

Abstract
Patients with chronic kidney disease (CKD) exhibit a myriad of metabolic derangements including dyslipidemia characterized by low plasma concentrations of high-density lipoprotein (HDL)-associated cholesterol. However, the effects of kidney disease on HDL composition have not been comprehensively determined. Here we used a targeted mass spectrometric approach to quantify 38 proteins contained in the HDL particles within a CKD cohort of 509 participants with a broad range of estimated glomerular filtration rates (eGFR) (CKD stages I-V, and a mean eGFR of 45.5 mL/min/1.73m2). After adjusting for multiple testing, demographics, comorbidities, medications, and other characteristics, eGFR was significantly associated with differences in four HDL proteins. Compared to participants with an eGFR of 60 mL/min/1.73m2 or more, those with an eGFR under 15 mL/min/1.73m2 exhibited 1.89-fold higher retinol binding protein 4 (95% confidence interval 1.34-2.67), 1.52-fold higher apolipoprotein C-III (1.25-1.84),0.70-fold lower apolipoprotein L1 (0.55-0.92), and 0.64-fold lower vitronectin (0.48- 0.85). Although the HDL apolipoprotein L1 was slightly lower among African-American than among Caucasian individuals, the relationship to eGFR did not differ by race. After adjustment, no HDL-associated proteins associated with albuminuria. Thus, modest changes in the HDL proteome provide preliminary evidence for an association between HDL proteins and declining kidney function, but this needs to be replicated. Future analyses will determine if HDL proteomics is indeed a clinical predictor of declining kidney function or cardiovascular outcomes.
Keywords: chronic kidney disease, cardiovascular disease, proteomic analysis, albuminuria, lipids, apolipoprotein L1
Introduction
Chronic kidney disease (CKD) leads to marked metabolic derangements, including a distinct profile of dyslipidemia that worsens with progressive loss of kidney function. The dyslipidemia of CKD is classically characterized by hypertriglyceridemia and low circulating concentrations of high-density lipoprotein-associated cholesterol (HDL-C).1-3 Notably, whereas serum HDL-C levels exhibit a strong, inverse relationship with cardiovascular disease (CVD) risk in the general population, this relationship is attenuated and eventually abrogated as glomerular filtration rate (GFR) declines.4-6 Further, the clinical efficacy of statin therapy is no longer observed among patients with end-stage renal disease (ESRD). These findings suggest that kidney disease confers complex changes in lipid and lipoprotein metabolism that are not fully captured by standard serum lipid measurements.
High-density lipoproteins (HDL) encompass a complex group of heterogeneous particles that play important roles in vascular function and CVD risk. Previous studies have suggested alterations in specific HDL proteins among chronic dialysis patients.7-11 However, these studies are limited by small sample sizes, comparison of ESRD patients to healthy controls, untargeted measurement strategies, and the potential for confounding by co-morbidities that are associated with kidney disease. To date, no studies have characterized potential differences in HDL composition across the spectrum of kidney function in a well-characterized clinical cohort with CKD not undergoing dialysis.
We employed a targeted mass spectrometric approach to quantify HDL-associated proteins in 509 CKD patients across the full spectrum of kidney function. The identification of key changes in HDL composition that occur with loss of GFR could be critical for better understanding changes in lipoprotein metabolism that result from impaired kidney function and thereby generate a more comprehensive understanding of the metabolic derangements consequent to CKD. Moreover, this line of investigation could yield novel insights into the unique dyslipidemia in CKD, as well as into the lack of clinical efficacy of standard lipid-lowering therapy among patients with advanced CKD. It might also promote the development of more effective treatment strategies. We hypothesized that progressive changes in the HDL proteome would be evident across a spectrum of GFR in patients with CKD.
Results
Study participants
Among the 509 Seattle Kidney Study participants who were included in this study, the mean age was 58 ±13.9 years and the mean estimated GFR (eGFR) was 45.5±26.4 mL/min/1.73m (Table 1)’. The prevalence of diabetes ranged from 41.3% among participants who had an eGFR ≥ 60 mL/min/1.73m2 to 61.8% among participants who had an eGFR <15 mL/min/1.73m2. Study participants exhibited a mild dyslipidemia, with a mean serum HDL-C concentration of 41.6±17.1 mg/dL and mean serum triglyceride concentration of 162.2±118.7 mg/dL. Serum HDL-C concentrations were modestly lower for the lowest category of eGFR; however, this distinction was not statistically significant (Figure 1). There was no association between serum triglycerides and eGFR (Supplemental Figure 1).
Table 1. Subject characteristics by estimated GFR.
| eGFR 60+ | eGFR 45-60 | eGFR 30-45 | eGFR 15-30 | eGFR < 15 | |
|---|---|---|---|---|---|
| N | 92 | 91 | 106 | 102 | 34 |
| Age, years | 49.9 (±12.1) | 57.3 (±11.6) | 63.1 (±13.3) | 60.6 (±14.9) | 61.0 (±12.6) |
| BMI, kg/m2 | 30.8 (±8.3) | 31.9 (±6.8) | 31.2 (±8.3) | 31.3 (±7.9) | 30.2 (±6.2) |
| eGFR (ckdepi), mL/min/1.73m2 | 82.8 (±20.6) | 53.9 (±9.0) | 39.0 (±6.7) | 22.7 (±4.7) | 11.5 (±4.1) |
| Female gender | 25 (27.2) | 19 (20.9) | 30 (28.3) | 25 (24.5) | 6 (17.6) |
| Prevalent Diabetes | 38 (41.3) | 47 (51.6) | 58 (54.7) | 50 (49.0) | 21 (61.8) |
| Prevalent CAD | 19 (20.7) | 29 (31.9) | 41 (38.7) | 42 (41.2) | 7 (20.6) |
| Race | |||||
| White | 56 (60.9) | 59 (64.8) | 74 (69.8) | 60 (58.8) | 22 (64.7) |
| Black | 29 (31.5) | 24 (64.8) | 23 (21.7) | 31 (30.4) | 6 (17.6) |
| Other | 7 (7.6) | 8 (64.8) | 9 (8.5) | 11 (10.8) | 6 (17.6) |
| Statin use | 43 (46.7) | 53 (58.2) | 59 (56.2) | 63 (62.4) | 17 (51.5) |
| Current smoking | 22 (24.2) | 22 (24.7) | 16 (15.5) | 20 (20.2) | 7 (21.9) |
| HDL cholesterol,mg/dL | 42.1 (±20.6) | 38.7 (±14.9) | 42.0 (±15.9) | 39.2 (±14.5) | 35.3 (±12.1) |
| LDL cholesterol,mg/dL | 113.6(±55.1) | 92.6 (±33.7) | 101.4 (±41.8) | 100.3 (±41.7) | 90.9 (±35.9) |
| Triglycerides, mg/Dl | 157.9(±126.7) | 168.4(±110.5) | 152.1(±109.5) | 164.0(±133.1) | 167.1 (±115.0) |
| Total cholesterol, mg/Dl | 183.5 (±67.3) | 161.9 (±41.9) | 174.9 (±54.5) | 173.4 (±61.1) | 158.1 (±42.8) |
| Urine ACR (median, IQR) | 65.3 (9.2, 451.5) | 30.4 (3.7, 337.2) | 73.1 (11.7, 452.1) | 189.8 (43.5, 783.3) | 484.0 (179.1,1510.3) |
| CRP (median, IQR) | 1.9 (0.7, 4.4) | 3.0 (1.2, 6.4) | 2.8 (0.8, 7.7) | 3.0 (1.0, 7.3) | 2.0 (0.7, 6.4) |
Clinical and demographic characteristics of subjects classified by eGFR. Normally distributed data are presented as mean(±SD), and non-normally distributed data are presented as median(interquartile range). Diabetes and CAD prevalence, race, gender, smoking status, and statin use are presented as number of subjects (%). CAD is defined as any history of myocardial infarction, cardiac arrest, coronary artery bypass graft, or percutaneous coronary intervention. eGFR=estimated glomerular filtration rate, BMI=body mass index, CAD=coronary artery disease, HDL=high-density lipoprotein, ACR=albumin:creatinine ratio, CRP=C-reactive protein
Figure 1. Serum HDL cholesterol concentrations in the Seattle Kidney Study cohort.
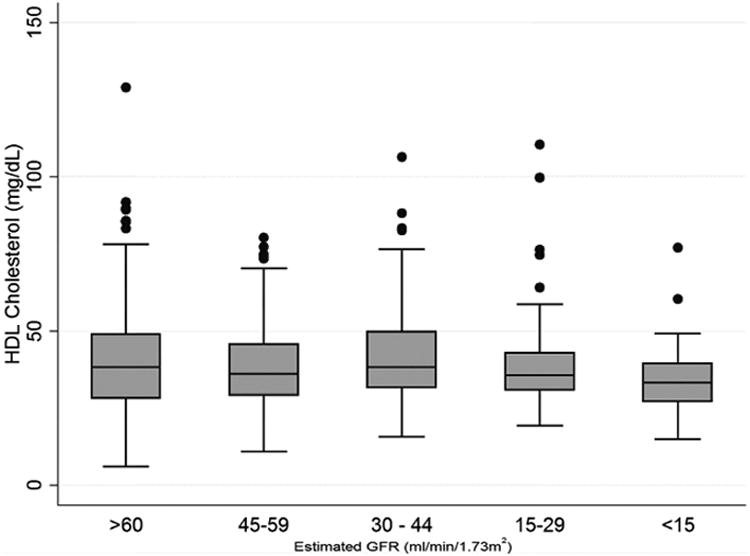
Box and whisker plots illustrate the serum high-density lipoprotein (HDL) cholesterol concentrations for each range of estimated glomerular filtration rate (GFR) noted. Top, middle, and bottom of boxes are 75th, 50th, and 25th percentile respectively, whiskers illustrate the range with outliers (greater than 1.5x the interquartile range from the median) shown as circles.
Associations of eGFR with HDL-associated proteins
After accounting for multiple testing, eGFR was significantly associated with differences in 5 HDL- associated proteins (p<0.0015, Figure 2, Supplemental Table 1). Specifically, lower eGFR was associated with higher HDL concentrations of apolipoprotein C-III (apoC-III) and retinol-binding protein 4 (RBP4) and with lower HDL concentrations of cholesteryl ester transfer protein (CETP), vitronectin, and apolipoprotein L1 (apoL1).
Figure 2. Alterations in HDL protein abundance associated with changes in eGFR.
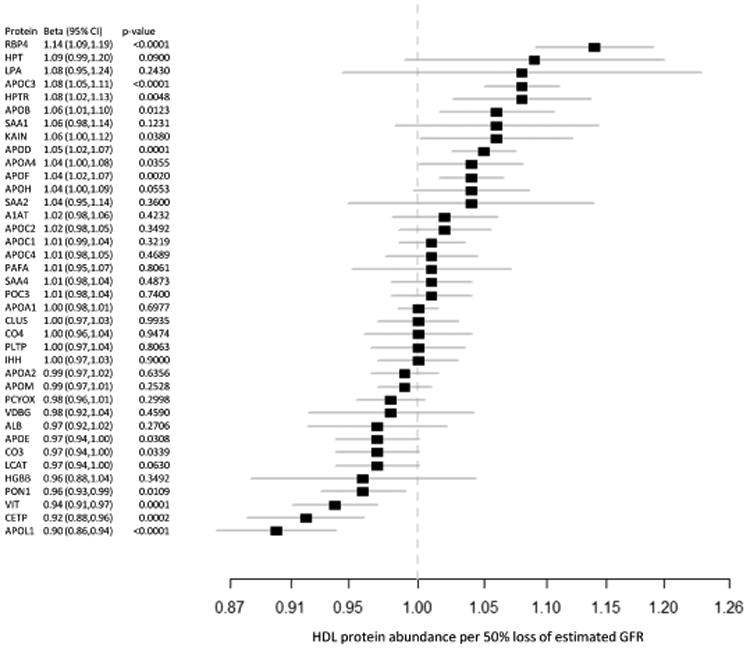
Fold enrichment for each protein in high-density lipoprotein (HDL) vs. loss of estimated glomerular filtration rate (eGFR) is shown with a square. Solid lines denote 95% CI. RBP4=retinol-binding protein 4, HPT=haptoglobin, LPA=apolipoprotein(a), APO=apolipoprotein, HPTR=haptoglobin-related protein, SAA=serum amyloid A, KAIN=kallistatin, A1AT=α-1-antitrypsin, PAFA=platelet-activating factor acetylhydrolase, PON3=paraoxonase 3, CLUS=clusterin, CO4=complement C4, PLTP=phospholipid transfer protein, IHH=Indian hedgehog protein, PCYOX=prenylcysteine oxidase 1, VDBG=vitamin D-binding globulin, ALB=albumin, CO3=complement C3, LCAT= phosphatidylcholine-sterol acyltransferase, HGBB=hemoglobin beta, PON1=paraoxonase 1, VIT=vitronectin, CETP=cholesteryl ester transfer protein
Associations of eGFR with these HDL-associated proteins remained significant after adjustment for demographics, BMI, smoking, diabetes, statin use, and serum lipid concentrations (Table 2). Associations of eGFR with apoC-III, RBP4, CETP, vitronectin, and apoL1 were not materially altered by additional adjustment for the urine albumin:creatinine ratio. However, the association with CETP no longer met statistical significance (p=0.0023). In the fully adjusted models, associations of eGFR with HDL protein concentrations appeared to be generally linear, with progressive differences in each HDL- associated protein across CKD stage (Figure 3).
Table 2. HDL-associated proteins that differed across estimated GFR categories.
| Protein | Model | eGFR 60+ | eGFR 45-60 | eGFR 30-45 | eGFR 15-30 | eGFR < 15 | p |
|---|---|---|---|---|---|---|---|
| RBP4 | Median(IQR) | 0.38(0.24,0.57) | 0.42(0.30,0.66) | 0.61(0.40,0.90) | 0.67(0.39,0.93) | 0.79(0.50,1.25) | |
| M1 | 1.00 (ref) | 1.05(0.83,1.33) | 1.54(1.22,1.96) | 1.59(1.25,2.01) | 1.94(1.40,2.70) | <0.0001 | |
| M2 | 1.00 (ref) | 1.05(0.83,1.33) | 1.53(1.20,1.94) | 1.60(1.26,2.04) | 1.89(1.34,2.67) | <0.0001 | |
| APOC3 | Median(IQR) | 1.36(1.02,1.82) | 1.56(1.11,2.07) | 1.73(1.21,2.42) | 1.82(1.29,2.38) | 2.16(1.38,3.16) | |
| M1 | 1.00 (ref) | 1.10(0.96,1.26) | 1.24(1.09,1.42) | 1.22(1.07,1.39) | 1.51(1.25,1.82) | <0.0001 | |
| M2 | 1.00 (ref) | 1.10(0.96,1.26) | 1.25(1.09,1.43) | 1.24(1.08,1.42) | 1.52(1.25,1.84) | <0.0001 | |
| CETP | Median(IQR) | 2.11(1.29,3.55) | 2.15(1.21,3.43) | 1.97(1.35,2.80) | 1.85(1.09,2.87) | 1.26(0.81,2.35) | |
| M1 | 1.00 (ref) | 0.91 (0.73,1.13) | 0.86 (0.69,1.08) | 0.74 (0.60,0.93) | 0.66 (0.48,0.89) | 0.0008 | |
| M2 | 1.00 (ref) | 0.90 (0.72,1.13) | 0.86 (0.69,1.08) | 0.77 (0.61,0.96) | 0.64 (0.46,0.88) | 0.0023 | |
| VIT | Median(IQR) | 0.94 (0.70,1.30) | 0.86 (0.62,1.18) | 0.85 (0.55,1.27) | 0.74 (0.53,1.05) | 0.71 (0.52,0.92) | |
| M1 | 1.00 (ref) | 0.85 (0.71,1.02) | 0.83 (0.70,1.00) | 0.73 (0.61,0.87) | 0.70 (0.55,0.90) | 0.0001 | |
| M2 | 1.00 (ref) | 0.85 (0.71,1.01) | 0.84 (0.70,1.00) | 0.75 (0.62,0.90) | 0.71 (0.55,0.92) | 0.0008 | |
| APOL1 | Median(IQR) | 1.11 (0.63,1.72) | 1.14 (0.72,1.75) | 0.99 (0.54,1.47) | 0.87 (0.37,1.32) | 0.78 (0.54,1.29) | |
| M1 | 1.00 (ref) | 1.02 (0.83,1.24) | 0.84 (0.68,1.02) | 0.70 (0.57,0.85) | 0.59 (0.45,0.78) | <0.0001 | |
| M2 | 1.00 (ref) | 1.02 (0.83,1.24) | 0.88 (0.72,1.07) | 0.73 (0.59,0.89) | 0.64 (0.48,0.85) | <0.0001 |
Relative abundance of HDL-associated proteins that varied as a function of eGFR. Absolute protein abundance is presented as well as adjusted models normalized to subject group with eGFR>60 mL/min/1.73m2. M1: adjusted for age, race (black vs. other), sex, prevalent diabetes, BMI, current smoking status, statin use, HDL, total cholesterol, and triglycerides. M2: Adjusted for all variables in M1 and albuminuria. Data are presented as median (interquartile range). HDL=high-density lipoprotein, GFR=glomerular filtration rate, RBP4=retinol-binding protein 4, APOC3=apolipoprotein C-III, CETP=cholesteryl ester transfer protein, VIT=vitronectin,APOL1 =apolipoprotein L1
Figure 3. Associations between eGFR and HDL protein abundance.
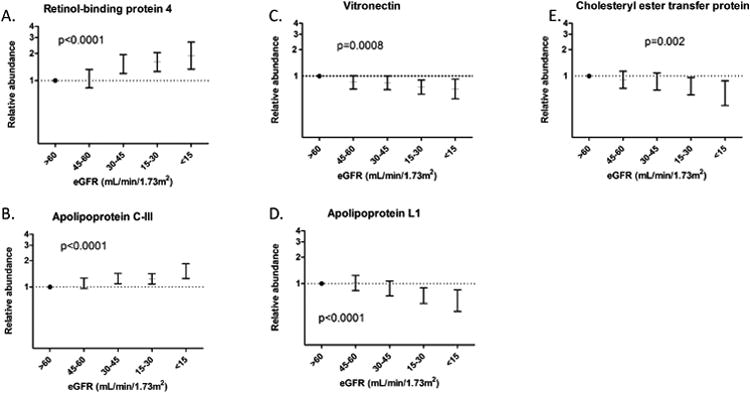
Relative concentrations in high- density lipoprotein (HDL) [Mean (95% CI)] are shown for each range of estimated glomerular filtration rate (eGFR) for retinol-binding protein (A), apolipoprotein C-III (B), vitronectin (C), apolipoprotein L1 (D), and cholesteryl ester transfer protein (E). Subject group with an eGFR >60 mL/min/1.73m2 is used as the reference, and data are adjusted for age, race, sex, race (black vs. other), prevalent diabetes, BMI, current smoking status, statin use, HDL, total cholesterol, triglycerides, and albuminuria. P-values represent results of linear regression analysis with a threshold for significance of p<0.0015.
To assess possible interactions between eGFR and other clinical variables, subgroup analyses were performed to compare associations between eGFR and HDL protein abundance for subjects with and without current statin use, existing diabetes, or a history of coronary artery disease (CAD) (Supplemental Table 2). No significant interactions were found between these subgroups and the associations between eGFR and HDL protein abundance.
Further characterization of apoLl associations by race
The concentration of apoL1 in HDL was lower in black compared to non-black participants (Figure 4A). However, the association of lower eGFR with lower HDL-associated apoL1 did not differ as a function of race (Figure 4B). There was no statistical interaction between race and the association of eGFR with the amount of apoL1 in HDL.
Figure 4. HDL-associated apoL1 in blacks and whites.
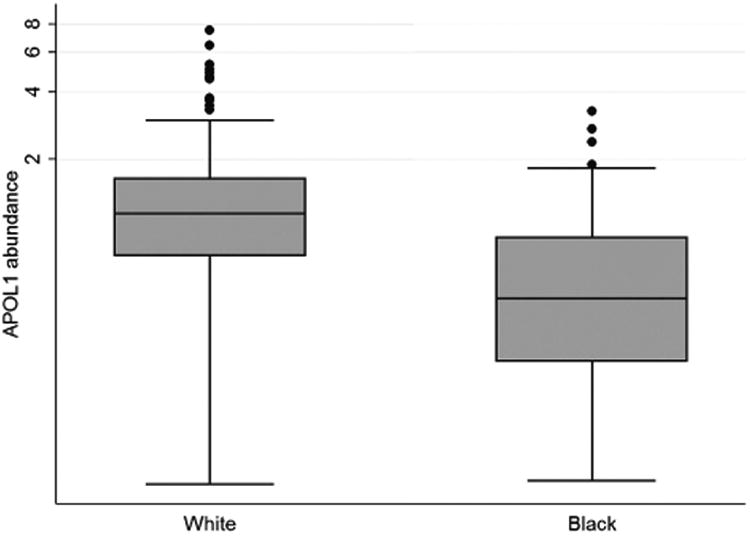
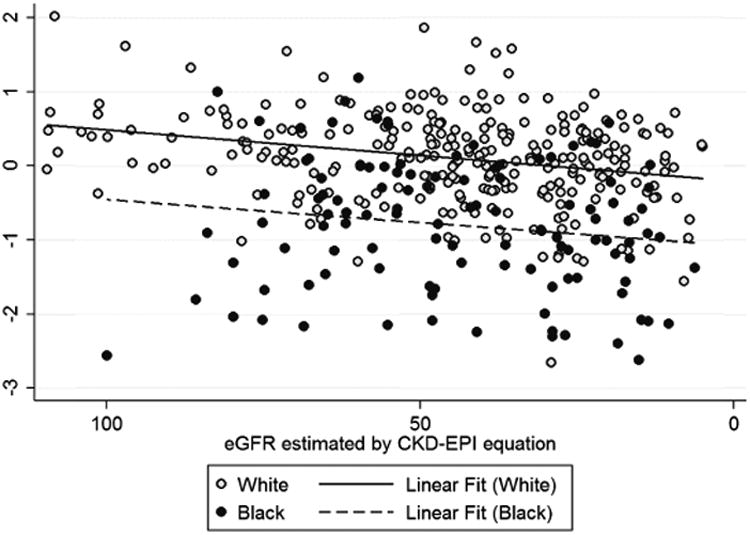
Box and whisker plots are shown for apolipoprotein L1 (apoL1) in high-density lipoprotein (HDL) for self-identified blacks and whites (A). Correlation between apoL1 in HDL and estimated glomerular filtration rate (eGFR) is shown for the Seattle Kidney Study (SKS) population, with the results of linear regression shown for blacks (dotted line) and for other races (solid line) (B).
Associations of albuminuria with proteins in HDL
On further analysis of the targeted quantification of HDL-associated proteins, evaluating for associations with albuminuria, only apoL1 met statistical significance after accounting for multiple testing (Figure 5). Specifically, each doubling of the urinary albumin:creatinine ratio was associated with a 4% lower apoL1 concentration in HDL (95% CI 2-6% lower, p<0.001). This association remained unchanged in magnitude and statistically significant (p=0.0008) after adjustment for demographic variables, BMI, smoking, diabetes, statin use, and serum lipid levels. However, the association lost significance after correction for eGFR (0.98, 95% CI 0.96-1.00, p=0.0525).
Figure 5. Alterations in HDL protein abundance associated with changes in albumin: creatinine ratio.
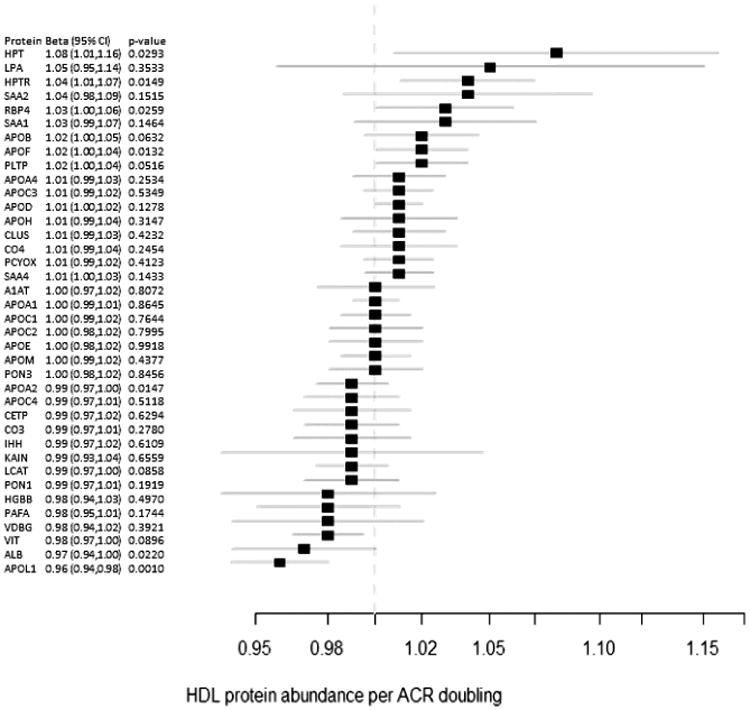
Fold enrichment for each protein in high-density lipoprotein (HDL) vs. increase in alubuminxreatinine ratio (ACR) is shown with a square. Solid lines denote 95% CI. RBP4=retinol- binding protein 4, HPT=haptoglobin, LPA=apolipoprotein(a), APO=apolipoprotein, HPTR=haptoglobin- related protein, SAA=serum amyloid A, KAIN=kallistatin, A1AT=α-1-antitrypsin, PAFA=platelet- activating factor acetylhydrolase, PON3=paraoxonase 3, CLUS=clusterin, CO4=complement C4, PLTP=phospholipid transfer protein, IHH=indian hedgehog protein, PCYOX=prenylcysteine oxidase 1, VDBG=vitamin D-binding globulin, ALB=albumin, CO3=complement C3, LCAT= phosphatidylcholine- sterol acyltransferase, HGBB=hemoglobin beta, PON1=paraoxonase 1, VIT=vitronectin, CETP=cholesteryl ester transfer protein
To further evaluate possible associations between HDL protein abundance and severity of albuminuria, subjects were classified as having no significant albuminuria (albumin:creatinine ratio <30 mg/gm), moderate albuminuria (albumin:creatinine ratio of 30-300 mg/gm), or severe albuminuria (albumin:creatinine ratio >300 mg/gm). After adjustment for demographic variables, BMI, smoking, diabetes, statin use, and serum lipid levels, no associations between albuminuria and HDL protein abundance achieved statistical significance using the modified p-value threshold of <0.0015 (Supplemental Table 3).
Discussion
Among patients with a broad range of eGFR, lower eGFR was associated with higher abundance of apoC- III and RBP4 and lower abundance of apoLl and vitronectin in HDL particles. These associations were graded across the spectrum of eGFR, independent of important clinical characteristics, and observed in the absence of significant differences in serum HDL cholesterol concentration, as measured using routine clinical assays. Black participants had a lower abundance of apoLl in HDL particles compared with white participants, but the association of eGFR with apoLl was evident regardless of race. Albuminuria was associated with decreased HDL apoLl content, but this association was not significant after adjusting for eGFR, and albuminuria was not significantly associated with other HDL protein constituents.
Prior work has examined plasma levels and enzyme activity of HDL-associated proteins in patients with ESRD,11-14 but only a few studies to date have quantified proteins from isolated HDL, and most have focused on patients undergoing dialysis. Relative to healthy controls, HD patients exhibited HDL enrichment in serum amyloid A1 (SAA1), apolipoprotein A-IV (apoA-IV), and apoC-III in association with reduced HDL cholesterol efflux capacity.9 HDL enrichment in SAA1 was shown in 2 other cohorts of hemodialysis (HD) patients15, 16 and associated with adverse cardiovascular outcomes in a population of HD patients with type 2 diabetes mellitus (T2DM).17 Some changes in HDL composition and function appear to vary as a consequence of dialysis modality10 and may fail to normalize even after kidney transplant.18 In another prior study that compared HD patients and healthy controls, HDL enrichment in apoC-III, apoA-IV, RBP4 and haptoglobin-related protein were evident in conjunction with depletion of apoL1 and paraoxonase-1 (PON 1),19 consistent with our results. Our analyses employed a validated assay that differed from the one used in the prior study and included a much larger sample size, enabling analysis of a broad range of pre-dialysis CKD with adjustment for clinical covariates and parallel assessment of both eGFR and albuminuria.
Of HDL-associated proteins, apoC-III is among the best studied to date. ApoC-III acts as an inhibitor of lipoprotein lipase and thereby increases plasma triglyceride levels through decreased catabolism and clearance of triglyceride-rich lipoproteins. The potential for a pro-atherogenic function of apoC-III recently was supported by findings that loss-of-function mutations in apoC-III were associated with lower serum triglyceride levels and reduced CVD risk.20 These findings lend further evidence that changes in apoC-III metabolism and function may contribute to dyslipidemia in CKD patients, including the significant changes observed with respect to triglyceride-rich lipoprotein particles and HDL,21, 22 and, further, could be a candidate mediator in the accelerated atherosclerosis seen in this patient population. Notably, subgroup analysis did not show an interaction between eGFR and CVD prevalence for HDL apoC-III abundance. Whereas this analysis supports an association between eGFR and HDL-associated apoC-III independent of clinical CVD status, it does not directly address whether an association exists between HDL apoC-III and CVD. To specifically address this possible relationship, prospective studies are essential to determine whether HDL apoC-III abundance predicts atherosclerotic plaque progression and/or incident cardiovascular events in patients with CKD.
The roles of RBP4 in lipid and lipoprotein metabolism have not been clearly defined, and both atherogenic and cardioprotective effects of this protein have been posited. In aggregate findings from 5 inbred mouse strains, HDL-associated RBP4 exhibited a strong, positive correlation with HDL cholesterol efflux23 from macrophages via non-ABCAl-dependent pathways.24 However, in rats, RBP4 has been shown to increase proliferation of arterial smooth muscle cells, findings that could suggest a contributory role in atherosclerosis.25,26 Recent clinical data demonstrated that among patients with T2DM, higher plasma RBP4 levels associated with the presence of carotid artery atherosclerosis (CAD).27 Higher serum RBP4 levels also were found among patients with CAD and hyperinsulinemia compared to healthy controls,28 and a polymorphism in the RBP4 gene associated with the presence though not severity of CAD.29 Thus, further work is needed to better assess the possible atherogenic roles of HDL- associated RBP4 in patients both with and without CKD.
ApoL1 is a widely expressed protein synthesized in endothelial cells, liver, pancreas, lung, and placenta.30, 31 In plasma, apoL1 predominantly associates with HDL particles,31 specifically the HDL3 subfraction,32 and functions as part of host defense against trypanosome infection.33,34 The role of apoL1 in lipid and lipoprotein metabolism is not well understood. Plasma concentrations of apoL1 have been shown to correlate with plasma triglyceride concentrations,35, 36 and genetic variants in apoL1 may associate with HDL particle size distribution, though findings have been inconsistent to date.37, 38 ApoL1 signaling also may be dependent on its lipid-binding activity.39
Single nucleotide polymorphisms in apoL1 have been identified that confer markedly increased risk of progressive nondiabetic CKD and ESRD in patients of West African descent.40-43 Interestingly, whereas CKD risk conferred by apoL1 variants appears exclusive to patients of African descent,42 our analyses showed no interaction between race and the association between eGFR and apoL1 abundance in HDL.
In prior studies, plasma apoL1 levels did not correlate with apoL1 genotype, severity of CKD, or albuminuria.44, 45 In one of these studies, plasma levels of apoL1 were actually higher in black participants than in those of European or Hispanic descent.45 Importantly, our study differs from these previous studies because we quantified the amount of apoL1 in isolated HDL, rather than the total amount of apoL1 in plasma. Therefore, we have captured apoL1 abundance as a fraction of total HDL protein mass. As a result, these previous studies indicate that our observations for HDL-associated apoL1 cannot be explained as simply a reflection of differences in total plasma apoL1 levels. A potential explanation for all of these findings is that genetic variation in apoL1 could alter its interaction with HDL particles. Both the G1 and G2 apoL1 genotypes change the conformation of the apoL1 protein near the C-terminus and prevent apoL1 binding by the serum resistance antigen (SRA), a parasite protein that abolishes apoLl-mediated trypanosome lysis.46 Therefore, apoLl genetic variants may produce conformational changes that specifically reduce HDL association without changing total plasma levels.
The possibility of a direct, causal relationship between apoLl and kidney function is supported by preclinical data. For example, in a zebrafish model, apoLl knockdown resulted in increased glomerular permeability mediated at least in part by reduced nephrin expression.47 Further, transgenic mice that expressed 2 apoLl high-risk alleles developed loss of podocytes with age,48 and high-risk variants caused podocyte necrosis at a lower threshold concentration.49 An additional study has demonstrated that apoLl signaling can stimulate cell death irrespective of genetic variants.50 A direct relationship between apoLl and kidney function is further supported by the fact that donor but not recipient apoLl genotype predicts graft failure in patients who have undergone kidney transplant.51,52 Collectively, these findings underscore the need for clearer delineation of the relationship between apoLl protein abundance and genotype, as well as their respective effects on protein function and kidney disease progression. Thus, a pivotal facet of future work will be determining apoLl genotype for the SKS subject cohort and evaluating associations among HDL apoLl content, apoLl genotype, and kidney disease progression over the study duration.
Future work also will be needed to elucidate the mechanisms underlying these changes in HDL protein cargo. The possibility exists that these changes reflect altered HDL metabolism and changes in lipid content of HDL particles; thus, smaller, dense HDL particles have been observed in the setting of CKD, and this change in HDL particle density could change the population of HDL particles isolated through ultracentrifugation, with loss of very small, apoA-I containing particles. This loss of apoA-I could lead to increases in the abundance of other proteins including RBP4 and apoC-III. However, if the findings were solely due to loss of apoA-I, we would expect nearly uniform increases in other HDL-associated proteins, which we did not observe. The selective increase in HDL RBP4 may reflect that plasma levels of this protein are elevated in CKD roughly 2-fold,53 a comparable difference to what we observed in HDL- associated RBP4. HDL enrichment in apoC-III could reflect changes in the apoC-III exchange among different lipoprotein species, specifically transfer from VLDL to HDL, as has been observed in lipoprotein lipase- and hepatic lipase-mediated lipolysis of VLDL.54, 55 Another potential explanation for our observed differences in HDL proteins is that the accumulation of urea, advanced glycation endproducts, and other small molecules due to the metabolic derangements of CKD alter the binding affinity of some apolipoproteins for HDL. This could account, for example, for the decrease in HDL- associated apoLl we observed. Notably, no significant associations were observed between HDL protein abundance and albuminuria, a finding potentially in contrast with prior descriptions of extensive HDL protein remodeling in the setting of nephrotic syndrome.56, 57 It is possible, therefore, that changes in HDL protein composition only become evident with nephrotic range proteinuria. Alternatively, HDL remodeling in nephrotic syndrome may not solely be due to albuminuria but also may result from hypoalbuminemia or other components of this syndrome, as has been previously suggested.56
A primary limitation of these analyses is that they entail observational, cross-sectional data and, therefore, do not clearly demonstrate causality in the relationship between eGFR and HDL protein composition. Nonetheless, the observed progressive differences in the abundance of several HDL-associated proteins with lower eGFR after adjustment for important clinical covariates supports reduced GFR as a key determinant of these observed changes in HDL composition. Critically, too, the functional significance of these compositional differences in HDL must be determined. Finally, as HDL phospholipid and sphingolipid cargo similarly may influence particle function and vary across disease states,58-60 quantifying changes in the HDL lipidome across the spectrum of clinical CKD is an important area of future investigation. Nonetheless, strengths of our analyses include use of a validated, targeted mass spectrometry platform to quantify the protein content of isolated HDL; the size of the cohort that permitted adjustments to reduce the risk of confounding demographic and clinical variables; and stringent criteria for statistical significance through a modified Bonferroni correction.
In aggregate, our findings suggest that modest alterations in HDL protein composition are another facet of the metabolic derangements attendant to loss of eGFR. Given the magnitude of the alterations, it is unlikely that they are causal with respect to declining kidney function. Additional work is required to reproduce these findings and to determine if our observed changes in HDL protein composition reflect a mechanism underlying increased triglycerides and reduced HDL cholesterol in CKD. Further, additional studies are necessary to establish if the observed modest changes in the HDL proteome are associated with HDL function, for example cholesterol efflux from lipid-laden macrophages, or if they are predictive of incident cardiovascular disease in patients with CKD.
Methods
Study subjects
Secondary analyses were performed for plasma samples collected through the Seattle Kidney Study (SKS).61 The SKS recruited 590 patients from outpatient clinics in the Seattle, WA, area with either an eGFR ≤ 90 mL/min/1.73m2 or a urinary albumin:creatinine ratio ≥ 30 mg/g. Anticipated initiation of dialysis within 3 months was an exclusion criterion. Complete inclusion and exclusion criteria for both studies have been described elsewhere.61 The creatinine-cystatin C equation (CKI-EPI 2012) was used to calculate eGFR.62 Complete data for all covariates were available for 425 subjects.
Laboratory assessments
Serum concentrations of creatinine, total cholesterol, HDL cholesterol, triglycerides, and C-reactive protein, as well as urine concentrations of albumin and creatinine were determined using a standard clinical chemistry analyzer at the University of Washington Kidney Research Institute (Beckman Coulter DxC).
HDL isolation and quantification of HDL-associated proteins
Complete details for the purification of HDL particles and the quantification of proteins in purified HDL by targeted liquid chromatography-tandem mass spectrometry are provided in the Supplemental Material.
Briefly, the HDL fraction of plasma (d=1.063-1.210 g/mL) was purified using a two-step density gradient ultracentrifugation with potassium bromide (KBr). First, all lipoproteins were floated using a KBr solution of 1.210 g/mL and transferred to a new tube. Second, all lipoproteins less dense than HDL were floated using a KBr solution of 1.063 g/mL. The lipoproteins at the bottom of each sample after this second step were dialyzed and frozen at -80°C before use. Median storage time for all samples was 6.03 years with a range of 2.45-8.36 years. The significant associations found between eGFR and the abundance of 5 HDL-associated proteins were not affected by adjustment for freeze time (Supplemental Table 4). No associations were evident between sample freeze time and HDL protein abundance (Supplemental Table 5). Internal standard (15N-labeled apoA-I) was added to each sample and the lipoproteins were denatured using Rapigest (Waters), reduced with dithiothreitol, alkylated using iodoacetamide, and digested with trypsin. The Rapigest, particulates, and phospholipids were removed with a phospholipid removal plate (Phenomenex) prior to being analyzed by nanoflow-liquid chromatography-tandem mass spectrometry on a Q-Exactive mass spectrometer (Thermo).
Data reduction
Peak areas for each endogenous peptide were normalized to internal standard peptides from stable isotope labeled internal standard protein to generate a peak area ratio for each peptide in each sample. Peak area ratios for each protein were averaged and protein peak area ratios were normalized to protein peak area ratios for calibrator samples included in each digestion batch. The resulting calibrated protein peak area ratios were used as relative concentrations of each protein in HDL in each sample.
Statistical analyses
To evaluate the relationship between HDL protein abundance and eGFR, linear regression analyses were performed with each 15 mL/min/1.73m2 decrement in eGFR as the exposure and log-transformed HDL protein abundance as the outcome. The p-value threshold for statistical significance was established as p<0.0015, based on a Bonferroni correction with 33.7 effective tests as determined by the Cheverud method.63
Supplementary Material
Acknowledgments
Sources of Support: This work was supported by NIH/NHLBI (R01HL111375 to ANH) and the University of Washington Nutrition Obesity Research Center P30DK035816.
Footnotes
Disclosures: The authors have no competing financial interests to disclose.
Supplementary information is available at Kidney International’s website.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harper CR, Jacobson TA. Managing dyslipidemia in chronic kidney disease. J Am Coll Cardiol. 2008;51:2375–2384. doi: 10.1016/j.jacc.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 2.Krane V, Wanner C. Statins, inflammation and kidney disease. Nat Rev Nephrol. 2011;7:385397. doi: 10.1038/nrneph.2011.62. [DOI] [PubMed] [Google Scholar]
- 3.de Boer IH, Astor BC, Kramer H, et al. Mild elevations of urine albumin excretion are associated with atherogenic lipoprotein abnormalities in the Multi-Ethnic Study of Atherosclerosis (MESA) Atherosclerosis. 2008;197:407–414. doi: 10.1016/j.atherosclerosis.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Boer IH, Brunzell JD. HDL in CKD: how good is the “good cholesterol?”. J Am Soc Nephrol. 2014;25:871–874. doi: 10.1681/ASN.2014010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zewinger S, Speer T, Kleber ME, et al. HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. J Am Soc Nephrol. 2014;25:1073–1082. doi: 10.1681/ASN.2013050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowe B, Xie Y, Xian H, et al. High Density Lipoprotein Cholesterol and the Risk of All-Cause Mortality among U.S. Veterans. Clin J Am Soc Nephrol. 2016 doi: 10.2215/CJN.00730116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shroff R, Speer T, Colin S, et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J Am Soc Nephrol. 2014;25:2658–2668. doi: 10.1681/ASN.2013111212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speer T, Rohrer L, Blyszczuk P, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity. 2013;38:754–768. doi: 10.1016/j.immuni.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Holzer M, Birner-Gruenberger R, Stojakovic T, et al. Uremia alters HDL composition and function. J Am Soc Nephrol. 2011;22:1631–1641. doi: 10.1681/ASN.2010111144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holzer M, Schilcher G, Curcic S, et al. Dialysis Modalities and HDL Composition and Function. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2014030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cacciagiú LD, González AI, Gomez Rosso L, et al. HDL-associated enzymes and proteins in hemodialysis patients. Clin Biochem. 2012;45:243–248. doi: 10.1016/j.clinbiochem.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 12.Pahl MV, Ni Z, Sepassi L, et al. Plasma phospholipid transfer protein, cholesteryl ester transfer protein and lecithin:cholesterol acyltransferase in end-stage renal disease (ESRD) Nephrol Dial Transplant. 2009;24:2541–2546. doi: 10.1093/ndt/gfp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlitt A, Heine GH, Jiang XC, et al. Phospholipid transfer protein in hemodialysis patients. Am J Nephrol. 2007;27:138–143. doi: 10.1159/000099943. [DOI] [PubMed] [Google Scholar]
- 14.Dirican M, Akca R, Sarandol E, et al. Serum paraoxonase activity in uremic predialysis and hemodialysis patients. J Nephrol. 2004;17:813–818. [PubMed] [Google Scholar]
- 15.Weichhart T, Kopecky C, Kubicek M, et al. Serum amyloid A in uremic HDL promotes inflammation. J Am Soc Nephrol. 2012;23:934–947. doi: 10.1681/ASN.2011070668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mangé A, Goux A, Badiou S, et al. HDL proteome in hemodialysis patients: a quantitative nanoflow liquid chromatography-tandem mass spectrometry approach. PLoS ONE. 2012;7:e34107. doi: 10.1371/journal.pone.0034107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopecky C, Genser B, Drechsler C, et al. Quantification of HDL proteins, cardiac events, and mortality in patients with type 2 diabetes on hemodialysis. Clin J Am Soc Nephrol. 2015;10:224231. doi: 10.2215/CJN.06560714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopecky C, Haidinger M, Birner-Grünberger R, et al. Restoration of renal function does not correct impairment of uremic HDL properties. J Am Soc Nephrol. 2015;26:565–575. doi: 10.1681/ASN.2013111219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao B, de Boer I, Tang C, et al. A Cluster of Proteins Implicated in Kidney Disease Is Increased in High-Density Lipoprotein Isolated from Hemodialysis Subjects. J Proteome Res. 2015;14:2792–2806. doi: 10.1021/acs.jproteome.5b00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. doi: 10.1056/NEJMoa1307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan DT, Dogra GK, Irish AB, et al. Chronic kidney disease delays VLDL-apoB-100 particle catabolism: potential role of apolipoprotein C-III. J Lipid Res. 2009;50:2524–2531. doi: 10.1194/jlr.P900003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ooi EM, Chan DT, Watts GF, et al. Plasma apolipoprotein C-III metabolism in patients with chronic kidney disease. J Lipid Res. 2011;52:794–800. doi: 10.1194/jlr.M011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng C, Khoo C, Furtado J, et al. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation. 2010;121:1722–1734. doi: 10.1161/CIRCULATIONAHA.109.875807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pamir N, Hutchins P, Ronsein G, et al. Proteomic analysis of HDL from inbred mouse strains implicates APOE associated with HDL in reduced cholesterol efflux capacity via the ABCA1 pathway. J Lipid Res. 2016;57:246–257. doi: 10.1194/jlr.M063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li F, Xia K, Sheikh MS, et al. Retinol binding protein 4 promotes hyperinsulinism-induced proliferation of rat aortic smooth muscle cells. Mol Med Rep. 2014;9:1634–1640. doi: 10.3892/mmr.2014.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li F, Xia K, Sheikh SA, et al. Involvement of RBP4 in hyperinsulinism-induced vascular smooth muscle cell proliferation. Endocrine. 2015;48:472–482. doi: 10.1007/s12020-014-0304-0. [DOI] [PubMed] [Google Scholar]
- 27.Feng S, Zhu Y, Yan C, et al. Retinol binding protein 4 correlates with and is an early predictor of carotid atherosclerosis in type 2 diabetes mellitus patients. J Biomed Res. 2015;29 doi: 10.7555/JBR.29.20140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li F, Xia K, Li C, et al. Retinol-binding protein 4 as a novel risk factor for cardiovascular disease in patients with coronary artery disease and hyperinsulinemia. Am J Med Sci. 2014;348:474–479. doi: 10.1097/MAJ.0000000000000347. [DOI] [PubMed] [Google Scholar]
- 29.Wan K, Zhao J, Deng Y, et al. A genetic polymorphism in RBP4 is associated with coronary artery disease. Int J Mol Sci. 2014;15:22309–22319. doi: 10.3390/ijms151222309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monajemi H, Fontijn RD, Pannekoek H, et al. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics. 2002;79:539–546. doi: 10.1006/geno.2002.6729. [DOI] [PubMed] [Google Scholar]
- 31.Duchateau PN, Pullinger CR, Cho MH, et al. Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J Lipid Res. 2001;42:620–630. [PubMed] [Google Scholar]
- 32.Davidson WS, Silva RA, Chantepie S, et al. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29:870–876. doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanhollebeke B, Pays E. The trypanolytic factor of human serum: many ways to enter the parasite, a single way to kill. Mol Microbiol. 2010;76:806–814. doi: 10.1111/j.1365-2958.2010.07156.x. [DOI] [PubMed] [Google Scholar]
- 34.Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422:83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 35.Albert TS, Duchateau PN, Deeb SS, et al. Apolipoprotein L-I is positively associated with hyperglycemia and plasma triglycerides in CAD patients with low HDL. J Lipid Res. 2005;46:469–474. doi: 10.1194/jlr.M400304-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Duchateau PN, Movsesyan I, Yamashita S, et al. Plasma apolipoprotein L concentrations correlate with plasma triglycerides and cholesterol levels in normolipidemic, hyperlipidemic, and diabetic subjects. J Lipid Res. 2000;41:1231–1236. [PubMed] [Google Scholar]
- 37.Gutiérrez OM, Judd SE, Irvin MR, et al. APOL1 nephropathy risk variants are associated with altered high-density lipoprotein profiles in African Americans. Nephrol Dial Transplant. 2016;31:602–608. doi: 10.1093/ndt/gfv229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freedman BI, Langefeld CD, Murea M, et al. Apolipoprotein L1 nephropathy risk variants associate with HDL subfraction concentration in African Americans. Nephrol Dial Transplant. 2011;26:3805–3810. doi: 10.1093/ndt/gfr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan G, Zhaorigetu S, Liu Z, et al. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283:21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoLl variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peralta CA, Bibbins-Domingo K, Vittinghoff E, et al. APOL1 Genotype and Race Differences in Incident Albuminuria and Renal Function Decline. J Am Soc Nephrol. 2016;27:887–893. doi: 10.1681/ASN.2015020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol. 2013;24:1484–1491. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bruggeman LA, O’Toole JF, Ross MD, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol. 2014;25:634–644. doi: 10.1681/ASN.2013070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozlitina J, Zhou H, Brown PN, et al. Plasma Levels of Risk-Variant APOL1 Do Not Associate with Renal Disease in a Population-Based Cohort. J Am Soc Nephrol. 2016 doi: 10.1681/ASN.2015101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog. 2009;5:e1000685. doi: 10.1371/journal.ppat.1000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kotb AM, Simon O, Blumenthal A, et al. Knockdown of ApoL1 in Zebrafish Larvae Affects the Glomerular Filtration Barrier and the Expression of Nephrin. PLoS ONE. 2016;11:e0153768. doi: 10.1371/journal.pone.0153768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruggeman LA, Wu Z, Luo L, et al. APOL1-G0 or APOL1-G2 Transgenic Models Develop Preeclampsia but Not Kidney Disease. J Am Soc Nephrol. 2016 doi: 10.1681/ASN.2015111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lan X, Jhaveri A, Cheng K, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307:F326–336. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhaorigetu S, Wan G, Kaini R, et al. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant. 2012;12:19241928. doi: 10.1111/j.1600-6143.2012.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11:1025–1030. doi: 10.1111/j.1600-6143.2011.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jing J, Isoherranen N, Robinson-Cohen C, et al. Chronic Kidney Disease Alters Vitamin A Homeostasis via Effects on Hepatic RBP4 Protein Expression and Metabolic Enzymes. Clin Transl Sci. 2016;9:207–215. doi: 10.1111/cts.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murdoch SJ, Breckenridge WC. Influence of lipoprotein lipase and hepatic lipase on the transformation of VLDL and HDL during lipolysis of VLDL. Atherosclerosis. 1995;118:193212. doi: 10.1016/0021-9150(95)05606-8. [DOI] [PubMed] [Google Scholar]
- 55.Murdoch SJ, Breckenridge WC. Effect of lipid transfer proteins on lipoprotein lipase induced transformation of VLDL and HDL. Biochim Biophys Acta. 1996;1303:222–232. doi: 10.1016/0005-2760(96)00105-1. [DOI] [PubMed] [Google Scholar]
- 56.Vaziri ND. Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int. 2016;90:41–52. doi: 10.1016/j.kint.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaziri ND. HDL abnormalities in nephrotic syndrome and chronic kidney disease. Nat Rev Nephrol. 2016;12:37–47. doi: 10.1038/nrneph.2015.180. [DOI] [PubMed] [Google Scholar]
- 58.Annema W, von Eckardstein A. High-density lipoproteins Multifunctional but vulnerable protections from atherosclerosis. Circ J. 2013;77:2432–2448. doi: 10.1253/circj.cj-13-1025. [DOI] [PubMed] [Google Scholar]
- 59.Kontush A, Lhomme M, Chapman MJ. Unraveling the complexities of the HDL lipidome. J Lipid Res. 2013;54:2950–2963. doi: 10.1194/jlr.R036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stahlman M, Fagerberg B, Adiels M, et al. Dyslipidemia, but not hyperglycemia and insulin resistance, is associated with marked alterations in the HDL lipidome in type 2 diabetic subjects in the DIWA cohort: impact on small HDL particles. Biochim Biophys Acta. 2013;1831:16091617. doi: 10.1016/j.bbalip.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 61.Bosworth CR, Levin G, Robinson-Cohen C, et al. The serum 24,25-dihydroxyvitamin D concentration, a marker of vitamin D catabolism, is reduced in chronic kidney disease. Kidney Int. 2012;82:693–700. doi: 10.1038/ki.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Inker LA, Schmid CH, Tighiouart H, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012;367:20–29. doi: 10.1056/NEJMoa1114248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheverud JM, Vaughn TT, Pletscher LS, et al. Genetic architecture of adiposity in the cross of LG/J and SM/J inbred mice. Mamm Genome. 2001;12:3–12. doi: 10.1007/s003350010218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.