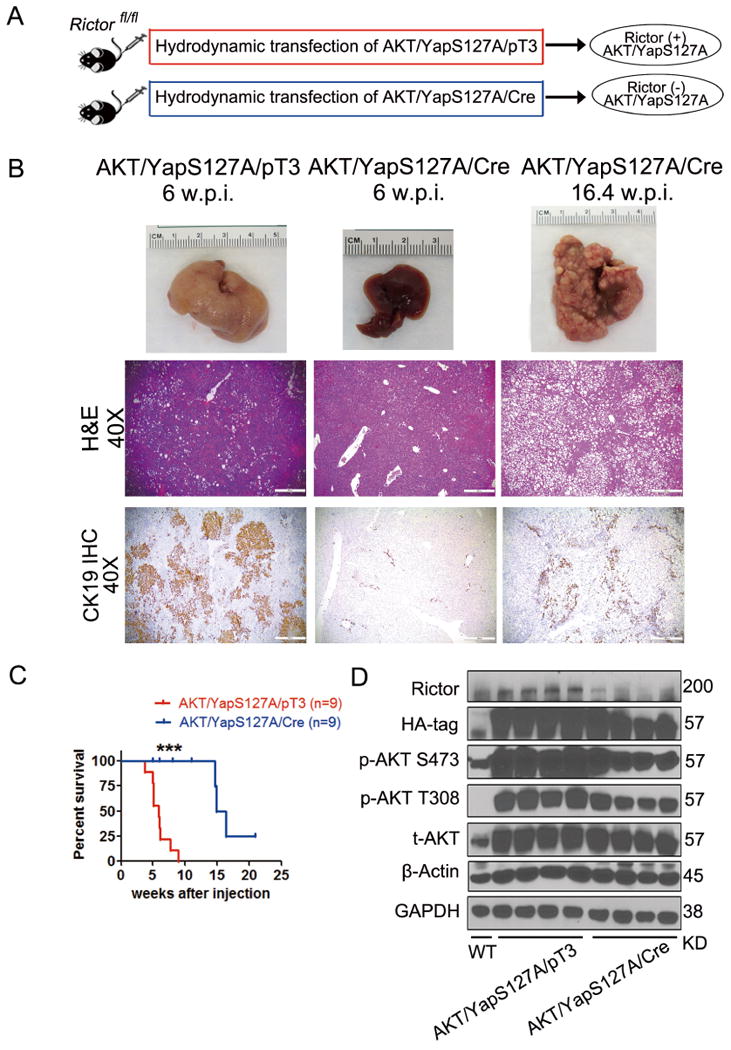
(A) Study design. (B) Gross images, H&E and CK19 staining of AKT/YapS127A/pT3 and AKT/YapS127A/Cre Rictorflox/flox mouse livers at indicated time points. (Scale bar: 500μm). (C) Survival curve of AKT/YapS127A/pT3 and AKT/YapS127A/Cre Rictorflox/flox mice. ***P < 0.001. (D) Representative immunoblotting in wild-type (WT), AKT/YapS127A/pT3 and AKT/YapS127A/Cre liver tissues. GAPDH and β-Actin were used as loading controls.
Therapeutic efficacy of Gemcitabine/Oxaliplatin in the treatment of AKT/YapS127A ICC
Since Gemcitabine/Oxaliplatin regimen is the standard chemotherapy for advanced ICC [3], we first tested the therapeutic effect of Gemcitabine plus Oxaliplatin in the AKT/YapS127A ICC model. We started the therapy 3.5 weeks post injection when the tumor nodules start to emerge (Supplementary Fig. 1). Within 5.2 to 6.5 weeks post injection, all vehicle-treated mice (n=5) developed lethal burden of tumor and had to be euthanized, while the Gemcitabine/Oxaliplatin-treated mice were in good conditions. The ICC burden was significantly decreased in Gemcitabine/Oxaliplatin-treated mice than that in vehicle-treated mice based on liver weight and mouse survival (Supplementary Fig. 4B and C). Histological examination confirmed the results, indicating that Gemcitabine/Oxaliplatin is effective against early stage AKT/YapS127A ICC. Since most ICC patients are diagnosed at late stage [2], we next determined the efficacy of this treatment for advanced ICC in AKT/YapS127A mice. Administration of Gemcitabine plus Oxaliplatin was started 5.5 weeks post injection, when mice have moderate tumor burden, for a total of 3 weeks (Supplementary Fig. 5A). ICC continued to grow, although at a slower pace (Supplementary Fig. 5B and C), and mouse survival was prolonged by ∼2 weeks upon chemotherapy (Supplementary Fig. 5C). Thus, Gemcitabine/Oxaliplatin has relatively mild efficacy in treating late stage AKT/YapS127A ICC.
MLN0128 inhibits ICC cell growth in vitro
Since both mTORC1 and mTORC2 cascades are crucial for AKT/YapS127A induced cholangiocarcinogenesis (Fig. 3 and 4), we investigated the therapeutic effect of the second generation mTOR inhibitor MLN0128 in antagonizing ICC development. We screened 8 human CCA cell lines and selected out 6 cell lines with high expression of both mTORC1 (p-RPS6 and p-4EBP1) and mTORC2 (p-AKT) cascades (Fig. 5A). Next, we determined the in vitro effect of MLN0128 and treated CCA cells with MLN0128 for 48 hours. MLN0128 inhibited the growth of all 6 cell lines at nanomolar concentrations (Fig. 5B). At the biochemical level, as fast as 2 hours after MLN0128 treatment, AKT/mTOR signaling was suppressed, as indicated by decreased expression of p-AKT, p-mTOR, p-RPS6 and p-4E-BP1 in ICC cells (Fig. 5C). While MLN0128 could suppress the expression of Cyclin D1/E in the KMCH cell line, it had no effect on the expression of other proliferation-associated proteins, including PCNA and Cyclin A/B1/D1/E, in HuCCT1 cells. However, the expression of cleaved caspase-3/7 and PARP, well known markers of apoptosis induction, were elevated in both cell lines starting 24 hours after MLN0128 treatment (Fig. 5C). Pretreatment of the cells with the pan-Caspase inhibitor V-ZAD-FMK blunted MLN0128 induced apoptosis (Supplementary Fig. 6). Altogether, our data suggest that MLN0128 effectively inhibits ICC cell growth, mainly due to induction of apoptosis.
Figure 5. MLN0128 inhibits ICC cell growth in vitro.
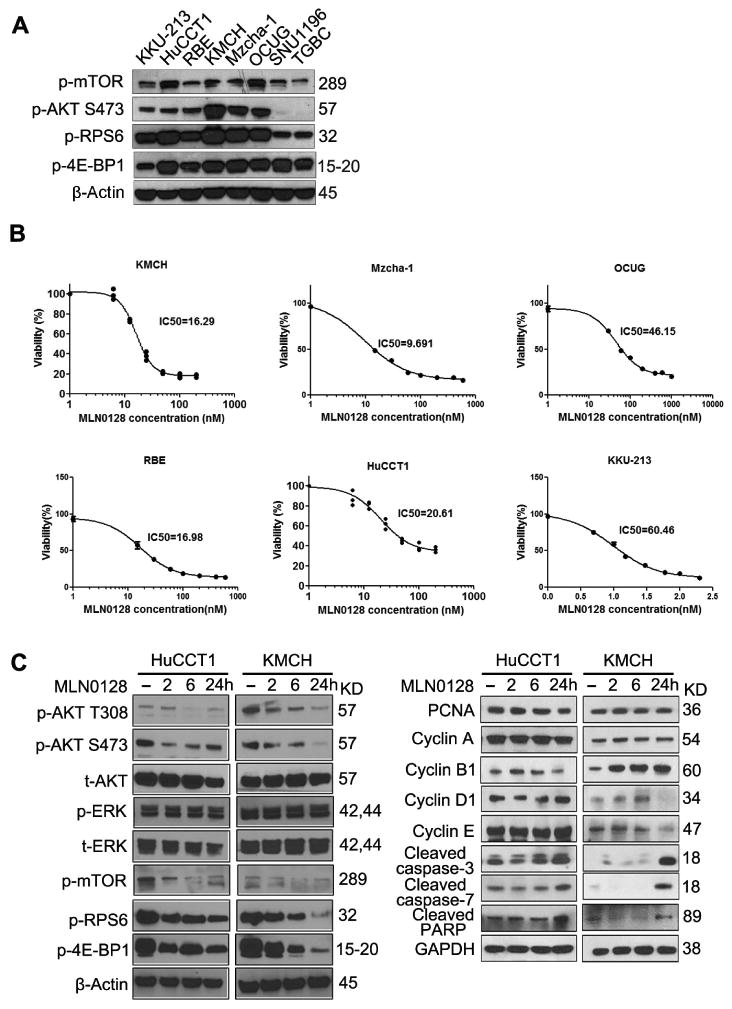
(A) The expression of key components in both mTORC1/2 cascades was detected in 8 CCA cell lines by western blotting for screening. (B) 6 cell lines selected out with high mTORC1/2 cascades were treated with escalating concentrations of MLN0128, and IC50 was determined after 48 hours treatment. (C) Representative immunoblotting in HuCCT1 and KMCH cells treated with vehicle or IC50 concentration of MLN0128 for different time points. GAPDH and β-Actin were used as loading controls.
MLN0128 leads to stable disease in early stage AKT/Yap127A ICC mice
Next, the therapeutic efficacy of MLN0128 in vivo was investigated. We treated AKT/YapS127A mice with MLN0128 at 1mg/kg/day according to previous studies [12,14,17,18,25]. Our experiment confirmed that mTOR signaling was effectively suppressed after 5 days of MLN0128 administration, with no obvious toxicity (Supplementary Fig. 7). We first started the MLN0128 therapy in early stage of ICC development, namely at 3.5 weeks post injection. Vehicle or MLN0128 was orally administered by gavage to the mice (Fig. 6A). Lethal tumor burden occurred 6.5 weeks post injection in vehicle-treated mice, when MLN0128-treated mice appeared instead healthy with normal abdominal size (Fig. 6B). ICC burden in MLN0128-treated mice was significantly smaller than that in vehicle-treated mice as shown by gross and histological evidence, and by liver weight (Fig. 6C and D). MLN0128 administration significantly improved AKT/YapS127A mouse survival (Fig. 6E). CK19 staining also provided consistent evidence (Fig. 6F). Importantly, MLN0128 showed significantly superior efficacy to that of Gemcitabine/Oxaliplatin in early stage AKT/YapS127A ICC when comparing liver weight data, as the measurement of tumor burden (Supplementary Fig. 4D). Histological analysis revealed that AKT/YapS127A mice after MLN0128 treatment for 3 weeks had a similar tumor burden to AKT/YapS127A mice at 3.5 weeks post injection, the time point when MLN0128 therapy started. In addition, there was no significant difference in liver weight between the two groups (P = 0.518; Supplementary Fig. 8), indicating that MLN0128 treatment leads to stable disease when applied to early stage tumors. Unexpectedly, tumor cells still were highly proliferative upon MLN0128 treatment, as shown by Ki-67 staining, with only mild decrease of proliferation index from vehicle treated tumors (Fig. 6F). In striking contrast, MLN0128 administration induced massive apoptosis in AKT/YapS127A ICC cells, as detected by TUNEL assay. Specifically, apoptosis index was ∼5 times higher in MLN0128 treated tumor samples when compared with the vehicle treated group (Fig. 6F).
Figure 6. MLN0128 significantly inhibits tumor growth in early stage AKT/YapS127A ICC.
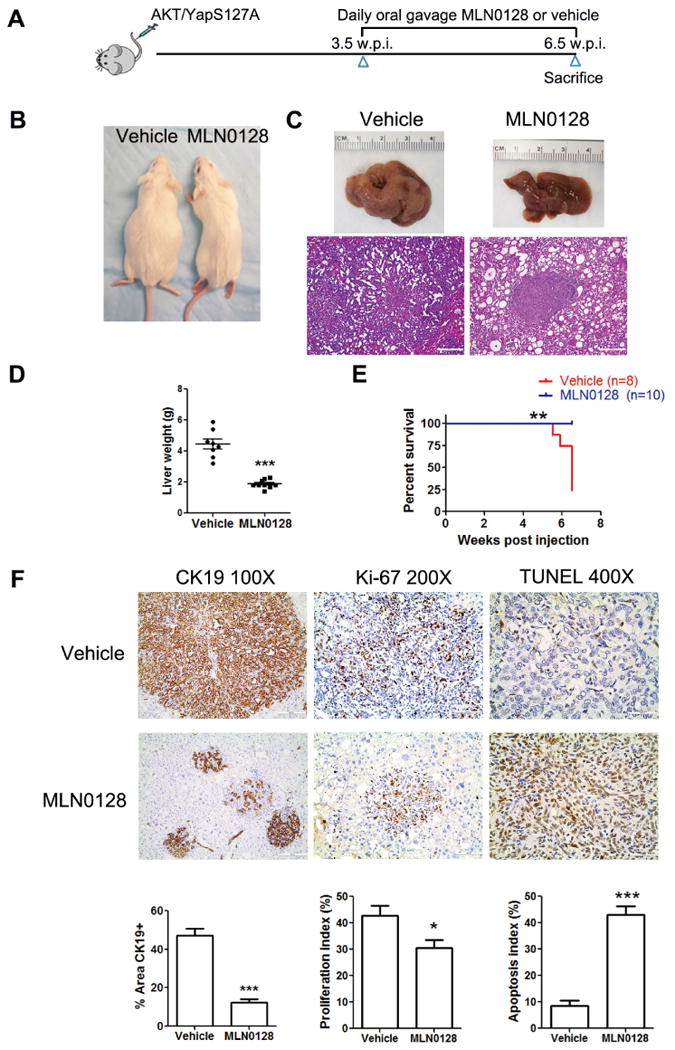
(A) Scheme of the experiment. (B) Phenotype of AKT/YapS127A ICC mice 6.5 w.p.i. after 3 weeks treatment with either vehicle or MLN0128. (C) Gross images and H&E staining of livers from vehicle- and MLN0128-treated AKT/YapS127A mice. (Magnification: 100×, Scale bar: 200μm). (D) Liver weight of vehicle- and MLN0128-treated AKT/YapS127A mice. ***P < 0.001. (E) Survival curve of AKT/YapS127A mice treated with vehicle or MLN0128. **P < 0.01. (F) CK19, Ki-67 and TUNEL staining in livers from AKT/YapS127A mice with either vehicle or MLN0128 treatment. Representative staining images were shown in the upper panel. CK19 IHC staining was quantified and demonstrated as the percentage of positive staining area of the whole area. Ki-67 and TUNEL positive cells were counted and quantified as proliferation and apoptosis index. Quantified data are presented as mean ± SE in lower panel. *P < 0.05; ***P < 0.001.
To further investigate the molecular mechanisms underlying therapeutic efficacy of MLN0128, we evaluated the protein expression of major signaling pathways in vehicle or MLN0128 treated tumor samples. We first used tumor samples treated with vehicle or MLN0128 harvested after 3 weeks of treatment. However, because of the significant differences of tumor burden (Fig. 6), we found difficult to determine whether the changes were due to tumor burden difference or changes in signaling pathways (Supplementary Fig. 9). We therefore chose to use ICC tumor samples from mice at 3.5 weeks post injection and compare them with tumor harvested after 3 weeks of MLN0128 therapy, as we have shown that tumor burdens are similar in the pre-treatment and MLN0128 treated cohorts (Supplementary Fig. 8). Indeed, HA tag represented the expression of transfected AKT, and there was no difference in HA expression, indicating equal tumor burden from the two groups of ICC tissues.
We found that MLN0128 could effectively inhibit AKT/mTOR signaling, while had no influence on Yap and Jag1 expression. There was no difference in p-ERK expression between the two groups, suggesting that no feedback activation of the Ras/MAPK pathway occurs after AKT signaling inhibition by MLN0128 (Fig. 7A and Supplementary Fig. 10A). Consistent with in vitro findings, MLN0128 treatment had no effects on cell proliferation as indicated by equal expression level of PCNA and Cyclin A/B1/D1/E in tissues from the two groups (Fig. 7B and Supplementary Fig. 10B). The expressions of cleaved caspase-3/7 increased in ICC after MLN0128 treatment, indicating induction of apoptosis by MLN0128 (Fig. 7C and Supplementary Fig. 10C). Next, we investigated the mechanisms underlying the increased apoptosis. The expression of major survival and apoptosis related genes, including Survivin, MCL-1, BCL-XL, BIM and BCL-2 did not show significant difference in control and MLN0128 treated tumor samples (Fig. 7C and Supplementary Fig. 10C). BAD proapoptotic protein was mildly upregulated in MLN0128 samples (Fig. 7C and Supplementary Fig. 10C). Importantly, ER stress markers, including Bip and CHOP, were highly upregulated in MLN0128 treated ICC tumor samples (Fig. 7C and Supplementary Fig. 10C). Overall, the data suggest that MLN0128 achieves disease control in early stage AKT/YapS127A ICC by triggering apoptosis, most likely via inducing ER stress.
Figure 7. MLN0128 suppresses both mTORC1 and mTORC2 signaling, leading to apoptosis in AKT/YapS127A ICC.
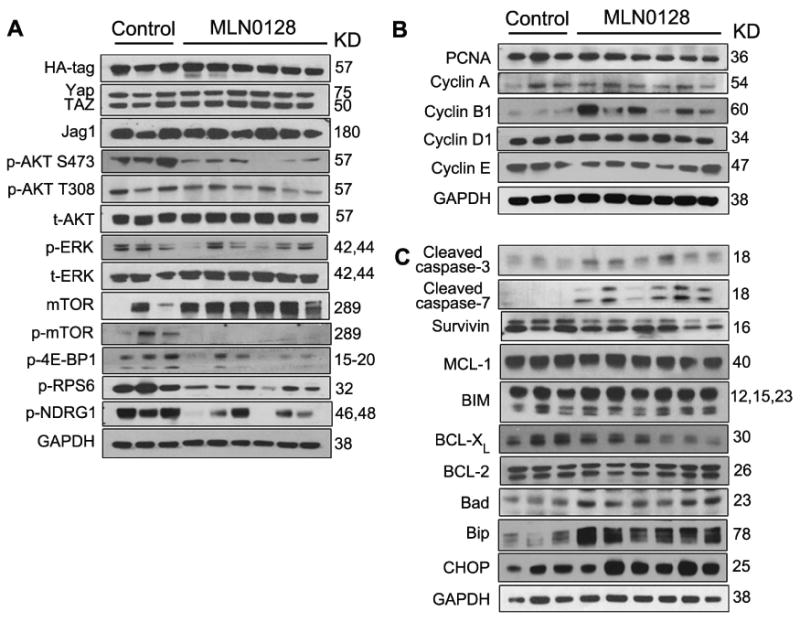
Western blotting was performed to analyze mTORC1 and mTORC2 signaling (A), proliferation (B) and apoptosis (C) in ICC tissues from control AKT/YapS127A mice 3.5 w.p.i and AKT/YapS127A mice 6.5 w.p.i after 3 weeks of MLN0128 treatment.
MLN0128 leads to tumor regression in late stage AKT/YapS127A ICC
Next, we evaluated the efficacy of MLN0128 in the treatment of advanced AKT/YapS127A ICC. MLN0128 administration was started 5.5 weeks post injection. Mice were euthanized 3 days, 1, 2 or 3 weeks after MLN0128 administration (Fig. 8A). During the first 3 days, a striking shrinkage of the mouse abdomen was observed (Supplementary Fig. 11A). The liver burden and liver weight gradually reduced after 1, 2 or 3 weeks of MLN0128 therapy (Fig. 8B and Supplementary Fig. 11B). MLN0128 treatment greatly improved survival, with mice at 8.5 weeks post injection being in good conditions (Fig. 8B), although low to moderate tumor burden could still be observed (Fig. 8B and Supplementary Fig. 11B). Consistent with the observation in early stage ICC treatment, MLN0128 had limited influence on cancer cell proliferation as indicated by Ki-67 staining, but promoted massive apoptosis, as shown by TUNEL assay (Fig. 8C and D). Overall, the data demonstrate that MLN0128 induces cancer cell apoptosis and leads to partial tumor regression in late stage AKT/YapS127A ICC mice.
Figure 8. MLN0128 leads to tumor regression in late stage AKT/YapS127A ICC mice.
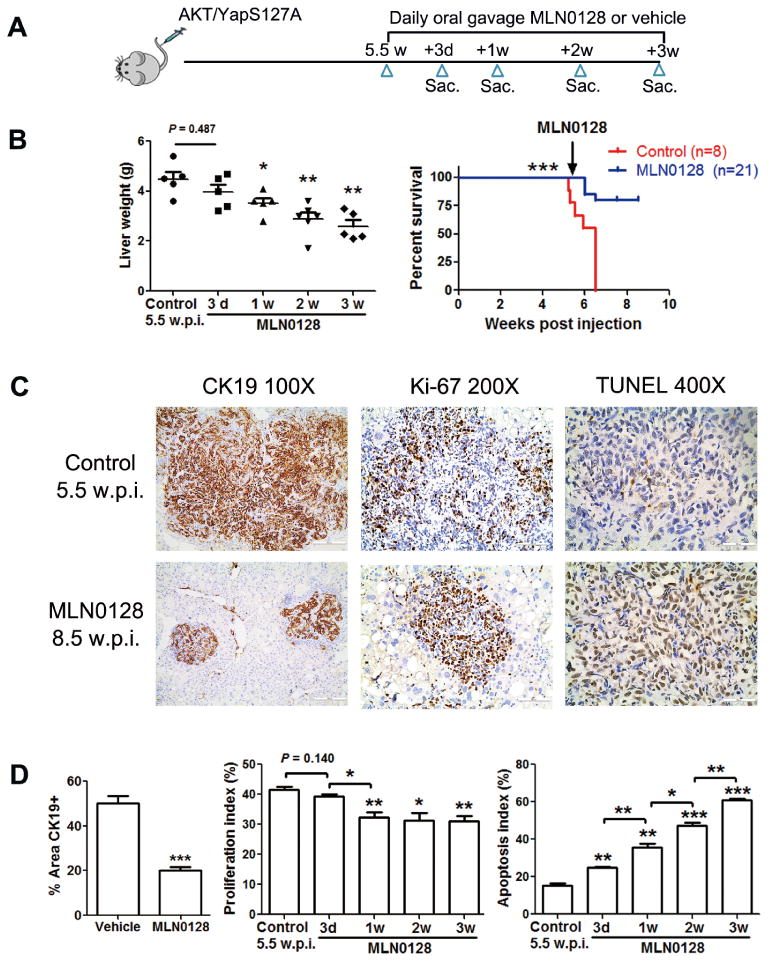
(A) Study design. (B) Liver weight of control AKT/YapS127A mice 5.5 w.p.i. and MLN0128-treated AKT/YapS127A mice at different time points. Survival curve of AKT/YapS127A mice treated with vehicle or MLN0128. (C) CK19, Ki-67, and TUNEL staining of livers from control AKT/YapS127A mice 5.5 w.p.i and AKT/YapS127A mice 8.5 w.p.i. after 3 weeks of MLN0128 treatment. (Scale bar: 200μm for 100×; 100μm for 200×; 50μm for 400×). (D) CK19 staining was quantified and demonstrated as the percentage of positive staining area of whole area. Ki-67 and TUNEL positive cells from indicated liver tissues were counted and quantified as proliferation and apoptosis index. Data are presented as mean ± SE. *P < 0.05; **P < 0.01; ***P < 0.001. Sac, sacrifice.
Discussion
ICC is a deadly malignancy with limited treatment options. Murine models are critical to study the molecular mechanisms of tumorigenesis and to evaluate drug therapeutic potential. However, ICC mouse models are still limited and difficult to generate, which limits the use of these preclinical models. Here, we show that hydrodynamic transfection of activated forms of AKT (myr-AKT) and Yap (YapS127A) into the mouse liver rapidly induces ICC formation. Yamada et al. established an ICC murine model by intrabiliary transduction of AKT and Yap that was coupled with bile duct ligation followed by IL-33 administration (AKT/Yap/IL-33) [26]. Histological analyses indicated that the two ICC models are highly similar morphologically (Supplementary Fig. 12A). We performed microarray analysis of AKT/YapS127A ICC and normal liver, and identified 1320 and 799 mRNAs that were upregulated and downregulated, respectively, in AKT/YapS127A ICC. We compared the differentially regulated genes in AKT/YapS127A ICC with those identified in AKT/Yap/IL-33 ICC, and found that up- and down-regulated genes overlapped significantly in the two datasets (p < 1E-200 for both up- and down-regulated genes) (Supplementary Fig. 12B). Thus, the myr-AKT/YapS127A mouse model represents a reliable and reproducible system to study ICC pathogenesis in vivo.
It is important to note that we applied hydrodynamic transfection of AKT and YapS127A plasmids to induce ICC in mice. Hydrodynamic injection delivers gene(s) into hepatocytes around the pericentral region of the liver [20,27]. Lineage tracing experiment demonstrated that AKT/YapS127A ICCs derived from hepatocytes (Wang J, unpublished data). Recent studies suggest that hepatocytes can reprogram into BECs in various chronic liver diseases, such as chronic HCV infection [28]. In Western and East Asian countries, where biliary tract inflammation is rare, HBV and HCV infection and alcohol abuse are the major risk factors for ICC, as for HCC [29]. Thus, ICC derived from hepatocytes might be a highly relevant model to study human cholangiocarcinogenesis, especially in the context of chronic liver injury where hepatocytes are presumably the targets of transformation. The precise mechanisms whereby activated AKT/mTOR and Yap pathways synergize to promote ICC development remain to be determined. It has been reported that Yap activation induces ductal fate differentiation in mature hepatocytes [30]. We hypothesize that YapS127A induces the de-differentiation of hepatocytes towards biliary cell lineage, and AKT/mTOR signaling promotes malignant transformation to eventually form ICC. Clearly, additional experiments are required to elucidate the functional contribution of AKT/mTOR and Yap signaling in cholangiocarcinogenesis.
Our genetic studies demonstrated that both mTORC1 and mTORC2 cascades are required for AKT/YapS127A driven ICC development. As mTORC1 is a major cascade downstream of AKT, as expected, silencing of mTORC1 strongly inhibited AKT/YapS127A induced tumorigenesis. Interestingly, ablation of mTORC2 inhibited AKT/S127A ICC development, leading instead to HCC formation in the Rictor KO genetic background. Our preliminary data suggest that Notch signaling, a key determinant of ICC development, is suppressed by the loss of mTORC2 (Xu M, unpublished data). It would be important to further investigate the biochemical crosstalk between mTORC2 and Notch cascades and how they differentially regulate ICC and HCC development.
Using AKT/YapS127A mice as preclinical model, we tested the therapeutic efficacy of multiple drugs, including Gemcitabine/Oxaliplatin, MLN0128, and Everolimus. Everolimus, a Rapalogue, has been widely tested in clinical trials for cancer treatment, with unsatisfactory results. This is likely due to the fact that Everolimus is a partial mTORC1 inhibitor and has no activity against mTORC2. Indeed, when we tested Everolimus for the treatment of AKT/YapS127A ICC, we found that it has rather limited efficacy (Supplementary Fig. 13). Currently, multiple efforts are focusing on the development of more efficient mTOR inhibitors, including pan-mTOR inhibitors or dual PI3K/mTOR inhibitors, for cancer therapeutics. Because our genetic studies suggest that both mTORC1 and mTORC2 are required for cholangiocarcinogenesis, we chose to test the therapeutic efficacy of the dual mTOR inhibitor MLN0128, which has not been tested in ICC. Here, we show that MLN0128 efficiently inhibits the growth of a panel of human ICC cells with high AKT/mTOR activity, mainly via inducing apoptosis. Furthermore, we found that MLN0128 leads to stable disease in AKT/YapS127A mice when administered in early stage of tumorigenesis and partial regression when given at late stage. Importantly, MLN0128 exhibited superior therapeutic efficacy than Gemcitabine/Oxaliplatin and Everolimus in the treatment of late stage AKT/YapS127A ICC (Supplementary Fig. 13B). Mechanistically, we demonstrated that the in vivo anti-tumor activity of MLN0128 is mainly due to apoptosis induction. Further evidence indicates that MLN0128 promotes ER stress in AKT/YapS127A ICC cells, resulting in the expression of the pro-apoptotic gene CHOP. The role of ER stress in the induction of apoptosis by mTOR inhibitors has been recently shown also in colon cancer cells, where DR5 was reported to be the potential downstream target of CHOP [31]. Differently from this study, however, no changes in DR5 mRNA expression were detected in MLN0128 treated AKT/YapS127A ICC (Supplementary Fig. 10D). ER stress is induced by several types of stressors, leading to unfolded protein response (UPR). ER stress/UPR has three different arms, one controlled by PERK, another by IRE1α, and a third by ATF6 [32]. It would be important to identify the arm of ER Stress/UPR modulated by MLN0128 in ICC tumor cells and mechanisms contributing to MLN0128 induced apoptosis.
Altogether, our in vitro and in vivo data support further investigation on MLN0128 and other pan-mTOR inhibitors for ICC treatment. Additional studies using patient derived xenograft (PDX) models will be of high importance. Furthermore, combined therapy of MLN0128 with other drugs should be evaluated to establish the usefulness of mTOR inhibitors for the treatment of human ICC.
Supplementary Material
Highlights.
A novel preclinical mouse model of intrahepatic cholangiocarcinoma (ICC) was established via hydrodynamic transfection of activated forms of AKT and Yap, two protooncogenes frequently activated in human ICC.
This model recapitulates many morphological and molecular features of human ICC.
The anti-neoplastic potential of MLN0128 may be superior to Gemcitabine/Oxaliplatin based chemotherapy for the treatment of ICC, especially in the ICCs exhibiting activated AKT/mTOR cascade. Strong induction of apoptosis was identified as a major mechanism triggering growth restraint of ICC cells.
Acknowledgments
Financial Support statement: This work is supported by NIH grants R01CA136606 and R01CA190606 to XC; P30DK026743 for UCSF Liver Center; Grants from National Natural Science Foundation of China (Grant No. 81402026, 81472278, 81521091, 31360220, 81602424); Grant from Science and Technology Commission of Shanghai Municipality (Grant No. 14ZR1409200); Scholarship from China Scholarship Council (contract 201406580003, 201506350124, 201408525080); Grant from the Deutsche Forschungsgemeinschaft DFG (Grant No. RI2695/1-1) to SR; JDG is supported by an American Cancer Society Postdoctoral Fellowship; and grant from the Italian Association Against Cancer (AIRC; grant number IG 18737) to GG.
List of Abbreviations
- ICC
intrahepatic cholangiocarcinoma
- AKT
v-akt murine thymoma viral oncogene homolog
- mTOR
mammalian target of rapamycin
- RAPTOR
regulatory-associated protein of mTOR
- mTORC1
mTOR complex 1
- RICTOR
rapamycin-insensitive companion of mTOR
- mTORC2
mTOR complex 2
- Yap
Yes-associated protein
- WT
Wild-type
- IHC
immunohistochemistry
- W.P.I
weeks post injection
- Sac
sacrifice
Footnotes
Shanshan Zhang, Xinhua Song, Dan Cao and Zhong Xu contributed equally to this work.
Conflict of interest statement: none.
Author Contributions: Shanshan Zhang, Xinhua Song, Dan Cao, Zhong Xu, Junjie Hu, Biao Fan, Bin Chen, Mingjie Dong, Maria G Pilo, Antonio Cigliano, Silvia Ribback, Katja Evert, Frank Dombrowski, Antonio Cossu, Alberto Porcu, Gianpaolo Vidili, Rosa M Pascale, Maria M. Simile, Giovanni M. Pes, Gianluigi Giannelli, and Matthias Evert acquired experimental data. Li Che provided administrative, technical, or material support. John Gordan and Lixin Wei assisted in study design and interpretation. Shanshan Zhang drafted the manuscript. Xin Chen, Diego F. Calvisi and Wenming Cong were involved in study design, drafting of the manuscript, study supervision and obtaining funding.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Author names in bold designate shared co-first authorship.
- 1.Bridgewater J, Galle PR, Khan SA, Llovet JM, Park JW, Patel T, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. Journal of hepatology. 2014;60:1268–1289. doi: 10.1016/j.jhep.2014.01.021. [DOI] [PubMed] [Google Scholar]
- 2.Benson AB, 3rd, D'Angelica MI, Abrams TA, Are C, Bloomston PM, Chang DT, et al. Hepatobiliary cancers, version 2.2014. Journal of the National Comprehensive Cancer Network: JNCCN. 2014;12:1152–1182. doi: 10.6004/jnccn.2014.0112. [DOI] [PubMed] [Google Scholar]
- 3.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polivka J, Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacology & therapeutics. 2014;142:164–175. doi: 10.1016/j.pharmthera.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature reviews Drug discovery. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz KJ, Lang H, Wohlschlaeger J, Sotiropoulos GC, Reis H, Schmid KW, et al. AKT and ERK1/2 signaling in intrahepatic cholangiocarcinoma. World journal of gastroenterology. 2007;13:6470–6477. doi: 10.3748/wjg.v13.i48.6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung JY, Hong SM, Choi BY, Cho H, Yu E, Hewitt SM. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:660–667. doi: 10.1158/1078-0432.CCR-08-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho C, Wang C, Mattu S, Destefanis G, Ladu S, Delogu S, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833–845. doi: 10.1002/hep.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang C, Cigliano A, Jiang L, Li X, Fan B, Pilo MG, et al. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and NRas proto-oncogenes in mice. Hepatology. 2015;61:200–213. doi: 10.1002/hep.27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nature reviews Drug discovery. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes & development. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang YJ, Duan Y, Zheng XF. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug discovery today. 2011;16:325–331. doi: 10.1016/j.drudis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slotkin EK, Patwardhan PP, Vasudeva SD, de Stanchina E, Tap WD, Schwartz GK. MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma. Molecular cancer therapeutics. 2015;14:395–406. doi: 10.1158/1535-7163.MCT-14-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassan B, Akcakanat A, Sangai T, Evans KW, Adkins F, Eterovic AK, et al. Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget. 2014;5:8544–8557. doi: 10.18632/oncotarget.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:2603–2612. doi: 10.1158/1078-0432.CCR-11-2750. [DOI] [PubMed] [Google Scholar]
- 17.Janes MR, Vu C, Mallya S, Shieh MP, Limon JJ, Li LS, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2013;27:586–594. doi: 10.1038/leu.2012.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingels A, Zhao H, Thong AE, Saar M, Valta MP, Nolley R, et al. Preclinical trial of a new dual mTOR inhibitor, MLN0128, using renal cell carcinoma tumorgrafts. International journal of cancer. 2014;134:2322–2329. doi: 10.1002/ijc.28579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nature reviews Drug discovery. 2011;10:868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 20.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. The American journal of pathology. 2014;184:912–923. doi: 10.1016/j.ajpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014;147:690–701. doi: 10.1053/j.gastro.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62:1497–1510. doi: 10.1002/hep.27992. [DOI] [PubMed] [Google Scholar]
- 23.Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140:1071–1083. doi: 10.1053/j.gastro.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu J, Che L, Li L, Pilo MG, Cigliano A, Ribback S, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Scientific reports. 2016;6:20484. doi: 10.1038/srep20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11988–11993. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamada D, Rizvi S, Razumilava N, Bronk SF, Davila JI, Champion MD, et al. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology. 2015;61:1627–1642. doi: 10.1002/hep.27687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang G, Gao X, Song YK, Vollmer R, Stolz DB, Gasiorowski JZ, et al. Hydroporation as the mechanism of hydrodynamic delivery. Gene therapy. 2004;11:675–682. doi: 10.1038/sj.gt.3302210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes & development. 2013;27:719–724. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer WC, Patel T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. Journal of hepatology. 2012;57:69–76. doi: 10.1016/j.jhep.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He K, Zheng X, Li M, Zhang L, Yu J. mTOR inhibitors induce apoptosis in colon cancer cells via CHOP-dependent DR5 induction on 4E-BP1 dephosphorylation. Oncogene. 2016;35:148–157. doi: 10.1038/onc.2015.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harbor perspectives in biology. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.