

Abstract
Therapy-related acute myeloid leukemias (t-AML) with translocations of the MLL gene are associated with the use of topoisomerase II inhibitors. We established the emergence of the malignant clone in a child who developed t-AML with a t(11;19) (q23;p13.3) during treatment for acute lymphoblastic leukemia (ALL). The MLL-ENL and the reciprocal ENL-MLL genomic fusions and their chimeric transcripts were characterized from samples collected at the time of t-AML diagnosis. We used PCR with patient-specific genomic primers to establish the emergence of the MLL-ENL fusion in serially obtained DNA samples. The MLL-ENL fusion was not detectable in bone marrow at the time of ALL diagnosis or after 2 months of chemotherapy (frequency <8.3 × 10−7 cells−1). The genomic fusion was first detected in bone marrow after 6 months of treatment at a frequency of one in 4,000 mononuclear bone marrow cells; the frequency was one in 70 cells after 20 months of therapy. At the first detection of MLL-ENL, the only topoisomerase II inhibitors the patient had received were one dose of daunorubicin and two doses of etoposide. The MLL-ENL fusion was not detectable in blood at the time of ALL diagnosis or after 0.7, 2, 8, 10, and 12 months of therapy but was detectable in blood at 16 months (one in 2.3 × 104 cells). Recombinogenic Alu sequences bracketed the breakpoints in both fusions. These data indicate that the malignant clone was not present before therapy, arose early during chemotherapy, and was able to proliferate even during exposure to antileukemic therapy.
Therapy-related acute myeloid leukemia (t-AML) is a devastating complication of antineoplastic chemotherapy. There are two recognized categories of therapy-related leukemia: one associated with alkylating agents and the other associated with topoisomerase II inhibitors (1). Topoisomerase II inhibitors are widely used to treat leukemias, lymphomas, testicular, breast, and ovarian cancers. t-AML associated with topoisomerase II inhibitors has a short latency [median 24–34 months (2, 3), as short as 10 months (4)], and a predominance of myelomonoblastic and monocytic morphologic features. Despite the epidemiological link, the roles of schedule and dose for topoisomerase II inhibitors in the pathogenesis of t-AML remain unclear.
Interestingly, over 70% of the cases of t-AML that are associated with exposure to topoisomerase II inhibitors have translocations involving the MLL gene on chromosome 11-band q23 (5, 6), resulting in the in-frame fusion of MLL with any one of more than 40 partner genes (7). The resulting MLL chimeric products may act through a gain-of-function mechanism and are important for leukemic transformation (8); haploinsufficiency for wild-type MLL allele may also play a role during malignant transformation (9). t-AML occurs in up to 10% of children treated for acute lymphoblastic leukemia (ALL) (1, 2, 10), thereby constituting a significant proportion of treatment failures.
Therapy of childhood ALL is unique in that it comprises hundreds of doses of chronically administered chemotherapy, given weekly or daily, for 2–3 years. The use of topoisomerase II inhibitors to treat ALL is controversial, particularly the leukemogenic epipodophyllotoxins etoposide and teniposide, especially in view of improved event-free survival (≈80%) in some contemporary clinical trials (11). The incorporation of the anthracycline-type topoisomerase II inhibitors into ALL remission induction and reinduction regimens, a common approach (11), is also controversial because they have also been implicated in t-AML (12–14). t-AML has not been clearly linked to cumulative doses of topoisomerase II inhibitors (15, 16), having been reported after regimens that contain only a few doses of the agents (14, 17). Thus, the timing of the induction of MLL rearrangements, relative to the few doses of topoisomerase II inhibitors that may be used for lower-risk ALL, is of considerable importance in devising ALL treatment regimens.
Herein, we report the molecular emergence of t-AML during treatment for ALL. We sequenced the MLL-ENL fusion and the reciprocal ENL-MLL from DNA samples at the time of t-AML diagnosis. Using patient-specific primers, we backtracked the emerging malignant clone in bone marrow and peripheral blood serially collected during ALL therapy. Our data document that there was no evidence of AML before starting ALL therapy (frequency <8.3 × 10−7 cells−1) and demonstrate the early development of the MLL-ENL fusion, occurring after only three low doses of topoisomerase II inhibitors.
Case.
A 16-year-old boy with B-lineage ALL presented with 98% lymphoblasts in bone marrow and a leukemic cell DNA index of 1.22. He had no detectable blasts in cerebrospinal fluid and no signs of testicular involvement. The bone marrow karyotype at diagnosis of ALL was hyperdiploid: 58, XY,+X,+X,+4,+8,+10,+12,+17,+18,+20,+21,+21,+22. Immunophenotyping was as described (18). The presence of extra chromosomes was confirmed by fluorescence in situ hybridization (Chromoprobe Interphase, Rainbow Scientific, Windsor, CT). The bone marrow was negative for the following translocations and their fusion transcripts: t(9;22) [p210 BCR-ABL and p190 BCR-ABL], t(1;19) E2A-PBX1, t(4;11) MLL-AF4, and t(12;21) TEL-AML1 (19).
The patient was enrolled on the Total Therapy Study XIII B at St. Jude Children's Research Hospital. Informed consent was obtained from the patient and his guardian. He was assigned to the low-risk treatment arm and received window therapy with methotrexate (1 g/m2 i.v.) for 24 h and 6-mercaptopurine (1 g/m2 i.v.) over 6 h. He received 6 weeks of remission induction therapy with prednisone 40 mg/m2 daily, daunorubicin 25 mg/m2 on day 1, vincristine 1.5 mg/m2 i.v. weekly, asparaginase 10,000 units/m2 i.m. three times per week × six doses, and etoposide 300 mg/m2 plus cytarabine 300 mg/m2 on days 35 and 41 (Fig. 1). Because of a preexisting infection and severe mucositis and neutropenia, the patient did not receive two other scheduled doses of topoisomerase II inhibitors, and he received five doses of granulocyte colony-stimulating factor (G-CSF) 5 μg/kg during remission induction. On attaining clinical remission, he received weekly methotrexate 2 g/m2 i.v. × 2 and daily oral mercaptopurine 75 mg/m2. Continuation therapy consisted of weekly blocks of chemotherapy: daily oral mercaptopurine 75 mg/m2 and weekly methotrexate 40 mg/m2, i.v. or i.m.; daily oral dexamethasone 8 mg/m2/day plus vincristine 1.5 mg/m2 every 4 weeks; and methotrexate 2 g/m2 i.v. and daily oral mercaptopurine 75 mg/m2 every 8 weeks during the first year. Reinduction therapy, given from weeks 28 to 34, was the same as induction therapy, except that two doses of daunorubicin (days 1 and 8) and only one dose of etoposide plus cytarabine (day 22) were given. The patient received 14 doses of intrathecal chemotherapy during the first year.
Figure 1.

Treatment protocol (St. Jude Children's Research Hospital Total XIII B) for ALL in a patient who developed t-AML (Upper). The time of t-AML diagnosis is indicated by an arrow. The interval corresponding to each therapy block is indicated in gray scale boxes (Upper) and corresponds with the time line in days (Lower). The times of administration of topoisomerase II inhibitors (etoposide, 300 mg/m2 ▿, and daunorubicin, 25 mg/m2 Δ), asparaginase, 10,000 units/m2 thrice weekly × six doses (■), and G-CSF (|) are indicated. MLL-ENL genomic clone frequencies (Lower) for bone marrow (■) and for peripheral blood samples (□). N.D., not detected.
Twenty-three months into therapy, he was diagnosed with t-AML (FAB M4), confirmed by extensive immunophenotyping (18). The bone marrow karyotype at the time of diagnosis of t-AML was 46,XY,t(11;19)(q23;p13.3) in 60% of the metaphase cells; blasts were positive for MLL-ENL fusion transcripts by reverse transcription–PCR (20) and negative for MLL-AF4, MLL-AF9 and MLL-ELL transcripts.
He received an allogeneic stem cell transplant and died of complications (graft-versus-host disease) 7 months later.
Pharmacokinetics and Pharmacogenetics.
The patient was genotyped for the presence of mutations TPMT *2, *3A, *3B, and *3C in thiopurine methyltransferase (TPMT) (21). TPMT activity was measured in red blood cells (22). Blood DNA was also genotyped for a polymorphism in the CYP3A4 promoter (23) and in NAD(P)H/quinone oxidoreductase (NQO1) (24). DNA from bone marrow at the time of diagnosis of t-AML was analyzed for point mutations in the cytoplasmic domain of the G-CSF receptor (25).
Plasma clearance of etoposide and area under the plasma concentration vs. time curve of etoposide and its catechol metabolite were measured during reinduction therapy, as described (26).
Samples.
DNA and RNA were isolated by personnel not involved in further PCR analysis in a laboratory free of PCR amplification products.
Blood and bone marrow mononuclear cells were collected by centrifugation on a density gradient (Lymphoprep, Nycomed, Oslo). DNA and RNA were isolated by using TriReagent (Molecular Research Center, Cincinnati). DNA was dissolved in Tris-EDTA buffer and stored at 4°C until use. RNA was dissolved in water and stored at −70°C until use.
Cloning MLL-ENL and ENL-MLL Breakpoints.
Long-distance inverse PCR (27) was used to amplify the MLL-ENL breakpoint from 100 ng of DNA from blood (43% blasts) at the time of diagnosis of t-AML, by using the Expand Long Template PCR system (Roche Molecular Biochemicals). MLL “inverse” primers were: primer A, 5′-ATACATCCCTGAGAAATGGCAGAGAAAC-3′ (position 708–681, GenBank no. U04737) and primer B, 5′-AGCACCAACTGGGGGAATGAATAAGAAC-3′ (position 2576–2603) (Fig. 2). Conditions (MJ Research Thermal Cycler, Watertown, MA) were: 95°C × 5 min; 45 cycles at 95°C × 30 s, 58°C × 1 min, and 68°C × 12 min; extension at 70°C × 10 min. The PCR products spanning the MLL-ENL breakpoint were sequenced (ABIprism 3700, Applied Biosystems) and analyzed with gcg Ver. 10.1. Sequence motifs (e.g., Alu repeats, topoisomerase II sites) were analyzed [e.g., findpatterns (www.gcg.com), censor (http://www.girinst.org/Censor_Server.html), mar finder (http://www.futuresoft.org/MAR-Wiz/)]. The reciprocal ENL-MLL breakpoint was amplified by using 100 ng of DNA from bone marrow at the time of t-AML, by using the Expand Long Template PCR system (Roche Molecular Biochemicals). Patient-specific primers to cover the putative breakpoint were: 5′-ATCAGCCCACTACAACCTCCAC-3′ (forward) and 5′-AAAACAGACACCCTCCCTTCAC-3′ (reverse). Touchdown PCR conditions were 92°C × 2 min, 5 cycles of 92°C × 15 s, 65°C (−1°C per cycle) × 35 s and 68°C × 3 min; then 30 cycles of 92°C × 15 s, 60°C × 30 s and extension at 68°C. PCR products were analyzed as for the MLL-ENL genomic fusion.
Figure 2.

(A) Long-distance inverse PCR amplification of a 1.9-kb product containing the MLL-ENL fusion from peripheral blood DNA at the time of diagnosis of t-AML (Upper) and of a 1.8-kb product containing the ENL-MLL fusion from bone marrow at the time of t-AML diagnosis (Lower). W, water control. DNA marker: 1-kb ladder. (B) Schematic of MLL (Top), indicating exons 5–9, the location of the MLL breakpoint in this case, and the location of an in vivo topoisomerase II cleavage site (46) by an arrow. The MLL-ENL breakpoint region amplified by long-distance inverse PCR (Middle) is indicated. The positions of the MLL inverse primers (A and B), the Eco57-I restriction sites, the Alu Y repeats, and two in vitro topoisomerase II recognition sites (black arrows located below the fusion product) are indicated. Details of the MLL, der (11), ENL, and der (19) DNA sequences bracketing the fusion points are depicted (Bottom). The triplet GTA in bold depicts the fusion points in both parental alleles and in the fusions. (C) Genomic organization of ENL, covering ≈58 kb, derived from alignment of genomic and cDNA sequences (GenBank no. L04285). Boxes show 11 exons. The ENL breakpoint corresponds to position 46,181 (GenBank no. NT_011316).
MLL-ENL and ENL-MLL Fusion Transcripts.
One microgram of RNA was reverse transcribed with Superscript (GIBCO/BRL) by using oligo(dT)12–18 primers.
For amplification of the MLL-ENL transcripts, primers were: forward 5′-CAATAAGCAGGAGAATGCAGG-3′ (position 2370–2390, GenBank no. U04737) and reverse 5′-GGAATTGTGGGTAACATGGGG-3′ (position 494–474, GenBank no. L04285). For amplification of the ENL-MLL transcripts, primers were: forward 5′-GTTAGAGCTGGGGCATCGC-3′ (position 30–48, GenBank no. L04285) and reverse 5′-TTGTGGGTTTGGTGGGGTAG-3′ (position 8045–8026, GenBank no. U04737). Each 50-μl PCR contained 2 μl of the cDNA conversion mixture, nuclease-free water, 50 pmol of each primer, 2.5 units of Amplitaq Gold DNA polymerase (Perkin–Elmer), 250 μM each dNTP (Invitrogen), and Gene Amp PCR buffer (Perkin–Elmer). Amplification conditions were: 95°C × 1 min; 45 cycles of 95°C × 30 s, 53°C × 30 s, and 70°C × 45 s; and final extension at 70°C × 10 min. Reverse transcription–PCR products were sequenced by using the PCR primers.
PCR Analysis in Serially Collected Samples.
Reagents, equipment, and samples were isolated from potential sources of contamination by use of separate laboratories and Clean Spot PCR work stations (Coy Laboratory, Grass Lake, MI). Nested PCR was used to amplify MLL-ENL in DNA samples collected during ALL therapy. First-round primers were: 5′-AGCAGCAGTTATTTTTGGACTC-3′ (forward) and 5′-GCCTCCCTTACTAGATACCCAC-3′ (reverse); second round primers were: 5′-GAAAATGTGTGGGAGATGGGAG-3′ (forward) and 5′-CAAGTGTGGCAAAGGGTTTCAG-3′ (reverse). Each first-round 50-μl PCR reaction contained 100 ng of DNA, nuclease-free water, 50 pmol of each primer, 1.25 units of Amplitaq Gold, 250 μM each dNTP, and Gene Amp PCR buffer. The frequency of the MLL-ENL rearrangement can be estimated by using the principles of limiting dilution and Poisson statistics (28). Assuming 100 ng of DNA represents 16,000 cells, a single PCR with 100 ng of DNA comprising a 104 dilution of MLL-ENL-carrying cells in MLL-ENL-negative cells would have 80% of replicate PCRs positive, and a 105 dilution would have only 15% positive (28). These frequencies agree with those observed (data not shown), substantiating the assumption that each PCR amplified MLL-ENL if it was present. Amplification conditions were 95°C × 5 min; 45 cycles of 95°C × 45 s, 53°C × 1 min, 70°C × 1 min; and a final extension at 70°C × 10 min. One microliter of the first-round PCR was reamplified by using identical conditions with the nested primers. The integrity of each DNA sample for PCR was confirmed by using internal MLL primers: forward 5′-AGCACCAACTGGGGGAATGAATAAGAAC-3′ (position 2576–2603, GenBank no. U04737) and reverse 5′-CTCAGACACGGACTATTAAAAGGCTCAC-3′ (position 2875–2848), with the same PCR conditions as for MLL-ENL.
Results
Sequencing of long-distance inverse PCR products at diagnosis of t-AML (Fig. 2) revealed that the breakpoint occurred at position 3131 of the MLL breakpoint cluster region (GenBank no. U04737). The breakpoint is in exon 8, 14 bases upstream from its 3′ end (Fig. 2). The 3′ portion of the breakpoint mapped to chromosome 19 (GenBank no. NT_011316), with the breakpoint at position 46,181. To obtain some insight about the genomic organization and the location of the breakpoint, we compared the cDNA sequence of ENL (GenBank no. L04285) and the partial genomic sequence of chromosome 19. ENL exon positions were confirmed and refined by the use of National Center for Biotechnology Information ace view (Fig. 2). The analysis indicated that the ENL gene contains 11 exons and that the breakpoint is located in intron 3, which contains 31,512 bp and is the largest intron in ENL (Fig. 2).
The MLL-ENL breakpoint is flanked by two Alu Y repeats (29) (Fig. 2). One Alu Y repeat (290-bp long, DNA-positive strand) is in MLL, 1,422 bp 5′ from the breakpoint. An Alu Y repeat (290-bp long, DNA-negative strand) on the ENL side is located 630 bp 3′ of the MLL-ENL breakpoint. Both Alu Y segments contain the core sequence 5′-CCTGTAATCCCAGCACTTTGGGAGGC-3′ (30); one G→A transition (position 25) was found in the core element located in ENL. The breakpoint is flanked by two topoisomerase II sites 5′-A/GNT/CNNCNNGT/CNGG/TTNT/CNT/C-3′ (31); the site in MLL (9/10 matches, DNA-positive strand) is located 523 bp from the breakpoint, and the site in ENL (9/10 matches, DNA-positive strand) is 101 bp 3′ of the breakpoint (Fig. 2).
The derivative ENL-MLL genomic fusion was amplified from bone marrow DNA at the time of t-AML diagnosis (Fig. 2). Interestingly, the ENL-MLL breakpoint is also flanked by two Alu Y repeats containing the core element. One Alu Y repeat is in ENL (288 bp long, DNA-negative strand) 758 bp 5′ from the breakpoint; the other (290 bp long, DNA-positive strand) is in MLL 842 bp 3′ from the fusion point. Three topoisomerase II sites (9/10 matches, DNA-positive strand) were found in ENL at a distance of 56, 153, and 351 bp 5′ from the fusion point, respectively.
The triplet GTA is present on both MLL-ENL and ENL-MLL genomic fusion points and on the sequences of both parental genes. Both fusions indicate that the fusion event occurred in a conservative manner without gain or loss of DNA from either MLL or ENL (Fig. 2).
The MLL-ENL transcript has an in-frame fusion of MLL exon 7 to ENL exon 3. The ENL-MLL transcript contains ENL exon 2 fused in frame to MLL exon 9. No MLL exon 8 was found in either the MLL-ENL or ENL-MLL transcripts, indicating that the genomic translocation prevented exon 8 expression.
The sensitivity of the PCR reaction to track the emergence of the MLL-ENL fusion during therapy was tested by diluting the t-AML diagnostic blood DNA in normal leukocyte DNA (100 ng). The PCR reaction was sensitive enough to detect one cell carrying the MLL-ENL fusion per PCR reaction, which was tested against a background of 16,000 cells without the fusion (Fig. 3A). Specificity was demonstrated by the absence of MLL-ENL fusions in DNA samples from five normal volunteers (data not shown).
Figure 3.

(A) Limit of detection of the MLL-ENL genomic PCR reaction. Second-round PCR amplification of a 245-bp MLL-ENL breakpoint from t-AML diagnostic blood DNA (100 ng–0.01 pg) serially diluted in normal blood DNA. W, water. 100 bp = DNA ladder. (B) DNA integrity control for samples serially collected during ALL therapy. PCR amplification of a 300-bp segment of MLL gene. Lanes 1–5, bone marrow DNA; lanes 6–13, blood DNA. W, water. 100 bp = DNA ladder. (C) Second-round PCR of a 245-bp MLL-ENL breakpoint from bone marrow DNA at 608 days of ALL therapy. All of the replicates (100 ng each) were positive and were run with positive and negative controls (see details in the text). (D) Second-round PCR of a 245-bp MLL-ENL breakpoint from blood DNA collected after 497 days of ALL therapy. Four of 10 replicates (100 ng each) were positive and were run with positive and negative controls.
DNA samples from bone marrow (n = 5) and blood (n = 8) collected during ALL therapy were analyzed for the MLL-ENL fusion (Fig. 3 C and D). Each experiment included at least as many PCR negative controls (water) as it did PCRs with patient DNA, and negative controls of 100 ng of DNA from normal volunteers were included in each experiment. We tested 10 replicates for each of the blood DNA samples at days 21, 65, and 246 of ALL therapy; for the remainder of the blood and bone marrow samples, the average number of replicates tested per sample was 40, ≈6.4 × 105 cells per sample.
The MLL-ENL genomic fusion was not detectable in bone marrow at diagnosis of ALL or in bone marrow 56 days after the start of ALL therapy. As each PCR reaction could detect the MLL-ENL fusion if it was present (see Methods, PCR Analysis in serially collected samples), by increasing the number of replicates (75 × of 100 ng at both times), we confirmed the absence of the MLL-ENL fusion in an amount of DNA corresponding to ≈1.2 × 106 bone marrow cells for each of the early samples, indicating that the frequency was likely to be less than 8.3 × 10−7 cells−1 at diagnosis and at the end of remission induction.
The MLL-ENL genomic fusion was detected in bone marrow after 6 months (day 187) of chemotherapy, just before remission reinduction. The frequency of the MLL-ENL clone was estimated by limiting dilution (28) at 2.7 × 10−4 cells−1 (1 in 4,000 cells) (Fig. 1). The fusion was also detectable in bone marrow after 20 months (day 608, Fig. 3C) at a frequency of 0.0142 cells−1 (1 in 70 cells) and at the time of t-AML diagnosis at 0.46 cells−1.
The MLL-ENL fusion was not detected in DNA from blood at the time of diagnosis of ALL or after 0.7, 2, 8, 10, and 12 months of ALL therapy (Fig. 1). The MLL-ENL fusion was detected in a blood sample after 16 months (day 497, Fig. 3D) at 4.3 × 10−5 cells−1 (1 in 2.3 × 104). The clone frequency at the time of diagnosis of t-AML in peripheral blood was 0.52, consistent with the 43% of blast cells established by morphology.
The CYP3A4 promoter genotype was wild type; inactivating mutations in TPMT (nucleotides 238, 460, and 719) were not present; and there were no point mutations in the cytoplasmic domain of the G-CSF receptor. The genotype for NQO1 (a C→T change at nucleotide 609) was homozygous variant. The clearance for etoposide was 2.2 l/h/m2, and the areas under the plasma concentration vs. time curve for etoposide and its catechol metabolite were 229 μM⋅h and 1.88 μM⋅h, respectively, comparable to those of other patients treated at the same time point in therapy.
Discussion
The present study demonstrates the emergence of a t-AML-specific MLL-ENL genomic fusion after only three low doses of topoisomerase II inhibitors in a child with low-risk ALL. This represents, to our knowledge, the first temporal characterization and quantitative evaluation of the emergence of a t-AML-related fusion during ALL therapy. The early onset of a t-AML-related fusion was also demonstrated in a child with neuroblastoma (14).
The use of the MLL-ENL gene fusion as a specific leukemia-associated marker is supported by evidence showing that the resulting MLL-ENL chimeric proteins are etiologically important in leukemias (32). The reciprocal ENL-MLL fusion occurred with no loss or gain of genetic material relative to the MLL-ENL fusion, and the reciprocal ENL-MLL transcripts were also present. The biological relevance of these reciprocal fusion transcripts remains to be elucidated.
We studied the timing of the fusion in DNA samples spanning 700 days of therapy for ALL. The MLL-ENL genomic fusion was not detected in either bone marrow or blood at the time of diagnosis of ALL nor was it detected at the end of remission–induction therapy. The final “doubling time” of the MLL-ENL bearing clone was 18.3 and 15.0 days in the bone marrow and peripheral blood, respectively, in the few months preceding diagnosis of t-AML, whereas the growth appeared to be slower (doubling time: 73.6 days) between days 187 and 608 of therapy. It is conceivable that the intense chemotherapy given during reinduction (starting at day 199) had an antiproliferative effect on the MLL-ENL clone. The increase in the MLL-ENL clone frequency during continuation chemotherapy indicates that the regimen was ineffective to stop the growth of the malignant clone. Whether the earlier detection in bone marrow relative to blood is because of the predilection of myeloid leukemia to localize in the bone marrow or because of a chemotherapy-induced suppression of migration of the myeloid cells from bone marrow to the periphery cannot be ascertained.
There are at least two theories to explain the early onset of the MLL-ENL clone in the context of this ALL regimen. One is that the topoisomerase II inhibitors directly caused the translocation event. That 70% of t-AML cases associated with the use of topoisomerase II inhibitors have MLL translocations compared with almost no cases of “alkylator-associated” AML supports the notion that the topoisomerase II inhibitors cause the rearrangements. However, we cannot rule out the possibility that a cell carrying the translocation was already present at the time of ALL diagnosis at a frequency of less than one in 1.2 × 106 cells, and that the clone was “selected for” by the chemotherapy. In another case of early-onset of t-AML (14), the translocation was not detected in pretherapy bone marrow slides, although the number of cells tested was far fewer than the numbers we were able to test. That many effective antileukemic regimens that do not contain topoisomerase II inhibitors are not able to “select for” such MLL rearrangements, and that the t-AML remains responsive to topoisomerase II inhibitors, argue against the theory of clonal selection (3).
Other risk factors such as concurrent chemotherapy and host-related factors might contribute to the development of t-AML (1, 33). For example, the use of asparaginase has been linked to an increased risk for t-AML (34, 35), and this patient had asparaginase concomitantly with or before the topoisomerase II inhibitors (Fig. 1).
Pharmacogenetic polymorphisms can account for interindividual variability in response to anticancer drugs (36), which may contribute to the risk of t-AML. A paucity of polymorphisms in the CYP3A4 promoter was associated with the risk of t-AML (23), and in accord with this finding, the patient had a wild-type CYP3A4 promoter genotype. A polymorphism at position 609 of NQO1 has been associated with an increased risk of leukemias with MLL fusions (37) and also with therapy-related myeloid leukemia (24). The patient had a genotype that dictates the absence of NQO1 protein and enzymatic activity (38). Thus, the patient may have been particularly susceptible to the mutagenic effects of the chemotherapeutic agents.
The patient's pharmacokinetic parameters (clearance and area under the plasma concentration vs. time curve) for etoposide and its catechol were close to the median for this population, in agreement with the lack of differences in etoposide disposition between patients who did and did not develop t-AML (26). Patients with etoposide-related AML tend to have low TPMT activity (26, 39), but this patient had wild-type TPMT activity and genotype.
Point mutations in the G-CSF receptor gene have been reported in patients who developed myeloid leukemia after chronic use of G-CSF (40). Because this patient received therapy with G-CSF during remission induction (Fig. 1), we sequenced this region but found no point mutations in the germline or in t-AML blast DNA. The contribution of G-CSF to the risk of t-AML is controversial (41).
Chromosomal breakage results from the stabilization of DNA/topoisomerase II complexes by topoisomerase II inhibitors (42). The sequences of the MLL-ENL and ENL-MLL breakpoints indicate that the fusion process occurred in a conservative manner, with a total preservation of the sequence corresponding to each one of the partners. Both MLL-ENL and ENL-MLL breakpoints are flanked by Alu Y repeats that are symmetrically positioned relative to each fusion point (Fig. 4). The Alu Y repeats on ENL are in an antiparallel orientation with respect to the Alu Y repeats of MLL. The Alu repeat elements contain the Alu core associated with recombination. The pentanucleotide motif CCAGC, which was contained in all four core elements, may stimulate the recombination processes (30). Alu repetitive elements have been found in other MLL gene rearrangements (14, 43–45). Whether topoisomerase II recognition sites near the breakpoints in fusions contributed to their genesis is not clear, as none of the sites precisely correspond to the breakpoints, and the function of such in vitro sequences has been questioned. An in vivo topoisomerase II cleavage site (46) is depicted in Fig. 2, and although it is well 3′ of the breakpoint, such cleavage has been hypothesized to participate in the genesis of MLL fusions. Assuming that cleavage occurred at the fusion breakpoint in both genes, the antiparallel Alu Y repetitive elements could have facilitated the alignment of MLL and ENL. The resulting reciprocal fusion may have occurred as an attempt to resolve the DNA cleavage caused by drug action (Fig. 4). That there are no insertions or deletions in either the MLL-ENL or ENL-MLL fusions indicates that both DNA breaks must have been remarkably stabilized, and that cellular recombination machinery in joining the nonhomologous partners was relatively uncomplicated, without the “nibbling” or insertions associated with other translocations. The GTA triplet present at the MLL and ENL breakpoints suggests the ultimate role of DNA end-joining, as has been proposed for other MLL translocations (14, 47).
Figure 4.
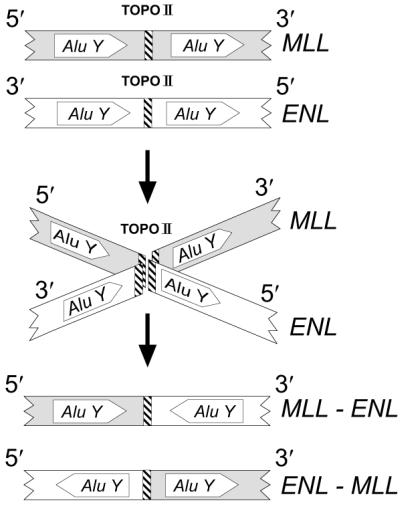
Model for the genesis of the MLL-ENL and ENL-MLL fusions during ALL treatment with topoisomerase II inhibitors. Both MLL and ENL gene fragments originate by DNA double-strand breaks and are positioned by Alu Y repeats (Top). Double-strand DNA cleavage in MLL and ENL (Middle) is catalyzed by topoisomerase II (TOPO II) and stabilized by topoisomerase II inhibitors. Recombination processes resolve the breaks (Bottom), with MLL-ENL and ENL-MLL partners aligned regionally by Alu Y elements and locally by a region of short homology (the GTA triplet in the parental alleles and in the fusions is indicated by a crosshatched vertical bar). The resulting derivative fusions contribute to leukemogenesis and are present at the time of t-AML diagnosis.
Therapy-related malignancies are devastating complications that account for a significant proportion of ALL treatment failures in children. The evidence presented here indicates that the malignant clone was present in bone marrow early during ALL chemotherapy. We hypothesize that the use of topoisomerase II inhibitors triggered the fusion event, a necessary step for malignant transformation. Our data are consistent with the notion that modest exposure to topoisomerase II inhibitors, in a permissive setting, can be sufficient to induce t-AML.
Acknowledgments
The assistance of Pamela McGill, Miriam Tamaño and Amanda Taylor is gratefully acknowledged. This work was supported by National Institutes of Health Grants CA51001, CA36401, and CA21765, by a Center of Excellence grant from the State of Tennessee, and by American Lebanese Syrian Associated Charities.
Abbreviations
- t-AML
therapy-related acute myeloid leukemia
- ALL
acute lymphoblastic leukemia
- G-CSF
granulocyte colony-stimulating factor
- TPMT
thiopurine methyltransferase
- NQO1
NAD(P)H/quinone oxidoreductase
Footnotes
References
- 1.Pui C H, Relling M V. Br J Haematol. 2000;109:13–23. doi: 10.1046/j.1365-2141.2000.01843.x. [DOI] [PubMed] [Google Scholar]
- 2.Winick N J, Smith S D, Shuster J, Lauer S, Wharam M D, Land V, Buchanan G, Rivera G. J Clin Oncol. 1993;11:271–278. doi: 10.1200/JCO.1993.11.2.271. [DOI] [PubMed] [Google Scholar]
- 3.Pui C H, Relling M V, Rivera G K, Hancock M L, Raimondi S C, Heslop H E, Santana V M, Ribeiro R C, Sandlund J T, Mahmoud H H. Leukemia. 1995;9:1990–1996. [PubMed] [Google Scholar]
- 4.Kushner B H, Heller G, Cheung N-K V, Wollner N, Kramer K, Bajorin D, Polyak T, Meyers P A. J Clin Oncol. 1998;16:3016–3020. doi: 10.1200/JCO.1998.16.9.3016. [DOI] [PubMed] [Google Scholar]
- 5.Pui C H, Behm F G, Raimondi S C, Dodge R K, George S L, Rivera G K, Mirro J, Jr, Kalwinsky D K, Dahl G V, Murphy S B. N Engl J Med. 1989;321:136–142. doi: 10.1056/NEJM198907203210302. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen-Bjergaard J, Rowley J D. Blood. 1994;83:2780–2786. [PubMed] [Google Scholar]
- 7.Rowley J D. Semin Hematol. 1999;36:59–72. [PubMed] [Google Scholar]
- 8.Corral J, Lavenir I, Impey H, Warren A J, Forster A, Larson T A, Bell S, McKenzie A N J, King G, Rabbitts T H. Cell. 1996;85:853–861. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- 9.Yu B D, Hess J L, Horning S E, Brown G A, Korsmeyer S J. Nature (London) 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 10.Pui C H, Ribeiro R C, Hancock M L, Rivera G K, Evans W E, Raimondi S C, Head D R, Behm F G, Mahmoud M H, Sandlund J T, Crist W M. N Engl J Med. 1991;325:1682–1687. doi: 10.1056/NEJM199112123252402. [DOI] [PubMed] [Google Scholar]
- 11.Pui C H, Evans W E. N Engl J Med. 1998;339:605–615. doi: 10.1056/NEJM199808273390907. [DOI] [PubMed] [Google Scholar]
- 12.Pedersen-Bjergaard J, Daugaard G, Hansen S W, Philip P, Larsen S O, Rorth M. Lancet. 1991;338:359–363. doi: 10.1016/0140-6736(91)90490-g. [DOI] [PubMed] [Google Scholar]
- 13.Hawkins M M, Wilson L M K, Stovall M A, Marsden H B, Potok M H N, Kingston J E, Chessells J M. Br Med J. 1992;304:951–958. doi: 10.1136/bmj.304.6832.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Megonigal M D, Cheung N-K V, Rappaport E F, Nowell P C, Wilson R B, Jones D H, Addya K, Leonard D G, Kushner B H, Williams T M, et al. Proc Natl Acad Sci USA. 2000;97:2814–2819. doi: 10.1073/pnas.050397097. . (First Published March 7, 2000; 10.1073/pnas.050397097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stine K C, Saylors R L, Sawyer J R, Becton D L. J Clin Oncol. 1997;15:1583–1586. doi: 10.1200/JCO.1997.15.4.1583. [DOI] [PubMed] [Google Scholar]
- 16.Smith M A, Rubinstein L, Anderson J R, Arthur D, Catalano P J, Freidlin B, Heyn R, Khayat A, Krailo M, Land V J, Miser J, Shuster J, Vena D. J Clin Oncol. 1999;17:569–577. doi: 10.1200/JCO.1999.17.2.569. [DOI] [PubMed] [Google Scholar]
- 17.Leblanc T, Hillion J, Derre J, Le Coniat M, Baruchel A, Daniel M T, Berger R. Leukemia. 1994;8:1646–1648. [PubMed] [Google Scholar]
- 18.Behm F G, Smith F O, Raimondi S C, Pui C H, Bernstein I D. Blood. 1996;87:1134–1139. [PubMed] [Google Scholar]
- 19.Scurto P, Hsu Rocha M, Kane J R, Williams W K, Haney D M, Conn W P, Shurtleff S A, Downing J R. Leukemia. 1998;12:1994–2005. doi: 10.1038/sj.leu.2401224. [DOI] [PubMed] [Google Scholar]
- 20.Rubnitz J E, Behm F G, Curcio-Brint A M, Pinheiro V R P, Carroll A J, Raimondi S C, Shurtleff S A, Downing J R. Blood. 1996;87:4804–4808. [PubMed] [Google Scholar]
- 21.Yates C R, Krynetski E Y, Loennechen T, Fessing M Y, Tai H L, Pui C H, Relling M V, Evans W E. Ann Intern Med. 1997;126:608–614. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 22.McLeod H L, Lin J S, Scott E P, Pui C H, Evans W E. Clin Pharmacol Ther. 1994;55:15–20. doi: 10.1038/clpt.1994.4. [DOI] [PubMed] [Google Scholar]
- 23.Felix C A, Walker A H, Lange B J, Williams T M, Winick N J, Cheung N-K V, Lovett B D, Nowell P C, Blair I A, Rebbeck T R. Proc Natl Acad Sci USA. 1998;95:13176–13181. doi: 10.1073/pnas.95.22.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larson R A, Wang Y, Banerjee M, Wiemels J, Hartford C, Le Beau M M, Smith M T. Blood. 1999;94:803–807. [PubMed] [Google Scholar]
- 25.Dong F, van Paassen M, van Buitenen C, Hoefsloot L H, Löwenberg B, Touw I P. Blood. 1995;85:902–911. [PubMed] [Google Scholar]
- 26.Relling M V, Yanishevski Y, Nemec J, Evans W E, Boyett J M, Behm F G, Pui C H. Leukemia. 1998;12:346–352. doi: 10.1038/sj.leu.2400928. [DOI] [PubMed] [Google Scholar]
- 27.Wiemels J L, Greaves M. Cancer Res. 1999;59:4075–4082. [PubMed] [Google Scholar]
- 28.Sykes P J, Neoh S H, Brisco M J, Hughes E, Condon J, Morley A A. BioTechniques. 1992;13:444–449. [PubMed] [Google Scholar]
- 29.Jurka J, Klonowski P, Dagman V, Pelton P. Comput Chem. 1996;20:119–121. doi: 10.1016/s0097-8485(96)80013-1. [DOI] [PubMed] [Google Scholar]
- 30.Rüdiger N S, Gregersen N, Kielland-Brandt M C. Nucleic Acids Res. 1995;23:256–260. doi: 10.1093/nar/23.2.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller M T, Spitzner J R, DiDonato J A, Mehta V B, Tsutsui K, Tsutsui K. Biochemistry. 1988;27:8369–8379. doi: 10.1021/bi00422a012. [DOI] [PubMed] [Google Scholar]
- 32.Rubnitz J E, Morrissey J, Savage P A, Cleary M L. Blood. 1994;84:1747–1752. [PubMed] [Google Scholar]
- 33.Smith M A, McCaffrey R P, Karp J E. J Natl Cancer Inst. 1996;88:407–418. doi: 10.1093/jnci/88.7.407. [DOI] [PubMed] [Google Scholar]
- 34.Pui C H, Relling M V, Behm F G, Hancock M L, Boyett J M, Raimondi S C, Krance R A, Mahmoud H H, Ribeiro R C, Sandlund J T. Leukemia. 1995;9:1680–1684. [PubMed] [Google Scholar]
- 35.Amylon M D, Shuster J, Pullen J, Berard C, Link M P, Wharam M, Katz J, Yu A, Laver J, Ravindranath Y, et al. Leukemia. 1999;13:335–342. doi: 10.1038/sj.leu.2401310. [DOI] [PubMed] [Google Scholar]
- 36.Evans W E, Relling M V. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 37.Wiemels J L, Pagnamenta A, Taylor G M, Eden O B, Alexander F E, Greaves M F. Cancer Res. 1999;59:4095–4099. [PubMed] [Google Scholar]
- 38.Traver R D, Horikoshi T, Danenberg K D, Stadlbauer T H, Danenberg P V, Ross D, Gibson N W. Cancer Res. 1992;52:797–802. [PubMed] [Google Scholar]
- 39.Bo Thomsen J, Schrøder H, Kristinsson J, Madsen B, Szumlanski C, Weinshilboum R, Andersen J B, Schmiegelow K. Cancer. 1999;86:1080–1086. doi: 10.1002/(sici)1097-0142(19990915)86:6<1080::aid-cncr26>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Tidow N, Pilz C, Teichmann B, Müller-Brechlin A, Germeshausen M, Kasper B, Rauprich P, Sykora K W, Welte K. Blood. 1997;89:2369–2375. [PubMed] [Google Scholar]
- 41.Locasciulli A, Arcese W, Locatelli F, Di Bona E, Bacigalupo A. Lancet. 2001;357:43–44. doi: 10.1016/s0140-6736(00)03574-1. [DOI] [PubMed] [Google Scholar]
- 42.Strick R, Strissel P L, Borgers S, Smith S L, Rowley J D. Proc Natl Acad Sci USA. 2000;97:4790–4795. doi: 10.1073/pnas.070061297. . (First Published April 11, 2000; 10.1073/pnas.070061297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schichman S A, Caligiuri M A, Strout M P, Carter S L, Gu Y, Canaani E, Bloomfield C D, Croce C M. Cancer Res. 1994;54:4277–4280. [PubMed] [Google Scholar]
- 44.So C W, Ma Z G, Price C M, Dong S, Chen S J, Gu L J, So C K C, Wiedemann L M, Chan L C. Cancer Res. 1997;57:117–122. [PubMed] [Google Scholar]
- 45.Whitman S P, Strout M P, Marcucci G, Freud A G, Culley L L, Zeleznik-Le N J, Mrózek K, Theil K S, Kees U R, Bloomfield C D, Caligiuri M A. Cancer Res. 2001;61:59–63. [PubMed] [Google Scholar]
- 46.Strissel P L, Strick R, Rowley J D, Zeleznik-Le N J. Blood. 1998;92:3793–3803. [PubMed] [Google Scholar]
- 47.Super H G, Strissel P L, Sobulo O M, Burian D, Reshmi S C, Roe B, Zeleznik-Le N J, Diaz M O, Rowley J D. Genes Chromosomes Cancer. 1997;20:185–195. [PubMed] [Google Scholar]