

Abstract
Bimetallic complexes are important sites in metalloproteins but are often difficult to prepare synthetically. We have previously introduced an approach to form discrete bimetallic complexes with MII–(μ-OH)–FeIII (MII = Mn, Fe) cores using the tripodal ligand N,N′,N″-[2,2′,2″-nitrilotris(ethane-2,1-diyl)]tris(2,4,6-trimethylbenzenesulfonamido) ([MST]3−). This series is extended to include the rest of the late 3d transition metal ions (MII = Co, Ni, Cu, Zn). All the bimetallic complexes have similar spectroscopic and structural properties that reflect little change despite varying the MII centers. Magnetic studies performed on the complexes in solution using electron paramagnetic resonance spectroscopy showed that the observed spin states varied incrementally from S = 0 through S = 5/2; these results are consistent with the antiferromagnetic coupling between the high-spin MII and FeIII centers. However, the difference in the MII ion occupancy yielded only slight changes in the magnetic exchange coupling strength, and all complexes had J-values ranging from +26(4) to +35(3) cm−1.
Keywords: Bimetallic complexes
Graphical Abstract
Discrete bimetallic complexes with MII–(μ-OH)–FeIII (MII = Mn, Fe, Co, Ni, Cu, Zn) cores have been prepared and X-ray diffraction studies found that each had similar overall structures. The properties of the complexes were examined which allowed for an evaluation on the effects of the divalent metal ions. For instance, electron paramagnetic resonance spectroscopy investigations revealed that the observed overall spin states (Sc) varied incrementally from Sc = 0 through Sc = 5/2 that is consistent with antiferromagnetic coupling between the high-spin MII and FeIII centers.
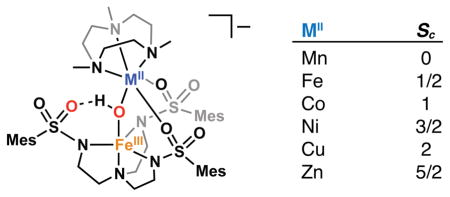
INTRODUCTION
Metalloenzymes containing bimetallic transition metal active sites perform many important functions and reactions in biology. Examples of such active sites include the MnFe site of ribonucleoide reductase which mediates the synthesis of deoxyribonucleotides,1 the FeFe site of hemerytherin which transports O2 in certain invertebrates,2 the NiFe site of [NiFe] hydrogenase which reversibly forms H2 from protons,3 and the ZnFe site of purple acid phosphatase which degrades organophosphates.4 The two metal centers in bimetallic active sites often have different primary coordination spheres and are bridged by one or more oxido or hydroxido ligands. Synthetic bimetallic species have been prepared as structural or functional models for these types of metalloenzymes. Though many of these synthetic bimetallic compounds contain symmetrical coordination sites for the two metals centers due to the use of symmetric dinucleating ligands,5–16 several groups have been able to prepare synthetically challenging bimetallic complexes with unsymmetrical coordination sites.17–29
We have previously shown that the tripodal ligand N,N′,N″-[2,2′,2″-nitrilotris(ethane-2,1-diyl)]tris(2,4,6-trimethylbenzenesulfonamido) ([MST]3−) can be used to prepare monometallic complexes with various metals.30–36 These monomeric species can be oxidized with O2 in the presence of a second metal ion (MII) and a capping ligand (L) to form bimetallic species of the formulation [(L)MII–(μ-OH)–MIIIMST]+ (Figure 1). The two metal centers have different coordination environments and are bridged by a hydroxido ligand formed via the activation of O2. In our initial reports, we showed that redox-inactive metals capped by crown ethers could be installed into the auxiliary coordination site to form heterobimetallic cores of the formulation MII–(μ-OH)–MIII (MII = Ca, Sr, Ba and MIII = Mn, Fe, Figure 1A).30,31,37 Formation of these complexes provided a means to examine the effects of redox inactive metal ions on the chemical properties of Mn and Fe complexes. For instance, similar reduction potentials were uncovered for heterobimetallic complexes containing CaII and SrII ions, contrasting with the more negative potential for the system containing the BaII ion. We have further demonstrated that transition metal ions may also be bound in the auxiliary coordination site when capped with the ligand 1,4,7-trimethyl-1,4,7-triazacyclononane (TMTACN) to form MII–(μ-OH)–MIII cores (MIIMIII = MnIIMnIII, MnIIFeIII, FeIIFeIII, Figure 1B).38
Figure 1.

Examples of bimetallic systems using the ligand [MST]3− with (A) redox-inactive metal ions and (B) transition metal ions.
In this report, we incorporated several other late first-row transition metal ions into the auxiliary coordination site, to prepare a series of [(TMTACN)MII–(μ-OH)–FeIIIMST]+ complexes (MII = Co, Ni, Cu, Zn, denoted as [MII(OH)FeIII]+). This family of compounds, including the previously synthesized [MnII(OH)FeIII]+ and [FeII(OH)FeIII]+ complexes, have similar bimetallic molecular structures as determined by X-ray diffraction methods. These [MII(OH)FeIII]+ complexes thus allow us to systematically probe the effects of binding an additional transition metal ion to an FeIII–OH complex. Our findings indicate that the [MII(OH)FeIII]+ cores are maintained in solution and that the complexes have similar optical, vibrational and electrochemical properties. Furthermore, magnetic studies show that each metal ion is high spin with the total ground spin states varying from S = 0 for [MnII(OH)FeIII]+ to S = 5/2 for [ZnII(OH)FeIII]+ owing to a moderate antiferromagnetic coupling between the two metal centers. These results demonstrate that the binding of a second transition metal has little change in the properties of the [MII(OH)FeIII]+ complexes, including the magnetic coupling within the [MII(μ-OH)FeIII] core.
EXPERIMENTAL SECTION
General Methods
All reagents were purchased from commercial sources and used as received, unless otherwise noted. Solvents were sparged with argon and dried over columns containing Q-5 and molecular sieves. Sodium hydride (NaH) as a dispersion in mineral oil was filtered with a medium porosity glass-fritted funnel and washed 5 times each with pentane and diethyl ether (Et2O). Solid NaH was dried under vacuum and stored under inert atmosphere (Caution: NaH is flammable when contacted with air or water). The synthesis of the ligand was carried out in the air and preparations of the metal complexes were conducted in a Vacuum Atmospheres, Co. drybox under an argon atmosphere. Dioxygen was dried on a Drierite gas purifier purchased from Fischer Scientific. CoII(OTf)2·2MeCN,39 NiII(OTf)2·5MeCN,39 CuII(OTf)2·2MeCN,39 ZnII(OTf)2·2MeCN,39 1,4,7-trimethyl-1,4,7-triazacyclononane (TMTACN),40 NMe4[FeIIMST],37 [MnII(OH)FeIII]OTf,38 and [FeII(OH)FeIII]OTf38 were synthesized according to previous reports.
Complex Synthesis
[CoII(OH)FeIII]OTf
This salt was prepared using literature procedures for the analogous [MnII(OH)FeIII]OTf salt38 using CoII(OTf)2·2MeCN (69.3 mg, 158 mmol), TMTACN (27 mg, 16 mmol), and NMe4[FeIIMST] (132 mg, 158 mmol) to produce the desired salt with yields that range from 20–25%. Orange crystals of [CoII(OH)FeIII]OTf suitable for X-ray diffraction were grown from concentrated MeCN solutions by the vapor diffusion method with Et2O. Elemental analysis calcd. for [CoII(OH)FeIII]OTf·0.75CH2Cl2, C43.75H68.5Cl1.5CoF3FeN7O10S4: C, 43.58; H, 5.73; N, 8.13%, found: C, 43.44; H, 6.14; N, 8.00%. UV-vis (CH2Cl2, λmax, nm (εmax, M−1cm−1)) 275 (6560), 389 (4330), 470 (sh). FTIR (KBr disc, cm−1, selected bands): 3263(OH), 3023, 2974, 2936, 2914, 2866, 2827, 1603, 1560, 1492, 1467, 1400, 1383, 1364, 1337, 1270, 1221, 1146, 1123, 1098, 1074, 1054, 1030, 1010, 980, 966, 954, 935, 892, 853, 824, 781, 742, 719, 666, 637, 611, 579, 535, 517. Electrospray ionization mass spectrometry (ESI-MS) (CH2Cl2, ES+, m/z): Exact mass calcd for [CoII(OH)FeIII]+, C42H67CoFeN7O7S3: 992.3 found: 992.3. E1/2 (CH2Cl2, V versus [FeCp2]+/0): −0.89.
[NiII(OH)FeIII]OTf
This salt was prepared using the method described above for [CoII(OH)FeIII]OTf using NiII(OTf)2·5MeCN (50.0 mg, 0.0889 mmol), TMTACN (15.2 mg, 0.0887 mmol), and NMe4[FeIIMST] (72.8 mg, 0.0888 mmol) to produce the desired salt with yields ranging from 30–35%. Dark red crystals of [NiII(OH)FeIII]OTf suitable for X-ray diffraction were grown from a concentrated MeCN solution by the vapor diffusion method with Et2O. Elemental analysis calcd. for [NiII(OH)FeIII]OTf·CH2Cl2, C44H69Cl2F3FeN7NiO10S4: C, 43.08; H, 5.67; N, 7.99%, found: C, 42.92; H, 5.39; N, 8.11%. UV-vis (CH2Cl2, λmax, nm (εmax, M−1cm−1)) 275 (6570), 381 (4190), 470 (sh). FTIR (KBr disc, cm−1, selected bands): 3277(OH), 3019, 2974, 2934, 2925, 2866, 2827, 1604, 1566, 1494, 1466, 1401, 1383, 1364, 1273, 1223, 1151, 1123, 1101, 1076, 1056, 1031, 1013, 983, 967, 955, 932, 897, 852, 819, 789, 747, 720, 666, 637, 612, 581, 535, 517. ESI-MS (CH2Cl2, ES+, m/z): Exact mass calcd for [NiII(OH)FeIII]+, C42H67NiFeN7O7S3: 991.3 found: 991.3. E1/2 (CH2Cl2, V versus [FeCp2]+/0): −0.99.
[CuII(OH)FeIII]OTf
This salt was prepared using the method described above for [CoII(OH)FeIII]OTf using CuII(OTf)2·2MeCN (64.2 mg, 0.178 mmol), TMTACN (20.6 mg, 0.120 mmol), and NMe4[FeIIMST] (145.6 mg, 0.178 mmol) to produce the desired salt with yields from 20–25%. Dark red crystals of [CuII(OH)FeIII]OTf suitable for X-ray diffraction were grown by layering pentane over a concentrated CH2Cl2 solution of the salt. Elemental analysis calcd. for [CuII(OH)FeIII]OTf·0.25CH2Cl2, C43.25H67.5Cl0.5CuF3FeN7O10S4: C, 44.48; H, 5.83; N, 8.40%, found: C, 44.49; H, 5.97; N, 8.23%. UV-vis (CH2Cl2, λmax, nm (εmax, M−1cm−1)) 275 (14300), 383 (7480), 470 (sh). FTIR (KBr disc, cm−1, selected bands): 3255(OH), 3023, 2972, 2936, 2924, 2866, 2827, 1603, 1560, 1492, 1466, 1405, 1383, 1364, 1271, 1223, 1150, 1126, 1102, 1075, 1056, 1031, 1009, 981, 966, 954, 935, 893, 852, 821, 785, 743, 721, 662, 638, 611, 574, 536, 517. ESI-MS (CH2Cl2, ES+, m/z): Exact mass calcd for [CuII(OH)FeIII]+, C42H67CuFeN7O7S3: 996.3 found: 996.3. E1/2 (CH2Cl2, V versus [FeCp2]+/0): −0.87.
[ZnII(OH)FeIII]OTf
This salt was prepared using the method described above for [CoII(OH)FeIII]OTf using ZnII(OTf)2·2MeCN (79.1 mg, 0.178 mmol), TMTACN (26.6 mg, 0.155 mmol), and NMe4[MnIIMST] (146 mg, 0.178 mmol) to produce the desired salt with yields range between 25–30%. Dark red crystals of [ZnII(OH)FeIII]OTf suitable for X-ray diffraction were grown by layering pentane over a concentrated CH2Cl2 solution of the salt. Elemental analysis calcd. for [ZnII(OH)FeIII]OTf ·0.5CH2Cl2, C43.5H68ClZnF3FeN7O10S4: C, 43.87; H, 5.76; N, 8.23%, found: C, 43.45; H, 6.02; N, 8.13%. UV-vis (CH2Cl2, λmax, nm (εmax, M−1cm−1)) 275 (9410), 391 (6320), 470 (sh). FTIR (KBr disc, cm−1, selected bands): 3238(OH), 3025, 2968, 2934, 2926, 2868, 2824, 1603, 1561, 1492, 1466, 1405, 1380, 1366, 1265, 1223, 1153, 1126, 1115, 1093, 1061, 1031, 1013, 988, 969, 957, 928, 889, 850, 817, 775, 743, 721, 657, 638, 613, 574, 537, 518. ESI-MS (CH2Cl2, ES+, m/z): Exact mass calcd for [ZnII(OH)FeIII]+, C42H67ZnFeN7O7S3: 997.3 found: 997.2. E1/2 (CH2Cl2, V versus [FeCp2]+/0): −0.89.
Physical Methods
Elemental analyses were performed on a Perkin-Elmer 2400 CHNS analyzer. Electronic absorbance spectra were recorded with a Cary 50 using a 1.00 cm or 0.10 cm quartz cuvette. Fourier transform infrared (FTIR) spectra were collected on a Varian 800 Scimitar Series FTIR spectrometer in air. High-resolution mass spectra were collected using Waters Micromass LCT Premier Mass Spectrometer. Cyclic voltammetry (CV) experiments were conducted using a CH1600C electrochemical analyzer and measured on CH2Cl2 solutions of the complexes. A 2.0 mm glassy carbon electrode was used as the working electrode at a scan velocity of 0.1 V s−1 and a Pt wire were used as the auxiliary electrode. A ferrocenium/ferrocene couple ([FeCp2]+/0) was used as an internal reference to monitor the reference electrode (Ag0/+). Tetrabutylammonium hexafluorophosphate (TBAP) was use as the supporting electrolyte at a concentration of 0.1 M.
Data Collection and Analysis of EPR Measurements
X-band EPR spectra were recorded on a Bruker ELEXSYS spectrometer equipped with an Oxford ESR-910 liquid helium cryostat and a Bruker bimodal cavity for generation of microwave fields parallel and transverse to the applied magnetic field. The quantification of all signals was measured relative to a CuEDTA spin standard prepared from a copper atomic absorption standard (Sigma-Aldrich). The microwave frequency was calibrated with a frequency counter and the magnetic field with a NMR gaussmeter. The temperature was calibrated with a carbon glass resistor (Lakeshore CGR-1-1000) mounted inside an EPR tube to provide an accurate measure of the sample temperature under various conditions. A modulation frequency of 100 kHz was used for all EPR spectra. Mössbauer spectra were recorded with a Janis Research Super-Varitemp dewar. Isomer shifts are reported relative to Fe metal at 298 K.
The simulation software SpinCount was written by one of the authors.41 The software diagonalizes the electronic terms of the spin Hamiltonian:
| (1) |
with Hi = βSi·gi·B + Si·Di·Si (i=1,2) and Si·Di·Si = Di[Szi2 − Si(Si + 1)/3 + (E/D)i(Sxi2 − Syi2)] where the parameters have the usual definitions,42 and performs least-squares fitting of simulations to the spectra. The spin Si refers to individual FeIII (S = 5/2) or MII sites depending on the MII ion incorporated into the ligand. Di is assumed coaxial with gi unless specified.43 The quantitative simulations are generated with consideration of all intensity factors, which allows computation of simulated spectra for a specified sample concentration.
Crystallography
A Bruker SMART APEX II diffractometer and the APEX2 program package was used to determine the unit-cell parameters and for data collection. Crystallographic details are summarized in Supporting Information, and in Table S1.
RESULTS AND DISCUSSION
Preparation Route for the [MII(OH)FeIII]+ complexes
The preparation of the bimetallic complexes, [MII(OH)FeIII]+ was achieved via the synthetic route outlined in Scheme 1. In a typical reaction, a CH2Cl2 suspension of MII(OTf)2·xMeCN (x = 2 for all except NiII which had x = 5), TMTACN, and NMe4[FeIIMST] was treated with 0.5 equiv. of O2 for 1 h. The [MII(OH)FeIII]+ products were purified via recrystallization as microcrystalline or needle-shaped crystals from CH2Cl2–pentane mixtures, with yields of 20–50%. The formulations of these crystals as [MII(OH)FeIII]+ complexes was supported by ESI-MS, in which the molecular weight and experimental isotope patterns matched those calculated for [MII(OH)FeIII]+ (Figure 2). All the complexes have similar optical properties with a characteristic absorbance band with λmax between 380–390 nm and extension coefficients ranging from 4000–8000 M−1cm−1 (Figure 2). These bands are similar to previously reported bands observed for comparable [L⊃MII–(μ-OH)–FeIIIMST]+ complexes (L⊃MII = 15-crown-5⊃CaII, 15-crown-5⊃SrII, or 18-crown-6⊃ BaII, denoted as [C⊃MII(OH)FeIII]+).37 The mechanism that produced the [MII(μOH)FeIII] cores from dioxygen in these complexes is unknown. It is notable that all other bimetallic complexes of [MST]3− that followed this synthetic route produced similar hydroxido species.30,37,38 In these other cases, we were also not able to locate the source of the proton on hydroxido ligand but determined the source of the oxygen atom was from dioxygen.30,37
Scheme 1.

Preparation of [(TMTACN)MII–(μ-OH)–FeIIIMST]+ complexes. MII = Mn, Fe, Co, Ni, Cu, Zn, x = 2 for all syntheses except NiII where x = 5.
Figure 2.
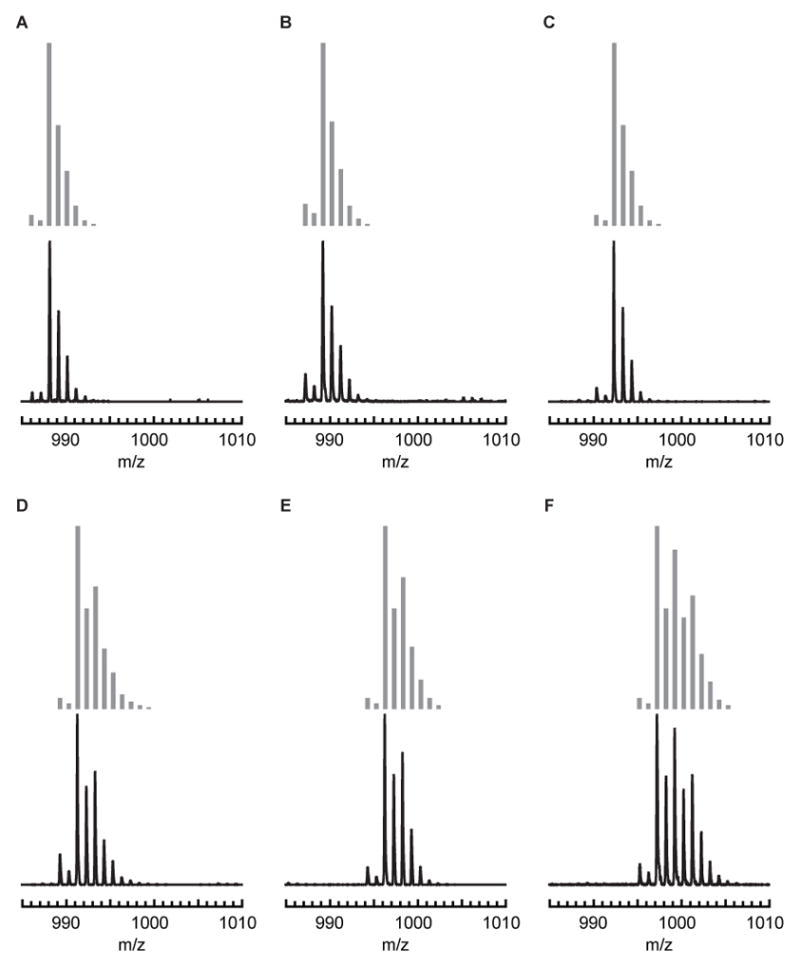
ESI-MS spectra of (A)[MnII(OH)FeIII]+,38 (B) [FeII(OH)FeIII]+,38 (C) [CoII(OH)FeIII]+, (D) [NiII(OH)FeIII]+, (E) [CuII(OH)FeIII]+, (F) [ZnII(OH)FeIII]+, with the simulated spectra in grey.
Vibrational Properties
The solid-state vibrational properties of the [MII(OH)FeIII]OTf salts were assessed using FTIR spectroscopy. As this series of complexes only differed by the identity of the MII ion, the FTIR spectra are nearly identical with ν(OH) bands observed between 3200–3300 cm−1 (Figure 3B). The energies and relative broadness of the bands suggests the presence of intramolecular H-bonds between the bridging hydroxido ligands and one of the sulfonamido oxygen atoms from [MST]3−.44,45 Within the series of [MII(OH)FeIII]+ complexes we do not observe a significant change in the energies of ν(OH) or the shape of this band (Table 1). These results imply that changes in the auxiliary transition metal ion has a limited effect on the properties of the FeIII–O(H) unit. The small range in ionic radii for the divalent metal ions46 should correlate with each having similar Lewis acidities, which can explain the minimal changes in the energy of the ν(OH). Note that [C⊃CaII(OH)FeIII] and [C⊃SrII(OH)FeIII] complexes also have a small difference in the energies of their ν(OH) bands (3369 and 3389 cm−1).37 However, energies of the ν(OH) bands for the [MII(OH)FeIII]+ complexes are all smaller than those found for [C⊃MII(OH)FeIII] complexes. We attributed this difference to the [MII(OH)FeIII]+ complexes having stronger intramolecular H-bonds than those found in [C⊃MII(OH)FeIII]. This result can be explained by the auxiliary transition metal ions in the [MII(OH)FeIII]+ complexes being stronger Lewis acids relative to alkaline earth metal ions found in [C⊃MII(OH)FeIII]+. An increase in the Lewis acidity should weaken the O–H bonds, which make the hydroxido ligands more acidic to produce stronger intramolecular H-bonds.
Figure 3.

(A) Absorbance and (B) FTIR spectra for [MnII(OH)FeIII]+ (black solid line),38 [FeII(OH)FeIII]+ (black dashed line),38 [CoII(OH)FeIII]+ (black dotted line), [NiII(OH)FeIII]+ (grey solid line), [CuII(OH)FeIII]+ (grey dashed line), (F) [ZnII(OH)FeIII]+ (grey dotted line). Absorbance measurements were performed on 0.1 mM CH2Cl2 solutions at room temperature, and FTIR measurements were performed in KBr discs.
Table 1.
Physical properties for the [MII(OH)FeIII]+ complexes.
Solid-state molecular structures of [MII(OH)FeIII]+ complexes
The molecular structures of the [MII(OH)FeIII]+ complexes were determined by X-ray diffraction methods and revealed the bimetallic core structures (Figure 3). Selected metrical parameters and calculated values are shown in Table 2. As expected, the FeIII and MII centers have different coordination geometries. The FeIII centers have a five-coordinate N4O primary coordination sphere, formed by the [MST]3− ligand and a bridging hydroxido ligand, and adopt a distorted trigonal bipyramidal (tbp) geometry. This coordination geometry was assessed using the trigonality structural parameter τ5 in which ideal tbp geometry has τ5 = 1 and ideal square pyramidal geometry has τ5 = 0:48 the τ5-values for the FeIII centers range from 0.863 to 0.745 for the series of [MII(OH)FeIII]+ complexes (Table 2). In contrast, the MII centers have a six-coordinate, N3O3 primary coordination sphere formed by two sulfonamido groups of the [MST]3− ligand, a bridging hydroxido ligand, and the TMTACN ligand. The MII centers adopt a distorted octahedral geometry based on the octahedral quadratic elongation parameter λoct = 1.010–1.036, where ideal octahedral geometry has λoct = 1 and higher values reflect greater distortion from this idealized geometry (Table 1).49 The coordination geometries of the complexes with FeII, CoII, and CuII ions exhibited a Jahn-Teller distortion, with the strongest observed in [CuII(OH)FeIII]+ with λoct = 1.036. This effect also caused elongated axial Cu1–O4 and Cu1–N5 bond distances of 2.579(5) and 2.286(6) Å that are significantly longer than the other bond distances within the CuII primary coordination sphere. In contrast, the λoct-values of 1.021 for [FeII(OH)FeIII]+ and 1.013 for [CoII(OH)FeIII]+ are intermediate compared to the rest of the series and no significantly elongated bond lengths are observed for either complex. These observations are consistent with the Jahn-Teller distortions occurring within the t2g orbitals for the FeII and CoII ions, which are not appreciably involved in bonding to the ligands.
Table 2.
Selected metrical parameters for [MII(OH)FeIII]+ complexes.
| [MII(OH)FeIII]+ | MII | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| MnII,a | FeII,a | CoII | NiII | CuII | ZnII | |
| Bond distances (Å) | ||||||
| Fe1–N1 | 2.193(2) | 2.194(2) | 2.225(1) | 2.229(2) | 2.210(6) | 2.218(3) |
| Fe1–N2 | 2.030(2) | 2.019(2) | 2.002(1) | 2.001(2) | 2.010(5) | 2.000(3) |
| Fe1–N3 | 2.007(2) | 2.001(2) | 1.994(1) | 1.994(2) | 1.993(6) | 1.990(3) |
| Fe1–N4 | 2.036(2) | 2.033(2) | 2.031(1) | 2.031(2) | 2.036(6) | 2.029(3) |
| Fe1–O1 | 1.888(1) | 1.899(2) | 1.888(1) | 1.884(2) | 1.892(5) | 1.888(3) |
| O1···O6 | 2.646(2) | 2.649(2) | 2.635(2) | 2.644(3) | 2.622(7) | 2.619(4) |
| M1–O1 | 2.048(1) | 1.986(2) | 1.968(1) | 1.970(2) | 1.905(5) | 1.945(3) |
| M1–O2 | 2.196(1) | 2.202(2) | 2.165(1) | 2.148(2) | 2.049(5) | 2.213(3) |
| M1–O4 | 2.187(1) | 2.201(2) | 2.218(1) | 2.204(2) | 2.579(5) | 2.362(3) |
| M1–N5 | 2.268(2) | 2.202(2) | 2.163(1) | 2.102(2) | 2.286(6) | 2.164(3) |
| M1–N6 | 2.276(2) | 2.207(2) | 2.138(1) | 2.084(2) | 2.028(6) | 2.119(3) |
| M1–N7 | 2.294(2) | 2.221(2) | 2.157(1) | 2.106(2) | 2.058(6) | 2.148(4) |
| Fe1···M1 | 3.447(1) | 3.425(1) | 3.412(1) | 3.416(1) | 3.422(1) | 3.441(1) |
| Avg. Fe1–Neq | 2.024(2) | 2.018(2) | 2.009(1) | 2.009(2) | 2.013(6) | 2.006(3) |
| Avg. M1–NTMTACN | 2.279(2) | 2.210(2) | 2.153(1) | 2.097(2) | 2.124(6) | 2.144(3) |
| d[Fe1–Neq-MST] | 0.359 | 0.356 | 0.366 | 0.37 | 0.359 | 0.337 |
| d[M1–NTMTACN] | 1.552 | 1.471 | 1.387 | 1.317 | 1.345 | 1.362 |
| Bond angles (°) | ||||||
| O1–Fe1–N1 | 171.45(6) | 171.50(7) | 173.0(1) | 173.2(1) | 174.4(2) | 173.0(1) |
| N2–Fe1–N3 | 108.05(7) | 108.58(8) | 107.4(1) | 107.1(1) | 109.5(2) | 108.5(1) |
| N3–Fe1–N4 | 126.73(7) | 126.37(8) | 124.1(1) | 124.2(1) | 122.6(2) | 122.4(1) |
| N2–Fe1–N4 | 115.82(7) | 115.74(8) | 118.7(1) | 118.6(1) | 118.4(2) | 119.7(1) |
| Fe1–O1–M1 | 122.21(7) | 124.27(9) | 124.5(1) | 124.8(1) | 128.1(3) | 127.8(2) |
| O2–M1–O4 | 95.80(5) | 94.25(6) | 91.9(1) | 92.4(1) | 96.2(2) | 93.2(1) |
| N5–M1–N6 | 78.79(6) | 80.46(7) | 82.9(1) | 84.7(1) | 82.4(2) | 83.9(1) |
| N5–M1–N7 | 79.21(6) | 80.87(7) | 82.0(1) | 84.0(1) | 82.8(2) | 83.0(1) |
| N6–M1–N7 | 78.25(6) | 80.29(7) | 83.8(1) | 85.5(1) | 85.9(2) | 84.8(1) |
| Calculated values | ||||||
| τ5b | 0.745 | 0.752 | 0.815 | 0.817 | 0.863 | 0.843 |
| λoctc | 1.024 | 1.021 | 1.013 | 1.010 | 1.036 | 1.019 |
From reference 38
trigonality parameter, τ5 = (β–a)/60. β is the largest bond angle observed, and α is the second largest bond angle.48
octahedral quadratic elongation, λoct = Σ16(li/l0)2/6.λoct = 1 for an ideal octahedron. l0 represents the center-to-vertex distance of an octahedron with Oh symmetry whose volume is equal to that of the distorted octahedron with distances li. λ’oct is the octahedral quadratic elongation toward one axis.49
Displacement of the MII center from the N5–N6–N7 plane of the TMTACN ligand (d[M1–NTMTACN]) were correlated to the ionic radius46 of the MII ion. The NiII ion has the shortest ionic radius in this series and has the shortest d[M1–NTMTACN] of 1.317 Å, whereas the MnII ion has the longest ionic radius and a d[M1–NTMTACN] of 1.552 Å. Additionally, the bond distance between the MII metal center and O1 of the bridging hydroxido ligand also follows the trend of ionic radii, in which shorter distances generally correspond to the MII ions with shorter ionic radii (Figure S2A). The [CuII(OH)FeIII]+ complex does not follow the trend because of Jahn-Teller distortion that results in a significantly shorter Cu1–O1 bond distance of 1.905(5) Å (Figure S2A). We also examined whether there were correlations between the experimental parameters and the pKa values of the corresponding [MII(H2O)x]2+ complexes (Figure S2B).47 These pKa values have been shown to be good indicators of the Lewis acidity of the MII ions, and are useful predictors especially when used in comparing complexes with similar structural properties.50–52 A correlation was found between the MII–O1 bond distances and the pKa values of the corresponding [MII(H2O)6]2+ complexes (Figure S2B) with the Cu1–O1 bond distance now fitting the trend; however, correlations were not found to other structural or spectroscopic measurements.
The other metrical parameters in the series do not vary significantly between complexes. For instance, the Fe1–O1 bond distances are statistically identical within the series. The O1···O6 distances generally decrease from MnII to ZnII, but these differences are small with changes that only span 0.03 Å. The solid-state data agree with the observation drawn from the optical and vibrational studies that in this series of bimetallic complexes the identity of the divalent metal ion does not seem to alter physical properties at the FeIII center.
The O1···O6 distances ranging from 2.619(4)–2.649(2) Å in the [MII(OH)FeIII]+ indicate that intramolecular H-bonds are formed between the bridging hydroxido ligand and the O1 atom of the [MST]3− ligand.53 These distances are shorter than the reported distances for the comparable [C⊃MII(OH)FeIII]+ complexes, which have O1···O6 distances ranging from 2.685(6)–2.700(6) Å.37 The O1···O6 distances for the [MII(OH)FeIII]+ complexes suggest that stronger H-bonds are formed in the heterobimetallic complexes containing two transition metal ions. Moreover, these structural data corroborate findings from the vibrational studies that stronger Lewis acids within the [MII(OH)FeIII]+ core promote stronger intramolecular H-bonds.
Electrochemical properties of [MII(OH)FeIII]+ complexes
The electrochemical properties of the series of complexes were probed using cyclic voltammetry (Figure S1 and Table 1). The previously reported [MnII(OH)FeIII]+ and [FeII(OH)FeIII]+ complexes exhibited two quasi-reversible one-electron redox events which were assigned to the MIIFeII/MIIFeIII and MIIFeIII/MIIIFeIII couples.38 However, only one quasi-reversible one-electron redox event, assigned to the MIIFeII/MIIFeIII couple, was observed in the cases of [CoII(OH)FeIII]+, [NiII(OH)FeIII]+, [CuII(OH)FeIII]+, and [ZnII(OH)FeIII]+. Most of the MIIFeII/MIIFeIII redox potentials for the [MII(OH)FeIII]+ complexes range from −0.89 to −0.85 V versus [FeCp2]+/0; the potential for the [NiII(OH)FeIII]+ complex is slightly outside this range at −0.99 V. These data support the spectroscopic and structural findings that the additional transition metal ion does not have a large impact on the properties of the Fe center. The lack of a second oxidation event is consistent with the tendency of late transition metal ions to have more positive oxidization potentials that may be difficult to observe. It is thus not surprising that the MIIFeIII/MIIIFeIII couples could not be detected under the condition of the CV experiments. The separation between MIIFeIII/MIIIFeIII and the MIIFeIII/MIIIFeIII (M = Co, Ni, Cu, Zn) couples (ΔE1/2) should be even larger than the 1.54 and 1.21 V observed for the [MnII(OH)FeIII]+ or [FeII(OH)FeIII]+ complexes, respectively. The expected increase in ΔE1/2 suggest that the series of [MII(OH)FeIII]+ species should be stable toward disproportionation.
Electron paramagnetic resonance properties of [MII(OH)FeIII]+ complexes
We have investigated the EPR properties for each [MII(OH)FeIII] complex and the results are found in Table 3. All the complexes had observable signals except [CoII(OH)FeIII]+ which showed no EPR features over a temperature range of 10 to 80 K in both parallel- and perpendicular-modes. The lack of detectable signals is presumably caused by the antiferromagnetically spin-coupling of the S = 3/2 CoII and S = 5/2 FeIII centers to give a ground S = 1 state. Because of the large spin-orbit coupling constant of CoII ions, the resulting S = 1 and S = 2 states would have large D-values that prevent the complex from having detectable signals with X-band EPR spectroscopy.
Table 3.
Total Spin of the [MII(OH)FeIII]+ complexes and parameters used to simulate the EPR spectra.
| [MII(OH)FeIII]+ | MII | ||||
|---|---|---|---|---|---|
|
| |||||
| MnII | FeII | NiII | CuII | ZnII | |
| ST | 0 | 1/2 | 3/2 | 2 | 5/2 |
| SM(II) | 5/2 | 2 | 1 | 1/2 | 0 |
| gM(II) | 2,2,2 | 2, 2.25, 2.25 | 2.18, 2.18, 2.23 | 2.07, 2.07, 2.36 | 1.99, 1.99, 2.00a |
| DM(II), cm−1 | -0.13 | +2(1) | 16(2) | -2.5(1)a | |
| E/DM(II) | 0.08 | 0.10 | 0.03 | 0.213a | |
| J, cm−1 | +35(3) | +26(4) | +35(5) | +33(5) | |
parameters for FeIII center
We used the [ZnII(OH)FeIII]+ complex to evaluate the properties of the FeIII center in the absence of any magnetic coupling. The complex shows a perpendicular-mode signal with intense resonances near g = 4.97 and 3.84, and weaker resonances near g =10 that are indicative of a rhombic S = 5/2 system (Figure 5). Quantitative simulations of the signal at variable temperatures matched the spectra for zero-field splitting parameters of D = −2.5 cm−1 and E/D = 0.21, and agreed with the concentration of the complex. These parameters are similar to those previously reported for the mononuclear [FeIIIH3buea(OH)] − complex ([H3buea]3− = tris[(N′-tert-butylureaylato)-N-ethyl)]aminato, D = −2.4 cm−1, E/D = 0.16)54 which has a similar coordination geometry to the Fe center in [ZnII(OH)FeIII]. Moreover, the addition of the [ZnII(TMACN)]2+ fragment via the hydroxido bridging ligands affects the rhombicity of the Fe portion of the complex, but not the value for the axial D parameter.
Figure 5.
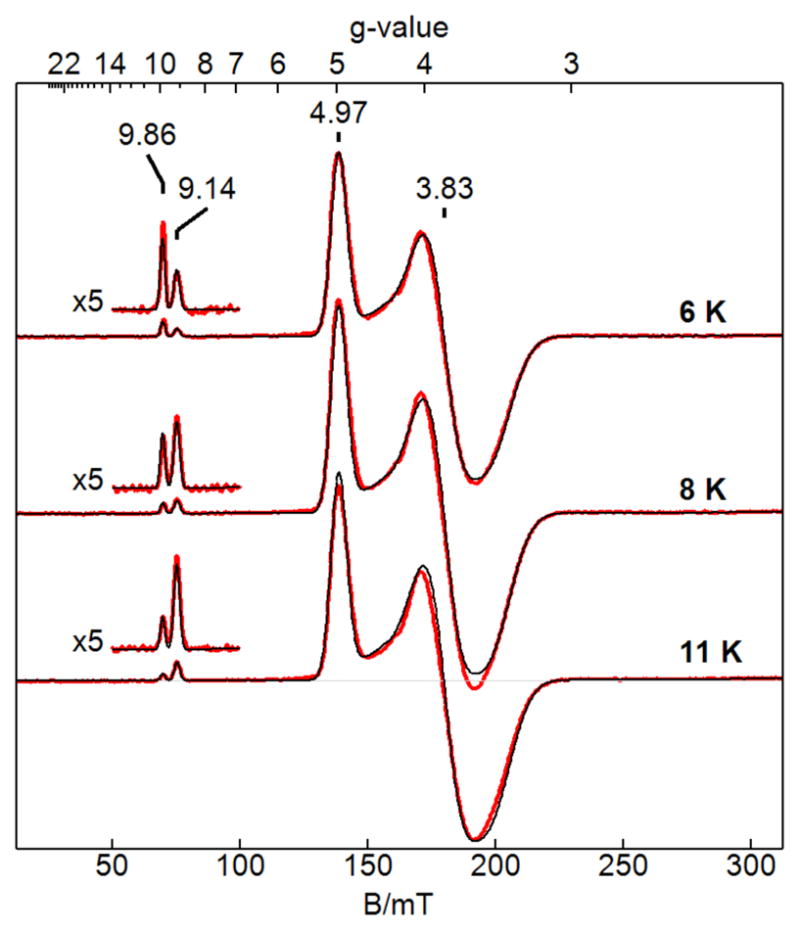
EPR spectra (red) and simulations (black) of ZnIIFeIII at the temperatures listed. Microwave parameters: 9.647 GHz, 0.02 mW, B1 ⊥ B. See Table 3 for simulation parameters.
The remaining [MII(OH)FeIII]+ complexes (MII = Mn, Fe, Ni, Cu) were analyzed using variable temperature EPR measurement and each showed antiferromagnetic coupling between the FeIII center and the other divalent metal ion. The magnetic exchange coupling did not vary significantly with J-value for the heterobimetallic complexes ranging from 26(4) to 35(3) cm−1 (Table 3).55 The EPR spectra for [MnII(OH)FeIII]+ and [FeII(OH)FeIII]+ have been previously described38 and the methods used to determine their exchange-coupling J-values are found in ESI (Figures S3, S4). The spectra and magnetic properties for [NiII(OH)FeIII]+ and [CuII(OH)FeIII]+ are discussed below.
[NiII(OH)FeIII]+
EPR spectra of [NiII(OH)FeIII]+ showed a perpendicular-mode signal with prominent features at g = 5.45 and 5.05 that are indicative of a rhombic spin-coupled SC = 3/2 ground state (Figure 6). The signals at g = 5.45 and 5.05 originate from the Ms = ±3/2 and ±1/2 doublets, respectively. This ground spin state results from the antiferromagnetic exchange coupling of the S = 5/2 FeIII center to S = 1 NiII site to give rise to an effective spin-coupled SC = 3/2 state. The relative intensities of these two sharp peaks changed significantly between temperatures of 2 to 30 K: the g = 5.45 feature is most intense at 2 K which indicates that D < 0 for SC = 3/2. At higher temperatures, a signal near g = 8 grows in intensity consistent with it originating from the excited state SC = 5/2 manifold. The temperature dependence of the signals was measured from 2 to 100 K with representative spectra are shown in Figure 6.
Figure 6.

EPR spectra (red) and simulations (black) of [NiII(OH)FeIII]+ at the temperatures listed, showing changes in the relative intensities of the signals (g = 5.45, 5.05, 2.34, 1.65) from the SC = 3/2 state and growth of the g = 8 signal from the SC = 5/2 state. Microwave parameters: 9.663 GHz, 0.2 mW, B1 ⊥ B. The simulations matched the temperature dependence of the signals and used a concentration that was in quantitative agreement with that measured for the complex.
The effect of temperature on each feature is plotted as signal intensity × temperature versus temperature (Figure 7) in which the ordinate axis is scaled to reflect the percentage population of the appropriate spin manifold.56,57 The fits to the temperature dependence for temperature less than 30 K gave D = −3.4 cm−1 for the SC = 3/2 manifold. The D-value for [NiII(OH)FeIII]+ is related to D-values for each metal-ion site through the spin-coupling relation, DS=3/2 = 28/15 DFe(III) − 1/15 DNi(II). 58 A zero-field splitting value of DNi(II) = 16 cm−1 was calculated from this relationship and using the DFe(III)-value (Table 3) determined for [ZnII(OH)FeIII]+. With these parameters, the temperature dependences of the signals were fit using eq 1 to determine the exchange coupling constant, J = +35(5) cm−1 (Figure 7). Above 30 K, both signals from the SC = 3/2 manifold decreased in intensity with concomitant increase in the g = 8 signal because the higher excited-state manifolds in the spin coupled system are populated.
Figure 7.
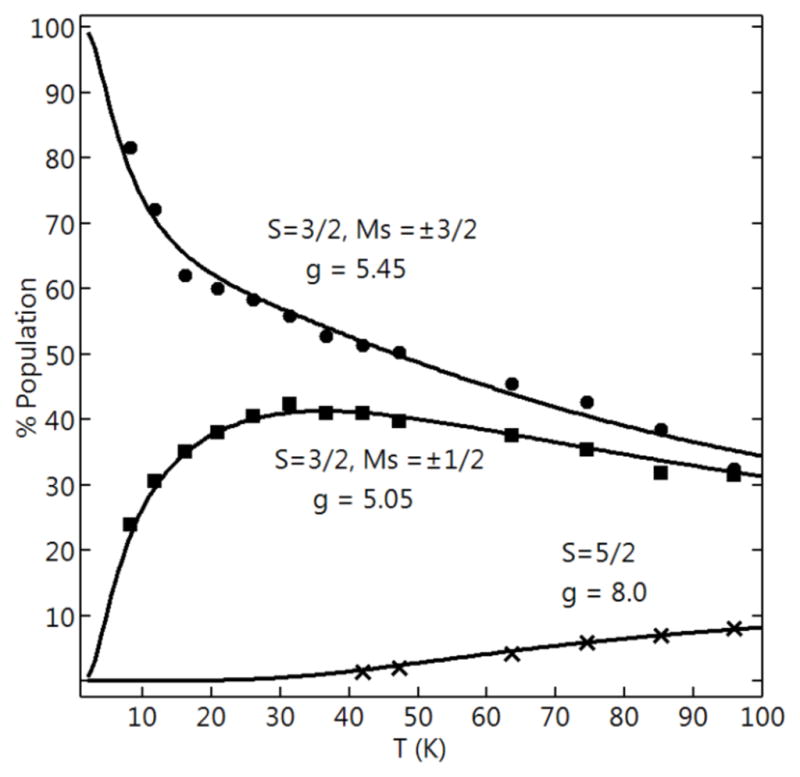
Signal intensity × temperature versus temperature of the signals at g = 5.45(●), 5.05(■), 8.0(×) observed from [NiII(OH)FeIII]+. The solid traces are % population of the indicated doublets calculated from eq 1 for J = 35 cm−1 and appropriate parameters in Table 3.
[CuII(OH)FeIII]+
This complex showed a parallel-mode EPR signal at g = 10.1 (Figure 8) that was most intense at the lowest temperatures allowed by the cryostat (2 K). The temperature dependence of the signal was recorded and a plot of intensity × temperature versus temperature is shown in the inset of Figure 8. The signal originates from the ground SC = 2 state of S = 5/2 FeIII site that is antiferromagnetically exchange-coupled to S = 1/2 state of the CuII center. Based on eq 1, the percent population of the doublet producing the g = 10 signal was plotted to give a J-value of +33(5) cm−1 (Figure 8, inset). There was no evidence of a signal from the SC = 3 state at temperatures > 60 K presumably because of broadening. The simulation overlaid on the spectra were again obtained using eq 1 and the D-value obtained for [ZnII(OH)FeIII]+ (Figure 8, Table 3). The deviations of the g-values from 2.0 for the CuII site and the hyperfine A-tensor are reduced by a factor of 1/6 because of the spin interaction with the FeIII center which results in only minor dependence of the signal to variations in the CuII parameters. Nevertheless, simulations of the signal for a set of parameters in agreement with for [ZnII(OH)FeIII]+ and a tetragonal CuII center provided an accurate fit to the observed signal and its temperature-dependence the concentration of the complex.
Figure 8.
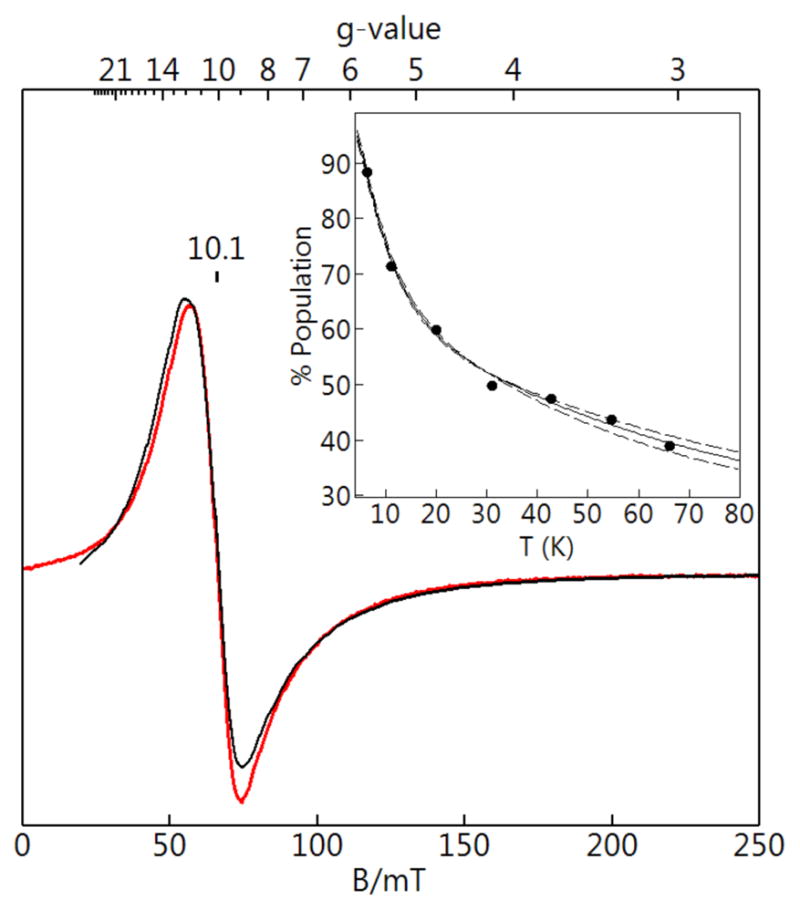
EPR spectrum (red) and simulation (black) of [CuII(OH)FeIII]+ attemperature of 7 K. Microwave parameters: 9.339 GHz, 0.02 mW, B1 || B. See Table 3 for simulation parameters. The inset shows signal intensity × temperature and the corresponding percent population of the corresponding doublet for J = +33(3) cm−1 with the error represented by the dashed lines.
CONCLUSIONS
This work has described the preparation and characterization of four new complexes in a series of bimetallic complexes of the formulation [(TMTACN)MII–(μ-OH)–FeIIIMST]+, where MII = Co, Ni, Cu, Zn. All compounds in this series have similar optical, vibrational, and electrochemical features compared to the previously published [MnII(OH)FeIII]+ and [FeII(OH)FeIII]+ complexes.38 The solid state structures determined by X-ray diffraction methods showed that all the complexes contain a MII–(μ-OH)–FeIII core. The two metal centers have different coordination environments, with the FeIII centers having five-coordinate, distorted tbp primary coordination spheres and the MII center having six-coordinate, distorted octahedral primary coordination spheres. The metrical parameters of the FeIII site remain consistent throughout the series, while the metrical parameters around the MII site vary predictably depending on the ionic radii of the MII center. The [CuII(OH)FeIII]+ complex has metrical parameters that do not follow the trend of ionic radii, but these differences are attributed to the strong Jahn-Teller distortion expected for a d9 CuII metal center. Taken together, our results show that the identity of the auxiliary divalent metal ion has only small effects on the properties of the FeIII–OH unit.
The magnetic properties of the complexes were investigated by EPR spectroscopy. The two high-spin metal centers exhibit antiferromagnetic coupling, allowing the series of complexes to access spin states ranging from S = 0 for the [MnII(OH)FeIII]+ complex to S = 5/2 for the [ZnII(OH)FeIII]+ complex. The EPR results suggest that the complexes remain assembled in solution and the magnitude of the J-values is consistent with a bridging hydroxido ligand. This series of heterobimetallic complexes allows a comparison of electronic properties for a range of transition metal ions without significant structural changes. The magnitude of J was found to be nearly invariant of the spin-state of the MII ions. Spin exchange interactions typically occur through the d orbitals of the metal ions and those of the bridging ligand. There are potentially many orbital pathways contributing to spin-spin exchange interactions, and the d orbitals containing the unpaired electrons would presumably influence the magnitude of J. The d-electron population of the MII center increased in our series of complexes that range from [MnII(OH)FeIII]+ to [CuII(OH)FeIII]+ and with this increase the number of unpaired electrons within the d orbitals changes. An explanation for the near invariance in the values of J, despite different exchange pathways through the d orbitals, is still unknown and requires further investigation.
Supplementary Material
Figure 4.

Thermal ellipsoid diagrams depicting the molecular structures of (A) [MnII(OH)FeIII]+,38 (B) [FeII(OH)FeIII]+,38 (C) [CoII(OH)FeIII]+, (D) [NiII(OH)FeIII]+, (E) [CuII(OH)FeIII]+, and (F) [ZnII(OH)FeIII]+. Ellipsoids are drawn at the 50% probability level, and only the hydroxido H atoms are shown for clarity.
Acknowledgments
We thank the NIH (Grant GM050781 to A.S.B., Grant GM 77387 to M.P.H.) for financial support of this work. Funding for the EPR spectrometer was from National Science Foundation grant CHE1126268.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. Crystallographic data in CIF formation, ESI-MS, FTIR spectra, and CV for all complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jiang W, Yun D, Saleh L, Barr E, Xing G, Hoffart LM, Maslak MA, Krebs C, Bollinger JM., Jr A manganese (IV)/iron (III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science. 2007;316:1188–1191. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 2.Kurtz DM. Comprehensive Coordination Chemistry II. Elsevier; 2003. Dioxygen-binding Proteins; pp. 229–260. [Google Scholar]
- 3.Fontecilla-Camps JC, Volbeda A. Encyclopedia of Metalloproteins. Springer; New York: 2013. pp. 1535–1544. [Google Scholar]
- 4.Schenk G, Mitić N, Gahan LR, Ollis DL, McGeary RP, Guddat LW. Binuclear Metallohydrolases: Complex Mechanistic Strategies for a Simple Chemical Reaction. Acc Chem Res. 2012;45:1593–1603. doi: 10.1021/ar300067g. [DOI] [PubMed] [Google Scholar]
- 5.Diril H, Chang HR, Zhang X, Larsen SK, Potenza JA, Pierpont CG, Schugar HJ, Isied SS, Hendrickson DN. Binuclear Mixed-Valence MnIIMnIII Complexes: Insight About the Resolution of Hyperfine Structure in the EPR Spectrum. J Am Chem Soc. 1987;109:6207–6208. [Google Scholar]
- 6.Borovik AS, Papaefthymiou V, Taylor LF, Anderson OP, Que L., Jr Models for Iron-Oxo Proteins. Structures and Properties of FeIIFeIII ZnIIFeIII and FeIIGaIII Complexes with (μ-Phenoxo)bis(μ-carboxylato)dimetal Cores. J Am Chem Soc. 1989;111:6183–6195. [Google Scholar]
- 7.Buchanan RM, Mashuta MS, Richardson JF, Webb RJ, Oberhausen KJ, Nanny MA, Hendrickson DN. Synthesis, Structure, and Properties of a Novel Heterobimetallic FeIIIMnII Complex Containing a Septadentate Polyimidazole Ligand. Inorg Chem. 1990;29:1299–1301. [Google Scholar]
- 8.Bossek U, Hummel H, Weyhermüller T, Bili E, Wieghardt K. The First μ(OH)-Bridged Model Complex for the Mixed-Valent FeIIFeIII Form of Hemerythrin. Angew Chem, Int Ed Engl. 1996;34:2642–2645. [Google Scholar]
- 9.Bossek U, Hummel H, Weyhermüller T, Wieghardt K, Russell S, van der Wolf L, Kolb U. The[Mn2IV(μ-O)(μ-PhBO2)2]2+ Unit: A New Structural Model for Manganese-Containing Metalloproteins. Angew Chemie Int Ed English. 1996;35:1552–1554. [Google Scholar]
- 10.Ng GKY, Ziller JW, Borovik AS. Structural Diversity in Metal Complexes with a Dinucleating Ligand Containing Carboxyamidopyridyl Groups. Inorg Chem. 2011;50:7922–7924. doi: 10.1021/ic200881t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng GKY, Ziller JW, Borovik AS. Preparation and structures of dinuclear complexes containing MII–OH centers. Chem Commun. 2012;48:2546–2548. doi: 10.1039/c2cc16277f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchler S, Meyer F, Kaifer E, Pritzkow H. Tunable TACN/pyrazolate hybrid ligands as dinucleating scaffolds for metallobiosite modeling—dinickel(II) complexes relevant to the urease active site. Inorganica Chim Acta. 2002;337:371–386. [Google Scholar]
- 13.Isaac JA, Gennarini F, López I, Thibon-Pourret A, David R, Gellon G, Gennaro B, Philouze C, Meyer F, Demeshko S, Le Mest Y, Réglier M, Jamet H, Le Poul N, Belle C. Room-Temperature Characterization of a Mixed-Valent μ-Hydroxodicopper(II,III) Complex. Inorg Chem. 2016;55:8263–8266. doi: 10.1021/acs.inorgchem.6b01504. [DOI] [PubMed] [Google Scholar]
- 14.Dunn TJ, Ramogida CF, Simmonds C, Paterson A, Wong EWY, Chiang L, Shimazaki Y, Storr T. Non-Innocent Ligand Behavior of a Bimetallic Ni Schiff-Base Complex Containing a Bridging Catecholate. Inorg Chem. 2011;50:6746–6755. doi: 10.1021/ic200785g. [DOI] [PubMed] [Google Scholar]
- 15.Clarke RM, Hazin K, Thompson JR, Savard D, Prosser KE, Storr T. Electronic Structure Description of a Doubly Oxidized Bimetallic Cobalt Complex with Proradical Ligands. Inorg Chem. 2016;55:762–774. doi: 10.1021/acs.inorgchem.5b02231. [DOI] [PubMed] [Google Scholar]
- 16.Alliger GE, Müller P, Do LH, Cummins CC, Nocera DG. Family of Cofacial Bimetallic Complexes of a Hexaanionic Carboxamide Cryptand. Inorg Chem. 2011;50:4107–4115. doi: 10.1021/ic200143b. [DOI] [PubMed] [Google Scholar]
- 17.Neves A, Lanznaster M, Bortoluzzi AJ, Peralta RA, Casellato A, Castellano EE, Herrald P, Riley MJ, Schenk G. An Unprecedented FeIII(μ-OH)ZnIIComplex that Mimics the Structural and Functional Properties of Purple Acid Phosphatases. J Am Chem Soc. 2007;129:7486–7487. doi: 10.1021/ja071184l. [DOI] [PubMed] [Google Scholar]
- 18.de Souza B, Kreft GL, Bortolotto T, Terenzi H, Bortoluzzi AJ, Castellano EE, Peralta RA, Domingos JB, Neves A. Second-Coordination-Sphere Effects Increase the Catalytic Efficiency of an Extended Model for FeIIIMII Purple Acid Phosphatases. Inorg Chem. 2013;52:3594–3596. doi: 10.1021/ic400025j. [DOI] [PubMed] [Google Scholar]
- 19.Wong JL, Higgins RF, Bhowmick I, Cao DX, Szigethy G, Ziller JW, Shores MP, Heyduk AF, Gagliardi L, Lu CC. Bimetallic iron–iron and iron–zinc complexes of the redox-active ONO pincer ligand. Chem Sci. 2016;7:1594–1599. doi: 10.1039/c5sc03006d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenkoetter KE, Ziller JW, Heyduk AF. A Heterobimetallic W–Ni Complex Containing a Redox-Active W[SNS]2 Metalloligand. Inorg Chem. 2016;55:6794–6798. doi: 10.1021/acs.inorgchem.6b01164. [DOI] [PubMed] [Google Scholar]
- 21.Rudd PA, Liu S, Gagliardi L, Young VG, Lu CC. Metal–Alane Adducts with Zero-Valent Nickel, Cobalt, and Iron. J Am Chem Soc. 2011;133:20724–20727. doi: 10.1021/ja2099744. [DOI] [PubMed] [Google Scholar]
- 22.Clouston LJ, Siedschlag RB, Rudd PA, Planas N, Hu S, Miller AD, Gagliardi L, Lu CC. Systematic Variation of Metal-Metal Bond Order in Metal–Chromium Complexes. J Am Chem Soc. 2013;135:13142–13148. doi: 10.1021/ja406506m. [DOI] [PubMed] [Google Scholar]
- 23.Eisenhart RJ, Clouston LJ, Lu CC. Configuring Bonds between First-Row Transition Metals. Acc Chem Res. 2015;48:2885–2894. doi: 10.1021/acs.accounts.5b00336. [DOI] [PubMed] [Google Scholar]
- 24.Serrano-Plana J, Garcia-Bosch I, Company A, Costas M. Structural and Reactivity Models for Copper Oxygenases: Cooperative Effects and Novel Reactivities. Acc Chem Res. 2015;48:2397–2406. doi: 10.1021/acs.accounts.5b00187. [DOI] [PubMed] [Google Scholar]
- 25.Delgado M, Ziegler JM, Seda T, Zakharov LN, Gilbertson JD. Pyridinediimine Iron Complexes with Pendant Redox-Inactive Metals Located in the Secondary Coordination Sphere. Inorg Chem. 2016;55:555–557. doi: 10.1021/acs.inorgchem.5b02544. [DOI] [PubMed] [Google Scholar]
- 26.Barton BE, Rauchfuss TB. Hydride-Containing Models for the Active Site of the Nickel–Iron Hydrogenases. J Am Chem Soc. 2010;132:14877–14885. doi: 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenwood BP, Forman SI, Rowe GT, Chen CH, Foxman BM, Thomas CM. Multielectron Redox Activity Facilitated by Metal–Metal Interactions in Early/Late Heterobimetallics: Co/Zr Complexes Supported by Phosphinoamide Ligands. Inorg Chem. 2009;48:6251–6260. doi: 10.1021/ic900552b. [DOI] [PubMed] [Google Scholar]
- 28.Krogman JP, Thomas CM. Metal–metal multiple bonding in C3-symmetric bimetallic complexes of the first row transition metals. Chem Commun. 2014;50:5115. doi: 10.1039/c3cc47537a. [DOI] [PubMed] [Google Scholar]
- 29.Wu B, Wilding MJT, Kuppuswamy S, Bezpalko MW, Foxman BM, Thomas CM. Exploring Trends in Metal–Metal Bonding, Spectroscopic Properties, and Conformational Flexibility in a Series of Heterobimetallic Ti/M and V/M Complexes (M = Fe, Co, Ni, and Cu) Inorg Chem. 2016;55:12137–12148. doi: 10.1021/acs.inorgchem.6b01543. [DOI] [PubMed] [Google Scholar]
- 30.Park YJ, Ziller JW, Borovik AS. The Effects of Redox-Inactive Metal Ions on the Activation of Dioxygen: Isolation and Characterization of a Heterobimetallic Complex Containing a MnIII–(μ-OH)–CaII Core. J Am Chem Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lacy DC, Park YJ, Ziller JW, Yano J, Borovik AS. Assembly and Properties of Heterobimetallic CoII/III/CaII Complexes with Aquo and Hydroxo Ligands. J Am Chem Soc. 2012;134:17526–17535. doi: 10.1021/ja304525n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sickerman NS, Henry RM, Ziller JW, Borovik AS. Preparation and structural properties of InIII–OH complexes. Polyhedron. 2013;58:65–70. doi: 10.1016/j.poly.2012.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sickerman NS, Peterson SM, Ziller JW, Borovik AS, Mäemets V, Leito I, Koppel IA, Borovik AS. Synthesis, structure and reactivity of FeII/III–NH3 complexes bearing a tripodal sulfonamido ligand. Chem Commun. 2014;50:2515. doi: 10.1039/c3cc48804g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cook SA, Ziller JW, Borovik AS. Iron(II) Complexes Supported by Sulfonamido Tripodal Ligands: Endogenous versus Exogenous Substrate Oxidation. Inorg Chem. 2014;53:11029–11035. doi: 10.1021/ic501531g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lau N, Ziller JW, Borovik AS. Sulfonamido tripods: Tuning redox potentials via ligand modifications. Polyhedron. 2015;85:777–782. doi: 10.1016/j.poly.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lau N, Sano Y, Ziller JW, Borovik AS. Terminal NiII–OH/–OH2 complexes in trigonal bipyramidal geometries derived from H2O. Polyhedron. 2017;125:179–185. doi: 10.1016/j.poly.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park YJ, Cook SA, Sickerman NS, Sano Y, Ziller JW, Borovik AS. Heterobimetallic complexes with MIII-(μ-OH)-MII cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): structural, kinetic, and redox properties. Chem Sci. 2013;4:717–726. doi: 10.1039/C2SC21400H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sano Y, Weitz AC, Ziller JW, Hendrich MP, Borovik AS. Unsymmetrical Bimetallic Complexes with MII–(μ-OH)–MIII Cores (MIIMIII = FeIIFeIII, MnIIFeIII, MnIIMnIII): Structural, Magnetic, and Redox Proper. Inorg Chem. 2013;52:10229–10231. doi: 10.1021/ic401561k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heintz RA, Smith JA, Szalay PS, Weisgerber A, Dunbar KR. Homoleptic transition metal acetonitrile cations with tetrafluoroborate or trifluoromethanesulfonate anions. Inorg Synth. 2002;33:75–83. [Google Scholar]
- 40.Schoenfeldt NJ, Ni Z, Korinda AW, Meyer RJ, Notestein JM. Manganese Triazacyclononane Oxidation Catalysts Grafted under Reaction Conditions on Solid Cocatalytic Supports. J Am Chem Soc. 2011;133:18684–18695. doi: 10.1021/ja204761e. [DOI] [PubMed] [Google Scholar]
- 41.Petasis DT, Hendrich MP. Quantitative Interpretation of Multifrequency Multimode EPR Spectra of Metal Containing Proteins, Enzymes, and Biomimetic Complexes. Methods Enzymol. 2015;563:171–208. doi: 10.1016/bs.mie.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abragam A, Bleaney B. Electron Paramagnetic Resonance of Transition Metal Ions. Clarendon Press; Oxford: 1970. [Google Scholar]
- 43.Brink DM, Satchler GR. Angular momentum. 3. Clarendon Press; Oxford: 1994. [Google Scholar]
- 44.Aaron HS. Conformational Analysis of Intramolecular-Hydrogen-Bonded Compounds in Dilute Solution by Infrared Spectroscopy. In: Allinger NL, Eliel EL, editors. Topics in Stereochemistry. Vol. 11. John Wiley & Sons, Inc; Hoboken, NJ, USA: 1979. [Google Scholar]
- 45.Gellman SH, Dado GP, Liang GB, Adams BR. Conformation-directing effects of a single intramolecular amide-amide hydrogen bond: variable-temperature NMR and IR studies on a homologous diamide series. J Am Chem Soc. 1991;113:1164–1173. [Google Scholar]
- 46.Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Sect A. 1976;32:751–767. [Google Scholar]
- 47.Jackson VE, Felmy AR, Dixon DA. Prediction of the pKa’s of Aqueous Metal Ion +2 Complexes. J Phys Chem A. 2015;119:2926–2939. doi: 10.1021/jp5118272. [DOI] [PubMed] [Google Scholar]
- 48.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J Chem Soc Dalt Trans. 1984;(7):1349. [Google Scholar]
- 49.Robinson K, Gibbs GV, Ribbe PH. Quadratic Elongation: A Quantitative Measure of Distortion in Coordination Polyhedra. Science. 1971;172:567–570. doi: 10.1126/science.172.3983.567. [DOI] [PubMed] [Google Scholar]
- 50.Tsui EY, Tran R, Yano J, Agapie T. Redox-inactive metals modulate the reduction potential in heterometallic manganese–oxido clusters. Nat Chem. 2013;5:293–299. doi: 10.1038/nchem.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herbert DE, Lionetti D, Rittle J, Agapie T. Heterometallic triiron-oxo/hydroxo clusters: effect of redox-inactive metals. J Am Chem Soc. 2013;135:19075–19078. doi: 10.1021/ja4104974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin PH, Takase MK, Agapie T. Investigations of the Effect of the Non-Manganese Metal in Heterometallic-Oxido Cluster Models of the Oxygen Evolving Complex of Photosystem II: Lanthanides as Substitutes for Calcium. Inorg Chem. 2015;54:59–64. doi: 10.1021/ic5015219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley J. Very strong hydrogen bonding. Chem Soc Rev. 1980;9:91. [Google Scholar]
- 54.Gupta R, Lacy DC, Bominaar EL, Borovik AS, Hendrich MP. Electron Paramagnetic Resonance and Mössbauer Spectroscopy and Density Functional Theory Analysis of a High-Spin FeIV–Oxo Complex. J Am Chem Soc. 2012;134:9775–9784. doi: 10.1021/ja303224p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kurtz DM. Oxo- and Hydroxo-Bridged Diiron Complexes: A Chemical Perspective on a Biological Unit. Chem Rev. 1990;90:585–606. [Google Scholar]
- 56.Golombek AP, Hendrich MP. Quantitative analysis of dinuclear manganese(II) EPR spectra. J Magn Reson. 2003;165:33–48. doi: 10.1016/j.jmr.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 57.Holman TR, Juarez-Garcia C, Hendrich MP, Que L, Munck E. Models for Iron-Oxo Proteins. Moessbauer and EPR Study of an Antiferromagnetically Coupled FeIIINiII Complex. J Am Chem Soc. 1990;112:7611–7618. [Google Scholar]
- 58.Bencini A, Gatteschi D. Electron Paramagnetic Resonance of Exchange Coupled Systems. 1. Springer Berlin Heidelberg; Berlin, Heidelberg: 1990. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.