

Abstract
Carbohydrate fuel augmentation following traumatic brain injury may be a viable treatment to improve recovery when cerebral oxidative metabolism of glucose is depressed. We performed a primed constant Sodium L-Lactate infusion in eleven moderate to severely brain injured adults. Blood was collected before and periodically during the infusion study. We quantified global cerebral uptake of glucose and lactate and other systemic metabolites associated with energy metabolism. Our hypothesis was that cerebral lactate uptake, as measured by the arteriovenous difference of lactate (AVDlac), would increase in severely injured TBI patients in the neurocritical care unit. Infusion of Sodium L-Lactate changed net cerebral lactate release, where the arteriovenous difference of lactate is negative, to net cerebral lactate uptake. Results from a mixed effects model of AVDlac with the fixed effects of infusion time, arterial lactate concentration, arterial glucose concentration and arteriovenous difference of glucose shows that doubling arterial lactate concentration (from 0.92 to 1.84 mM) results in an increase in AVDlac from −0.078 mM to 0.090 mM. We did not detect changes in systemic glucose during the course of the infusion study and observed significant changes in alanine (30% [20 39]), glutamine (34% [24 43]), acetate (87% [60 113]), valine (40% [28 51]), and leucine (24% [16 32]) from baseline levels. Further studies are required to establish the impact of lactate supplementation on cerebral and systemic flux of lactate, on gluconeogenesis, and on the impact on cerebral energetics following injury.
Keywords: traumatic brain injury, lactate infusion, metabolism
Introduction
Immediately following traumatic brain injury (TBI), the global cerebral metabolic rate of glucose (CMRglc) is elevated and, over the first week, falls below levels expected in healthy adults (Bergsneider et al., 1997; Bergsneider et al., 2001; Yoshino et al., 1991). Depressed cerebral glucose metabolism occurs simultaneous with a depressed cerebral metabolic rate of oxygen (CMRO2). Overall, cerebral metabolic dysfunction can exacerbate problems such as intracranial pressure (ICP) or recovery from coma (Obrist et al., 1984; Brodersen and Jorgensen, 1974) and is not a result of decreased supply to cerebral tissue. Blood glucose, the primary cerebral carbohydrate energy substrate, is routinely monitored to ensure sufficient glucose is available and to avoid hyperglycemic episodes, yet no treatment has been shown to improve cerebral glucose metabolism.
Non-glucose carbohydrates could provide an alternative and supplemental source for energy production following TBI. Lactate has been shown to contribute to oxidative cerebral metabolism following TBI through direct cerebral uptake (Glenn et al., 2015a) and indirectly as a gluconeogenic precursor (Glenn et al., 2015b). Outside the neurocritical care unit, lactate is a cerebral energy source for healthy tissue (Boumezbeur et al., 2010); is the primary cerebral fuel during exercise (van Hall et al., 2009); displaces cerebral glucose uptake at normoglycemia (Smith et al., 2003); and fuels the brain during hypoglycemic episodes (Herzog et al., 2013). Lactate supplementation in the critically ill population suggest that cerebral glucose is spared when the brain converts exogenous lactate into pyruvate to be used aerobically (Bouzat et al., 2014; Quintard et al., 2016).
This study aims to better understand the impact of exogenous lactate supplementation on cerebral uptake of lactate and on systemic glucose, lactate, and other metabolites. We performed a primed, constant infusion of Sodium L-Lactate in 11 severely injured TBI patients, beginning at a rate of 3.4 mg/kg/min for 5 minutes and continuing at 1.1 mg/kg/min for 3 hours. We collected arterial and jugular venous blood from patients before initiating the primed constant infusion and hourly during the infusion study and quantified metabolite concentrations with NMR spectroscopy. We hypothesized that increasing systemic lactate delivery through exogenous infusion would increase cerebral lactate uptake, quantified by the arteriovenous difference of lactate (AVDlac), and that systemic glucose levels would not change. Additionally, we collected data on other systemic metabolites to explore how supplementing lactate was impacting pyruvate, alanine, and more.
Methods
Eleven patients consented to the University of California at Los Angeles (UCLA) Brain Injury Research Center (BIRC) study of moderate to severe head injuries, approved by the UCLA Medical Institutional Review Board for human research, were included between 2011 and 2015. Eligible patients for UCLA BIRC studies include all mechanically ventilated head-injured patients, aged 16 years and older, admitted to the UCLA Medical Center within 24 hours of injury. Eligible patients have admission Glasgow Coma Scale (GCS) scores ≤8 or, if higher, deteriorate to ≤8 prior to emergency department discharge or present with positive gross CT findings. Exclusion criteria include pre-existing neurologic illness, acute complete spinal cord injury, prior head injuries, and any history of diabetes mellitus.
Patients admitted to the ICU and treated in accordance with the brain Trauma Foundation and American Association of Neurological Surgeons (AANS)/Congress of Neurological Surgeons (CNS) TBI Guidelines. The general management protocol of TBI patients included maintenance of ICP <20 mmHg and/or CPP >60 mmHg. Patients were initially treated with a standard approach to control blood glucose, including subcutaneous regular insulin (Humulin; Eli Lilly, Indianapolis, IN) for admission blood glucose above 8.3 mM and a standard insulin sliding scale protocol to keep blood glucose between 5.6 and 8.3 mM.
Sodium L-Lactate infusion study design
Following consent of the patient, the UCLA BIRC research nurse contacted the UCLA research pharmacy and scheduled the Sodium L-Lactate infusion study. The research pharmacy produced 100 mg/mL Sodium L-Lactate (0.89 mol/L and 1.78 osmol/L) and the following equations were used to calculate dose:
| Bolus: | 17 mg/kg × patient weight = bolus in mg. |
| Infusate: | 203.5 mg/kg × patient weight = dose in mg. |
The pharmacy then prepared a syringe containing the correct volume of 100 mg/mL Sodium L-Lactate corresponding to the bolus dose and a bag containing the infusate dose. The concentration of the infusate changed based on patient weight (see Table). The bolus volume was pushed intravenously over 5 minutes. This corresponds to providing a bolus at approximately 3.4 mg/kg/min (30 mmol/kg/hr), followed by constant infusion at 1.1 mg/kg/min (9.8 mmol/kg/hr) for the remaining 3 hours.
|
| ||
| Weight (kg) |
IV Bag volume (mL) |
Infusion rate (mL/hr) |
|
| ||
| < 88.4 | 180 | 60 |
|
| ||
| > 88.4 | 270 | 90 |
|
| ||
At the end of the infusion study, blood serum was collected and sent to clinical laboratory for serum sodium analysis.
Blood was collected from arterial and jugular venous catheters prior to initiating the infusion study for baseline measurements of glucose and lactate. Subsequently, samples were collected at least every 60 minutes during the infusion study. Blood samples were collected, centrifuged, and transferred to tubes for storage at −80°C.
Blood metabolite measurements
Glucose and lactate concentrations were determined with a desktop Analox analyzer (Analox GM7, Analox Instruments, Stourbridge, UK) that uses a Clark-type amperometric oxygen electrode to quantify Glucose Oxidase oxygen usage following sample injection. In order to reliably quantify the AVDlac and arteriovenous difference of glucose (AVDglc), we collected 12 readings of the metabolite concentrations in both arterial and jugular venous blood to overcome the analytical imprecision of our equipment and reliably quantify the arteriovenous difference between the mean arterial and mean jugular venous concentrations.
Concentrations of glucose, lactate, alanine, etc. in plasma were determined by 1H NMR spectroscopy. The NMR sample preparation protocol for plasma was developed from the methods described by Gowda et. al. (Gowda et al., 2010) and had been published previously (Wolahan et al., 2016). Following solvent removal, plasma samples were reconstituted in 650 μL of 0.1 M phosphate buffered solution in deuterium oxide at pH 7.4, prepared with 0.1 mM 13C-labeled sodium formate (isotopic enrichment 99%, Cambridge Isotope Laboratories, Andover, MA) as an NMR internal standard. All NMR spectra were collected using standard Bruker cpmgpr1d pulse program on a Bruker AVANCE 600 MHz NMR Spectrometer equipped with an inverse broadband probe (parameter settings NS 32, SW 8 kHz, RG 128, O1 4.70 ppm, D1 5s, 90° pulse 13 μs, and a 200 ms CPMG decay period). Processed spectra were imported into R, version 3.1.3 (R Core Team, 2013), using the rNMR package (rNMR version 1.1.8) (Lewis et al., 2009). Metabolites of interest were quantified by integration of a particular chemical shift range (Wishart et al., 2008; Ulrich et al., 2008). We normalized plasma metabolite concentrations to the volume of plasma included in each sample and to glucose concentrations determined with the Analox glucose analyzer. Data were removed if no peak was detected at 4 standard deviations above the noise.
Statistical analysis
Due to nonnormalities in data distributions, we used robust statistical analytic methods (Wilcox, 1997). In general, we present Harrell-Davis estimates of medians and interquartile ranges (IQR) when summarizing cohort demographics and we present 20% trimmed means followed by the 95% confidence interval when describing the primed constant infusion of Sodium L-Lactate. We used the dependent Yuen test to compare changes in serum sodium and changes in intracranial pressure. Correlation coefficients presented are the percentage bend correlation, a robust analogue of Pearson’s correlation, followed by the 95% confidence interval and p-value.
To model repeated measures of glucose and lactate, we used mixed effects models as implemented in the nlme package (Pinheiro and Bates, 2000; Pinheiro et al., 2013). The fixed effects of interest were infusion time (baseline defined as 0 hours), glucose concentration, AVDglc, lactate concentration, and sex. The random effect included was subject. We modeled the logarithmic-transform of metabolite concentrations to control heteroscedasticity; the AVDlac was translated to positive values prior to logarithmic transformation by the equation log(AVDlac−min(AVDlac)+1), where min(AVDlac) is the minimum value across all patient data. The concentration fixed effects were mean centered prior to modeling. The best approximating model (Table 2) was selected by multimodel inference, an information-theoretic approach, such that inferences about model precision are made across the entire set of models with the Akaike information criteria (AIC) (Burnham and Anderson, 2002; Barton, 2013).
Table 2.
Summary of mixed effects model coefficients of arteriovenous difference (AVD) lactate as a function of four fixed effects: post infusion time, log transformed arterial lactate concentration (mean centered at 0.92 mM), log transformed glucose concentration (mean centered at 7.67 mM), and log transformed arteriovenous difference glucose (mean centered at 0.37 mM).
log(AVDlac−min(AVDlac)+1) = β0 + β1*time + β2*log(artlac) + β3*log(artglc) + β4*log(AVDglc), where min(AVDlac) ≡ −0.186 mM.
| Model Coefficients | Coefficient units | Estimate [95% CI] | t value (DF=37) | p value |
|---|---|---|---|---|
|
β0 Intercept |
log(mM) | 0.103 [0.066 0.140] |
8.5 | |
| β1 Time post infusion |
log(mM)/hr | 0.024 [0.004 0.045] |
2.4 | 0.022 |
| β2 Arterial lactate |
unitless | 0.098 [0.044 0.151] |
3.7 | <0.001 |
| β3 Arterial glucose |
unitless | −0.079 [−0.173 0.014] |
−1.7 | 0.092 |
| β4 AVD glucose |
unitless | 0.018 [−0.001 0.036] |
2.0 | 0.059 |
In order to investigate the fixed effect of percent change of lactate concentrations on other metabolites with significant inter-patient variability, we converted all concentrations to the percent change from baseline using the following equation, Δmetabolite = ([metabolite]/[metabolite]baseline−1)×100. To model repeated measures of plasma metabolites from NMR spectroscopy, we used mixed effects models, excluding baseline time points, with the fixed effect of percent change in lactate and the random effect of subject.
Results
Study participant demographic descriptors
The patient cohort included 11 severe TBI patients (8 male; median age 33 [IQR 24 48]), as characterized by the median post-resuscitation Glasgow Coma Scale (GCS) score of 4 [IQR 36] (Table 1). Median Glasgow Outcome Scale extended (GOSe) scores were 3.0 [IQR 2.7 3.6] at discharge and 4.5 [IQR 3.3 6.3] at 6-month follow-up (2 lost to follow-up). All patient injuries included diffuse axonal injury (DAI) and subarachnoid hemorrhage (SAH).
Table 1.
Cohort description of eleven TBI patients, including sex, age in years, weight at admission in kg, body mass index (BMI), Glasgow coma scale (GCS), and Glasgow Outcome Scale extended (GOSe).
| ID | Sex | Age (yr) |
Weight (kg) |
BMI (kg/m2) |
GCS* | GOSe* | Injury | PIH at infusion study start | |
|---|---|---|---|---|---|---|---|---|---|
| Discharge | 6-mo | ||||||||
| A | M | 54 | 95 | 30.9 | 8 | 3 | 1 | Bilateral SDH, DAI, SAH | 69 |
| B | M | 24 | 70 | 23.5 | 3 | 3 | 4 | Left SDH, DAI, SAH | 145 |
| C | F | 30 | 70 | 24.2 | 3 | 2 | 4 | DAI, SAH | 64 |
| D | M | 21 | 80 | 26 | 3 | 2 | 3 | DAI, SAH | 94 |
| E | M | 30 | 80 | ― | 3 | 4 | LTFU | DAI, SAH | 84 |
| F | F | 40 | 97.5 | 35.8 | 3 | 4 | 5 | Right SDH, DAI, SAH | 167 |
| G | M | 36 | 64.8 | 23.1 | 5 | 3 | LTFU | Left SDH, DAI, SAH, Contusion | 211 |
| H | M | 24 | 75 | 24.4 | 6 | 4 | 4 | DAI, Left EDH, SAH | 267 |
| I | M | 52 | 90.7 | 31.3 | 6 | 3 | 6 | Right SDH, DAI, SAH | 182 |
| J | M | 67 | 90 | 28.4 | 3 | 3 | 7 | Right SDH, DAI, SAH | 211 |
| K | F | 20 | 72 | 24.9 | 9 | 3 | 8 | DAI, SAH | 94 |
GCS* is the scoring system used to describe the level of consciousness ranging from 3 (severe) to 15 (mild). Scoring is determined by ratings in Eye Opening, Verbal Response, and Motor Response. GOSe describes the functional outcome ranging from 1 (Death) to 8 (Upper good recovery). GOSe are provided at Discharge and 6 months. Two patients Lost to Follow-Up (LFTU). Injury descriptions: subdural hemorrhage (SDH), diffuse axonal injury (DAI), subarachnoid hemorrhage (SAH), epidural hematoma (EDH). Post injury hour (PIH) at the start of infusion study.
Primed constant infusion of Sodium L-Lactate
The concentration of the sodium L-lactate infusate was 77.5 mg/mL [95%CI 70.6 84.5] on average (0.69 mol/L [95%CI 0.63 0.75]). The median time post-injury for beginning the infusion study was 137 hours [IQR 86 199]. Blood glucose and lactate concentrations at infusion start were variable between patients, with arterial glucose averaging 7.5 mM [95%CI 6.5 8.6] and arterial lactate 0.9 mM [95%CI 0.6 1.2] (Figure 1 at hour 0). All subjects’ blood lactate increased above lactate concentrations before the infusion study. The majority of patients (8 of 11) reached a maximum arterial lactate concentration before the end of the constant infusion; the trimmed mean of the average time to each subjects’ maximum arterial lactate concentration was 1.6 hours ([95%CI 0.9 2.4]). The average maximum arterial concentration achieved was 1.9 mM [95%CI 1.3 2.4], which corresponds to an average increase of 210% [95%CI 160 260] above baseline levels.
Figure 1.
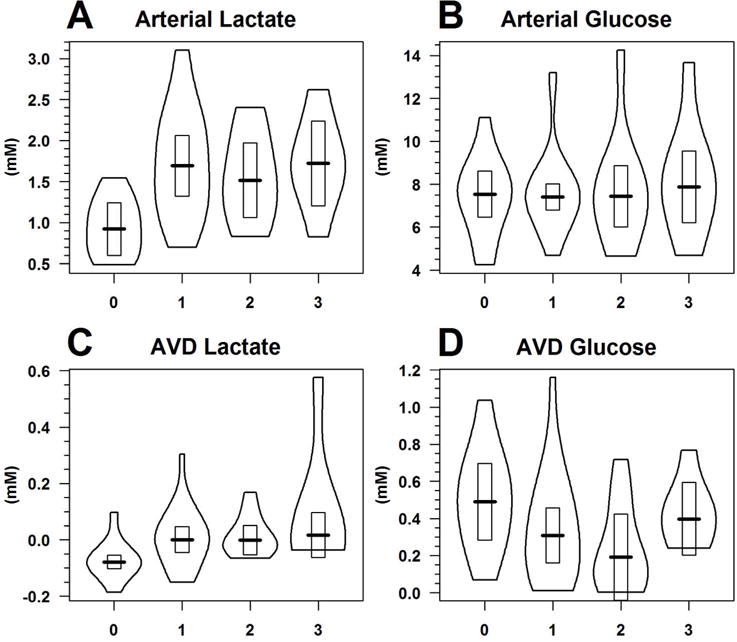
Violin plots of (A) arterial lactate, (B) arterial glucose, (C) AVD lactate, and (D) AVD glucose over the 3 hour lactate supplementation infusion study, where hour 0 summarizes baseline metabolite concentrations before starting the infusion. The outside border of the violin plot is an estimate of the distribution of the data; the dark horizontal line the 20% trimmed mean; and the rectangle the 95% confidence interval of the trimmed mean.
All patients had serum sodium measured before and after the infusion study. On average, there was a 3.0 mM [95%CI 1.0 4.8, p=0.009] increase in serum sodium from 145.9 mM [95%CI 141.6 150.2] to 148.9 mM [95%CI 144.9 152.8]. Sodium levels were transiently increased; post-hoc analysis of the next clinical serum sodium test, taken on average 6.7 hours [95%CI 3.3 10.1] following infusion study end, shows patient sodium levels decreased to levels not discernibly different than before the infusion study, of 146.6 mM [95%CI 141.7 151.5] (p=0.47).
Eight subjects had extraventricular drains and ICP monitoring at the time of the infusion study. ICP was well controlled in these patients ahead of the infusion study at 11.4 mmHg [8.5 14.3]. ICP was transiently decreased following the infusion study. The average ICP in the hour following the infusion study was 7.7 mmHg [6.0 9.5], corresponding to a 3.6 mmHg ([1.3 6.0], p=0.010) decrease from baseline levels.
Mixed effects modeling of systemic lactate and glucose concentrations
There was no discernible change in arterial glucose concentration (p=0.93) nor in the AVDglc (p=0.60) over the infusion study time using mixed effects models with the fixed effect of post-infusion time and random effect of subject (model results not presented; data shown in Figure 1).
The AVDlac was moderately correlated to arterial lactate concentrations (r=0.51 [0.28 0.69], p<0.001). Multimodel inference revealed the best approximating model of AVDlac was an additive model including all four fixed effects of post-infusion time, arterial lactate, arterial glucose, and AVDglc (Table 2). AVDlac was slightly negative at the beginning of the infusion study (−0.078 mM, 95%CI [−0.118 −0.036]) at arterial glucose concentration of 7.6 mM, AVDglc of 0.3 mM, and arterial lactate concentration of 0.92 mM (Table 3 or the solution to exp(β0)+min(AVDlac)-1 from Table 2). The fixed effect of time increased AVDlac at an average rate of 0.028 mM/hr independent of changes in arterial lactate concentration (Table 3); because of log transforms, the β1 value is difficult to interpret with units log(mM)/hr).
Table 3.
Mixed effects model results for arteriovenous difference (AVD) lactate at four conditions; arterial glucose and AVDglc are constant at 7.6 mM and 0.3 mM, respectively. The AVDlac model estimate and 95% confidence intervals are back transformed to units of mM from the equation presented in Table 2.
| Infusion time (hr) |
Arterial lactate (mM) |
Model estimate and 95%CI of AVDlac (mM) |
|---|---|---|
| 0 | 0.92 | −0.078 [−0.118 −0.036] |
| 0 | 1.84 | 0.000 [−0.047 0.048] |
| 3 | 0.92 | 0.006 [−0.054 0.069] |
| 3 | 1.84 | 0.090 [0.037 0.145] |
The strongest effect in the model comes from arterial lactate concentration (t value 3.7). When adding the effect of the 3 hour infusion to an increasing arterial lactate concentration, doubling the arterial lactate concentration (from 0.92 to 1.84 mM) after 3 hours would increase AVDlac from −0.078 mM to 0.090 mM [0.037 0.145]. The fixed effects of arterial glucose and AVDglc do not reach the 0.05 significance level but indicate important trends within the context of this model. An increase in arterial glucose was associated with a decrease in AVDlac and an increase in AVDglc was associated with an increase in AVDlac.
Systemic metabolites
All 11 subjects had plasma drawn at baseline and a median of three draws during the primed constant infusion study, corresponding to a total of 48 blood plasma metabolite measurements obtained with quantitative NMR spectroscopy. All glucose and lactate peaks were above the peak picking threshold of 4 standard deviations above noise. There was no discernible difference between arterial and jugular venous blood metabolite concentrations and all metabolites of interest were strongly correlated between arterial and venous blood; jugular venous blood metabolites are not presented.
Arterial lactate concentrations determined with NMR spectroscopy are highly correlated to arterial lactate concentrations determined with the desktop analyzer (rpb=0.92, p<0.001) and the analysis of metabolites from NMR spectroscopy is performed with NMR-determined lactate concentrations. There was no significant change in arterial glucose based on changes in lactate yet there were significant increases in alanine, glutamine, acetate, valine, and leucine (Table 4). A Bonferroni correction for multiple comparisons requires p<0.0056 for significance and no changes were observed in pyruvate, acetoacetate, or β-hydroxybutyrate.
Table 4.
Summary of systemic metabolite concentrations at baseline (mM) and of mixed effects model results. The model coefficient results are presented as a percent change from baseline concentrations for each metabolite assuming a 100% increase in baseline lactate, following the t-value, degrees of freedom (DF), p-value, and number of patients and number of readings available for the analysis.
| Metabolite | Baseline concentration (mM) |
Mixed effects model | |||
|---|---|---|---|---|---|
| Coefficient (% change from baseline) |
t-value (DF) |
p-value | # patients (# readings) | ||
| Glucose | 7.54 [6.46 8.62] |
2 [−1 6] |
1.5 (36) | 0.13 | 11 (48) |
| Alanine | 0.17 [0.12 0.21] |
30 [20 39] |
6.4 (33) | <0.001 | 11 (45) |
| Pyruvate | 0.030 [NA 0.061] |
33 [−1 68] |
2.1 (11) | 0.057 | 5 (17) |
| Glutamine | 0.26 [0.18 0.33] |
34 [24 43] |
7.1 (30) | <0.001 | 10 (41) |
| Acetate | 0.063 [0.042 0.085] |
87 [60 113] |
6.8 (23) | <0.001 | 9 (33) |
| Valine | 0.18 [0.12 0.24] |
40 [28 51] |
7.1 (32) | <0.001 | 11 (44) |
| Leucine | 0.10 [0.08 0.13] |
24 [16 32] |
6.1 (33) | <0.001 | 11 (45) |
| Acetoacetate | 0.075 [NA 0.172] |
−2 [−17 12] |
−0.3 (17) | 0.76 | 7 (25) |
| β-hydroxybutyrate | 0.149 [NA 0.382] |
16 [−11 42] |
1.2 (18) | 0.23 | 7 (26) |
Discussion
Lactate supplementation by primed constant infusion of Sodium L-Lactate increased the arteriovenous difference of lactate (AVDlac), shifting the net balance from negative to positive. The brain both takes up and releases lactate into the blood stream at all times, and a positive AVDlac is representative of net cerebral uptake. Arterial lactate concentrations increased above baseline in all patients; arterial glucose and AVDglc were not impacted by the exogenous lactate infusion. Mixed effects modeling of AVDlac revealed the strongest effect came from arterial lactate concentration (Table 2), indicating that by doubling arterial lactate concentration over the 3 hour study, AVDlac increased from −0.078 mM to 0.090 mM (Table 3). Quantitative analysis of plasma metabolites showed there were significant increases in alanine (30% [20 39]), glutamine (34% [24 43]), acetate (87% [60 113]), valine (40% [28 51]), and leucine (24% [16 32]). These results show that exogenous lactate supplementation leads to increased cerebral uptake of lactate and increased concentrations of systemic metabolites.
Supplementing carbohydrate fuel delivery to the brain without increasing systemic glucose could be an effective therapy in the acute recovery phase. Cerebral lactate uptake was associated with improved neurological outcomes (Glenn et al., 2003) and has been shown to occur even when cerebral lactate levels are high (Jalloh et al., 2013). Infusion of stable isotope-labeled tracers of lactate revealed no difference between the percent lactate uptake oxidized between TBI (91%) and healthy subjects (92%) (Glenn et al., 2015a). This study protocol was designed to target 2.0 mM systemic lactate concentrations and, on average, the maximum concentration achieved was 1.9 mM [1.3 2.4]. This is the first report of a primed constant lactate infusion study targeting moderate lactate concentration elevations. Our findings are consistent with existing examples of the positive effects lactate therapy on ICP reduction (Ichai et al., 2013). In eight patients with ICP monitoring at the time of lactate supplementation, intracranial pressure decreased 3.6 mmHg [95%CI 1.3 6.0].
The additive mixed effects model of AVDlac shows that increased cerebral lactate uptake increases with systemic lactate concentrations and with study infusion time. Even in the absence of an increase in arterial lactate concentration, the model indicates that primed constant infusion will increase AVDlac from −0.078 mM to 0.006 mM over the 3 hour study (Table 3). There was variability between individuals in both baseline lactate concentrations and in the dynamics of blood lactate concentration changes during the study, suggesting that investigating lactate supplementation as a cerebral metabolic therapy requires point-of-care blood lactate readings. As the size of our cohort is small, further studies are required to confirm the generality of our results. No differences in the response to lactate supplementation were identified based on patient’s sex.
Active changes in blood metabolite concentrations could change cerebral blood flow, and active monitoring of blood flow changes will be key to compare metabolic rates between patients and between studies. Although evidence from isotopically labeled tracer infusion studies suggest the vast majority of labeled lactate taken up by the brain is oxidized and released as isotope-labeled CO2 (Glenn et al., 2015a), we cannot definitively say that lactate taken up by the brain in the present study was oxidized. Quantification of cerebral glucose and lactate flux during lactate supplementation is needed to definitively show increased lactate oxidation.
In the attempt to provide alternative fuels to the brain, monitoring systemic changes is important to consider because of the pivotal role lactate plays in systemic signaling (Leverve, 2001; Mosienko et al., 2015; Proia et al., 2016; Mason, 2017). One limitation of our blood plasma metabolite analysis is that we were only able to report on 9 metabolites due to sensitivity limitations of NMR spectroscopy and limited blood plasma for NMR analysis. The baseline concentrations of branched-chain amino acids (valine and leucine) are consistent with previous reports in TBI patients (Robertson et al., 1988) and increases in these amino acids may effect neurochemistry (Fernstrom, 2013). Glutamine has been studied as a nutritional treatment following TBI (Scrimgeour and Condlin, 2014).
Traumatic brain injury is a dynamic disease process that results in depressed cerebral glucose and oxygen metabolism, both of which have been associated with poor outcomes. Supplementing carbohydrate fuel to the brain may provide benefits, but supplementing glucose alone can induce hyperglycemia. Thus, investigation of lactate as a supplemental fuel source for the injured brain has shown improvements in neurochemistry (Bouzat et al., 2014; Quintard et al., 2016), and could also benefit the brain and body through changes to other systemic metabolites. We have shown that at moderate infusion rates and variable changes to patient’s systemic lactate that the net balance of cerebral lactate uptake and release shifts toward uptake, which could improve cerebral neuroenergetics by generating additional ATP to fuel the cellular recovery processes.
Significance Statement.
Following traumatic brain injury, the brain tissue has high energy needs to recovery from the injury and yet is not able to take up fuel to create energy at the same rate as healthy adults brains. This study investigates the impact of moderate lactate supplementation on cerebral uptake of glucose and lactate. We show that supplementing Sodium L-Lactate to approximately 2 mM by constant intravenous infusion results in an increase in total cerebral lactate uptake without changing glucose availability or uptake. Lactate infusion could have therapeutic potential during the recovery phase when glucose does not generate sufficient energy.
Acknowledgments
The authors would like to thank Maria Etchepare, R.N., Rachael Sturtevant, and Matthew P. MacFarlane for their skilled assistance with all ICU studies. We also thank the nurses and staff of the UCLA Neurointensive Care Unit for their help and support. Finally, we thank the UCLA Brain Injury Research Center, the National Institute of Neurological Diseases and Stroke, the University of California Neurotrauma Initiative, the Baskin and Bronson Family Foundation, Evelyn Freed, and the McDonald Family Foundation for their financial support.
Support or Grant Information
National Institute of Neurological Diseases and Stroke (NS055902, NS057252, NS30308, NS058489)
Footnotes
Associate Editor: Dr. Sharon Juliano, Associate Editor, Journal of Neuroscience Research Dr. Eric Prager, Editor-in-Chief, Journal of Neuroscience Research
Conflict of Interest Statement:
No conflicting financial interests exist.
Role of Authors: All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Stephanie M. Wolahan, Howard C. Mao, Thomas C. Glenn. Acquisition of data: Courtney Real, Stephanie M. Wolahan, Howard C. Mao. Analysis and interpretation of data: Stephanie M. Wolahan, Howard C. Mao. Drafting of the manuscript: Stephanie M. Wolahan, Howard C. Mao. Critical revision of the manuscript for important intellectual content: Thomas C. Glenn, Paul M. Vespa. Statistical analysis: Stephanie M. Wolahan. Obtained funding: UCLA Brain Injury Research Center: TCG, SMW, PV, CR, HCM. Administrative, technical, and material support: Courtney Real, Paul M. Vespa, Thomas C. Glenn. Study supervision: Thomas C. Glenn, Courtney Real, Paul M. Vespa.
Data Accessibility:
Please email corresponding author.
Foot Notes
Not applicable.
References
- [Barton, 2013].Barton K. MuMIn: Multi-model inference. 2013. (R package version 1.9.5). [Google Scholar]
- [Bergsneider et al., 2001].Bergsneider M, Hovda D, McArthur D, Etchepare M, Huang SC, Sehati N, Satz P, Phelps M, Becker D. Metabolic recovery following human traumatic brain injury based on FDG-PET: Time course and relationship to neurological disability. J Head Trauma Rehabil. 2001;16:135–148. doi: 10.1097/00001199-200104000-00004. [DOI] [PubMed] [Google Scholar]
- [Bergsneider et al., 1997].Bergsneider M, Hovda D, Shalmon E, Kelly D, Vespa P, Martin N, Phelps M, McArthur D, Caron M, Kraus J, Becker D. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. J Neurosurg. 1997;86:241–251. doi: 10.3171/jns.1997.86.2.0241. [DOI] [PubMed] [Google Scholar]
- [Boumezbeur et al., 2010].Boumezbeur F, Petersen K, Cline G, Mason G, Behar K, Shulman G, Rothman D. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13c nuclear magnetic resonance spectroscopy. J Neurosci. 2010;30:13983–13991. doi: 10.1523/JNEUROSCI.2040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Bouzat et al., 2014].Bouzat P, Sala N, Suys T, Zerlauth JB, Marques-Vidal P, Feihl F, Bloch J, Messerer M, Levivier M, Meuli R, Magistretti P, Oddo M. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 2014;40:412–421. doi: 10.1007/s00134-013-3203-6. [DOI] [PubMed] [Google Scholar]
- [Brodersen and Jorgensen 1974].Brodersen P, Jorgensen E. Cerebral blood flow and oxygen uptake, and cerebrospinal fluid biochemistry in severe coma. J Neurol Neurosurg Psychiatry. 1974;37:384–391. doi: 10.1136/jnnp.37.4.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Burnham and Anderson 2002].Burnham K, Anderson D. Model selection and multimodel inference: A practical information-theoretic approach. second Springer; 2002. [Google Scholar]
- [Fernstrom 2013].Fernstrom J. Large neutral amino acids: dietary effects on brain neurochemistry and function. Amino Acids. 2013;45:419–430. doi: 10.1007/s00726-012-1330-y. [DOI] [PubMed] [Google Scholar]
- [Glenn et al., 2003].Glenn T, Kelly D, Boscardin W, McArthur D, Vespa P, Oertel M, Hovda D, Bergsneider M, Hillered L, Martin N. Energy dysfunction as a predictor of outcome after moderate or severe head injury: Indices of oxygen, glucose, and lactate metabolism. J Cerebr Blood Flow Metab. 2003;23:1239–1250. doi: 10.1097/01.WCB.0000089833.23606.7F. [DOI] [PubMed] [Google Scholar]
- [Glenn et al., 2015a].Glenn T, Martin N, Horning M, McArthur D, Hovda D, Vespa P, Brooks G. Lactate: Brain fuel in human traumatic brain injury. a comparison to normal healthy control subjects. J Neurotrauma. 2015a;32:820–832. doi: 10.1089/neu.2014.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Glenn et al., 2015b].Glenn T, Martin N, McArthur D, Vespa P, Horning M, Johnson M, Brooks G. Endogenous nutritive support following traumatic brain injury: peripheral lactate production for glucose supply via gluconeogenesis. J Neurotrauma. 2015b;32:811–819. doi: 10.1089/neu.2014.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Gowda et al., 2010].Gowda G, Tayyari F, Ye T, Suryani Y, Wei S, Shanaiah N, Raftery D. Quantitative analysis of blood plasma metabolites using isotope enhanced NMR methods. Anal Chem. 2010;82:8983–8990. doi: 10.1021/ac101938w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Herzog et al., 2013].Herzog R, Jiang L, Herman P, Zhao C, Sanganahalli B, Mason G, Hyder F, Rothman D, Sherwin R, Behar K. Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J Clin Invest. 2013;123:1988–1998. doi: 10.1172/JCI65105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Ichai et al., 2013].Ichai C, Payen JF, Orban JC, Quintard H, Roth H, Legrand R, Francony G, Leverve X. Half-molar sodium lactate infusion to prevent intracranial hypertensive episodes in severe traumatic brain injured patients: a randomized controlled trial. Intensive Care Med. 2013;39:1413–1422. doi: 10.1007/s00134-013-2978-9. [DOI] [PubMed] [Google Scholar]
- [Jalloh et al., 2013].Jalloh I, Helmy A, Shannon R, Gallagher C, Menon D, Carpenter K, Hutchinson P. Lactate uptake by the injured human brain: Evidence from an arteriovenous gradient and cerebral microdialysis study. J Neurotrauma. 2013;30:2031–2037. doi: 10.1089/neu.2013.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Leverve 2001].Leverve X. Inter-organ substrate exchanges in the critically ill. Curr Opin Clin Nutr Metab Care. 2001;4:137–142. doi: 10.1097/00075197-200103000-00010. [DOI] [PubMed] [Google Scholar]
- [Lewis et al., 2009].Lewis I, Schommer S, Markley J. rNMR: open source software for identifying and quantifying metabolites in NMR spectra. Magn Reson Chem. 2009;47:S123–S126. doi: 10.1002/mrc.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Mason 2017].Mason S. Lactate shuttles in neuroenergetics - homeostasis, allostasis and beyond. Front Neuroscience. 2017;11(11):43. doi: 10.3389/fnins.2017.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Mosienko et al., 2015].Mosienko V, Teschemacher A, Kasparov S. Is L-lactate a novel signaling molecule in the brain? J Cerebr Blood Flow Metab. 2015;35:1069–1075. doi: 10.1038/jcbfm.2015.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Obrist et al., 1984].Obrist W, Langfitt T, Jaggi J, Cruz J, Gennarelli T. Cerebral blood flow and metabolism in comatose patients with acute head injury. J Neurosurg. 1984;61:241–253. doi: 10.3171/jns.1984.61.2.0241. [DOI] [PubMed] [Google Scholar]
- [Pinheiro et al., 2013].Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team . nlme: Linear and Nonlinear Mixed Effects Models. 2013. (R package version 3.1-108). [Google Scholar]
- [Pinheiro and Bates, 2000].Pinheiro J, Bates D. Mixed-Effects Models in S and S-PLUS. Springer; 2000. [Google Scholar]
- [Proia et al., 2016].Proia P, Di Liegro C, Schiera G, Fricano A, Di Liegro I. Lactate as a metabolite and a regulator in the central nervous system. Int J Mol Sci. 2016;17:1450. doi: 10.3390/ijms17091450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Quintard et al., 2016].Quintard H, Patet C, Zerlauth JB, Suys T, Bouzat P, Pellerin L, Meuli R, Magistretti P, Oddo M. Improvement in neuroenergetic by hypertonic lactate therapy in patients with traumatic brain injury is dependent on baseline cerebral lactate/pyruvate ratio. J Neurotrauma. 2016;33:681–687. doi: 10.1089/neu.2015.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [R Core Team, 2013].R Core Team. R: A Language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. [Google Scholar]
- [Robertson et al., 1988].Robertson C, Clifton G, Grossman R, Ou CN, Goodman J, Borum P, Bejot S, Barrodale P. Alterations in cerebral availability of metabolic substrates after severe head injury. J Trauma. 1988;28:1523–1532. doi: 10.1097/00005373-198811000-00002. [DOI] [PubMed] [Google Scholar]
- [Scrimgeour and Condlin 2014].Scrimgeour A, Condlin M. Nutritional treatment for traumatic brain injury. J Neurotrauma. 2014;31:989–999. doi: 10.1089/neu.2013.3234. [DOI] [PubMed] [Google Scholar]
- [Smith et al., 2003].Smith D, Pernet A, Hallett W, Bingham E, Marsden P, Amiel S. Lactate: A preferred fuel for human brain metabolism in vivo. J Cerebr Blood Flow Metab. 2003;23:658–664. doi: 10.1097/01.WCB.0000063991.19746.11. [DOI] [PubMed] [Google Scholar]
- [Ulrich et al., 2008].Ulrich E, Akutsu H, Doreleijers J, Harano Y, Ioannidis Y, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte C, Tolmie D, Wenger R, Yao H, Markley J. BioMagResBank. Nucleic Acids Res. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [van Hall et al., 2009].van Hall G, Stromstad M, Rasmussen P, Jans O, Zaar M, Gam C, Quistorff B, Secher NH, Nielsen HB. Blood lactate is an important energy source for the human brain. J Cerebr Blood Flow Metab. 2009;29:1121–1129. doi: 10.1038/jcbfm.2009.35. [DOI] [PubMed] [Google Scholar]
- [Wilcox, 1997].Wilcox R. Introduction to robust estimation and hypothesis testing. Academic Press; 1997. [Google Scholar]
- [Wishart et al., 2008].Wishart D, Lewis M, Morrissey J, Flegel M, Jeroncic K, Xiong Y, Cheng D, Eisner R, Gautam B, Tzur D, Sawhney S, Bamforth F, Greiner R, Li L. The human cerebrospinal fluid metabolome. J Chromatogr B. 2008;871:164–173. doi: 10.1016/j.jchromb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- [Wolahan et al., 2016].Wolahan S, Prins M, McArthur D, Real C, Hovda D, Martin N, Vespa P, Glenn T. Influence of glycemic control on endogenous circulating ketone concentrations in adults following traumatic brain injury. Neurocrit care. 2016 doi: 10.1007/s12028-016-0313-3. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Yoshino et al., 1991].Yoshino A, Hovda D, Kawamata T, Katayama Y, Becker D, editors. Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats: Evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561:106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]