

Abstract
Lycium ruthenicum is a perennial shrub species that has attracted considerable interest in recent years owing to its nutritional value and ability to thrive in a harsh environment. However, only extremely limited transcriptomic and genomic data related to this species can be found in public databases, thereby limiting breeding research and molecular function analysis. In this study, we characterized the physiological and biochemical responses to saline-alkaline mixed stress by measuring photochemical efficiency, chlorophyll content, and protective enzyme activity. We performed global transcriptomic profiling analysis using the Illumina platform. After optimizing the assembly, a total of 68 063 unique transcript sequences with an average length of 877 bp were obtained. Among these sequences, 4096 unigenes were upregulated and 4381 unigenes were down-regulated after saline-alkaline mixed treatment. The most abundant transcripts and over-represented items were assigned to gene ontology (GO) terms or Kyoto Encyclopedia of Genes and the Genomes (KEGG) categories for overall unigenes, and differentially expressed unigenes were analyzed in detail. Based on this set of RNA-sequencing data, a total of 9216 perfect potential simple sequence repeats (SSRs) were identified within 7940 unigenes with a frequency of 1/6.48 kb. A total of 77 primer pairs were synthesized and examined in wet-laboratory experiments, of which 68 loci (88.3%) were successfully amplified with specific products. Eleven pairs of polymorphic primers were verified in 225 individuals from nine populations. The inbreeding coefficient and the polymorphism information content value ranged from 0.011 to 0.179 and from 0.1112 to 0.6750, respectively. The observed and expected heterozygosities ranged from 0.064 to 0.840 and from 0.115 to 0.726, respectively. Nine populations were clustered into three groups based on a genetic diversity study using these novel markers. Our data will be useful for functional genomic investigations of L. ruthenicum and could be used as a basis for further research on the genetic diversity, genetic differentiation, and gene flow of L. ruthenicum and other closely related species.
Keywords: De novo, Genetic diversity, Lycium ruthenicum, Molecular marker, Saline-alkaline mixed stress
1. Introduction
A total area of 831 million hectares in the world is affected by different kinds of salt stresses. Within this area, saline-alkaline mixed soils constitute 434 million hectares in comparison with 397 million hectares under neutral-saline soils. Furthermore, 7% of total farmland areas in China are saline soil area (Jin et al., 2006; Qiu et al., 2009). Saline-alkaline soil has become a major environmental factor that limits global crop productivity, which adversely hinders the growth and productivity of major crop plants. Alkali soil is characterized by high pH, high exchangeable sodium percentage, and poor fertility; high alkalinity is due to high Na2CO3 and NaHCO3 content, which exerts a markedly stronger destructive effect on plants than neutral salt soil (Babuin et al., 2014). However, soil salinization and alkalization are often combined in nature; therefore mixed saline-alkaline stresses are the main source of constraint for farming (Shi and Sheng, 2005). Plant responses to mixed saline-alkaline stress conditions are believed to be different from responses to a simple neutral salt, and the condition is considered an adverse environment that can affect the solubility of essential micronutrients such as iron and zinc (Alam et al., 1999).
Lycium ruthenicum belongs to the Lycium genus of the Solanaceae family. This family has served as a model for linking genomics and biodiversity (Knapp et al., 2004) owing to its numerous important species for the study of plant developmental processes, including tomato, potato, tobacco, and Petunia hybrida, which exhibit considerable variations in morphology (Wang L. et al., 2015). However, as an important traditional Chinese herbal plant with distinguished abiotic stress resistance, the Lycium genus is markedly less reported than other genera within the family. L. ruthenicum is widely distributed in the salinized and alkalinized desert of Northwest China (Liu et al., 2012), mainly in the Qinghai and Gansu Provinces and Xinjiang Uygur Autonomous Region of China. The species is often used for the treatment of heart disease, irregular menstruation, and menopause (Liu Y.L. et al., 2014). Its black fruit is primarily comprised of high concentrations of anthocyanin, essential oils, and polysaccharides, which are believed to perform functions in immunoregulation, anti-aging, and anti-fatigue activity (Altintas et al., 2006; Zheng et al., 2011; Peng et al., 2016). Due to its outstanding trait of abiotic stress resistance especially in mixed saline-alkaline soils, L. ruthenicum is an ideal plant for preventing soil desertification and alleviating the degree of soil salinity-alkalinity, which is highly important for the ecosystem and agriculture in remote areas (Guo et al., 2016).
Although L. ruthenicum plays an important role in the ecosystem and could be exploited as a functional food (Tang et al., 2017), limited genomic and transcriptomic information is publicly available. As of Dec. 2016, only 149 nucleotide and expressed sequence tag (EST) sequences of L. ruthenicum had been deposited in the GenBank database. Thus, studies on population genetics, such as the genetic diversity and genetic structure of this species, are scarce. RNA-sequencing, which can generate massive ESTs in a short time, has been widely used in plants. Furthermore, the sequencing results could be used to explore studies of global expression patterns and networks of genes that respond to diverse stresses, such as cold, drought, and salinity in a variety of plants (Chen et al., 2014). The transcriptome dataset is a valuable resource not only for the construction of expression profiles but also for novel gene discovery as well as for a large number of simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers, which are highly useful for variety identification, population structure analysis, and linkage map development, and will contribute to the acceleration of breeding programs. To date, certain important plants of the Solanaceae family have been sequenced, such as the tomato and the potato (Xu et al., 2011; Sato et al., 2012). The deep RNA-sequencing of L. ruthenicum will aid in understanding the key aspects of the biology and evolution of Lyceum, especially the molecular basis underlying its high saline-alkaline stress tolerance. We generated a clone of L. ruthenicum and performed RNA-sequencing using the Illumina platform in order to show the transcripts and pathways that are overrepresented in response to saline-alkaline mixed treatment. The identification and functional characterization of saline-alkaline stress responsive genes may yield advantageous candidate genes that can facilitate the improved tolerance of plants to saline-alkaline environments by genetic engineering. Furthermore, the genome-wide survey and microsatellite marker mining based on de novo transcriptome profiling of L. ruthenicum will be useful for characterizing the genetic diversity and population genetic structure of this highly interesting desert-distributed species.
2. Materials and methods
2.1. Sample preparation
For Illumina RNA-sequencing analysis, the seedling of “GL5” was provided by the National Laboratory of Seedling Bioengineering (Ningxia Hui Autonomous Region of China) and then subjected to tissue culture at the National Engineering Laboratory for Tree Breeding (Beijing, China). The sub-cultured seedlings were placed at 21 °C under a 16-h photoperiod and an 8-h dark period in an environmentally controlled glasshouse for tissue culture propagation before treatments. After approximately two weeks, when the seven-leaf stage was reached, these tissue culture seedlings were treated with or without mixed salt alkali (molar ratio of NaCl:Na2SO4:NaHCO3: Na2CO3, 1:9:9:1, soil pH 9.0, which approaches that of the natural habitat) for 6 h (Shi and Sheng, 2005), with the unstressed plants as a control. After treatment, 10 seedlings randomly selected from each growth condition were harvested and mixed, immediately frozen in liquid nitrogen, and stored at −70 °C for RNA isolation. These materials were used for Illumina sequencing or real-time quantitative polymerase chain reaction (qPCR) analysis.
For the genetic diversity study, nine L. ruthenicum provenances were collected from Qinghai and Gansu Provinces, Xinjiang Uygur and Inner Mongolia Autonomous Regions of China in 2016. Twenty-five individuals were sampled from each locality (Table 1). Fresh leaves were sampled and dried using silica gel in the field, prior to being transported to our laboratory.
Table 1.
Sampling information of L. ruthenicum populations in the present study
| No. | Locality* | Population | Lt (°) | Ln (°) | Collection date | Sample size |
| 1 | Akesu (Xinjiang) | AKS | 40.38 | 81.51 | 7 July, 2016 | 25 |
| 2 | Kuerle (Xinjiang) | KEL | 41.72 | 86.11 | 7 July, 2016 | 25 |
| 3 | Bachu (Xinjiang) | BC | 39.79 | 78.56 | 8 July, 2016 | 25 |
| 4 | Shaya (Xinjiang) | SY | 41.22 | 82.78 | 8 July, 2016 | 25 |
| 5 | Geermu (Qinghai) | GEM | 36.47 | 94.95 | 23 July, 2016 | 25 |
| 6 | Nuomuhong (Qinghai) | NMH | 36.45 | 96.45 | 24 July, 2016 | 25 |
| 7 | Guazhou (Gansu) | GZ | 40.52 | 95.78 | 3 August, 2016 | 25 |
| 8 | Yongjing (Gansu) | YJ | 36.05 | 103.18 | 5 August, 2016 | 25 |
| 9 | Alashan (Inner Mongolia) | ALS | 38.50 | 105.41 | 22 August, 2016 | 25 |
Lt: latitude; Ln: longitude.
Province or Autonomous Region of China
2.2. Chlorophyll fluorescence and chlorophyll content measurement
Photochemical efficiency was monitored using a Dual-PAM-100, P700 and chlorophyll fluorescence measuring system (Walz, Effeltrich, Germany) at room temperature before and after treatment as previously described (Chen et al., 2009). In our study, we represented our measured leaf chlorophyll content in the form of SPAD indices by using a SPAD-502 Plus chlorophyll meter. The value provided by the meter indicates the relative content of chlorophyll in the leaves (Liu Y.D. et al., 2014).
2.3. Antioxidant enzyme activity and maldonaldehyde content measurements
Leaf samples (about 0.5 g) were used for extraction of antioxidative enzymes (Diao et al., 2016). Superoxide (SOD) activity was measured following the method established by Spychalla and Desborough (1990). Peroxidase (POD) activity was determined following the method as described by Polle et al. (1994). The maldonaldehyde (MDA) content was measured using the thiobarbituric acid (TBA) reaction method (Hong et al., 2000). After measuring the absorbency of the solution at 450, 532, and 600 nm, represented as A 450, A 532, and A 600, respectively, MDA content (C MDA) was calculated according to the formula of C MDA=6.45×(A 532−A 600)−0.56×A 450.
2.4. RNA isolation and Illumina DNA library construction
Total RNA was extracted from frozen seedlings using the cetyltrimethyl ammonium bromide (CTAB)-based protocol (Chang et al., 1993). After treatment with RNase-free DNase, the RNA quality was examined using NanoDrop 2000 (Thermo Scientific, DE, USA) and A 260/A 280 absorbance ratios ranging from 1.9 to 2.1 were selected. The integrity of the RNA samples was also verified using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA) and their RNA integrity number (RIN) values were set to >8.5.
Illumina sequencing was performed using the HiSeq™ 2000 platform according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Briefly, 20 μg Poly(A) RNA samples extracted from five individual plants with a concentration of ≥750 ng/μl were used for each complementary DNA (cDNA) library construction. To avoid priming bias, mRNA was initially enriched with oligo(dT) and subsequently fragmented into small pieces by using divalent cations at 94 °C for 5 min. Based on these cleaved RNA fragments, random hexamer-primer and reverse transcriptase (Invitrogen, Carlsbad, CA, USA) were used to synthesize the double-strand cDNA. Two paired-end cDNA libraries were constructed with an insert size of 200 bp and subsequently sequenced using Illumina Genome Analyzer IIX (Illumina, San Diego, CA, USA) (Wang et al., 2010). The sequencing data were generated in Fastq format and deposited in the National Center for Biotechnology Information (NCBI) Sequence Reas Archive (SRA) database (Wheeler et al., 2002).
2.5. Data filtering and de novo assembly
Raw data generated from Solexa sequencing were preprocessed for data filtering. The non-sense sequences, including adapters, contamination, sequences with ambiguous bases >5%, and reads containing quality value <10 bases more than 20% were first removed using Perl script. The preprocessed sequences were then assembled using Trinity software (release-20130225) (Grabherr et al., 2011). Reads were first combined with overlap lengths by setting k-mer size (oligomers of fixed length) of 25 to form fragments known as contigs. These contigs were further realigned to construct sequences that could not be extended at either end by Trinity software. These sequences were defined as unigenes. After the gap was closed, we constructed a non-redundant unigene set from the two assembled datasets by TGICL program (Version 2.1) to achieve the longest possible length (Pertea et al., 2003). To determine the sequential orientation of each unigene, we performed a set of sequential BLASTx alignment (E<10−5) steps and predicted the coding DNA sequence (CDS) by BLAST against the non-redundant (Nr) database of GenBank, the Swiss-Prot protein database (http://www.expasy.ch/sprot), the Kyoto Encyclopedia of Genes and the Genomes (KEGG) pathway database, and the Clusters of Orthologous Groups of proteins (COG) database (http://www.ncbi.nlm.nih. gov/COG). For unigenes that cannot be aligned to any of these databases, ESTScan (Version 3.0.2) was used to determine the sequence orientation (Iseli et al., 1999).
2.6. Unigene annotation and function classification
For further annotation, all unigenes were searched using BLASTx against the databases of Nr and Swiss-Prot with an E-value cutoff of 10−5 to gain the most descriptive annotation. The protein with the highest sequence similarity was retrieved. Gene ontology (GO) information was obtained using the Blast2GO (Version 2.5.0) server based on BLASTx hits against the NCBI Nr database with an E-value cutoff of 10−5. GO functional classification was performed using the WEGO software with plant categories defined by molecular function, cellular component, and biological process ontologies (Conesa et al., 2005; Ye et al., 2006). The unigene sequences were also aligned to the COG database to predict and classify their functions as well as aligned to the KEGG database to obtain pathway assignments. For both alignments, an E-value threshold of 10−5 was used.
2.7. Gene expression analysis
The expression level of the unigene was normalized according to the number of fragments per kilobase of transcript per million fragments mapped (FPKM) (Mortazavi et al., 2008). After normalization was conducted, we determined the number of reads for each coding region in the control and treated libraries. Then, the ratio of the reads in the two libraries was calculated. The non-treated sample was considered the control in the differential gene expression analysis. The statistical significance of the differential expression value of each unigene was determined according to the Benjamini and Hochberg false discovery rate (FDR) correction (Benjamini and Hochberg, 1995). To eliminate the effect of the high sensitivity of the Illumina sequencing method, we defined DE unigenes as those with an absolute value of log2Ratio≥2, FDR<0.001, as well as those with an expression level of ≥1 FPKM in at least sample.
The GO term enrichment analysis of DE unigenes was performed on the genome background. The cutoff P-value after correction was 0.05 on a rigorous Bonferroni correction method (Chen et al., 2014). GO terms that meet this standard were defined as significantly enriched GO terms. The KEGG pathway enrichment analysis of DE unigenes was performed to determine the main biochemical and signal transduction pathways involving DE unigenes. Corrected P≤0.05 was used as a threshold to identify the overrepresented pathways.
2.8. Verification of differentially expressed genes by qPCR analysis
We verified the expression pattern of certain crucial genes associated with abiotic stress response through qPCR analysis. The qPCR was performed using a power SYBR Green PCR kit (ABI, Foster City, CA, USA) with a StepOnePlus™ Real-Time PCR System (ABI). The 2−∆∆ C T method was used to calculate the relative quantification value. The reactions were prepared and performed as described previously (Chen et al., 2014). The gene-specific primers used in the qPCR analysis are listed in Table S1. For the PCR analyses, three independent biological samples were treated in the same manner as those used for the deep sequencing. Three technical replicates were performed for each sample. To test the significance of the correlation between the qPCR and deep sequencing, we conducted Pearson’s correlation test using a P-value of 0.05.
2.9. SSR mining and primer design
MIcroSAtellite (MISA; http://pgrc.ipk-gatersleben.de/misa/misa.html) Perl script was used to identify microsatellites from the de novo assembled unigenes (Wei et al., 2014). cDNA-based SSRs defined as mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides were searched with a minimum of 12, 6, 5, 5, 4, and 4 repeat units, respectively. We first designed five primer pairs for each SSR locus by using the software Primer 3 program in the 150-bp flanking regions of the SSRs before filtering low-quality primers (Rozen and Skaletsky, 2000). Primers were designed based on the following criteria: (1) with a minimum length of 18 bp; (2) with a melting temperature between 46 and 55 °C and a maximum discrepancy within 4 °C between two primers, and (3) with a PCR product size ranging from 100 to 350 bp.
2.10. SSR marker development
To extract the genomic DNA, we used the modified CTAB with the blades of L. ruthenicum (Doyle and Doyle, 1987). We chose 77 primer pairs randomly and used them for amplification to test their accuracy. The repeat times of trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides were at least 6, 5, 4, and 3, respectively (Chagné et al., 2004). The PCR system consisted of 1 μl of DNA, 1 μl of upstream primer, 1 μl of downstream primer, 10 μl of amplification mix containing a Taq enzyme, and 7 μl of double-distilled H2O. PCR amplification involved pre-degeneration at 95 °C for 5 min, denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s, in 34 cycles. The PCR products were examined through 1% (0.01 g/ml) agarose gel electrophoresis to filter out ineffective primers. To develop polymorphic SSR primers, we used 8% (0.08 g/ml) native polyacrylamide gels in a 0.5×TBE buffer, stained the gels with silver, and photographed them with Bio-Rad DOC2000 (Hercules, CA, USA). The products amplified by the polymorphic primers were then sequenced through capillary electrophoresis, and the results were output in the form of allele sizes.
2.11. Genetic diversity analysis
In polyacrylamide gel analysis, the polymorphic locus is defined as more than one band detected at the same position in all materials (Bi et al., 2014). The inbreeding coefficient (F is), observed heterozygosity (H o), and expected heterozygosity (H e) were computed using WinArl35. The allele numbers (N A) and polymorphism information content (PIC) value were calculated using PIC-CALC. Data conversion was completed with Convert 1.3.1 (Rumeu et al., 2013). Nei (1978)’s unbiased measures of genetic distance and identity between all pairwise combinations of populations were calculated using POPGENE Version 1.31 (Yeh et al., 1999). An unweighted pair-group method with an arithmetic average algorithm was used based on the matrix of Nei’s unbiased genetic distance by utilizing NTSYS pc Version 2.1 after the data were pre-treated with NTEDIT (Rohlf, 2000; Ge et al., 2010).
3. Results
3.1. Physiological measurement
Given that salt stress may have a significant effect on photosynthesis and the chlorophyll fluorescence of L. ruthenicum (Luo et al., 2017), the growth state of seedlings was assessed in terms of the maximum photochemical efficiency of the photosystem II (PSII) of their chloroplasts (the ratio of variable to maximum fluorescence (F v/F m)) and chlorophyll content. The F v/F m was measured using seedling leaves at room temperature (25 °C) with a chlorophyll fluorometer (Dual-PAM-100, P700), following a 30-min dark adaptation. The results indicated that no evident variation was observed in control seedlings within 12 h. However, the F v/F m ratios of mixed salt-alkali-treated plants continuously decreased within the first 9 h and showed an increase at the 12 h point (Fig. 1a). The SPAD value measured synchronously for the same seedlings decreased during the first 6 h and remained stable within the 6 to 12 h period (Fig. 1b). Both experiments indicated that the short-term saline-alkaline mixed stress treatment influenced photosynthesis and the growth state began to change starting from an early time point of 6 h after treatment began.
Fig. 1.
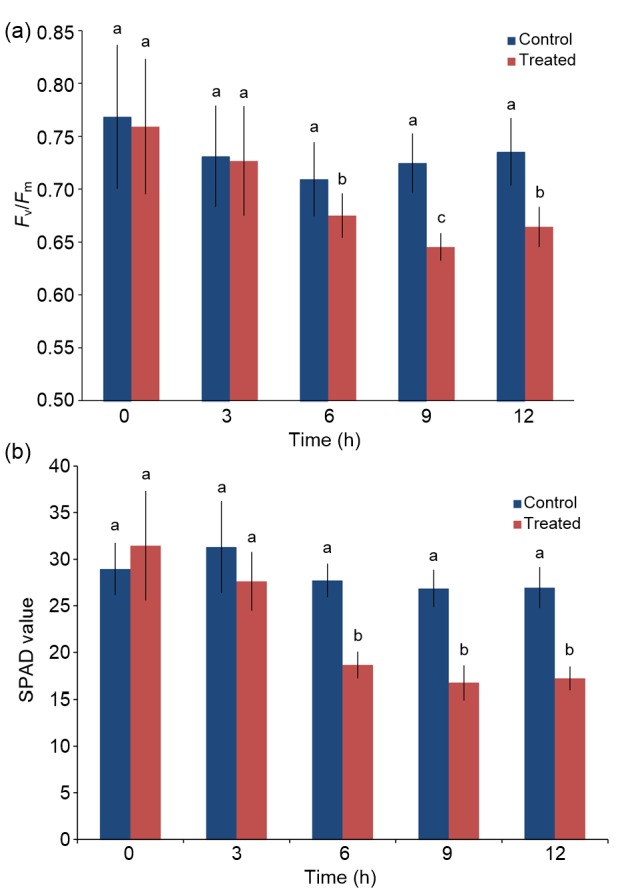
Effects of saline-alkaline mixed stress on chlorophyll fluorescence and chlorophyll content
(a) Time course of photosynthetic efficiency (F v/F m) for the control and treated samples. (b) Chlorophyll content (SPAD value) of the control and treated samples. Values are represented as mean±standard error (n=10). Different letters present statistical difference (P<0.05)
3.2. Antioxidant enzymes and malondialdehyde activity
Compared with the untreated control plants, plants treated with short-term saline-alkaline mixed stress exhibited enhanced SOD and POD activity within the 6 h after treatment start (Figs. 2a and 2b). However, the increment differed in the two antioxidant enzymes. Significant enhancement of SOD activity was observed in treated plants during all monitored time points. For POD activity, significant enhancement was only observed at the time points of 6 and 9 h and no significant change was observed at 12 h after treatment had started (Fig. 2b).
Fig. 2.
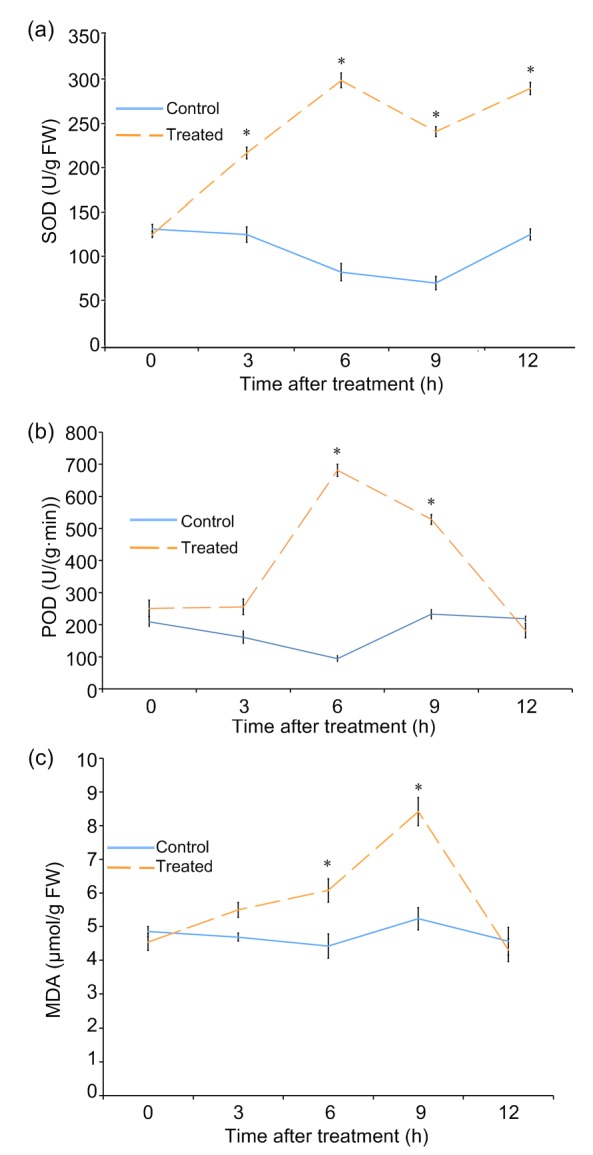
Effect of saline-alkaline mixed stress on the activity of SOD (a) and POD (b), and MDA content (c) in the leaves of L. ruthenicum seedlings
Values are represented as mean±standard error (n=10). * P<0.05 vs. 0 h
As a decomposition product of lipid peroxidation, the MDA content was often measured to evaluate the damage level of the plasma membrane during stress treatment. The MDA content in the treated leaves was significantly higher than that in the control leaves at 6 and 9 h with the highest level at 9 h (Fig. 2c). Subsequently, the MDA content decreased significantly with prolonged treatment. Compared with the protective enzyme activity of SOD and POD, which both achieve their highest level at 6 h, the highest level of MDA appeared to lag 3 h behind.
3.3. Illumina sequencing and de novo transcriptome assembly
To provide a comprehensive landscape of transcriptome and the saline-alkaline stress response for L. ruthenicum, we generated mixed cDNA libraries with mRNA from the control and the mixed saline-alkaline-treated seedlings, using Illumina sequencing. After removing adapters, low-quality sequences, and ambiguous reads, each library gained 52.5 million clean reads with a mean length of 90 bp. The Q20 (a quality threshold of 20) percentages for the control and the treated library, respectively, were 97.9% and 98.1%, whereas their GC percentages were 42.3% and 43.5%, respectively. Raw data were deposited in the NCBI SRA database under the accession number SRP059046 (which will be released immediately after this paper is accepted). All trimmed reads were de novo assembled into contigs using the Trinity method with an average contig size exceeding 350 bp in each of the two libraries (Table 2). This can be considered long compared with those reported in certain related studies (Wang et al., 2010; Wei et al., 2014). Among the contigs in each library, about 70% were shorter than 300 bp, about 28% ranged from 300 to 500 bp, and the remaining 2% were longer than 1000 bp (Fig. 3).
Table 2.
Length distribution of assembled contigs and unigenes
| Sample | Total number | Total length (bp) | Mean length (bp) | N50 (bp) | Total consensus sequence | Distinct cluster | Distinct singleton |
| Contig | |||||||
| Control | 121 361 | 42 900 816 | 353 | 651 | |||
| Treated | 117 483 | 41 211 110 | 351 | 655 | |||
| Unigene | |||||||
| Control | 67 901 | 44 901 911 | 661 | 1161 | 67 901 | 19 314 | 48 587 |
| Treated | 73 239 | 47 731 758 | 652 | 1133 | 73 239 | 21 311 | 51 928 |
| All unigenes | 68 063 | 59 724 269 | 877 | 1411 | 68 063 | 25 961 | 42 102 |
Fig. 3.

Length frequency distribution of contigs and unigenes obtained from de novo assembly
By using paired-end information, the contigs were then joined with assembled unigenes (Chen et al., 2014). A total of 68 063 unigenes were obtained with an average length of 877 bp and N50 (the shortest sequence length at 50% of the genome) of 1411 bp (Table 2), suggesting that this non-model organism has been effectively and accurately assembled. Among these unigenes, distinct clusters and singletons account for 38.14% and 61.86%, respectively. All unigenes were longer than 200 bp, with 68.80% between 200 and 1000 bp and 21.68% between 1000 and 2000 bp. Unigenes with a length over 1000 bp account for 9.52% of the total (Fig. 3). The generated unigenes may be useful for further research on L. ruthenicum functional genomics.
3.4. Annotation, CDS prediction, and function classification
Among the 68 063 high-quality unique sequences, 53 366 (78%) showed at least one significant match to an existing gene model in the BLAST search. Among which, the numbers (percentage) that could be hit to NCBI non-redundant database (Nr), Swiss-Prot, COG, and KEGG account for 50 803 (95.2%), 29 828 (55.89%), 16 432 (30.8%), and 25 550 (47.9%), respectively. Moreover, approximately 86.76% (46 303) were aligned with known proteins in the Nr database, and the top hits with a similarity greater than 80% against the Nr database accounted for 64.5%. From the distribution of Nr annotation results, the transcripts generated were most similar to Solanum tuberosum (60.9%), which possesses a well-characterized transcriptome database, followed by Solanum lycopersicum (25.2%). We observed markedly lower sequence similarity with Nicotiana tabacum (2.2%), which also belongs to Solanaceae but is of a different genus (Fig. 4). The CDS was predicted using BLASTx or BLASTn against the Nr database, and the Swiss-Prot, KEGG, and COG databases successively with an E-value cutoff of 10−5. As a result, 45 275 unigenes showed homologous matches to these databases. ESTScan could predict the other 1168 unigenes with no matches to these four databases. In total, 46 443 (68.24%) unigenes can be predicted by homology analysis using the BLAST or ESTScan prediction.
Fig. 4.

Characteristics of homology search of Illumina sequences against the Nr database
(a) E-value distribution of BLAST hits for each unique sequence with a cut-off E-value of 10–5. (b) Similarity distribution of the top BLAST hits for each sequence. (c) Species distribution shown as a percentage of the total homologous sequences with an E-value <10–5
Classification of GO terms was also performed according to the Nr annotation by using the Blast2GO software. As a result, a total of 29 102 unigenes were classified into 57 subcategories with 202 978 functional terms (Fig. S1). More than two-thirds of these terms (98 220, 69.84%) belonged to biological processes, followed by the categories of cellular components (70 845, 50.37%) and molecular function (33 913, 24.11%). In the biological processes category, the largest groups were metabolic processes (18 762, 19.1%), cellular processes (16 829, 17.1%), single-organism processes (14 116, 14.37%), and stimulus process response (7362, 7.5%). According to the COG database, 16 433 (24.14%) unigenes were categorized into 25 functional clusters (Fig. S2). Given that most of the unigenes could be annotated by more than one category, a total of 30 731 functional terms were obtained for 16 433 unigenes. The five largest categories were: (1) general function prediction only (5389, 17.54%); (2) transcription (2774, 9.00%), (3) replication, recombination, and repair (2732, 8.89%); (4) signal transduction mechanisms (2260, 7.35%); (5) posttranslational modification, protein turnover, and chaperones (2164, 7.04%).
3.5. Pathway assignment by KEGG analysis
Besides gene annotation, KEGG pathway analysis is another useful analytic method for predicting potential genes and their functions at the whole transcriptome level. To obtain a better understanding of the specific network of these unigenes, pathways were assigned based on the KEGG database using BLASTx with an E-value cutoff of <10−5. As a result, 25 551 (37.53%) unigenes with significant matches against this database were assigned to five main categories comprising 127 KEGG pathways. Most of the unigenes (21 619, 84.6%) were mapped to the metabolism category. In this main metabolism category, global map, lipid metabolism, nucleotide metabolism, carbohydrate metabolism, and biosynthesis of other secondary metabolites were the five most represented subcategories. Given that L. ruthenicum is a medicinal species and is rich in secondary metabolites, the secondary metabolites searched here may be useful for further investigation in future related studies. As for the environmental category, 2014 (7.88%) unigenes were involved, including the subcategories of signal transduction and membrane transport.
3.6. Global gene expression analysis
For the plant material treatment, ten L. ruthenicum plants generated from selected superior clones were exposed to mixed saline-alkaline stresses, leaving ten other seedlings untreated. Leaves were harvested 6 h after treatment. Capped and polyadenylated mRNA was purified from the harvested leaves, and hexanucleotide-primed cDNA was produced and sequenced. Based on the analyses of the combined data, 88.95% of the unigenes (60 544 of 68 063) were expressed with FPKM>1 in at least one sample, and they were considered “expressed genes” under these growth conditions. By contrast, 11.08% (7539 out of 68 063) of the unigenes were designated as poorly or non-expressed genes (FPKM<1 in all samples). Detailed analysis revealed that 4652 (61.70%) of these non-expressed unigenes could be annotated as serine/threonine-protein kinase, tubby-like F-box protein, and transcription factors by the Nr database. We noticed that several unigenes from these families were alternatively included in the lists of upregulated or most abundant transcripts. Therefore, we proposed that several of these lesser-expressed genes may result from gene redundancy in gene families.
The 100 most abundant transcripts with annotation in the two samples were analyzed. All these genes had an FPKM over 600 (Tables S2 and S3). The two samples possessed 77 of the most abundant transcripts in common, including cell wall structural involved proteins, chlorophyll a/b binding protein, photosystem I/II center subunit protein, and ribulosebisphosphate carboxylase mall chain. The most abundant transcripts in the treated sample, which were different from the control, included dehydration-induced proteins, glycine rich proteins, and certain heat shock proteins.
3.7. Identification of differentially expressed unigenes
According to our applied criteria (FDR<0.001, absolute value of log2Ratio≥1, and FPKM≥1 in at least one sample), approximately 4096 unigenes appeared to be upregulated after saline-alkaline mixed stress treatment, whereas 4381 unigenes were downregulated (Fig. 5, Tables S4 and S5). Detailed analysis on the identification of differentially expressed genes that related to log2 fold change revealed that 411 unigenes were upregulated at a level of more than five, while 710 unigenes were upregulated at a level between three and five. Among these highly inducible unigenes, 288 unigenes did not present any hit in the available database. These unigenes may play unique saline-alkaline stress response roles in L. ruthenicum.
Fig. 5.
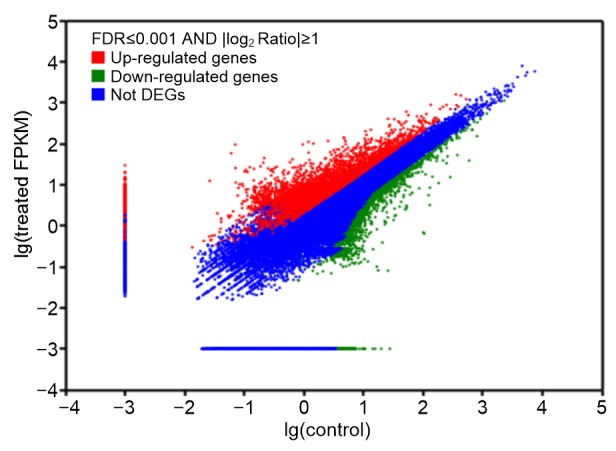
Distribution of differentially expressed genes between the control and stress-treated samples
We selected the top 300 most upregulated transcripts with annotation for further analysis (FDR<0.001; Table S6). As a result, unigenes encoding certain transcription factors including dehydration responsive element binding protein (DREB), myeloblastosis (MYB), and WRKY were overrepresented in the 300 unigenes. This result was consistent with the findings that high salinity and alkalinity could trigger the expression of specific families of transcription factors, which consequently activates the downstream stress-responsive genes correspondingly (Zhu, 2001; Wang et al., 2007). Kinases such as serine/threonine-protein kinase and cysteine-rich receptor-like protein kinase were overrepresented in the top 300 lists. The high percentage of the transcription factors and kinases in the most upregulated gene list suggested that they were the primary factors affecting the saline-alkaline mixed stress tolerance of L. ruthenicum. Moreover, unigenes encoding aquaporin, zinc finger proteins, and calcium-binding proteins were also overrepresented, indicating their important roles in such stress responses. The GO annotation of the top 300 most upregulated unigenes showed their significant involvement in the metabolic and cellular processes and in the stimulus process response, which indicates the efficacy of our treatments (Fig. 6).
Fig. 6.

Function classification of GO terms of the top 300 most abundant L. ruthenicum transcripts
Based on high-score BLASTx matches in the Nr plant protein database, 223 L. ruthenicum unigenes were classified into three main GO categories and 37 sub-categories. The left y-axis indicates the percentage of a specific category of genes in each main category. The right y-axis indicates the number of genes in the same category
3.8. Validation of gene expression by qPCR
To test the reliability of our deep sequencing results, we further randomly selected twenty differentially expressed transcripts that may be involved in abiotic stress regulation, and performed qPCR analysis. The qPCR results indicated that the expression of all selected unigenes exhibited similar tendencies between deep sequencing and qPCR results (Fig. 7a). Pearson’s correlation test showed that the expression fold change of these 20 unigenes exhibited significantly positive correlations between deep sequencing and qPCR results (R 2=0.8756 and P<0.001; Fig. 7b). The excellent correlation index indicated that the deep sequencing results are reliable for quantifying gene expression abundance in our study.
Fig. 7.

Validation and comparison of the expression of 20 unigenes between qPCR and deep sequencing
(a) Expression analyses of 20 putative differentially expressed unigenes by qPCR. (b) Gene expression correlation between qPCR and deep sequencing. The bar in (a) represents the standard deviation
3.9. Functional categorization and pathway assignment of differentially expressed genes
Both gene annotation and KEGG pathway analyses are useful for predicting potential genes and their functions at a whole transcriptome level. All DE unigenes were mapped to the GO database and the gene numbers were calculated from each GO term. As a result, 3619 DE unigenes could be assigned. Using a hypergeometric test, we identified the significantly enriched GO terms based on the genomic background (P≤0.05, after Bonferroni correction). As a result, the significant GO clusters for DE unigenes are “metabolic process”, “cellular process”, and “single-organism process” categories of the biological process, the “cell” and “cell part” categories of the cellular component category, and the “antioxidant activity” and “binding categories” of the molecular function category. Detailed analyses indicated 59 ontology terms of biological processes were significantly enriched (corrected P≤0.05), these terms included the oxygen-containing compound response process, water deprivation response process, and abiotic stimulus response process (Table 3). This result suggested that these processes are important in the saline-alkaline mixed stress response. The differentially expressed transcripts in these systems may indicate their important roles in the protection of L. ruthenicum under conditions of such stress.
Table 3.
Over-representative GO terms of biological process for DE unigenes between the control and the saline-alkaline-treated sample
| Gene ontology term | Cluster number in treated sample | Cluster frequency | Corrected P-value |
| Response to oxygen-containing compound | 377 | 0.127 | 3.87×10−15 |
| Response to chemical | 589 | 0.198 | 1.01×10−12 |
| Response to acid | 267 | 0.090 | 1.94×10−12 |
| Response to water | 108 | 0.036 | 7.44×10−11 |
| Response to water deprivation | 106 | 0.036 | 8.26×10−11 |
| Response to chitin | 54 | 0.018 | 5.17×10−9 |
| Response to organonitrogen compound | 63 | 0.021 | 6.99×10−9 |
| Response to endogenous stimulus | 294 | 0.099 | 7.14×10−9 |
| Response to inorganic substance | 277 | 0.093 | 1.89×10−8 |
| Response to stimulus | 1050 | 0.353 | 7.29×10−8 |
| Response to karrikin | 55 | 0.018 | 1.16×10−6 |
| Response to fungus | 92 | 0.031 | 2.14×10−6 |
| Multi-organism process | 295 | 0.099 | 4.07×10−6 |
| Nucleosome assembly | 43 | 0.014 | 4.46×10−6 |
| Nucleosome organization | 43 | 0.014 | 4.46×10−6 |
| Response to organic substance | 357 | 0.120 | 7.62×10−6 |
| Response to biotic stimulus | 223 | 0.075 | 1.17×10−5 |
| DNA packaging | 48 | 0.016 | 1.21×10−5 |
| Response to stress | 625 | 0.210 | 1.27×10−5 |
| Response to external biotic stimulus | 215 | 0.072 | 1.49×10−5 |
| Response to other organism | 215 | 0.072 | 1.49×10−5 |
| DNA conformation change | 62 | 0.021 | 2.37×10−5 |
| Benzene-containing compound metabolic process | 29 | 0.010 | 2.94×10−5 |
| Response to heat | 74 | 0.05 | 3.72×10−5 |
| Response to hormone | 256 | 0.086 | 5.26×10−5 |
| Chromatin assembly | 43 | 0.014 | 5.32×10−5 |
| Protein-DNA complex assembly | 43 | 0.014 | 6.69×10−5 |
| Protein-DNA complex subunit organization | 43 | 0.014 | 6.69×10−5 |
| Defense response to fungus | 65 | 0.022 | 7.49×10−5 |
| hromatin assembly or disassembly | 47 | 0.016 | 8.54×10−5 |
| Defense response | 208 | 0.070 | 9.45×10−5 |
| Anthocyanin-containing compound biosynthetic process | 12 | 0.004 | 1.30×10−4 |
| Response to abiotic stimulus | 28 | 0.144 | 4.70×10−4 |
| Sulfate transport | 24 | 0.008 | 9.90×10−4 |
| Response to external stimulus | 282 | 0.095 | 1.22×10−4 |
| Photoinhibition | 12 | 0.004 | 1.83×10−3 |
| Negative regulation of photosynthesis, light reaction | 12 | 0.004 | 1.83×10−3 |
| Defense response to other organism | 149 | 0.050 | 1.83×10−3 |
| Defense response, incompatible interction | 59 | 0.020 | 2.14×10−3 |
| Response to nitrogen compound | 82 | 0.028 | 2.24×10−3 |
| Respiratory burst involved in defense response | 15 | 0.005 | 3.22×10−3 |
| Response to temperature stimulus | 155 | 0.052 | 4.04×10−3 |
| Respiratory burst | 15 | 0.005 | 5.35×10−3 |
| Negative regulation of cellular protein metabolic process | 25 | 0.008 | 5.51×10−3 |
| Negative regulation of protein metabolic process | 25 | 0.008 | 5.51×10−3 |
| Phenol-containing compound metabolic process | 21 | 0.007 | 7.15×10−3 |
| Iorganic anion transport | 52 | 0.017 | 8.11×10−3 |
| Negative regulation of abscisic acid-activated signaling pathway | 19 | 0.006 | 1.22×10−2 |
| Phenol-containing compound biosynthetic process | 19 | 0.006 | 1.22×10−2 |
| Negative regulation of response to alcohol | 19 | 0.006 | 1.22×10−2 |
| Sulfur compound transport | 25 | 0.008 | 1.24×10−2 |
| Sulfate transmembrane transport | 20 | 0.007 | 2.10×10−2 |
| Cell proliferation | 31 | 0.010 | 2.23×10−2 |
| Systemic acquired resistance | 39 | 0.013 | 2.65×10−2 |
| Salicylic acid biosynthetic process | 17 | 0.006 | 2.91×10−2 |
| egative regulation of protein modification process | 18 | 0.06 | 3.71×10−2 |
| Response to osmotic stress | 161 | 0.054 | 3.97×10−2 |
| Salicylic acid metabolic process | 17 | 0.006 | 4.13×10−2 |
| Flavonoid biosynthetic process | 22 | 0.007 | 4.71×10−2 |
Pathways that were possibly affected by saline-alkaline mixed stress were checked by comparing them against the KEGG database. A total of 27 pathways changed significantly (P≤0.05) under mixed saline-alkaline stress (Table 4). Pathways involved in plant hormone signal transduction, glycerophospholipid metabolism, and zeatin biosynthesis were included. A total of 533 unigenes were associated with the biosynthesis of secondary metabolites. A number of pathways related to the biosynthesis of other secondary metabolites, such as phenylpropanoid biosynthesis, the flavonoid biosynthesis pathway, and glucosinolate biosynthesis, were enriched in differentially expressed unigenes. The overall top 10 significantly changed pathways included “phenylpropanoid biosynthesis”, “flavone and flavonol biosynthesis”, “flavonoid biosynthesis”, “biosynthesis of secondary metabolites”, “stilbenoid, diaryheptanoid and gingerol biosynthesis”, “plant-pathogen interaction”, “zeatin biosynthesis”, “plant hormone signal transduction”, “isoflavonoid biosynthesis”, and “cysteine and methionine metabolites”. Two pathways related to signal transduction, namely, the “phosphatidylinositol signaling system” and “plant hormone signal transduction”, both showed significant enrichment. Among the 275 DE unigenes involved in the plant hormone signal transduction pathway, 190 unigenes were shown to be upregulated, whereas 85 members were downregulated. The upregulated unigenes mainly included the ABA-induced protein phosphatase 2C, abscisic acid receptor PYL genes, and the ABA-responsive element binding protein, which all belong to ABA signaling pathway, and cytokinin-regulated transcription factor ARR1. The downregulated dunigenes mainly included auxin-induced proteins and ethylene-responsive transcription factors.
Table 4.
Pathway enrichment analyses for DE unigenes between the control and the saline-alkaline mixed treated samples
| Pathway ID | Level 1 | Level 2 | Pathway | Q-value | DEG No. | Up | Down |
| ko04075 | Environmental information processing | Signal transduction | Plant hormone signal transduction | 1.03×10−10 | 275 | 190 | 85 |
| ko02010 | Environmental information processing | Membrane transport | ABC transporters | 4.54×10−3 | 51 | 46 | 5 |
| ko03010 | Genetic information processing | Translation | Ribosome | 5.27×10−3 | 107 | 41 | 66 |
| ko00940 | Metabolism | Biosynthesis of other secondary metabolites | Phenylpropanoid biosynthesis | 3.51×10−19 | 131 | 97 | 34 |
| ko00944 | Metabolism | Biosynthesis of other secondary metabolites | Flavone and flavonol biosynthesis | 3.51×10−19 | 55 | 43 | 12 |
| ko00941 | Metabolism | Biosynthesis of other secondary metabolites | Flavonoid biosynthesis | 5.03×10−17 | 83 | 53 | 30 |
| ko01110 | Metabolism | Global map | Biosynthesis of secondary metabolites | 5.03×10−17 | 533 | 344 | 189 |
| ko00945 | Metabolism | Biosynthesis of other secondary metabolites | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 2.51×10−15 | 82 | 54 | 28 |
| ko00908 | Metabolism | Metabolism of terpenoids and polyketides | Zeatin biosynthesis | 1.25×10−11 | 75 | 60 | 15 |
| ko00943 | Metabolism | Biosynthesis of other secondary metabolites | Isoflavonoid biosynthesis | 1.68×10−8 | 29 | 26 | 3 |
| ko00270 | Metabolism | Amino acid metabolism | Cysteine and methionine metabolism | 6.62×10−8 | 56 | 30 | 26 |
| ko00903 | Metabolism | Metabolism of terpenoids and polyketides | Limonene and pinene degradation | 2.40×10−7 | 51 | 34 | 17 |
| ko00040 | Metabolism | Carbohydrate metabolism | Pentose and glucuronate interconversions | 1.22×10−5 | 54 | 24 | 30 |
| ko00073 | Metabolism | Lipid metabolism | Cutin, suberine and wax biosynthesis | 2.11×10−5 | 38 | 21 | 17 |
| ko01100 | Metabolism | Global map | Metabolic pathways | 3.98×10−5 | 813 | 491 | 322 |
| ko00360 | Metabolism | Amino acid metabolism | Phenylalanine metabolism | 4.45×10−5 | 46 | 35 | 11 |
| ko00966 | Metabolism | Biosynthesis of other secondary metabolites | Glucosinolate biosynthesis | 5.38×10−5 | 17 | 10 | 7 |
| ko00906 | Metabolism | Metabolism of terpenoids and polyketides | Carotenoid biosynthesis | 5.87×10−4 | 49 | 28 | 21 |
| ko00196 | Metabolism | Energy metabolism | Photosynthesis of antenna proteins | 7.63×10−4 | 14 | 0 | 14 |
| ko00920 | Metabolism | Energy metabolism | Sulfur metabolism | 4.27×10−3 | 15 | 8 | 7 |
| ko00250 | Metabolism | Amino acid metabolism | Alanine, aspartate and glutamate metabolism | 5.27×10−3 | 28 | 23 | 5 |
| ko00910 | Metabolism | Energy metabolism | Nitrogen metabolism | 8.73×10−3 | 23 | 16 | 7 |
| ko00904 | Metabolism | Metabolism of terpenoids and polyketides | Diterpenoid biosynthesis | 1.17×10−2 | 28 | 20 | 8 |
| ko00565 | Metabolism | Lipid metabolism | Ether lipid metabolism | 2.68×10−2 | 55 | 40 | 15 |
| ko00430 | Metabolism | Metabolism of other amino acids | Taurine and hypotaurine metabolism | 2.85×10−2 | 7 | 6 | 1 |
| ko00500 | Metabolism | Carbohydrate metabolism | Starch and sucrose metabolism | 3.57×10−2 | 84 | 45 | 39 |
| ko04626 | Organismal systems | Environmental adaptation | Plant-pathogen interaction | 1.04×10−12 | 280 | 197 | 83 |
3.10. Genome-wide SSR mining
All 68 083 unigenes generated in this study were used to mine potential microsatellites by using the MISA tool. The SSRs that were located only in one single read had been eliminated (Wei et al., 2014). As a result, a total of 9216 potential SSRs were identified within 7940 unigenes, which means every 6.48 kb of examined sequences composed one SSR on average. This frequency was slightly lower than the results for plants such as pomelo (1/5.6 kb) and wheat (1/5.46 kb). Among these SSRs, 1052 sequences contained more than one SSR, and 449 SSRs were present in compound form (Table 5).
Table 5.
Summary of SSR analysis results
| Item | Number |
| Total number of sequences examined | 68 063 |
| Total size of examined sequences (bp) | 59 724 269 |
| Total number of identified SSRs | 9216 |
| Number of SSR containing sequences | 7940 |
| Number of sequences containing more than 1 SSR | 1052 |
| Number of SSRs present in compound formation | 449 |
The frequency, type, and distribution of the potential 9216 SSRs were further analyzed. Among these SSRs, the mono-, di-, and tri-nucleotide repeat motifs were the most abundant types (4340, 47.09%; 2027, 21.99%; 2563, 27.81%, respectively), followed by hexa-(139, 1.51%), penta-(78, 0.85%), and quad-nucleotide (69, 0.75%) repeat motifs. The length distributions of all potential SSRs based on the number of repeat units were further analyzed (Table 6). The size of these SSRs was mostly distributed from 12 to 22 bp (8979, 88.27%), followed by 23 to 30 bp length range (311, 3.37%). A total of 412 SSRs (4.47%) were longer than 30 bp.
Table 6.
Length of distribution of SSRs based on the number of repeat units
| Number of repeat | Mono- | Di- | Tri- | Quad- | Penta- | Hexa- |
| 4 | 0 | 0 | 0 | 0 | 74 | 139 |
| 5 | 0 | 0 | 1460 | 64 | 4 | 0 |
| 6 | 0 | 695 | 661 | 5 | 0 | 0 |
| 7 | 0 | 423 | 398 | 0 | 0 | 0 |
| 8 | 0 | 339 | 43 | 0 | 0 | 0 |
| 9 | 0 | 220 | 1 | 0 | 0 | 0 |
| 10 | 0 | 184 | 0 | 0 | 0 | 0 |
| 11 | 0 | 151 | 0 | 0 | 0 | 0 |
| ≥12 | 4340 | 15 | 0 | 0 | 0 | 0 |
| Subtotal | 4340 | 2027 | 2563 | 69 | 78 | 139 |
Within the preliminarily identified SSRs, 261 motif types were detected, among which, mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide repeats had 2, 3, 10, 15, 23, and 61 types, respectively. The A/T mono-nucleotide and AG/CT di-nucleotide repeats were the most abundant motifs in our SSRs, accounting for 45.16% and 12.65% of the total SSRs, respectively, while the remaining 259 types of motif only accounted for 42.19% (Fig. 8).
Fig. 8.
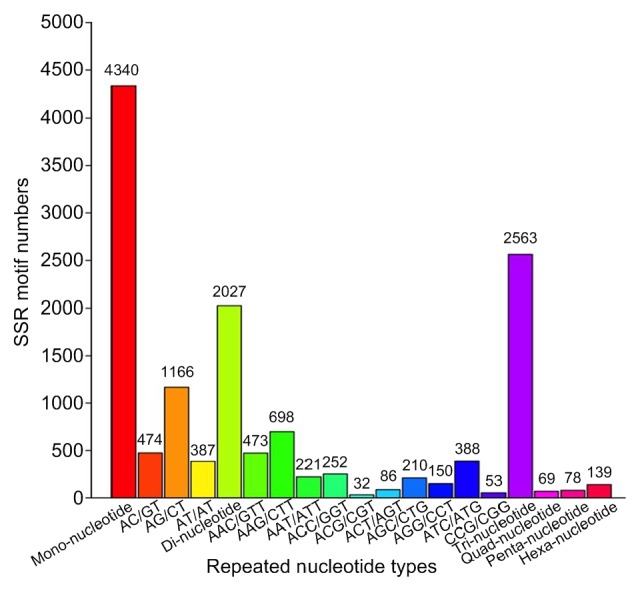
Frequency distribution of SSRs based on motif sequence types
3.11. SSR marker polymorphism
Using the transcription data, we initially selected 77 primer pairs and used them for amplification to evaluate accuracy. After the primers that appeared to match non-target loci or species were excluded through 1% (0.01 g/ml) agarose gel electrophoresis, 68 primer pairs were further tested with DNA from 225 individuals belonging to nine unrelated provenances. After screening, we observed that 11 of 68 primers yielded the predicted size with polymorphic loci. Thus, the polyacrylamide gel contributed to the development of 11 polymorphic SSR primers. The products amplified by the 11 polymorphic primers were then sequenced through capillary electrophoresis, and the results were in the form of allele sizes. All primers were submitted to GenBank. Their numbers, primer sequences, repeat motifs, sizes, and annealing temperatures (T a) are shown in Table 7. The inbreeding coefficient ranged from 0.011 to 0.179. The PIC value ranged from 0.1112 to 0.6750. The observed and expected heterozygosities were 0.064–0.840 and 0.115–0.726, respectively.
Table 7.
Characteristics of 11 microsatellite loci and their genetic diversity among the wild L. ruthenicum
| Primer | GenBank No. | Sequence (5'→3') | Repeat motif | Size (bp) | T a (°C) | F is | H o | H e | N A | PIC |
| U20 | KY284848 | F: CCCACTTCTCAAAAATGGTACAC | (ATT)6 | 88 | 60.0 | 0.058 | 0.544 | 0.481 | 4 | 0.4232 |
| R: ATAGTTGCCAACAAACCCTTCTT | ||||||||||
| U21 | KY284849 | F: GGATGAAGAAGAAGAGGATGACA | (AAT)6 | 126 | 59.9 | 0.042 | 0.324 | 0.458 | 5 | 0.4174 |
| R: CTTCTCAAAAATGGTACACTGCC | ||||||||||
| U23 | KY284850 | F: CTACTTCCATTTGTGGAAAGCTG | (TGC)6 | 164 | 60.0 | 0.179 | 0.576 | 0.667 | 4 | 0.6120 |
| R: TAGCCAGTCTAATCTTCGGTTTG | ||||||||||
| U25 | KY284851 | F: CAGGAAGGAGAAGAGTCTGATGA | (GCA)5 | 90 | 60.1 | 0.011 | 0.064 | 0.115 | 3 | 0.1112 |
| R: TTATCATTAACGGCTTCCATTTG | ||||||||||
| U26 | KY284852 | F: AATGGGGAAAGGTAAAGGAAGTT | (GTA)6 | 156 | 60.1 | 0.135 | 0.576 | 0.539 | 4 | 0.4701 |
| R: CCTTGTGGAATTTTACTTTCCAAT | ||||||||||
| U27 | KY284853 | F: CCACCCAGATAGTGGTGGTAATA | (GAA)6 | 108 | 59.8 | 0.072 | 0.380 | 0.379 | 5 | 0.3403 |
| R: GCTGATGTTTTCACATTTGTCAC | ||||||||||
| U31 | KY284854 | F: TAGGGTTTGAGGGTTTGAAGAAT | (CAC)5 | 109 | 59.4 | 0.076 | 0.164 | 0.202 | 3 | 0.1918 |
| R: ATTATTATGGCTTCTTCACCTGG | ||||||||||
| U36 | KY284855 | F: CTACCACTCCAACGTGTACCAAT | (CAA)6 | 138 | 60.1 | 0.099 | 0.536 | 0.607 | 4 | 05489 |
| R: TTCTTGCTCTAATTCTGAAACCG | ||||||||||
| U42 | KY284856 | F: GTCTCCATTTTACCCCTACCAAG | (ATT)6 | 150 | 60.2 | 0.135 | 0.316 | 0.726 | 4 | 0.6750 |
| R: TTTGCAAATAAAATGCGATTATTG | ||||||||||
| U46 | KY284857 | F: ATGAAGGCAATATTTAGGGCAGT | (TTG)7 | 153 | 60.3 | 0.049 | 0.132 | 0.270 | 3 | 0.2525 |
| R: CAATTTCATATTTGTGCTCTGCAT | ||||||||||
| D1 | KY284858 | F: TTCCAAGAACATTAGCACAAACA | (TTGGCT)4 | 136 | 59.7 | 0.027 | 0.840 | 0.576 | 4 | 0.4868 |
| R: TGGCACTTGTCCTAGTCCTAAAC |
T a: annealing temperature; F is: inbreeding coefficient; H o: observed heterozygosity; H e: expected heterozygosity; N A: allele numbers; PIC: polymorphism information content
3.12. Genetic diversity of wild Lycium ruthenicum population
Based on the band size of each primer pair, we could distinguish the nine provenances from one another. According to the primers shown in Table 8, the ALS, KEL, GZ, YJ, NMH, BC, and AKS groups could be determined by using single primer pairs. Conversely, SY and GEM could be differentiated with multiple primers.
Table 8.
Primers which can distinguish the 9 groups from each other
| Group | Primer |
| ALS | U20 |
| KEL | U21 |
| GZ | U25 |
| YJ | U26 |
| NMH | U31 |
| BC | U31 |
| AKS | U36 |
| SY | U20+U21+U25+U26+U31+U36+D1 |
| GEM | U20+U21+U25+U26+U31+U36+D1+U25 |
The phylogenetic tree (Fig. 9) revealed that AKS and KEL from Xinjiang (China) exhibited similar genetic diversities. The same phenomena were observed in BC and SY from Xinjiang (China) and GEM and NMH from Qinghai (China). High genetic similarity was found in YJ and GZ from Gansu (China). ALS from Inner Mongolia (China) was distinguished itself by diversity study. Genetic diversity results analyzed here indicated that our polymorphic microsatellite markers were applicable in the genetic diversity study.
Fig. 9.
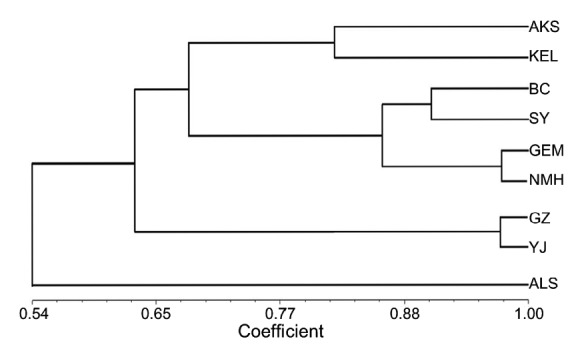
UPGMA dendrogram of 225 L. ruthenicum based on Nei (1978)’s genetic distances, indicating the clustering relationships of nine sampled populations
4. Discussion
4.1. Characterization of the Lycium ruthenicum transcriptome
Next generation sequencing is an efficient and low-cost method for genome or transcriptome study. This approach has been applied to various plant organisms in recent years and has greatly accelerated the research on understanding the mechanisms that underlie the growth and development of these organisms. In this study, we demonstrated a strategy of de novo assembly of a transcriptome by using short reads for a Chinese traditional medicinal plant, L. ruthenicum, whose sequence data are limited in the public databases at present. With the help of Illumina sequencing, 52.5 million paired-end reads were obtained for each sample and finally generated 68 063 unigenes with an average length of 877 bp and N50 of 1411 bp were finally generated. Thus, the number of genes/unigenes identified in L. ruthenicum achieved substantial progress from 77 to 68 063. The sequence similarities between L. ruthenicum and S. tuberosum, S. lycopersicum, N. tabacum were 60.9%, 25.2%, and 2.2%, respectively. The similarities observed here may present clues to determine the phylogenetic relationship between these species, which indicate that L. ruthenicum is closer to Solanum than Nicotiana within the Solanaceae family. A total of 53 366 (78%) unigenes showed a minimum of one significant match to an existing gene model, this ratio was relatively high compared with those in previous studies, indicating the efficiency of our data. The functions of the unigenes were classified by COG and GO, and the metabolic pathways were assigned using the KEGG database. The GO classification analysis showed that the predominant terms of metabolic and cellular processes in biological process were similar to those of numerous other plants, such as Populus euphratica, Boehmeria nivea, and Ipomoea batatas (Wang et al., 2010; Liu et al., 2013; Chen et al., 2014). The predominant term of “single-organism process” is similar to Halogeton glomeratus, another salt-tolerant plant species distributed in arid regions (Wang J. et al., 2015). Thus, we conclude that this process may perform a special function in the salt or salt-alkalinity stress response. The unigenes produced here can facilitate further research on L. ruthenicum functional genomics or molecular mechanism studies, especially those on response mechanisms under abiotic stresses.
4.2. Unigenes involved in saline-alkaline mixed stress
Analysis of the DE unigenes could reveal the tolerance mechanisms in response to saline-alkaline stress. After DE unigenes were identified, the GO categories and pathways involved in stress tolerance were analyzed. The predominant GO clusters for DE unigenes are involved in the metabolic process, cellular process, and single-organism process categories of the biological process, the cell and cell part categories of the cellular component domain, and the antioxidant activity and binding categories of the molecular function domain. Similar results were found in numerous plant species, except for the enrichment of unigenes that belong to the “single-organism process category”. This finding indicated the important or requisite functions of “single-organism process”-involved unigenes in the L. ruthenicum sodic stress response.
The top three pathways enriched in DE unigenes were “phenylpropanoid biosynthesis”, “flavone and flavonol biosynthesis”, and “flavonoid biosynthesis” pathways. Among these unigenes, “phenylpropanoid biosynthesis” and “flavonoid biosynthesis” have been reported in soybean under sodic stress (Ge et al., 2010). Phenylpropanoid biosynthesis often results in the synthesis of phenolic metabolites, such as tannins, hydroxycinnamate esters, and structural polymer lignin, which often play specific roles in plant protection. Previously, phenylpropanoid biosynthesis was enriched in the drought response in plants such as rice. Phenylpropanoid biosynthesis was supposed to represent an alternative pathway that is beneficial for enhancing the antioxidant capacity of the cells under environmental stresses (Grace and Logan, 2000). Flavonoids are a group of plant polyphenolic secondary metabolites, including flavonols that perform a wide range of functions, such as antioxidant activity, ultraviolet (UV)-light protection, and biotic and abiotic stress responses (Petrussa et al., 2013). Several studies reported the effects of temperature and water deficit on the biosynthesis of flavonoids. Here, we supposed that the saline-alkaline stress may also activate the flavonoid metabolism pathway for the number of DE unigenes significantly enriched in this pathway.
4.3. Establishment and application of novel microsatellite markers
SSR markers are widely used in the field of genetic mapping and diversity research and association analysis. For L. ruthenicum, the available molecular markers are insufficient, which makes it difficult to distinguish the individuals or populations of this genus. To date, the genetic diversity analysis of L. ruthenicum is restricted to sequence-related amplified polymorphism markers (Liu et al., 2012). EST sequences are important resources for the development of SSR markers. By means of Illumina sequencing, 9216 potential genome-wide SSRs were identified from the 7940 unigene sequences with a frequency of 1/6.48 kb. This finding will substantially facilitate genetic mapping and diversity research in the future. Furthermore, we developed 11 novel polymorphic SSR primers of L. ruthenicum from 77 chosen primers based on 225 individuals selected from nine provenances. Genomic backgrounds and resources could be distinguished by subjecting genetic similarity to cluster analysis with SSR markers. These novel and highly polymorphic SSR markers could be efficiently used for phylogenetic studies. L. ruthenicum is significant for economic and scientific research. Therefore, the proposed polymorphic EST-derived microsatellite loci could possibly facilitate marker-assisted selection in L. ruthenicum breeding and improve the ecological and economic environments in China.
5. Conclusions
In this study, we analyzed the global transcriptome profiling of L. ruthenicum by de novo sequencing without the presence of a reference genome using Illumina sequencing. With 68 083 high quality unigenes identified, we achieved a great leap in the knowledge of expressed genes for the L. ruthenicum species. Further comparison between the control and the saline-alkaline mixed samples revealed that thousands of unigenes, such as DREB transcription factor genes and calmodulin (CaM) genes, were significantly upregulated in the treated sample. GO terms for differentially expressed unigenes were significantly enriched in hormone-mediated signal, biological process regulation, and metabolic process regulation. For the first time, the large number of sequences generated for L. ruthenicum identified in this study provided valuable sequence information at the transcriptomic level of this species and will facilitate the understanding of the mechanisms underlying their growth and development, especially regarding the processes enabling L. ruthenicum to cope with saline-alkaline mixed stress. Moreover, based on these assembled sequences, we identified 9216 potential SSR markers and characterized some of them. Eleven microsatellite markers were established and applied in a genetic diversity study. The large numbers of sequences generated and SSR markers identified in this study provide valuable sequence information at the genome level of this species. The newly identified SSRs significantly contribute to the L. ruthenicum genomic resources and will facilitate a number of genetic and genomic studies, including high-density linkage mapping, genome-wide association analysis, comparative genomics analysis, and evolution analysis.
Acknowledgments
We sincerely thank Dr. Qiang-zhang DU of Beijing Forestry University, China for providing valuable suggestions for this study.
List of electronic supplementary materials
Table S1 Primer sequences used in qPCR analysis
Table S2 Top 100 most abundant transcripts in control sample
Table S3 Top 100 most abundant transcripts in salt-alkaline mixed treated sample
Table S4 Upregulated transcripts between the control and the saline-alkaline-treated sample
Table S5 Downregulated unigenes between the control and treated sample
Table S6 Top 300 most upregulated transcripts after treatment with annotation
Fig. S1 Function classifications of GO terms of all L.ruthenicum transcripts
Fig. S2 COG functional classification of the L.ruthenicum transcriptome
Footnotes
Project supported by the Fundamental Research Funds for the Central Universities (No. 2016ZCQ05), the Forestry Industry Research Special Funds for Public Welfare Projects (No. 201504101), and the Ningxia Goji Special Funds of Establishment and Application of Technical System of Molecular Breeding for Wolfberry, China
Electronic supplementary materials: The online version of this article (http://dx.doi.org/10.1631/jzus.B1700135) contains supplementary materials, which are available to authorized users
Compliance with ethics guidelines: Jin-huan CHEN, Dong-zhi ZHANG, Chong ZHANG, Mei-long XU, and Wei-lun YIN declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Alam SM, Naqvi SSM, Ansari R. Impact of soil pH on nutrient uptake by crop plants. In: Pessarakli M, editor. Handbook of Plant and Crop Stress. New York: Marcel Dekker, Inc.; 1999. pp. 51–60. [Google Scholar]
- 2.Altintas A, Kosar M, Kirimer N, et al. Composition of the essential oils of Lyceum barbarum and L. ruthenicum fruits. Chem Nat Compd. 2006;42(1):24–25. doi: 10.1007/s10600-006-0028-3. [DOI] [Google Scholar]
- 3.Babuin MF, Campestre MP, Rocco R, et al. Response to long-term NaHCO3-derived alkalinity in model Lotus japonicus ecotypes Gifu B-129 and Miyakojima MG-20: transcriptomic profiling and physiological characterization. PLoS ONE. 2014;9(5):e97106. doi: 10.1371/journal.pone.0097106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57(1):289–300. [Google Scholar]
- 5.Bi YH, Wu YY, Zhou ZG. Genetic diversity of wild population of Pyropia haitanensis based on SSR analysis. Biochem Syst Ecol. 2014;54:307–312. doi: 10.1016/j.bse.2014.02.010. [DOI] [Google Scholar]
- 6.Chagné D, Chaumeil P, Ramboer A, et al. Cross-species transferability and mapping of genomic and cDNA SSRs in pines. Theor Appl Genet. 2004;109(6):1204–1214. doi: 10.1007/s00122-004-1683-z. [DOI] [PubMed] [Google Scholar]
- 7.Chang S, Puryear J, Cairney J. A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Rep. 1993;11(2):113–116. doi: 10.1007/BF02670468. [DOI] [Google Scholar]
- 8.Chen JH, Xia XL, Yin WL. Expression profiling and functional characterization of a DREB2-type gene from Populuse uphratica . Biochem Biophys Res Commun. 2009;378(3):483–487. doi: 10.1016/j.bbrc.2008.11.071. [DOI] [PubMed] [Google Scholar]
- 9.Chen JH, Tian QQ, Pang T, et al. Deep-sequencing transcriptome analysis of low temperature perception in a desert tree, Populuse uphratica . BMC Genomics. 2014;15:326. doi: 10.1186/1471-2164-15-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conesa A, Gotz S, Garcia-Gomez JM, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 11.Diao QN, Song YJ, Shi DM, et al. Nitric oxide induced by polyamines involves antioxidant systems against chilling stress in tomato (Lycopersicon esculentum Mill.) seedling. J Zhejiang Univ-Sci B (Biomed & Biotechnol) 2016;17(12):916–930. doi: 10.1631/jzus.B1600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doyle J, Doyle JL. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem Bull. 1987;19(11):11–15. [Google Scholar]
- 13.Ge Y, Li Y, Zhu YM, et al. Global transcriptome profiling of wild soybean (Glycine soja) roots under NaHCO3 treatment. BMC Plant Biol. 2010;10:153. doi: 10.1186/1471-2229-10-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grace SC, Logan BA. Energy dissipation and radical scavenging by the plant phenylpropanoid pathway. Philos Trans R Soc B. 2000;355(1402):1499–1510. doi: 10.1098/rstb.2000.0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo YY, Yu HY, Kong DS, et al. Effects of drought stress on growth and chlorophyll fluorescence of Lycium ruthenicum Murr. seedlings. Photosynthetica. 2016;54(4):524–531. doi: 10.1007/s11099-016-0206-x. [DOI] [Google Scholar]
- 17.Hong Z, Lakkineni K, Zhang Z, et al. Removal of feedback inhibition of Δ1-pyrroline-5-carboxylate synthetase results in increased proline accumulation and protection of plants from osmotic stress. Plant Physiol. 2000;122(4):1129–1136. doi: 10.1104/pp.122.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iseli C, Jongeneel CV, Bucher P. ESTScan a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. ISMB. 1999;99:138–148. [PubMed] [Google Scholar]
- 19.Jin H, Plaha P, Park JY, et al. Comparative EST profiles of leaf and root of leymuschinensis, a xerophilous grass adapted to high pH sodic soil. Plant Sci. 2006;170(6):1081–1086. doi: 10.1016/j.plantsci.2006.01.002. [DOI] [Google Scholar]
- 20.Knapp S, Bohs L, Nee M, et al. Solanaceae a model for linking genomics with biodiversity. Comp Funct Genom. 2004;5(3):285–291. doi: 10.1002/cfg.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T, Zhu S, Tang Q, et al. De novo assembly and characterization of transcriptome using Illumina paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L. Gaud) BMC Genomics. 2013;14:125. doi: 10.1186/1471-2164-14-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu YD, Zhang GW, Liu DL. Simultaneous measurement of chlorophyll and water content in navel orange leaves based on hyperspectral imaging. Spectroscopy. 2014;29(4):40–44. [Google Scholar]
- 23.Liu YL, Zeng SH, Sun W, et al. Comparative analysis of carotenoid accumulation in two goji (Lycium barbarum L. and L. ruthenicum Murr.) fruits. BMC Plant Biol. 2014;14:269. doi: 10.1186/s12870-014-0269-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu ZG, Shu QY, Wang L, et al. Genetic diversity of the endangered and medically important Lycium ruthenicum Murr. revealed by sequence-related amplified polymorphism (SRAP) markers. Biochem Syst Ecol. 2012;45:86–97. doi: 10.1016/j.bse.2012.07.017. [DOI] [Google Scholar]
- 25.Luo J, Huang C, Peng F, et al. Effect of salt stress on photosynthesis and related physiological characteristics of Lycium ruthenicum Murr. Acta Agric Scand B. 2017;67(8):1–13. doi: 10.1080/09064710.2017.1326521. [DOI] [Google Scholar]
- 26.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 27.Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89(3):583–590. doi: 10.1093/genetics/89.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng Q, Liu H, Lei H, et al. Relationship between structure and immunological activity of an arabinogalactan from Lyceum ruthenicum . Food Chem. 2016;194:595–600. doi: 10.1016/j.foodchem.2015.08.087. [DOI] [PubMed] [Google Scholar]
- 29.Pertea G, Huang XQ, Liang F, et al. TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 30.Petrussa E, Braidot E, Zancani M, et al. Plant flavonoids–biosynthesis, transport and involvement in stress responses. Int J Mol Sci. 2013;14(7):14950–14973. doi: 10.3390/ijms140714950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polle A, Otter T, Seifert F. Apoplastic peroxidases and lignification in needles of norway spruce (Piceaabies L.) Plant Physiol. 1994;106(1):53–60. doi: 10.1104/pp.106.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu Y, Li X, Zhi H, et al. Differential expression of salt tolerance related genes in Brassica campestris L. ssp. chinensis (L.) Makino var. communis Tsen et Lee. J Zhejiang Univ-Sci B (Biomed & Biotechnol) 2009;10(11):847–851. doi: 10.1631/jzus.B0920098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rohlf FJ. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System, Version 2.1. Setauket, New York, USA: Exeter Software; 2000. [Google Scholar]
- 34.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Misener S, Krawetz SA, editors. Bioinformatics Methods and Protocols.Methods in Molecular Biology.Vol.132. Totowa, NJ: Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 35.Rumeu B, Sosa PA, Nogales M, et al. Development and characterization of 13 SSR markers for an endangered insular juniper (Juniperus cedrus Webb & Berth.) Conserv Genet Resour. 2013;5(2):457–459. doi: 10.1007/s12686-012-9827-y. [DOI] [Google Scholar]
- 36.Sato S, Tabata S, Hirakawa H, et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485(7400):635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi DC, Sheng YM. Effect of various salt-alkaline mixed stress conditions on sunflower seedlings and analysis of their stress factors. Environ Exp Bot. 2005;54(1):8–21. doi: 10.1016/j.envexpbot.2004.05.003. [DOI] [Google Scholar]
- 38.Spychalla JP, Desborough SL. Superoxide dismutase, catalase, and α-tocopherol content of stored potato tubers. Plant physiol. 1990;94(3):1214–1218. doi: 10.1104/pp.94.3.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang J, Yan Y, Ran L, et al. Isolation, antioxidant property and protective effect on PC12 cell of the main anthocyanin in fruit of Lycium ruthenicum Murray. J Funct Foods. 2017;30:97–107. doi: 10.1016/j.jff.2017.01.015. [DOI] [Google Scholar]
- 40.Wang J, Li B, Meng Y, et al. Transcriptomic profiling of the salt-stress response in the halophyte Halogeton glomeratus . BMC Genomics. 2015;16:169. doi: 10.1186/s12864-015-1373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Li J, Zhao J, et al. Evolutionary developmental genetics of fruit morphological variation within the Solanaceae. Front Plant Sci. 2015;6:248. doi: 10.3389/fpls.2015.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang YC, Chu YG, Liu GF, et al. Identification of expressed sequence tags in an alkali grass (Puccinellia tenuiflora) cDNA library. J Plant Physiol. 2007;164(1):78–89. doi: 10.1016/j.jplph.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Fang B, Chen J, et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas) BMC Genomics. 2010;11:726. doi: 10.1186/1471-2164-11-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei L, Li S, Liu S, et al. Transcriptome analysis of Houttuynia cordata Thunb. by Illumina paired-end RNA sequencing and SSR marker discovery. PLoS ONE. 2014;9(1):e84105. doi: 10.1371/journal.pone.0084105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wheeler DL, Church DM, Lash AE, et al. Database resources of the National Center for Biotechnology Information: 2002 update. Nucleic Acids Res. 2002;30(1):13–16. doi: 10.1093/nar/30.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X, Pan SK, Cheng SF, et al. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475(7355):189–195. doi: 10.1038/nature10158. [DOI] [PubMed] [Google Scholar]
- 47.Ye J, Fang L, Zheng HK, et al. WEGO a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Suppl. 2):W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeh FC, Yang RC, Boyle T. POPGENE Version 1. 31. Microsoft Window-Based Freeware for Population Genetic Analysis. University of Alberta and the Centre for International Forestry Research, CA.1999. [Google Scholar]
- 49.Zheng J, Ding CX, Wang LS, et al. Anthocyanins composition and antioxidant activity of wild Lycium ruthenicum Murr. from Qinghai-Tibet Plateau. Food Chem. 2011;126(3):859–865. doi: 10.1016/j.foodchem.2010.11.052. [DOI] [Google Scholar]
- 50.Zhu JK. Cell signaling under salt, water and cold stresses. Curr Opin Plant Biol. 2001;4(5):401–406. doi: 10.1016/S1369-5266(00)00192-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences used in qPCR analysis
Table S2 Top 100 most abundant transcripts in control sample
Table S3 Top 100 most abundant transcripts in salt-alkaline mixed treated sample
Table S4 Upregulated transcripts between the control and the saline-alkaline-treated sample
Table S5 Downregulated unigenes between the control and treated sample
Table S6 Top 300 most upregulated transcripts after treatment with annotation
Fig. S1 Function classifications of GO terms of all L.ruthenicum transcripts
Fig. S2 COG functional classification of the L.ruthenicum transcriptome