

Abstract
Activating mutations involving the members of the RAS signaling pathway, including KRAS, NRAS, and BRAF, have been reported in ovarian low-grade serous carcinoma and its precursor lesion, serous borderline tumor (SBT). Whether additional genetic alterations in the RAS oncogene family accumulate during the progression of serous borderline tumor (SBT) to invasive low grade serous carcinoma (LGSC) remains largely unknown. While mutations of KRAS and BRAF occur at a very early stage of progression, even preceding the development of SBT, additional driving events, such as NRAS mutations, have been postulated to facilitate progression. In this study, we analyzed NRAS exon 3 mutational status in 98 cases that were diagnosed with SBT/atypical proliferative serous tumor (SBT/APST), non-invasive LGSC (niLGSC), or invasive LGSC (iLGSC). Of the latter, NRAS Q61R (CAA to CGA) mutations were detected in only 2 of 56 (3.6%) cases. The same mutation was not detected in any of the SBT/APSTs or niLGSCs. Mutational analysis for hotspots in KRAS and BRAF demonstrated a wildtype pattern of KRAS and BRAF in one of the NRAS-mutated cases. Interestingly, another LGSC case with NRAS mutation harbored a concurrent BRAF V600L mutation. These findings indicate that, although recurrent NRAS mutations are present, their low prevalence indicates that NRAS plays a limited role in the development of LGSC. Further studies to identify other oncogenic drivers of LGSC progression is warranted.
Keywords: NRAS, Mutation, Low grade serous carcinoma
Introduction
Ovarian serous carcinoma has been classified as low- and high-grade based on distinct clinicopathological and molecular features [1]. Serous tubal intraepithelial carcinoma (STIC) is the presumable precursor lesion of high grade serous carcinoma (HGSC) and frequently harbors TP53 somatic mutations [2-4]. Most ovarian low grade serous carcinomas (LGSCs), in contrast, arise from a morphologically distinct precursor lesion, serous borderline tumor/atypical proliferative serous tumor (SBT/APST) [4-6]. There is a small subset of SBT/APSTs with micropapillary architecture that have a poor outcome; these are designated “noninvasive low-grade serous carcinoma (niLGSC)” [7]. Unlike invasive LGSC (iLGSC), niLGSC does not show a destructive growth pattern. The mechanisms by which a precursor lesion gives rise to invasive LGSC remain largely unknown. Targeted genetic analyses of candidate genes have detected somatic mutations in KRAS and BRAF in approximately 20-50% of SBT/APSTs and LGSCs (non-invasive and invasive) [8-12]. These mutations occur at a very early, pre-transformed stage, such as in cystadenoma, preceding the development of SBT/APST [13, 14]. Accordingly, additional genetic alterations other than KRAS and BRAF may accumulate as a driving force during the progression of SBT/APST to invasive LGSC.
It has been demonstrated that hemizygous or homozygous deletions of ch1p36 and ch9p21 are associated with the development of LGSC [15]. Moreover, deletions of both chromosomal regions occur more frequently in LGSCs than in SBT/APSTs [15]. Interestingly, our previous whole exome sequencing study showed that the mutation accumulation rate in bulk LGSCs was much lower than that in most adult tumors[16], consistent with the “indolent” nature of LGSCs and suggesting a low replication rate in this unique tumor type. Indeed, genome-wide analyses demonstrated very few recurrent somatic mutations in LGSCs [16].
More recently, recurrent NRAS mutations were reported in LGSCs, indicating an oncogenic driving role of NRAS in the development of LGSCs [17]. Consistent with this finding, another study identified several potential markers of progression and candidate driver genes of LGSC including NRAS, USP9X, and EIF1AX [18]. In this recent report, NRAS mutations were present in 26.3% (5/19) of LGSCs but were absent in SBT, and NRAS was thought to be a potent driver of LGSC tumorigenesis [18]. To further explore the role of NRAS in the development of LGSC, we investigated the mutational status of NRAS among our existing patient cohorts obtained from the Johns Hopkins Hospital and the Denmark National Patient Registry.
Materials and Methods
Selection of cases
A total of 98 cases were analyzed in this study, including 28 cases of SBT/APST, 14 cases of niLGSC, and 56 cases of iLGSC. Among these, 52 cases were obtained from the pathology archives at the Johns Hopkins Hospital, consisting of 15 SBT/APSTs, 10 niLGSCs, and 27 iLGSCs. The remaining cases (13 APSTs, 4 niLGSCs, and 29 iLGSCs) were obtained from the archives of the nationwide Denmark Pathology Data Bank [19, 20]. Histologic sections of these cases were reviewed by at least two pathologists (I.M.S./R.V. or I.M.S./D.X.). The study was approved by the Danish Data Protection Agency, the Danish Scientific Ethical Committee, and the Institutional Review Board at the Johns Hopkins Hospital.
DNA extraction and mutational analysis
Paraffin-embedded tumor tissues, identified by H&E staining of adjacent sections (tumor elements account for more than 70% of section area), were macrodissected, and genomic DNA was extracted using a QIAamp DNA FFPE Tissue Kit with an adapted protocol (Qiagen, Valencia, CA, USA). Briefly, slides bearing paraffin embedded tissue were baked at 68°C for 20 to 30 seconds; the tissue was deparaffinized 3 times with xylene, and residual xylene was removed by washing through serial dilutions of ethanol. Tumor tissue was separated from adjacent normal tissue and placed in a tube allowing for complete evaporation of residual ethanol. The tissue pellet was resuspended in Buffer ATL with added proteinase K. The rest of the procedure followed the manufacturer's instruction.
Mutational analysis was performed using conventional Sanger sequencing techniques at the NRAS mutational hotspot region on exon 3, including codon 61. The cases were also tested for other hotspot mutations of KRAS at exon 2, including codons 12-13, and BRAF at exon 15, including codon 600 as previously described [10, 21]. Briefly, PCR amplification was performed using genomic DNA from macro-dissected FFPE tissue with the following primers. For exon 3 of NRAS: forward 5′-CCCCCTTACCCTCCACAC-3′ and reverse 5′-TGGCAAATACACAGAGGAAGC-3′; for exon 15 of BRAF: forward 5′-TGCTTGCTCTGATAGGAAAATGA-3′ and reverse 5′-CCACAAAATGGATCCAGACAAC-3′; for exon 2 of KRAS: forward 5′-TAAGGCCTGCTGAAAATGACTG-3′ and reverse 5′-TGGTCCTGCACCAGTAATATGC-3′. Amplified PCR products were sequenced at Beckman Coulter, Inc., (Danvers, MA, USA), and analyzed using Mutation Surveyor DNA Variant Analysis Software.
Results
Representative histologic images of SBT/APST, niLGSC and iLGSC were present in Figure 1. Clinical factor analysis established that there was a similar age distribution between the cohort from Johns Hopkins Hospital and the cohort from the Denmark National Patient Registry. The age of patients with SBT/APSTs from the pathology files of the Johns Hopkins Hospital ranged from 24 to 67 years old (median = 43) (Table 1). The age of niLGSC patients ranged from 23 to 57 years (median = 32.5), and the age of patients with iLGSCs ranged from 31 to 72 years (median = 50). The median ages for patients from the Denmark cohort with SBT/APSTs, niLGSCs, and iLGSCs were 43, 37, and 49.5 years, respectively. Of the 98 patients, 92 (93.8%) were of Caucasian background.
Figure 1.
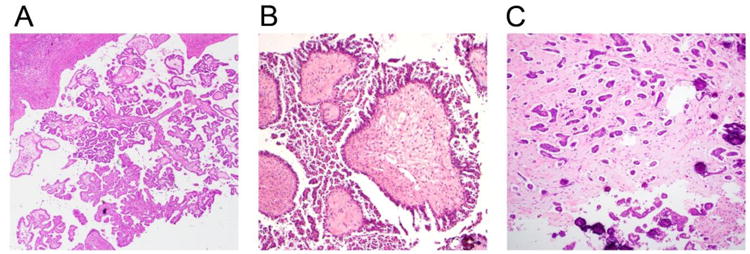
Representative histologic images of three different types of ovarian serous tumor. A, Serous borderline tumor/Atypical proliferative serous tumor (SBT/APST). B, Non-invasive low grade serous carcinoma (niLGSC). C. Invasive low grade serous carcinoma (iLGSC). H&E slides, Original magnification 100×.
Table 1.
Mutations of NRAS, KRAS, and BRAF in SBT/APSTs, niLGSCs, and iLGSCs.
| Pathologic Type | Patient Age | NRAS | KRAS | BRAF | ||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| Range | Median | Mutations | % | Mutations | % | Mutations | % | |
| Hopkins (n=52) | ||||||||
| SBT/APSTs | 24-67 | 43.0 | 0/15 | 0% | 6/15 (G 12D, 4; G12V, 2) | 40.0% | 8/15 (V600E, 7; D594G, 1) | 53.3% |
| niLGSCs | 23-57 | 32.5 | 0/10 | 0% | 1/10 (G12V, 1) | 10.0% | 2/10 (V600E, 1; T599I, 1) | 20.0% |
| iLGSCs | 31-72 | 50.0 | 2/27 (Q61R, 2) | 7.4% | 4/27 (G12D, 3; G12V, 1) | 14.8% | 7/27 (V600E, 6; V600L, 1) | 25.9% |
| Denmark (n=46) | ||||||||
| SBT/APSTs | 23-70 | 43.0 | 0/13 | 0% | 9/13 (G12D, 5; G12V, 3; G12A, 1) | 69.2% | 0/13 | 0% |
| niLGSCs | 30-54 | 37.0 | 0/4 | 0% | 1/4 (G12D, 1) | 25.0% | 0/4 | 0% |
| iLGSCs | 25-84 | 49.5 | 0/29 | 0% | 12/29 (G12D, 8; G12V, 3; G12A, 1) | 41.4% | 3/29 (V600E, 3) | 10.3% |
Note: SBT/APSTs, serous borderline tumor/atypical proliferative serous tumors; niLGSCs, non-invasive low grade serous carcinomas; iLGSCs, invasive low grade serous carcinomas.
NRAS mutational status was analyzed at the hotspot region of exon 3 in 28 SBT/APSTs, 14 niLGSCs, and 56 iLGSCs from both the Johns Hopkins Hospital and Denmark cohorts (Table 1). NRAS Q61R (CCA to CGA) mutations were detected in 2 of 27 (7.4%) Johns Hopkins Hospital invasive LGSC cases, with none were detected in the Denmark cohort (Figure 2). The overall mutation rate of detection of NRAS mutation was 3.6% (2 of 56 cases). The same hotspot mutation was not detected in any SBT/APSTs or niLGSCs. In one of the two NRAS-mutated cases, analysis of KRAS and BRAF hotspot mutations identified no alterations. Interestingly, while all KRAS and BRAF mutations were mutually exclusive, one NRAS-mutated iLGSC case harbored a concurrent BRAF V600L mutation. Among all patients, KRAS mutations were detected in 15 of 28 (53.6%) SBT/APSTs, 2 of 14 (14.3%) niLGSCs, and 16 of 56 (28.6%) iLGSCs. BRAF mutations were detected in 8 of 28 (28.6%) SBT/APSTs, 2 of 14 (14.3%) niLGSCs, and 10 of 56 (17.9%) iLGSCs. Rare mutations of BRAF T599I and BRAF D594G were detected in 1 niLGSC and 1 SBT/APST, respectively (Table 1). The combined frequency of KRAS and BRAF mutations in SBT/APSTs in the Johns Hopkins Hospital cohort appeared higher than that reported previously (Table 1), likely due to selection bias with limited case number (15 cases).
Figure 2.
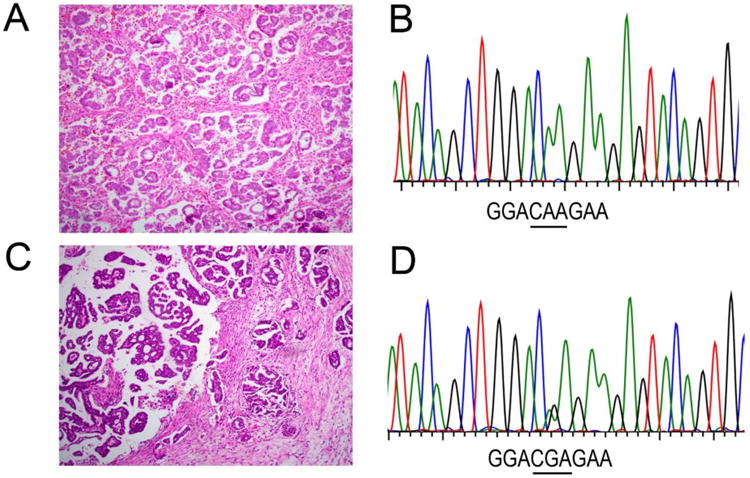
Histology and nucleotide sequences of NRAS in two representative invasive low grade serous carcinoma cases. Case 1, A. H&E staining of tumor. B. Chromatogram of nucleotide sequence shows wild-type pattern of NRAS containing codon 61 in a low grade serous carcinoma. Case 2, C. H&E staining of tumor. D. Chromatogram of nucleotide sequence shows a point NRAS Q61R mutation (CAA to CGA) in a low grade serous carcinoma. H&E slides, Original magnification 40×.
Discussion
In this study, we sought to investigate the mutational profile of NRAS in SBT/APSTs, non-invasive LGSCs, and invasive LGSCs. Similar to previous findings, our study, based on a large retrospective cohort, did not identify NRAS mutations in either SBT/APSTs or non-invasive LGSCs [17, 18]. We observed a relatively low mutation frequency (3.6%) of NRAS in invasive LGSCs. Our data suggests that NRAS may play a very limited role in promoting aggressive behavior of LGSCs.
It is well established that LGSCs develop from SBT/APSTs in a step-wise fashion. Molecular genetic studies have highlighted the significance of the MAPK signaling pathway in the pathogenesis of LGSCs, illustrated by the fact that activating mutations in codon 12 and codon 13 of KRAS or in codon 600 of BRAF occur in approximately half to two-thirds of SBT/APSTs and LGSCs [1, 6, 22]. Mutations of ERBB2, another key factor of the Ras/Raf/MEK/MAPK signaling pathway, occur in 6% of these tumors [23]. Identical KRAS or BRAF mutations have been detected in adjacent cystadenoma epithelium, the precursor lesion of SBT/APSTs, indicating that mutations of KRAS and BRAF are early genetic events associated with initiation of low-grade serous neoplasms, and that the small subset of serous cystadenomas that acquire these mutations may progress to SBT/APST [13, 14]. Our recent study also demonstrated that a substantial proportion of LGSCs most likely represent direct progression from SBTs, as illustrated by the detection of identical KRAS and BRAF mutations in both SBT/APSTs and in subsequent LGSCs of the same patient (in preparation). This observation implies that genetic alterations, in addition to KRAS and BRAF mutations, are required to drive the progression of SBT/APSTs to invasive LGSCs.
Progression of SBT/APSTs to LGSCs is thought to be associated with novel and distinct molecular events. Two studies demonstrated that NRAS is a critical oncogenic driver in the development of LGSCs. In one study, the frequency of activating NRAS mutations was 5 of 58 (9%) in invasive tumors with adjacent borderline tumor [17]. Given that a portion of these tumors had TP53 mutations that were consistent with HGSCs rather than LGSCs, the actual rate of NRAS mutation was higher. Whereas BRAF and KRAS mutations were found in both SBT/APSTs and invasive tumors, NRAS mutations were not found in any pure, non-ambiguous SBT/APST tumors tested. Another study demonstrated that NRAS mutations were detected in 26.3% of LGSCs, but none were detected in the SBT/APST cohort [18]. Consistent with previous findings, we did not observe NRAS mutations in either SBT/APSTs or niLGSCs. Mutations of NRAS Q61R were detected in 2 of 27 (7.4%) LGSCs in the Johns Hopkins Hospital cohort, and none were detected in the Danish cohort, a frequency lower than that of the previous study [18]. The precise reason for this difference is unclear. The possible explanations include selection bias due to limited sample sizes, geographic differences, and ethnic heterogeneity. Future studies with larger patient groups would provide more accurate information regarding NRAS mutation rates. Nonetheless, the overall mutation rate of NRAS appears to be low in LGSC patients. This result suggests that mutation of NRAS may be acquired at later stages in LGSCs, and that it may be involved in progression in a subset of tumors. It should be noted that only hotspot mutation in NRAS has been examined in this study. Mutation frequency of Ras/Raf/MEK/MAPK signaling pathway including NRAS could be underestimated due to incomplete gene sequencing.
Concurrent existence of NRAS and BRAF mutations in LGSC is unusual and contrasts with the mutually exclusive pattern of mutations among MAPK pathway components. These findings further suggest that, unlike KRAS or BRAF, NRAS mutations may function in a different context, such as in promoting invasiveness and aggressiveness of advanced tumors. [24-26].
In addition to mutations in genes of MAPK pathways, exome sequencing has identified mutations in novel candidate genes EIF1AX and USP9X, which are more common in LGSCs than in SBT/APSTs, suggesting that these genes play a vital role in the oncogenesis of LGSCs [18]. Mutations in EIF1AX and USP9X were almost exclusively identified in BRAF/KRAS/NRAS mutant tumors, suggesting a cooperative biological effect. Similarly, a most recent study demonstrated that recurrent mutations in EIF1AX significantly co-occurred with mutations in NRAS [27]. Since mutations involving MAPK pathways account for no more than two thirds of SBT/APST and LGSC cases, there remains a sizable proportion of cases with no known mutation. Accordingly, other genetic alterations, such as allelic deletions, loss of heterozygosity, or gene amplifications, may facilitate the development of LGSC. In fact, it has been found that frequent allelic deletions of 1p36 and 9p21 occur in LGSCs but not in APSTs [15]. The 1p36 region contains several candidate tumor genes that modulate cell cycle arrest and apoptosis. Chr9p21 harbors the CDKN2A locus, which encodes p16, p15, and p14/Arf. Similarly, loss of heterozygosity of 9p including CDKN2A has been observed in another study [18].
In summary, our study indicates that, although recurrent NRAS mutations are present, the low mutation rate suggests that NRAS itself plays a minor role in the development of LGSC. Further studies to identify other oncogenic drivers in the progression of LGSC are warranted.
Highlights.
NRAS mutation is postulated to facilitate the development of low grade serous carcinoma (LGSC).
NRAS hotspot (Q61R) mutation was detected in only 3.6% cases of LGSCs in this study.
Low prevalence of mutation indicates that NRAS plays a limited role in the development of LGSC.
Acknowledgments
Funding/Support: This study was partly funded by the Richard W. TeLinde Gynecologic Pathology program in the Department of Pathology, The Johns Hopkins University School of Medicine. This study was also partly supported by DoD W81XWH-11-2-0230 (T.-L. Wang), NCI UO1 CA200469 (I.-M. Shih), and NCI 1R01CA215483 (I.-M. Shih)
Footnotes
Conflict of interest: The authors have no competing interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kurman RJ, Shih Ie M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. The American journal of pathology. 2016;186(4):733–747. doi: 10.1016/j.ajpath.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlson JW, Miron A, Jarboe EA, Parast MM, Hirsch MS, Lee Y, Muto MG, Kindelberger D, Crum CP. Serous tubal intraepithelial carcinoma: its potential role in primary peritoneal serous carcinoma and serous cancer prevention. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(25):4160–4165. doi: 10.1200/JCO.2008.16.4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurman RJ. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Annals of oncology : official journal of the European Society for Medical Oncology. 2013;24(10):x16–21. doi: 10.1093/annonc/mdt463. [DOI] [PubMed] [Google Scholar]
- 4.Vang R, Shih Ie M, Kurman RJ. Fallopian tube precursors of ovarian low- and high-grade serous neoplasms. Histopathology. 2013;62(1):44–58. doi: 10.1111/his.12046. [DOI] [PubMed] [Google Scholar]
- 5.Kurman RJ, Shih Ie M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. 2010;34(3):433–443. doi: 10.1097/PAS.0b013e3181cf3d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nik NN, Vang R, Shih Ie M, Kurman RJ. Origin and pathogenesis of pelvic (ovarian, tubal, and primary peritoneal) serous carcinoma. Annual review of pathology. 2014;9:27–45. doi: 10.1146/annurev-pathol-020712-163949. [DOI] [PubMed] [Google Scholar]
- 7.Vang R, Hannibal CG, Junge J, Frederiksen K, Kjaer SK, Kurman RJ. Long-term Behavior of Serous Borderline Tumors Subdivided Into Atypical Proliferative Tumors and Noninvasive Low-grade Carcinomas: A Population-based Clinicopathologic Study of 942 Cases. Am J Surg Pathol. 2017;41(6):725–737. doi: 10.1097/PAS.0000000000000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singer G, Oldt R, 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, Shih Ie M. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. Journal of the National Cancer Institute. 2003;95(6):484–486. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 9.Wong KK, Tsang YT, Deavers MT, Mok SC, Zu Z, Sun C, Malpica A, Wolf JK, Lu KH, Gershenson DM. BRAF mutation is rare in advanced-stage low-grade ovarian serous carcinomas. The American journal of pathology. 2010;177(4):1611–1617. doi: 10.2353/ajpath.2010.100212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ardighieri L, Zeppernick F, Hannibal CG, Vang R, Cope L, Junge J, Kjaer SK, Kurman RJ, Shih Ie M. Mutational analysis of BRAF and KRAS in ovarian serous borderline (atypical proliferative) tumours and associated peritoneal implants. The Journal of pathology. 2014;232(1):16–22. doi: 10.1002/path.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heublein S, Grasse K, Hessel H, Burges A, Lenhard M, Engel J, Kirchner T, Jeschke U, Mayr D. KRAS, BRAF genotyping reveals genetic heterogeneity of ovarian borderline tumors and associated implants. BMC cancer. 2013;13:483. doi: 10.1186/1471-2407-13-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsang YT, Deavers MT, Sun CC, Kwan SY, Kuo E, Malpica A, Mok SC, Gershenson DM, Wong KK. KRAS (but not BRAF) mutations in ovarian serous borderline tumour are associated with recurrent low-grade serous carcinoma. The Journal of pathology. 2013;231(4):449–456. doi: 10.1002/path.4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng EJ, Kurman RJ, Wang M, Oldt R, Wang BG, Berman DM, Shih Ie M. Molecular genetic analysis of ovarian serous cystadenomas. Laboratory investigation; a journal of technical methods and pathology. 2004;84(6):778–784. doi: 10.1038/labinvest.3700103. [DOI] [PubMed] [Google Scholar]
- 14.Ho CL, Kurman RJ, Dehari R, Wang TL, Shih Ie M. Mutations of BRAF and KRAS precede the development of ovarian serous borderline tumors. Cancer Res. 2004;64(19):6915–6918. doi: 10.1158/0008-5472.CAN-04-2067. [DOI] [PubMed] [Google Scholar]
- 15.Kuo KT, Guan B, Feng Y, Mao TL, Chen X, Jinawath N, Wang Y, Kurman RJ, Shih Ie M, Wang TL. Analysis of DNA copy number alterations in ovarian serous tumors identifies new molecular genetic changes in low-grade and high-grade carcinomas. Cancer Res. 2009;69(9):4036–4042. doi: 10.1158/0008-5472.CAN-08-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones S, Wang TL, Kurman RJ, Nakayama K, Velculescu VE, Vogelstein B, Kinzler KW, Papadopoulos N, Shih Ie M. Low-grade serous carcinomas of the ovary contain very few point mutations. The Journal of pathology. 2012;226(3):413–420. doi: 10.1002/path.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emmanuel C, Chiew YE, George J, Etemadmoghadam D, Anglesio MS, Sharma R, Russell P, Kennedy C, Fereday S, Hung J, et al. Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(24):6618–6630. doi: 10.1158/1078-0432.CCR-14-1292. [DOI] [PubMed] [Google Scholar]
- 18.Hunter SM, Anglesio MS, Ryland GL, Sharma R, Chiew YE, Rowley SM, Doyle MA, Li J, Gilks CB, Moss P, et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget. 2015;6(35):37663–37677. doi: 10.18632/oncotarget.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hannibal CG, Vang R, Junge J, Frederiksen K, Kurman RJ, Kjaer SK. A nationwide study of ovarian serous borderline tumors in Denmark 1978-2002. Risk of recurrence, and development of ovarian serous carcinoma Gynecologic oncology. 2017;144(1):174–180. doi: 10.1016/j.ygyno.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannibal CG, Vang R, Junge J, Frederiksen K, Kjaerbye-Thygesen A, Andersen KK, Tabor A, Kurman RJ, Kjaer SK. A nationwide study of serous “borderline” ovarian tumors in Denmark 1978-2002: centralized pathology review and overall survival compared with the general population. Gynecologic oncology. 2014;134(2):267–273. doi: 10.1016/j.ygyno.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeppernick F, Ardighieri L, Hannibal CG, Vang R, Junge J, Kjaer SK, Zhang R, Kurman RJ, Shih Ie M. BRAF mutation is associated with a specific cell type with features suggestive of senescence in ovarian serous borderline (atypical proliferative) tumors. Am J Surg Pathol. 2014;38(12):1603–1611. doi: 10.1097/PAS.0000000000000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malpica A, Wong KK. The molecular pathology of ovarian serous borderline tumors. Annals of oncology : official journal of the European Society for Medical Oncology. 2016;27(1):i16–i19. doi: 10.1093/annonc/mdw089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anglesio MS, Arnold JM, George J, Tinker AV, Tothill R, Waddell N, Simms L, Locandro B, Fereday S, Traficante N, et al. Mutation of ERBB2 provides a novel alternative mechanism for the ubiquitous activation of RAS-MAPK in ovarian serous low malignant potential tumors. Molecular cancer research : MCR. 2008;6(11):1678–1690. doi: 10.1158/1541-7786.MCR-08-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matulonis UA, Hirsch M, Palescandolo E, Kim E, Liu J, van Hummelen P, MacConaill L, Drapkin R, Hahn WC. High throughput interrogation of somatic mutations in high grade serous cancer of the ovary. PloS one. 2011;6(9):e24433. doi: 10.1371/journal.pone.0024433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dokianakis DN, Varras MN, Papaefthimiou M, Apostolopoulou J, Simiakaki H, Diakomanolis E, Spandidos DA. Ras gene activation in malignant cells of human ovarian carcinoma peritoneal fluids. Clinical & experimental metastasis. 1999;17(4):293–297. doi: 10.1023/a:1006611220434. [DOI] [PubMed] [Google Scholar]
- 26.Anglesio MS, Wiegand KC, Melnyk N, Chow C, Salamanca C, Prentice LM, Senz J, Yang W, Spillman MA, Cochrane DR, et al. Type-specific cell line models for type-specific ovarian cancer research. PloS one. 2013;8(9):e72162. doi: 10.1371/journal.pone.0072162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etemadmoghadam D, Azar WJ, Lei Y, Moujaber T, Garsed DW, Kennedy C, Fereday S, Mitchell C, Chiew YE, Hendley J, et al. EIF1AX and NRAS mutations co-occur and cooperate in low-grade serous ovarian carcinomas. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-16-2224. [DOI] [PubMed] [Google Scholar]