

Abstract
Wolbachia endosymbionts may be acquired by horizontal transfer, by introgression through hybridization between closely related species, or by cladogenic retention during speciation. All three modes of acquisition have been demonstrated, but their relative frequency is largely unknown. Drosophila suzukii and its sister species D. subpulchrella harbor Wolbachia, denoted wSuz and wSpc, very closely related to wRi, identified in California populations of D. simulans. However, these variants differ in their induced phenotypes: wRi causes significant cytoplasmic incompatibility (CI) in D. simulans, but CI has not been detected in D. suzukii or D. subpulchrella. Our draft genomes of wSuz and wSpc contain full‐length copies of 703 of the 734 single‐copy genes found in wRi. Over these coding sequences, wSuz and wSpc differ by only 0.004% (i.e., 28 of 704,883 bp); they are sisters relative to wRi, from which each differs by 0.014%–0.015%. Using published data from D. melanogaster, Nasonia wasps and Nomada bees to calibrate relative rates of Wolbachia versus host nuclear divergence, we conclude that wSuz and wSpc are too similar—by at least a factor of 100—to be plausible candidates for cladogenic transmission. These three wRi‐like Wolbachia, which differ in CI phenotype in their native hosts, have different numbers of orthologs of genes postulated to contribute to CI; and the CI loci differ at several nucleotides that may account for the CI difference. We discuss the general problem of distinguishing alternative modes of Wolbachia acquisition, focusing on the difficulties posed by limited knowledge of variation in absolute and relative rates of molecular evolution for host nuclear genomes, mitochondria, and Wolbachia.
Keywords: cytoplasmic incompatibility loci, horizontal transmission, introgression, molecular clocks, relative rates, spotted‐wing Drosophila
1. Introduction
Drosophila suzukii Matsumura (Diptera Drosophilidae) is an invasive and destructive fruit fly native to southeast Asia that has recently invaded North America, South America, and Europe (Cini, Ioriatti, & Anfora, 2012; Hauser, 2011; Rota‐Stabelli, Blaxter, & Anfora, 2013). While most Drosophila species oviposit in fermenting fruits, D. suzukii and its close relative D. subpulchrella Takamori and Watabe use their atypical serrated ovipositors to pierce the skin of ripening soft fruits and lay eggs in them (Fig. 1, Atallah et al., 2014; McEvey, 2017a, 2017b). Leveraging the genetic resources of D. melanogaster, D. suzukii and D. subpulchrella (both members of the D. melanogaster species group) are becoming model species for fundamental and applied studies.
Figure 1.

Drosophila suzukii and D. subpulchrella, with males on the left. The photographs are from McEvey (2017a, 2017b); the composite image is courtesy of Shane McEvey
Wolbachia are obligately intracellular, maternally inherited alpha‐proteobacteria found in about half of all insect species and many other terrestrial arthropods and nematodes (Weinert et al., 2015). Wolbachia are often associated with reproductive manipulations, including cytoplasmic incompatibility (CI) (Hoffmann & Turelli, 1997), male killing (Hurst & Jiggins, 2000), feminization (Rousset et al., 1992), and parthenogenesis induction (Stouthamer et al., 1993), all of which enhance the relative fitness of infected females. But many Wolbachia infections, including those in D. suzukii and its sister species D. subpulchrella, cause no detectable reproductive manipulation and presumably persist by enhancing host fitness (Cattel et al., 2016; Hamm et al., 2014; Kriesner et al., 2013, 2016; Mazzetto, Gonella, & Alma, 2015). Indeed, it seems increasingly plausible that even infections that cause reproductive manipulations become established in new hosts because they enhance fitness, and hence tend to increase in frequency even when very rare (Kriesner et al., 2013). For example, the most common Wolbachia reproductive manipulation is CI, in which, embryos produced by uninfected females mated with infected males suffer increased mortality. Because CI is essentially irrelevant to the frequency dynamics of rare infections, initial spread of both CI‐causing infections and infections that do not manipulate reproduction is likely to be driven by mutualistic effects such as fecundity enhancement (Fast et al., 2011; Weeks et al., 2007), protection from viruses (Teixeira, Ferreira, & Ashburner, 2008), or metabolic provisioning (Brownlie et al., 2009).
To understand why Wolbachia are found in so many species, it is critical to know how Wolbachia infections are acquired and how long Wolbachia—host associations persist. As noted by Raychoudhury, Baldo, Oliveira, and Werren (2008), although Wolbachia are maternally transmitted, host lineages can acquire Wolbachia in three ways: by cladogenic transmission, in which, an infection persists through speciation; by introgression, in which, hybridization of closely related species leads to maternal cytoplasm transfer; or by horizontal transmission, in ways that remain indeterminate, in which, Wolbachia are transferred between closely or distantly related species through nonsexual mechanisms (such as predation or parasitism).
To complement an analysis of Wolbachia population biology and effects in Drosophila suzukii and its sister species D. subpulchrella, Hamm et al. (2014) presented a meta‐analysis of Wolbachia infections in Drosophila species that addressed the frequency of both reproductive manipulation and alternative modes of acquisition. However, we show that their informal methodology underestimated the relative frequencies of horizontal and introgressive transmission. Horizontal transmission of Wolbachia was first demonstrated by extreme discordance of the phylogenies of distantly related hosts and their infecting Wolbachia (O'Neill et al., 1992). In contrast, horizontal transmission seems negligible within the two species that have been examined most intensively, D. simulans (Turelli & Hoffmann, 1995) and D. melanogaster (Richardson et al., 2012). Hamm et al. (2014) implicitly assumed that if two closely related host species share closely related Wolbachia, the infections are likely to have been acquired by either cladogenic transmission or introgression. In particular, Hamm et al. (2014) postulated that because D. suzukii and its sister D. subpulchrella have concordant mitochondrial and nuclear phylogenies and harbor very similar Wolbachia, as indicated by identity at the multilocus sequence typing (MLST) loci used to classify Wolbachia (Baldo et al., 2006), cladogenic Wolbachia acquisition was likely. Here, we use comparative analyses of draft Wolbachia genomes, and extensive nuclear data from Drosophila and other insect hosts, to refute this hypothesis.
The three alternative modes of Wolbachia acquisition would be trivial to distinguish if reliable chronograms (dated phylogenies) were available for the nuclear, mitochondrial, and Wolbachia genomes. Under cladogenic transmission, without subsequent introgression or horizontal transmission, roughly concordant chronograms for all three genomes are expected. From the arguments of Gillespie and Langley (1979), we expect a slightly longer divergence time for nuclear than mitochondrial or Wolbachia given the greater intraspecific variation observed in nuclear DNA. However, for typical pairs of Drosophila species that diverged on the order of 106 years ago (Coyne & Orr, 1989, 1997), this discordance under cladogenic acquisition is unlikely to be as large as a factor of two. Under introgression without subsequent horizontal transmission, the mitochondrial and Wolbachia chronograms should be concordant (because they are simultaneously maternally transmitted) and show more recent divergence than the bulk of the nuclear genome. Finally, under horizontal transmission, more recent divergence is expected between infecting Wolbachia than between either the host mitochondrial or nuclear genomes. These simple criteria are difficult to apply because of uncertainty concerning the relative rates of nuclear, mitochondrial, and Wolbachia divergence. Here, using all available comparative data for Wolbachia and host divergence, we conclude that the Wolbachia in D. suzukii and D. subpulchrella are far too similar to make cladogenic transmission plausible. Our conclusion does not exclude the possibility that D. suzukii and D. subpulchrella retained a Wolbachia infection from their common ancestor. Our data indicate only that their current Wolbachia are too similar to have been diverging since the speciation of their hosts. In principle, one could establish cladogenic transmission followed by introgression or horizontal transmission if traces of historical infections could be found in host genomes (Hotopp et al., 2007). Unfortunately, as shown below, no such traces were found in our D. suzukii or D. subpulchrella genomes.
In addition to assessing Wolbachia acquisition, we examine patterns of molecular evolution by comparing the draft genomes for wSuz (Siozos et al., 2013) and wSpc (this paper) to the wRi reference genome (Klasson et al., 2009). We consider both a general pattern, namely the relative frequencies of nonsynonymous and synonymous substitutions, and sequence divergence for candidate loci associated with two Wolbachia‐induced phenotypes, life shortening and CI. The “Octomom” duplication, which distinguishes wMelPop (Min & Benzer, 1997) from wMel (Wu et al., 2004), contains the genes WD0507–WD0514 and is associated with extremely high Wolbachia titer and life shortening in D. melanogaster (Chrostek & Teixeira, 2015; but see Rohrscheib et al., 2016 for a critique and LePage et al., 2017 and Chrostek & Teixeira, 2017 for support of the hypothesis connecting these loci to life shortening or Wolbachia titer). Beckmann and Fallon (2013) used proteomics to identify the locus wPip_0282 in wPip, the Wolbachia found in Culex pipiens, as a candidate for producing CI. They found at least one homolog of this WO prophage locus in several CI‐causing Wolbachia, including wMel and wRi. Within wPip and other Wolbachia genomes, wPip_0282 and each homolog seemed to be part of two‐gene operons, with wPip_0282 adjacent to wPip_0283. This pair is orthologous to WD0631 and WD0632 in wMel, and there are three homologous/paralogous pairs in wRi. Beckmann, Ronau, and Hochstrasser (2017) and LePage et al. (2017) provide experimental and bioinformatic evidence that WD0631 and WD0632 contribute to CI (but LePage et al. (2017) argue against the operon hypothesis). We examine differences in homologs and paralogs of these loci among wSuz, wSpc, and wRi.
2. Materials and Methods
2.1. Sequence data
Genome data for D. suzukii and D. subpulchrella were generated by Edinburgh Genomics. The D. suzukii genome data were generated from an inbred Italian line (the Trento strain) as presented in Ometto et al. (2013), with the Wolbachia, wSuz, presented in Siozos et al. (2013). Illumina HiSeq2000 120‐base, paired‐end sequence data were generated from two libraries of 180 and 300 base pair (bp) inserts. The D. subpulchrella genome data were generated from a stock maintained at the Fondazione Edmund Mach laboratory that was established from San Diego Drosophila species stock center strain 14023‐0401.00, originally from Japan. Illumina HiSeq2000 125‐base, paired‐end sequence data were generated from two libraries of 350 and 550 bp inserts.
2.2. Assembly of Wolbachia in D. subpulchrella
To assemble wSpc, we initially cleaned, trimmed, and assembled reads for the Wolbachia‐infected D. subpulchrella using Sickle (https://github.com/najoshi/sickle) and SOAPdenovo v. 2.04 (Luo et al., 2012). For the assembly, K values of 31, 41, …, 101 were tried, and the best assembly (fewest contigs and largest N50) was kept. This preliminary assembly had over 100,000 contigs with a total length of 243 megabases (Mbp). Details of the D. subpulchrella assembly will be published elsewhere, together with a comparison to the D. suzukii genome. Most of the contigs were identified through BLAST search as deriving from Drosophila. Minor contamination from microbiota (such as Acetobacter spp.) was identified. Contigs with best nucleotide BLAST matches (with E‐values less than 10−10) to known Wolbachia sequences were extracted as the draft assembly for wSpc. We also attempted filtering the reads by alignment to wRi and assembling with SPAdes 3.0 (Bankevich et al. 2012). The assembly of wSpc is available from GenBank under accession number NTHL00000000 (project number PRJNA401169, Biosample SAMN07599555).
To assess the quality of our draft wSpc and wSuz assemblies, we used BUSCO v. 3.0.0 (Simão et al., 2015) to search for orthologs of the near‐universal, single‐copy genes in the BUSCO proteobacteria database. As a control, we performed the same search using the complete reference genomes for wRi (Klasson et al., 2009), wAu (Sutton, Harris, Parkhill, & Sinkins, 2014), wMel (Wu et al., 2004), wHa, and wNo (Ellegaard et al., 2013).
2.3. Phylogeny and estimates of divergence of wSpc and wSuz
The Wolbachia MLST loci gatB, hcpA, coxA, fbpA, and ftsZ (Baldo et al., 2006) were identified in the assemblies using BLAST. As reported in Hamm et al. (2014), the MLST sequences from wSpc and wSuz were identical both to each other and to those of the wRi reference genome from D. simulans (Klasson et al., 2009).
To distinguish these Wolbachia and determine their relationships, we extracted additional orthologous loci from the draft genomes. We annotated the genomes of wSuz and wSpc with Prokka v 1.11 (Seemann, 2014), which identifies orthologs to reference bacterial genes. To normalize our comparisons, we also annotated the genomes of wRi (Klasson et al., 2009), wAu (Sutton et al., 2014), and wMel (Richardson et al., 2012; Wu et al., 2004). We selected 512 genes present in full length and single copy in all five genomes, avoiding incomplete or pseudogenes and loci with paralogs. Genes were treated as single copy if no other gene in the genome was matched to the same reference bacterial gene by Prokka, and as full length if the orthologs in the other Wolbachia genomes all had the same length. The nucleotide sequences of the genes were aligned with MAFFT v. 7 (Katoh, 2013) and concatenated, giving an alignment of 480,831 bp. The strain phylogeny was estimated with a phylogram constructed with MrBayes v. 3.2 (Ronquist & Huelsenbeck, 2003) using the GTR+Γ model, partitioned by codon position. All model parameters for each partition were allowed to vary independently, except topology and branch length. We ran two independent chains, each with four incrementally heated subchains, for 1,000,000 generations. Trace files for each analysis were visualized in Tracer v. 1.6 (Rambaut, Suchard, & Drummond 2013) to ensure convergence of all continuous parameters. The first 25% of the generations were discarded as burn‐in. Only one topology had posterior probability >.001.
To estimate the divergence between wSuz and wSpc, 703 genes present in full length and single copy in wSuz, wSpc, and wRi (spanning a total of 704,883 bp) were extracted and aligned with MAFFT v. 7. As an additional assessment of the completeness of the wSuz and wSpc assemblies, we calculated the number of single‐copy genes in the wRi reference and found 734. The resulting alignments were concatenated. To estimate a chronogram, we assumed for simplicity that each partition evolved at a constant rate across the tree (allowing the rates to differ among codon positions). The constant‐rate chronogram was estimated with MrBayes v. 3.2, using the same procedure as our five‐sequence Wolbachia phylogram (which included wMel and wAu). The age of the wSuz–wSpc node was set at 1, as an arbitrary scaling of relative ages.
2.4. Nuclear divergence between D. subpulchrella and D. suzukii
Hamm et al. (2014) used Drosophila nuclear data extracted from Yang et al. (2012) to assess the relationships of D. suzukii, D. subpulchrella, and D. biarmipes, but these data have subsequently been shown to be unreliable (Catullo & Oakeshott, 2014). We reassessed these relationships and compared the Wolbachia and nuclear chronograms for D. suzukii and D. subpulchrella. We identified in FlyBase complete coding regions for D. melanogaster for the ten nuclear loci used by Hamm et al. (2014) (H2A, Adh, amylase, amyrel, cdc6, ddc, esc, hb, nucl, and ptc), plus ten additional nuclear loci (aconitase, enolase, glyp, glys, pepck, pgi, pgm, tpi, white, and wg). We used BLAST to identify orthologs in the D. suzukii assembly of Ometto et al. (2013), the unpublished draft D. subpulchrella assembly described above, a D. biarmipes assembly (Chen et al., 2014), and a second‐generation D. simulans assembly (Hu, Eisen, Thornton, & Andolfatto, 2013). Data for H2A and amylase were eliminated because H2A had multiple nonidentical paralogs in each species and homologs of D. melanogaster amylase could not be found in the assemblies. The coding sequences for the remaining 18 loci were aligned with MAFFT v. 7 and concatenated. (Our nuclear data from D. subpulchrella are available from GenBank under accession numbers MF908506–MF909523.) The alignment was analyzed with MrBayes v. 3.2 using the same model and procedures used for our Wolbachia analyses, except that we partitioned the data by both gene and codon position. We estimated both a phylogram and a constant‐rate chronogram. The latter assumed that each partition evolved at a constant rate over the tree. The age of the most recent common ancestor (MRCA) of D. suzukii and D. subpulchrella was set at 1, as an arbitrary scaling of relative ages. To test the robustness of our relative divergence‐time estimates for the host species, we also estimated the chronogram using a relaxed‐clock model in RevBayes (Hoehna et al., 2016). This analysis also partitioned the data by gene and codon position and used the GTR+Γ model, but it assumed uncorrelated lognormal rate variation across branches. Following the RevBayes tutorial (https://github.com/revbayes/revbayes_tutorial/blob/master/RB_BayesFactor_Tutorial/scripts/marginal_likelihood_GTR_Gamma_inv.Rev), we used a lognormal prior with mean and standard deviation parameters (−X 2/2, X) and a lognormal hyperprior on X with parameters (ln(2)/4, Sqrt[ln(2)/2]). To estimate k s and k a between D. suzukii and D. subpulchrella, we used DNAsp v. 5.10 (Rozas, 2009).
Following Hotopp et al. (2007), we looked for evidence of genetic transfer from wSuz and wSpc (or other Wolbachia) to these hosts. The D. suzukii and D. subpulchrella assemblies (including the Wolbachia contigs) were BLASTed against both all known melanogaster group nuclear sequences and all known Wolbachia sequences. We sought contigs for which part mapped to a Drosophila nuclear sequence and not to any Wolbachia sequence while another part mapped to a Wolbachia sequence and not to any Drosophila nuclear sequence.
2.5. Analysis of divergence between wSpc, wSuz, and wRi
The trimmed Illumina reads from D. suzukii and D. subpulchrella were aligned to the wRi reference (Klasson et al., 2009) with bwa v. 0.7.12 (Li & Durbin, 2009). As a control, we also aligned Illumina reads from Riv84 (Iturbe‐Ormaetxe et al., 2010), the D. simulans line used to make the wRi reference. Normalized read depth for each alignment was calculated over sliding 1,000‐bp windows by dividing the average depth in the window by the average depth over the entire genome. Putative copy‐number variant (CNV) locations were identified with ControlFREEC v. 8.0 (Boeva et al., 2012), using 500‐bp windows and the Riv84 alignment as a control. For the bulk of the genomes, we used an expected ploidy of one, but for variants involving sequences duplicated in wRi, we used a ploidy of two. We calculated p‐values for each putative CNV using the Kolmogorov–Smirnov test implemented in ControlFREEC.
Sequences for the “Octomom” genes WD0507–WD0514 (Chrostek & Teixeira, 2015; cf. Rohrscheib et al., 2016; Chrostek & Teixeira, 2017) were extracted from the wMel reference (Richardson et al., 2012; Wu et al., 2004). Using BLAST, we identified orthologs in the wRi reference (Klasson et al., 2009) and the draft assemblies for wSuz and wSpc.
Sequences homologous to loci putatively involved in CI in other Wolbachia strains (Beckmann & Fallon, 2013; Beckmann et al., 2017; LePage et al., 2017) were extracted from wRi (Klasson et al., 2009) and the draft assemblies for wSuz and wSpc. Differences among these three genomes at these loci were assessed by aligning the wSuz and wSpc reads to the wRi reference and calculating the percentage of reads with the non‐wRi base.
To identify a specific insertion of the transposable element ISWpi7, which occurs in 21 identical copies in wRi, and whose position differentiates wSpc and wSuz from wRi, an additional assembly step was required. The novel insertion occurs in the wSpc and wSuz orthologs of WRi_006720, one of the CI‐associated loci discussed below. The D. suzukii and D. subpulchrella reads were aligned to the wSpc assembly with bwa 0.7.12 (Li & Durbin, 2009). For both contigs that contain part of the WRi_006720 gene, reads mapping to the ISWpi7 transposable element plus the neighboring 500 bp were extracted and assembled with SOAPdenovo v. 2.04 (Luo et al., 2012), using a K value of 55. Both the D. suzukii and D. subpulchrella reads assembled into a single contig containing the two pieces of WRi_006720 interrupted by a single copy of ISWpi7. To test this bioinformatic result, we designed two pairs of PCR primers that spanned the hypothesized junctions between the ortholog of WRi_006720 and ISWpi7. For the first set of primers (forward: ATGGTCACATTGAACAGAGGAT, reverse: GTTGGTGCTGCAATGCGTAA), the forward primer attaches at 728945–728966, part of WRi_006720. For the second set of primers (forward: AGCGTTGTGGAGGAACTCAG, reverse: CGTCATGCTGCAGTGCTTAG), the reverse primer attaches at 729570–729589, part of WRi_006720. No detectable product is expected with either primer set in wRi, which does not contain the insert in WRi_006720, whereas each primer set is expected to produce a unique band with wSpc and wSuz.
3. Results
3.1. Draft genome assembly for wSpc, the Wolbachia from D. subpulchrella
We generated a draft assembly of wSpc by filtering contigs from a joint Wolbachia–D. subpulchrella assembly. The draft wSpc assembly was in 100 contigs with N50 length of 31,871 bp and total length of 1.42 Mbp. This length is close to the 1.45 Mbp wRi reference (Klasson et al., 2009), suggesting that it may represent a nearly complete genome. In contrast, the assembly produced by SPAdes 3.0 had N50 of 8,360 bp and total length of 1.20 Mbp.
Out of 221 near‐universal, single‐copy orthologs in proteobacteria, BUSCO 3.0.0 (Simão et al., 2015) found effectively the same number in all of the tested genomes (wRi, wAu, wMel, wHa, wNo, and the drafts of wSuz and wSpc). Our draft assemblies for wSpc and wSuz contain two BUSCO‐annotated genes not found in wRi and wMel. See Table S1 for detailed information.
3.2. Wolbachia divergence
We aligned and compared wSpc and wSuz at 703 protein‐coding loci (704,883 bp) and identified only 28 single‐nucleotide variants (SNV), an overall divergence of 0.004%. wSuz had 103 SNV compared to wRi (0.015% divergence), and wSpc had 99 SNV (0.014% divergence) (Table S2). Most (87) of these SNV are shared. There were too few differences to definitively determine whether these genomes are recombinant (Ellegaard et al., 2013), but the data were fully consistent with no recombination (i.e., with so few differences, we have no power to detect recombination). Bayesian phylogenetic analysis placed wSuz and wSpc as sisters relative to wRi (Fig. 2a). For wSuz and wSpc, we derived point estimates and 95% confidence intervals for divergence at each codon position, calculated as the rate multiplier for that position times the branch length (fixed to 1) (Table 1). The rate multipliers express the relative rate of evolution for each codon position. Hence, the expected number of substitutions for each codon position along each branch of the phylogram is the branch length times the rate multiplier for that position. The estimated chronogram (Fig. 2b) shows that the divergence time of wRi from its MRCA with wSpc and wSuz is 3.51 times the divergence time of wSpc and wSuz, with a 95% confidence interval of (2.41, 4.87). We found no difference in the rates of divergence for first‐, second‐, and third‐codon positions, as also observed in the codivergence of Wolbachia and mtDNA haplotypes in D. melanogaster (Richardson et al., 2012). Following from this, estimates of synonymous, k s, and nonsynonymous, k a, substitution rates were very similar (Table 1).
Figure 2.
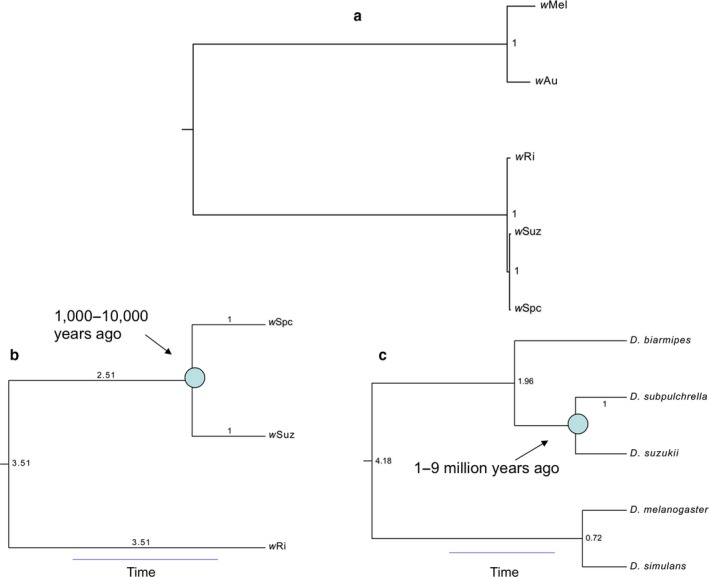
Phylogram and chronograms for the Wolbachia and hosts discussed. Clade posterior probabilities are shown. (a) Wolbachia phylogram. (b) Wolbachia chronogram with an estimate of the divergence time for wSuz and wSpc. Branch lengths relative to the wSpc–wSuz divergence are shown. All clade posterior probabilities are 1.0. (c) Host chronogram with an estimate of divergence time for Drosophila suzukii and D. subpulchrella. Branch lengths relative to the D. suzukii–D. subpulchrella divergence are shown. All clade posterior probabilities are 1.0
Table 1.
Estimated number of substitutions per site by codon position between wSuz and wSpc, plus estimates of synonymous (k s) and nonsynonymous (k a) substitution rates, see the text for details
| Position | Point estimates | 95% confidence interval |
|---|---|---|
| 1st | 5.0 × 10−5 | (3.0 × 10−5, 7.0 × 10−5) |
| 2nd | 3.2 × 10−5 | (1.6 × 10−5, 4.6 × 10−5) |
| 3rd | 4.0 × 10−5 | (2.4 × 10−5, 5.6 × 10−5) |
| Overall (k s, k a) | (3 × 10−5, 4 × 10−5) |
3.3. Host divergence
The host phylogram (data not shown) and chronogram (Fig. 2c) demonstrate that D. subpulchrella and D. suzukii are sisters relative to D. biarmipes, as reported by Hamm et al. (2014). The divergence time of D. biarmipes from its MRCA with D. subpulchrella and D. suzukii was estimated to be 1.96 times the divergence time for D. subpulchrella and D. suzukii, with 95% confidence interval (1.84, 2.08). The D. melanogaster and D. simulans divergence‐time estimate is 0.72 times as large as the estimated divergence time for D. subpulchrella–D. suzukii, with a 95% confidence interval of (0.65, 0.78). Point estimates and 95% confidence intervals for divergence at each codon position between D. subpulchrella and D. suzukii were calculated as the rate multiplier for that position times the branch length (fixed to 1) (Table 2). Our estimate of the third‐codon position substitutions per site, which we use to date D. subpulchrella–D. suzukii divergence, is 9.20 × 10−2 and a 95% confidence interval of (8.6 × 10−2, 9.80 × 10−2).
Table 2.
Estimated number of substitutions per site by codon position between Drosophila suzukii and D. subpulchrella for 18 nuclear loci, plus estimates of synonymous (k s) and nonsynonymous (k a) substitution rates, see the text for details
| Position | Point estimates | 95% confidence interval |
|---|---|---|
| 1st | 1.10 × 10−2 | (9.91 × 10−3, 1.21 × 10−2) |
| 2nd | 4.73 × 10−3 | (4.12 × 10−3, 5.28 × 10−3) |
| 3rd | 9.20 × 10−2 | (8.64 × 10−2, 9.80 × 10−2) |
| Overall (k s, k a) | (1.2 × 10−1, 5.3 × 10−3) |
Our RevBayes (Hoehna et al. 2016) relaxed‐clock chronogram (data not shown) was very similar to Fig. 2c. The divergence time of D. biarmipes from its MRCA with D. subpulchrella and D. suzukii was estimated to be 1.84 times the divergence time for D. subpulchrella and D. suzukii (instead of 1.96). Similarly, the D. melanogaster and D. simulans divergence‐time estimate is 0.76 times as large as the estimated divergence time for D. subpulchrella–D. suzukii (instead of 0.72). We note that the model underlying this analysis assumes for computational convenience that each partition undergoes proportional rate variation across each branch, that is, each partition speeds up or slows down by the same amount along each branch (but see Langley & Fitch, 1974).
We found no evidence for partial integration of any Wolbachia sequence into the nuclear genomes of either D. subpulchrella or D. suzukii.
3.4. Calibrations for Wolbachia versus host genome divergence and interpretation
We used estimates of relative divergence of the Wolbachia and Drosophila genomes to assess cladogenic versus lateral transmission of wSpc and wSuz. Our strategy was to compare our estimates of relative Wolbachia/host divergence to ratios obtained from published examples of cladogenic Wolbachia transmission. Table 3 summarizes our data and the data from two Nasonia wasp species (Raychoudhury et al., 2008; wNlonB1 versus wNgirB), and four Nomada bee species (Gerth & Bleidorn, 2016; plus unpublished data kindly provided by the authors). Our ratio of Wolbachia to host silent‐site divergence estimates is two or three orders of magnitude lower than found for Nasonia or Nomada. This strongly supports relatively recent Wolbachia transfer between D. suzukii and D. subpulchrella, being inconsistent with ratios observed under cladogenic Wolbachia acquisition. Given that we are looking at only single wSpc and wSuz sequences, their divergence time provides an upper bound for the time of interspecific transfer (Gillespie & Langley, 1979). Additional support for noncladogenic transmission comes from the analyses of Richardson et al. (2012), who inferred that Wolbachia substitution rates were roughly 10‐fold lower than the noncoding nuclear mutation rate for D. melanogaster, which is often considered a reasonable approximation for the rate of third‐position substitutions (at least for fourfold degenerate sites, Obbard et al. 2012). This is clearly inconsistent with the three‐order‐of‐magnitude difference we estimate (Table 3).
Table 3.
Estimated frequencies of synonymous (k s) versus nonsynonymous (k a) substitutions per site for Wolbachia in various hosts
| Data source | Species 1 | Species 2 | Host | Wolbachia | k s ratio | ||
|---|---|---|---|---|---|---|---|
| k s | k a | k s | k a | ||||
| This work | Drosophila suzukii | Drosophila subpulchrella | 1.2 × 10−1 | 6.8 × 10−3 | 3 × 10−5 | 4 × 10−5 | 0.00025 |
| Raychoudhury et al. (2008) | Nasonia giraulti | Nasonia longicornis | 1.22 × 10−2 | 5.4 × 10−3 | 3.7 × 10−3 | 2.2 × 10−3 | 0.30 |
| Gerth and Bleidorn (2016) | Nomada ferruginata | Nomada leucophthalma | 1.95 × 10−2 | 2.6 × 10−3 | 2.5 × 10−3 | 9 × 10−4 | 0.13 |
| Gerth and Bleidorn (2016) | N. ferruginata | N. flava | 1.92 × 10−2 | 2.7 × 10−3 | 2.5 × 10−3 | 9 × 10−4 | 0.13 |
| Gerth and Bleidorn (2016) | N. ferruginata | N. panzeri | 1.84 × 10−2 | 3.1 × 10−3 | 2.7 × 10−3 | 1.1 × 10−3 | 0.15 |
| Gerth and Bleidorn (2016) | N. leucophthalma | N. flava | 6.8 × 10−3 | 4 × 10−4 | 1 × 10−4 | 1 × 10−4 | 0.015 |
| Gerth and Bleidorn (2016) | N. leucophthalma | N. panzeri | 5.8 × 10−3 | 8 × 10−4 | 3 × 10−4 | 2 × 10−4 | 0.052 |
| Gerth and Bleidorn (2016) | N. flava | N. panzeri | 5.5 × 10−3 | 9 × 10−4 | 3 × 10−4 | 3 × 10−4 | 0.055 |
Comparing wSuz and wSpc, we found no difference in k s and k a (Table 1). This is also true for wMel variation in D. melanogaster (Richardson et al., 2012). M. Gerth and C. Bleidorn (2016, personal communication) find essentially identical estimates of k s and k a for all pairwise comparisons of the Wolbachia in the clade ([Nomada leucophthalma, Nomada flava], Nomada panzeri). In contrast, comparing wRi and wAu using the 429,765‐bp data set of single‐copy, full‐length genes (Table S3), we estimate k s of 4.34%, whereas the estimated k a is only 0.65% (or k s/k a = 6.7). Similarly, when comparing the Wolbachia of the outgroup host, Nomada ferruginata, to the Wolbachia of the three ingroup species, M. Gerth and C. Bleidorn (2016, personal communication) observed k s/k a values of 2.8, 2.8, and 2.5. In their comparisons of wNlonB1 and wNgirB from Nasonia longicornis and Nasonia giraulti, Raychoudhury et al. (2008) estimated k s/k a = 0.0037/0.0022 = 1.7. Our data and those from other very recently diverged Wolbachia are consistent with either accelerated adaptive Wolbachia evolution in a new host or a relaxation of constraints on nonsynonymous substitutions.
Estimating absolute divergence times (i.e., times to the MRCA) for wSuz and wSpc and their hosts is more difficult. Assuming 10 generations per year in Drosophila and using the wMel‐derived estimate of (2.88 × 10−10, 1.29 × 10−9) changes/site/host‐generation as the 95% confidence interval for the third‐position substitution rate of Wolbachia (Richardson et al., 2012), wSuz and wSpc diverged about 1,600–7,000 years ago. Using the 95% confidence interval for first‐ and second‐position substitution rates from Richardson et al. (2012) yields wSuz–wSpc divergence dates of 1,200–9,100 years. Given that D. suzukii and D. subpulchrella seem to be temperate species (Ometto et al., 2013; Takamori et al., 2006), the number of generations per year may be overestimated by a factor of two, which would inflate the Wolbachia divergence time by a factor of two. This does not affect our conclusions. Raychoudhury et al. (2008) estimated a Wolbachia k s of 4.7 × 10−9 changes/synonymous site/year in Nasonia. Using our k s from Table 1 with the Nasonia calibration, the estimated divergence for wSuz and wSpc is 6,400 years, which is consistent with our Drosophila calibration. These analyses suggest that wSuz and wSpc diverged on the order of 1,000–10,000 years ago, orders of magnitude shorter than typical time scales for Drosophila speciation (105–106 years, Coyne & Orr, 2004, p. 75; Obbard et al., 2012). Molecular estimates of Drosophila divergence times generally depend on speculative inferences from the phylogeography of the Hawaiian Drosophila radiation (Obbard et al., 2012). Using the Obbard et al. (2012) summary of available estimates for D. melanogaster and D. simulans divergence and our relative chronogram for D. subpulchrella and D. suzukii (Fig. 2c), we infer divergence times for D. subpulchrella and D. suzukii ranging from about 1 to 9 million years, two orders of magnitude larger than our estimates for wSuz versus wSpc. Hence, despite great uncertainties, our data clearly preclude cladogenic transmission of wSuz and wSpc. This conclusion is further supported in the Discussion by a review of variation in rates of bacterial molecular evolution.
3.5. Genome differences between wSpc, wSuz, and wRi: Structural variation and candidate genes
We identified CNV in wSuz and wSpc relative to the wRi reference sequence by plotting read depth along each genome (Fig. 3; Table 4). wSpc and wSuz share a deletion relative to wRi of 23,000 bp, between positions 733,000 and 756,000. wSuz has duplications 22,500 bp long from about 570,000 to 592,500 and 1,077,500 to 1,100,000. Both regions are part of the WO‐B prophage. In wRi, there are two nearly identical copies (99.4%) of WO‐B, from about 565,000 to 636,000 and from about 1,071,000 to 1,142,000 (Klasson et al., 2009). wSuz had an additional duplication between 1,345,000 and 1,347,500, outside of the WO prophage regions (Table 4).
Figure 3.
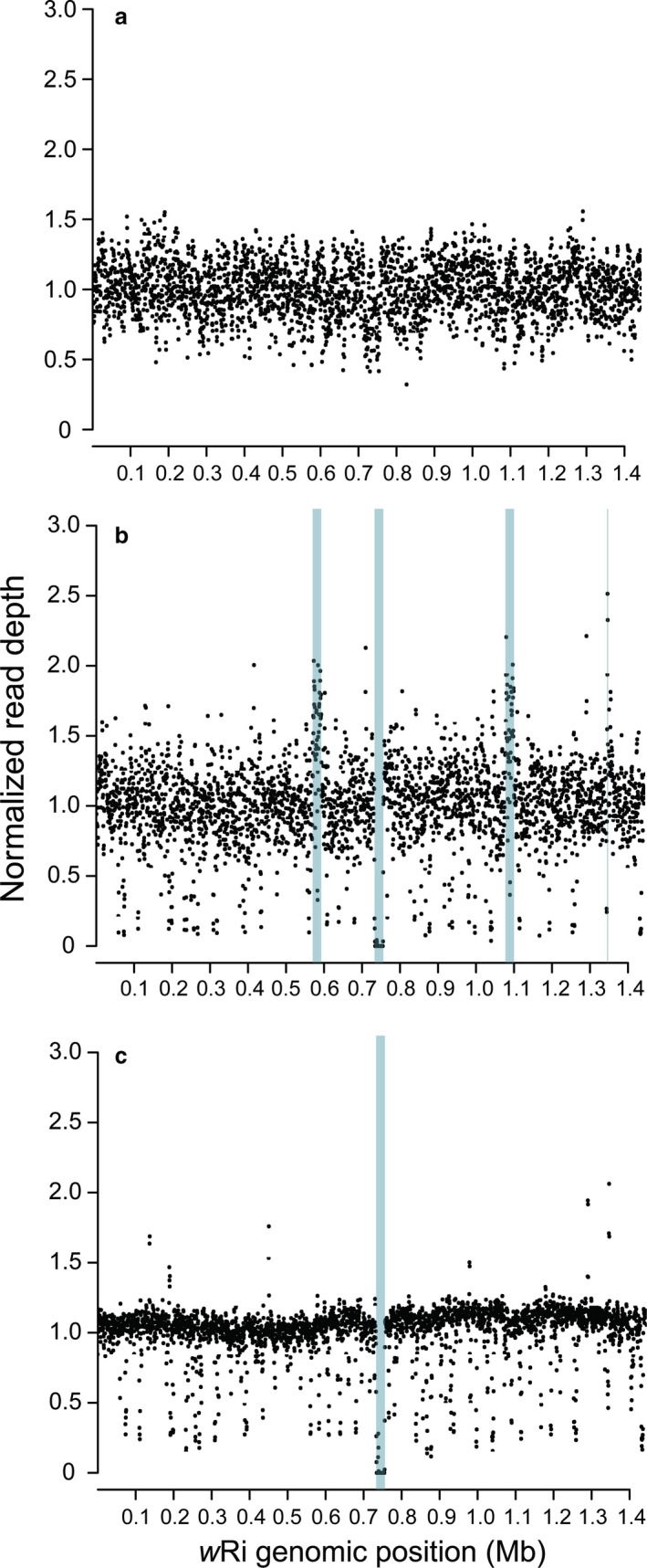
We compare normalized read density relative to the wRi reference sequence of Klasson et al. (2009) for: (a) the Illumina reads from the Riv84 version of wRi reported by Iturbe‐Ormaetxe et al. (2010), (b) the wSuz reads from Ometto et al. (2013), and (c) the wSpc reads from this study
Table 4.
Copy‐number variants in wSuz and wSpc relative to wRi. All positions are given relative to the wRi reference of Klasson et al. (2009)
| Start position | End position | Copy‐number change | Kolmogorov–Smirnov p‐value | Affected genomes |
|---|---|---|---|---|
| 570,000 | 592,500 | 2 → 3a | <.0001 | wSuz |
| 733,000 | 756,000 | 1 → 0 | <.0001 | wSuz, wSpc |
| 1,077,500 | 1,100,000 | 2 → 3a | <.0001 | wSuz |
| 1,345,000 | 1,347,500 | 1 → 2 | .016 | wSuz |
This sequence is duplicated in the wRi genome, so it was treated as diploid in our ControlFREEC v. 8.0 analysis.
We identified homologs in our target Wolbachia genomes of loci implicated in producing phenotypic effects. The Octomom phenotype of wMel (shortened life, high Wolbachia titer) has been associated with eight loci (WD0507–WD0514, Chrostek & Teixeira, 2015; but see also Chrostek & Teixeira, 2017; Rohrscheib et al., 2016). In the wRi reference, we found homologs of only WD0508 and WD0509. There were two WD0508‐like genes, at 632,500–633,385 and 1,138,959–1,139,844, within the wRi WO‐B prophages. A single WD0509‐like gene was present, from 1,419,589 to 1,421,396, not associated with WO‐B prophage. These two genes are not neighbors in wRi, wSpc, or wSuz, and they are not within regions that differentiate wSpc and wSuz from wRi.
Table 5 lists the orthologs and paralogs in wMel, wRi, wSuz, and wSpc of wPip_0282 and wPip_0283, the loci originally identified as CI‐causing by Beckmann and Fallon (2013) in wPip, the Wolbachia in Culex pipiens. These loci occur in pairs; and the “type I” pairs, orthologs of wPip_0282 and wPip_0283, may be a toxin–antidote operon (cf. Beckmann et al., 2017 with LePage et al., 2017). The orthologs in wMel are WD0631 and WD0632. As shown in Table 5, there are two copies of the type I pair in wRi, one copy in each of the two complete copies of the WO‐B prophage. As noted by Beckmann and Fallon (2013), in wRi, there is also a paralogous pair (wRi_006720 and wRi_006710), termed “type II” by LePage et al. (2017), that exists within what they term a “WO‐like island.”
Table 5.
Homologs of CI‐associated loci in wMel, wRi, wSuz, and wSpc. The gene designations in wSpc and wSuz reflect homology to loci identified in wMel and wRi
| Wolbachia | Gene paira | Gene 1 | Gene 2 | WO prophage associationb |
|---|---|---|---|---|
| wMel | I | WD0631 (cifA/cidA)c (antidote?) | WD0632 (cifB/cidB)c (toxin?) | Yes |
| wRi | I.1 | wRi_005370 | wRi‐p005380 d | Yes |
| I.2 | wRi_010030 | wRi_p010040 d | Yes | |
| II | wRi_006720 | wRi_006710 | No | |
| wSpc | I.1 | wSpc_0631.I.1 | wSpc_0632.I.1 | Yes |
| I.2 | wSpc_0631.I.2 | wSpc_0632.II.2 | Yes | |
| II | wSpc_6720 (disrupted) | wSpc_6710 | No | |
| wSuz | I.1 | wSuz_0631.I.1 | wSuz_0632.I.1 | Yes |
| I.2 | wSuz_0631.I.2 | wSuz_0632.II.2 | Yes | |
| I.3 | wSuz_0631.I.3 | wSuz_0632.II.3 | Partiale | |
| II | wSuz_6720 (disrupted) | wSuz_6710 | No |
Roman numerals follow the “type” designations in LePage et al. (2017).
This refers to location within an intact WO prophage, as opposed to a “WO‐like island” (cf. LePage et al., 2017).
Alternative designations (cif vs. cin) from LePage et al. (2017) and Beckmann et al. (2017), respectively. Beckmann et al. (2017) propose that WD0631 produces an antidote to the toxin produced by WD0632.
Annotated as pseudogenes, but see text.
This third copy in wSuz exists in the 1077500–1100000 CNV, noted in Table 4, which is a partial copy of the WO‐B prophage.
Table S5 lists genes included in the CNV regions of wSuz and wSpc relative to wRi. Notably, the orthologs of WD0631 and WD0632, implicated in causing CI (Beckmann & Fallon, 2013; Beckmann et al., 2017; LePage et al., 2017), are in a partial third copy of prophage WO‐B found in wSuz. Hence, wSuz contains three copies of these two loci, whereas wSpc has only two (see Table 5). The CNVs in wSuz or wSpc do not affect the type II loci.
Table 6 reports differences among wRi, wSuz, and wSpc at orthologs of the CI‐associated loci WD0631, WD0632, WRi_006710, and WRi_006720. The duplicate orthologs of WD0631 in wRi are WRi_005370 and WRi_010030. As noted by Beckmann and Fallon (2013), the (duplicate) orthologs of WD0632 in wRi have been annotated as pseudogenes, WRi_p005380 and WRi_p010040, because of premature stop codons, but they retain large, intact coding regions and may be functional (LePage et al., 2017 and Beckmann et al., 2017 provide evidence supporting function). Even with multiple orthologs of WD0631 and WD0632 in each genome (two in wRi, two in wSpc, and three in wSuz), all copies within each genome are identical and all interspecific comparisons consistently show the single‐nucleotide differences reported in Table 6. wSuz and wSpc share two missense substitutions in WD0631 and one in WD0632. As shown in Table 6, wSuz and wSpc also share one missense substitution in wRi_006710. This indicates that the duplications unique to wSuz occurred after the split of (wSuz, wSpc) from wRi. wSpc has a nonsense mutation at position 3,353 of WD0632, which results in a protein lacking the last 56 amino acids produced in wRi. These differences may account for the fact that while wRi causes appreciable CI in D. simulans and detectable CI in D. melanogaster, neither wSuz nor wSpc causes detectable CI in its native host (Hamm et al., 2014).
Table 6.
Comparisons between wRi, wSpc, and wSuz at the CI‐associated loci (type I, possible antidote, toxin), WD0631 and WD0632, from wMel, and the paralogous loci (type II), WRi_006710 and WRi_006720 from wRi. All reads from wSpc and wSuz are consistent with the differences shown
| Location (gene, amino acid) | wRi codon (codon, translation) | wSpc codon (codon, translation) | wSuz codon (codon, translation) |
|---|---|---|---|
| WD0631 a (antidote?) | |||
| 363 | AAA, Lys | GAA, Glu | GAA, Glu |
| 473 | AAA, Lys | AGA, Arg | AGA, Arg |
| WD0632 b (toxin?) | |||
| 91 | GGA, Gly | GGG, Gly | GGG, Gly |
| 176 | TAT, Tyr | GAT, Asp | GAT, Asp |
| 213 | TAT, Tyr | TAC, Tyr | TAC, Tyr |
| 1,118 | TTA, Leu | TGA, STOP | TTA, Leu |
| WRi_006710 | |||
| 663 | TAT, Tyr | CAT, His | CAT, His |
| WRi_006720 | |||
| 1–108 | Present | Disrupted, see text | Disrupted, see text |
The duplicate orthologs in wRi are WRi_005370 and WRi_010030.
The duplicate orthologs in wRi are WRi_p005380 and WRi_p010040.
Our bioinformatic and PCR data show that in both wSpc and wSuz (but not wRi), an IS element, identical to ISWpi7 of wRi (Klasson et al., 2009, Table S5), has inserted before base 323 of the ortholog to WRi_006720. There are 21 identical copies of the ISWpi7 transposon in wRi, each 1480 bp long with the transposase gene flanked on each side by about 200 bp. Clearly, this ISWpi7 insertion predates the divergence of wSpc and wSuz.
4. Discussion
4.1. Genomic data indicate noncladogenic acquisition of wSuz and wSpc
Despite considerable uncertainly in divergence‐time estimates for both wSuz and wSpc and their hosts, D. suzukii and D. subpulchrella, genomic data on relative rates of Wolbachia and host divergence contradict the conjecture by Hamm et al. (2014) that these species share similar Wolbachia because of cladogenic transmission. Based on this result, we must also revisit the Hamm et al. (2014) conclusion that cladogenic transmission of Wolbachia may be relatively common among Drosophila. That conclusion was based on the erroneous assumption that cladogenic transmission was the most plausible explanation for sister species sharing very similar Wolbachia. Given that on the order of half of Drosophila speciation events show evidence for reinforcement (i.e., accelerated rates of evolution for premating isolation associated with overlapping ranges) (Coyne & Orr, 1989, 1997; Turelli, Lipkowitz, & Brandvain, 2014), hybridization is apparently common among sister species of Drosophila. Introgression has been invoked to explain the closely related Wolbachia found within the simulans and yakuba clades in the D. melanogaster subgroup (Lachaise et al., 2000; Rousset & Solignac, 1995). In both cases, the introgression hypothesis is favored over horizontal transmission because the hosts also share essentially identical mitochondrial DNA. Wolbachia transmission within the yakuba clade is currently being reanalyzed using complete Wolbachia, mitochondrial, and nuclear genomes (Turelli, Conner, Turissini, Matute, and Cooper, in preparation).
Understanding the frequency of alternative modes of Wolbachia transmission is clearly related to determining how long Wolbachia infections typically persist in host lineages. Bailly‐Bechet et al. (2017) provide a meta‐analysis of more than 1,000 arthropod species from Tahiti that suggests average durations on the order of 7 million years. However, their molecular data, which involve only two Wolbachia loci and the CO1 mtDNA locus, do not have sufficient power to resolve the issue. Moreover, as they note, their analysis conflates imperfect maternal transmission with the gain and loss of Wolbachia infections within lineages. As our analyses indicate, nearly complete Wolbachia and mitochondrial genomes will often be needed to unravel the acquisition and retention of closely related Wolbachia within host clades.
4.2. Extremely variable rates of Wolbachia molecular evolution seem an implausible alternative
Gerth and Bleidorn (2016) proposed a time scale for Wolbachia evolution based on the apparent codivergence of Wolbachia and nuclear genomes in a clade of four Nomada bee species. Our discussion of their data emphasized comparisons between the outgroup host N. ferruginata and the three ingroup hosts, noting that the codivergence of these hosts and their Wolbachia produced relative rates of molecular divergence comparable to those inferred for a pair of Nasonia (Raychoudhury et al., 2008) and for D. melanogaster (Richardson et al., 2012). However, if we consider instead the sister species N. leucophthalma and N. flava from Gerth and Bleidorn (2016), we would infer much slower divergence of their Wolbachia (which recently acquired a biotin synthesis operon). For N. leucophthalma and N. flava, M. Gerth and C. Bleidorn (2016, personal communication) estimated synonymous nuclear substitution divergence of 6.8 × 10−3, with a corresponding Wolbachia synonymous substitution divergence of only 1.0 × 10−4 (Table 3). Under cladogenic transmission, this implies Wolbachia divergence that is roughly an order of magnitude slower than inferred from the three outgroup comparisons, with Wolbachia divergence at 1/68th the rate of the host nuclear genomes rather than 1/8. This indicates either 8.5‐fold rate variation for Wolbachia molecular evolution or that cladogenic transmission does not apply to this sister pair.
To explain our D. suzukii and D. subpulchrella data with cladogenic transmission and relative rate heterogeneity, we require that Wolbachia divergence is more than 1,000‐fold slower than third‐position nuclear divergence. This relative rate is 100‐fold slower than inferred for D. melanogaster and 30‐fold slower than the slow rate implied by cladogenic transmission between N. leucophthalma and N. flava. Such extreme heterogeneity seems implausible, but more examples of cladogenic Wolbachia transmission are needed to definitively rule this out.
Although there are relatively few taxa for which we can quantify the relative rates of nuclear versus Wolbachia molecular evolution, there are extensive data assessing the relative constancy of bacterial molecular evolution. Kuo and Ochman (2009) provide an overview, emphasizing that variation across taxa is too great for any locus or group of loci to provide a broadly applicable “molecular clock” for bacteria. Nevertheless, their analyses indicate that variation across lineages is typically much less than 10‐fold. Yet, if wSuz and wSpc were cladogenically inherited and we assume the implausibly short host divergence time of 500,000 years (half of our lowest plausible estimate, see Fig. 2), the inferred upper bound on the rate of Wolbachia silent‐site substitutions is about 1.0 × 10−11 per site per year. In contrast, the inferred rates of silent‐site substitutions from the Nasonia and Nomada data (Table 3) are at least two orders of magnitude faster. Such variation in Wolbachia substitution rates over many loci would be unprecedented among bacteria.
4.3. Comparative genomics and CI
Recent experiments strongly suggest that the wMel loci WD0631 and WD0632, contained within the WO‐B prophage, cause CI (Beckmann & Fallon, 2013; Beckmann et al., 2017; LePage et al., 2017). Despite having orthologs of both loci that are fairly similar to those in wRi, D. suzukii and D. subpulchrella show no apparent CI. There are two copies of these CI‐associated loci in wRi, two in wSpc, and three in wSuz. As argued above, the additional copy in wSuz was acquired after wSuz and wSpc diverged. The differences we document in Table 6 between wRi, wSuz, and wSpc at the CI‐associated loci may be informative about the portions of those loci essential to CI. Unpublished data (L. Mouton, personal communication) show that wRi causes detectable, but slight, CI when introduced into D. suzukii. Given the high level of CI that wRi causes in D. simulans, these data suggest that D. suzukii may suppress CI, perhaps indicating a relatively old association with CI‐causing Wolbachia (Hoffmann & Turelli, 1997; Turelli, 1994). We may be able to determine whether D. suzukii or D. subpulchrella was the donor of their closely related Wolbachia from population genomic analyses of their mtDNA and Wolbachia. Genomes from a geographically diverse sample of D. suzukii are currently being analyzed and may resolve the direction of Wolbachia transfer (J. C. Chiu, personal communication).
The published crossing studies in D. suzukii and D. subpulchrella, which found no statistically significant CI caused by wSuz or wSpc, are relatively small (Cattel et al., 2016; Hamm et al., 2014). They are comparable to the experiments that inferred no CI associated with the native Wolbachia infections in D. yakuba, D. teissieri, and D. santomea (Charlat, Ballard, & Mercot, 2004; Zabalou et al., 2004). However, larger experiments by Cooper, Ginsberg, Turelli, and Matute (2017) revealed consistent, albeit weak, CI in all three yakuba clade species—and interspecific CI between these species. More replicated assays for CI in D. suzukii and D. subpulchrella, as well as investigation of whether CI is produced when wSpc and wSuz, are transinfected into CI‐expressing hosts such as D. simulans, will indicate whether the differences described in Table 6 are candidates for disrupting the molecular processes underlying CI (Beckmann et al., 2017; LePage et al., 2017).
5. Conclusions and Open Questions
Understanding how host species acquire Wolbachia requires comparing divergence‐time estimates for closely related Wolbachia in host sister species to divergence‐time estimates for both their hosts' nuclear genes and mtDNA. To make confident inferences, we need better estimates of both the mean and variance of relative divergence rates for these three genomes. The variance for mtDNA divergence can be obtained from extant data, such as the many available Drosophila genomes. Estimates for nuclear, mitochondrial, and Wolbachia genomes can be obtained from groups like the filarial nematodes for which codivergence of the hosts and their obligate Wolbachia is well established (Bandi, Anderson, Genchi, & Blaxter, 1998). Our ability to infer processes of Wolbachia acquisition will be greatly enhanced by additional examples of cladogenic transmission among insects, besides Nasonia wasps (Raychoudhury et al., 2008) and Nomada bees (Gerth & Bleidorn, 2016). For D. suzukii and D. subpulchrella, distinguishing between introgression and horizontal transmission requires mtDNA sequences, which will be analyzed in our forthcoming D. subpulchrella genome paper.
It is a challenge to understand the pattern of molecular evolution between closely related Wolbachia whereby all three nucleotide positions evolve at similar rates, producing comparable rates of synonymous versus nonsynonymous substitutions. This is consistent with the pattern of variation seen for wMel within D. melanogaster (Richardson et al., 2012). In contrast, k s/k a increases to 2–3 for the cladogenically transmitted Wolbachia in Nasonia and Nomada, then increases to about 7 for the more distantly related wAu and wRi infecting D. simulans. Does Wolbachia “invasion” of a new host represent a relaxation of selective constraint or an opportunity for adaptation? The reigning paradigm for molecular evolution of endosymbionts involves the fixation of slightly deleterious mutations (Kuo & Ochman, 2009; Moran, 1996), consistent with relaxed constraints and reduced effective population size. However, we can test for rapid adaptation of Wolbachia to hosts by moving near‐identical Wolbachia between closely related hosts and comparing fitness (and reproductive) effects in native versus novel hosts.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
The genomic data for D. subpulchrella and wSpc were generated by O. R.‐S., L. O., M. B., and G. A. The bioinformatic analyses were performed by W. R. C. with input from M. T., M. B., and O. R.‐S. The first draft of the manuscript was produced by M. T., W. R. C., and M. B. with subsequent improvements by all authors.
DATA ACCESSIBILITY
Our nuclear data from D. subpulchrella are available from GenBank under accession numbers MF908506–MF909523. For wSpc, the Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession NTHL00000000. The version described in this paper is version NTHL01000000.
Supporting information
ACKNOWLEDGMENTS
We thank: John Beckmann, Seth Bordenstein, Brandon Cooper, Michael Gerth, Christian Schlötterer, and two anonymous reviewers for comments on previous drafts; Dave Begun, Chuck Langley, Mike May, Brian Moore, and Li Zhao for help with our analyses; Shane McEvey and Antoine Abrieux for photographs; and Joanna Chiu, Michael Gerth, and Laurence Mouton for sharing unpublished data.
Conner WR, Blaxter ML, Anfora G, Ometto L, Rota‐Stabelli O, Turelli M. Genome comparisons indicate recent transfer of wRi‐like Wolbachia between sister species Drosophila suzukii and D. subpulchrella . Ecol Evol. 2017;7:9391–9404. https://doi.org/10.1002/ece3.3449
Funding information
Our work was partially supported by US National Institutes of Health R01‐GM‐104325‐01 (MT) and UK Natural Environmental Research Council R8/H10/56 to Edinburgh Genomics (MB).
REFERENCES
- Atallah, J. , Teixeira, L. , Salazar, R. , Zaragoza, G. , & Kopp, A. (2014). The making of a pest: The evolution of a fruit‐penetrating ovipositor in Drosophila suzukii and related species. Proceedings of the Royal Society of London B, 281, 20132840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly‐Bechet, M. , Martins‐Simoes, P. , Szollosi, G. J. , Mialdea, G. , Sagot, M.‐F. , & Charlat, S. (2017). How long does Wolbachia stay on board? Molecular Biology and Evolution, 34, 1183–1193. [DOI] [PubMed] [Google Scholar]
- Baldo, L. , Dunning Hotopp, J. C. , Jolley, K. A. , Bordenstein, S. R. , Biber, S. A. , Choudhury, R. R. , Hayashi, C. , Maiden, M. C. , Tettelin, H. , & Werren, J. H . (2006). Multilocus sequence typing system for the endosymbiont Wolbachia pipientis . Applied Environmental Microbiology, 72, 7098–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandi, C. , Anderson, T. J. C. , Genchi, C. , & Blaxter, M. L. (1998). Phylogeny of Wolbachia in filarial nematodes. Proceedings of the Royal Society of London B, 265, 2407–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulokov, A. S. , … Pevzner, P. A. (2012). SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann, J. F. , & Fallon, A. M. (2013). Detection of the Wolbachia protein WPIP0282 in mosquito spermathecae: Implications for cytoplasmic incompatibility. Insect Biochemistry and Molecular Biology, 43, 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann, J. F. , Ronau, J. A. , & Hochstrasser, M. (2017). A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nature Microbiology, 2, 17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva, V. , Popova, T. , Bleakley, K. , Chiche, P. , Cappo, J. , Schleiermacher, G. , … Barillot, E. (2012). Control‐FREEC: A tool for assessing copy number and allelic content using next‐generation sequencing data. Bioinformatics, 28, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlie, J. C. , Cass, B. N. , Riegler, M. , Witsenburg, J. J. , Iturbe‐Ormaetxe, I. , McGraw, E. A. , & O'Neill, S. L. (2009). Evidence for metabolic provisioning by a common invertebrate endosymbiont, Wolbachia pipientis, during periods of nutritional stress. PLoS Pathogens, 5, e1000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattel, J. , Kaur, R. , Gilbert, P. , Martinez, J. , Fraimont, A. , Jiggins, F. , … Mouton, L. (2016). Wolbachia in European populations of the invasive pest Drosophila suzukii: Regional variation in infection frequencies. PLoS ONE, 11, e0147766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catullo, R. A. , & Oakeshott, J. G. (2014). Problems with data quality in the reconstruction of evolutionary relationships in the Drosophila melanogaster species group: Comments on Yang et al. (2012). Molecular Phylogenetics and Evolution, 78, 275–276. [DOI] [PubMed] [Google Scholar]
- Charlat, S. , Ballard, J. W. O. , & Mercot, H. (2004). What maintains noncytoplasmic incompatibility inducing Wolbachia in their hosts: A case study from a natural Drosophila yakuba population. Journal of Evolutionary Biology, 17, 322–330. [DOI] [PubMed] [Google Scholar]
- Chen, Z. X. , Sturgill, D. , Qu, J. , Jiang, H. , Park, S. , Boley, N. , … Richards, S. (2014). Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Research, 24, 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrostek, E. , & Teixeira, L. (2015). Mutualism breakdown by amplification of Wolbachia genes. PLoS Biology, 13, e1002065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrostek, E. , & Teixeira, L. (2017). Comment on Rohrscheib et al. 2016 “Intensity of mutualism breakdown is determined by temperature not amplification of Wolbachia genes”. PLoS Pathogens, 13, e1006540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cini, A. , Ioriatti, C. , & Anfora, G. (2012). A review of the invasion of Drosophila suzukii in Europe and a draft research agenda for integrated pest management. Bulletin of Insectology, 65, 149–160. [Google Scholar]
- Cooper, B. S. , Ginsberg, P. S. , Turelli, M. , & Matute, D. R. (2017). Wolbachia in the Drosophila yakuba complex: Pervasive frequency variation and weak cytoplasmic incompatibility, but no apparent effect on reproductive isolation. Genetics, 205, 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne, J. A. , & Orr, H. A. (1989). Patterns of speciation in Drosophila . Evolution, 43, 362–381. [DOI] [PubMed] [Google Scholar]
- Coyne, J. A. , & Orr, H. A. (1997). “Patterns of speciation in Drosophila” revisited. Evolution, 51, 295–303. [DOI] [PubMed] [Google Scholar]
- Coyne, J. A. , & Orr, H. A. (2004). Speciation. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Ellegaard, K. M. , Klasson, L. , Naslund, K. , Bourtziz, K. , & Andersson, S. G. E. (2013). Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genetics, 9, e1003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fast, E. M. , Toomey, M. E. , Panaram, K. , Desjardins, D. , Kolaczyk, E. D. , & Frydman, H. M. (2011). Wolbachia enhance Drosophila stem cell proliferation and target the germline stem cell niche. Science, 334, 990–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth, M. , & Bleidorn, C. (2016). Comparative genomics provides a timeframe for Wolbachia evolution and exposes a recent biotin synthesis operon transfer. Nature Microbiology, 2, 16241. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H. , & Langley, C. H. (1979). Are evolutionary rates really variable? Journal of Molecular Evolution, 13, 27–34. [DOI] [PubMed] [Google Scholar]
- Hamm, C. A. , Begun, D. J. , Vo, A. , Smith, C. C. R. , Saelao, P. , Shaver, A. O. , … Turelli, M. (2014). Wolbachia do not live by reproductive manipulation alone: Infection polymorphism in Drosophila suzukii and D. subpulchrella . Molecular Ecology, 23, 4871–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser, M. (2011). A historic account of the invasion of Drosophila suzukii (Matsumura) (Diptera: Drosophilidae) in the continental United States, with remarks on their identification. Pest Management Science, 67, 1352–1357. [DOI] [PubMed] [Google Scholar]
- Hoehna, S. , Landis, M. J. , Heath, T. A. , Boussau, B. , Lartillot, N. , Moore, B. R. , … Ronquist, F. (2016). RevBayes: Bayesian phylogenetic inference using graphical models and an interactive model‐specification language. Systematic Biology, 65, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, A. A. , & Turelli, M. (1997). Cytoplasmic incompatibility in insects In O'Neill S. L., Hoffmann A. A., & Werren J. H. (Eds.), Influential passengers: Inherited microorganisms and arthropod reproduction (pp. 42–80). New York, NY: Oxford University Press. [Google Scholar]
- Hotopp, J. C. D. , Clark, M. E. , Oliveira, D. C. S. G. , Foster, J. M. , Fischer, P. , Torres, M. C. M. , … Werren, J. H. (2007). Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science, 317, 1753–1756. [DOI] [PubMed] [Google Scholar]
- Hu, T. T. , Eisen, M. B. , Thornton, K. R. , & Andolfatto, P. (2013). A second‐generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage‐specific divergence. Genome Research, 23, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, G. D. , & Jiggins, F. M. (2000). Male‐killing bacteria in insects: Mechanisms, incidence, and implications. Emerging Infectious Diseases, 6, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturbe‐Ormaetxe, I. , Woolfit, M. , Rances, E. , Duplouy, A. , & O'Neill, S. L. (2010). A simple protocol to obtain highly pure Wolbachia endosymbiont DNA for genome sequencing. Journal of Microbiological Methods, 84, 134–136. [DOI] [PubMed] [Google Scholar]
- Katoh, S. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson, L. , Westberg, J. , Sapountzis, P. , Naslund, K. , Lutnaes, Y. , Darby, A. C. , … Andersson, S. G. E. (2009). The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans . Proceedings of the National Academy of Sciences of the United States of America, 106, 5725–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriesner, P. , Conner, W. R. , Weeks, A. R. , Turelli, M. , & Hoffmann, A. A. (2016). Persistence of a Wolbachia infection frequency cline in Drosophila melanogaster and the possible role of reproductive dormancy. Evolution, 70, 979–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriesner, P. , Hoffmann, A. A. , Lee, S. F. , Lee, S. F. , Turelli, M. , & Weeks, A. R. (2013). Rapid sequential spread of two Wolbachia variants in Drosophila simulans . PLoS Pathogens, 9, e1003607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C.‐H. , & Ochman, H. (2009). Inferring clocks when lacking rocks: The variable rates of molecular evolution in bacteria. Biology Direct, 4, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaise, D. , Harry, M. , Solignac, M. , Lemeunier, F. , Benassi, V. , & Cariou, M.‐L. (2000). Evolutionary novelties in islands: Drosophila santomea, a new melanogaster sister species from São Tomé. Proceedings of the Royal Society of London B, 267, 1487–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley, C. H. , & Fitch, W. M. (1974). An examination of the constancy of the rate of molecular evolution. Journal of Molecular Evolution, 3, 161–177. [DOI] [PubMed] [Google Scholar]
- LePage, D. P. , Metcalf, J. A. , Bordenstein, S. R. , On, J. , Perlmutter, J. I. , Shropshire, J. D. , … Bordenstein, S. R. (2017). Prophage WO genes recapitulate and enhance Wolbachia‐induced cytoplasmic incompatibility. Nature, 543, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Bianghang, L. , Yinlong, X. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , … Wang, J. (2012). SOAPdenovo2: An empirically improved memory‐efficient short‐read de novo assembler. GigaScience, 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzetto, F. , Gonella, E. , & Alma, A. (2015). Wolbachia infection affects female fecundity in Drosophila suzukii . Bulletin of Insectology, 68, 153–157. [Google Scholar]
- McEvey, S. (2017a). High resolution diagnostic images of Drosophila suzukii (Diptera: Drosophilidae). figshare. https://doi.org/10.6084/m9.figshare.4644793.v1
- McEvey, S. (2017b). High resolution diagnostic images of Drosophila subpulchrella (Diptera: Drosophilidae). figshare. https://doi.org/10.6084/m9.figshare.5363431.v1
- Min, K.‐T. , & Benzer, S. (1997). Wolbachia, normally a symbiont of Drosophila, can be virulent, causing degeneration and early death. Proceedings of the National Academy of Sciences of the United States of America, 94, 10792–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, N. A. (1996). Accelerated evolution and Muller's ratchet in endosymbiotic bacteria. Proceedings of the National Academy of Sciences of the United States of America, 93, 2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard, D. J. , Maclennan, J. , Kim, K. W. , Rambaut, A. , O'Grady, P. M. , & Jiggins, F. M. (2012). Estimating divergence dates and substitution rates in the Drosophila phylogeny. Molecular Biology and Evolution, 29, 3459–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto, L. , Cestaro, A. , Ramasamy, S. , Grassi, A. , Revadi, S. , Siozios, S. , … Rota‐Stabelli, O. (2013). Linking genomics and ecology to investigate the complex evolution of an invasive Drosophila pest. Genome Biology Evolution, 5, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill, S. L. , Giordano, R. , Colbert, A. M. , Colbert, A. M. E. , Karr, T. L. , & Robertson, H. M. (1992). 16S rRNA phylogenetic analysis of the bacterial endosymbionts associated with cytoplasmic incompatibility in insects. Proceedings of the National Academy of Sciences of the United States of America, 89, 2699–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Suchard, M. , & Drummond, A. J. (2014). Tracer v1.6. Retrieved from http://tree.bio.ed.ac.uk/software/tracer/
- Raychoudhury, R. , Baldo, L. , Oliveira, D. C. , & Werren, J. H. (2008). Modes of acquisition of Wolbachia: Horizontal transfer, hybrid introgression, and codivergence in the Nasonia species complex. Evolution, 63, 165–183. [DOI] [PubMed] [Google Scholar]
- Richardson, M. F. , Weinert, L. A. , Welch, J. J. , Linheiro, R. S. , Magwire, M. M. , Jiggins, F. M. , & Bergman, C. M. (2012). Population genomics of the Wolbachia endosymbiont in Drosophila melanogaster . PLoS Genetics, 8, e1003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrscheib, C. E. , Frentiu, F. D. , Horn, E. , Ritchie, F. K. , van Swinderen, B. , Weible, W. M. II , … Brownlie, J. C. (2016). Intensity of mutualism breakdown is determined by temperature not amplification of Wolbachia genes. PLoS Pathogens, 12, e1005888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F. , & Huelsenbeck, J. P. (2003). MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572–1574. [DOI] [PubMed] [Google Scholar]
- Rota‐Stabelli, O. , Blaxter, M. , & Anfora, G. (2013). Quick guide: Drosophila suzukii . Current Biology, 23, R8–R9. [DOI] [PubMed] [Google Scholar]
- Rousset, F. , Bouchon, D. , Pintureau, B. , Juchault, P. , & Solignac, M. (1992). Wolbachia endosymbionts responsible for various alterations of sexuality in arthropods. Proceedings of the Royal Society of London B, 250, 91–98. [DOI] [PubMed] [Google Scholar]
- Rousset, F. , & Solignac, M. (1995). Evolution of single and double Wolbachia symbioses during speciation in the Drosophila simulans complex. Proceedings of the National Academy of Sciences of the United States of America, 92, 6389–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J. (2009). DNA sequence polymorphism analysis using DnaSP In Posada D. (Ed.), Bioinformatics for DNA sequence analysis; Methods in Molecular Biology Series (Vol. 537, pp. 337–350). Clinton, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics, 30, 2068–2069. [DOI] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Siozos, S. , Cestaro, A. , Kaur, R. , Pertot, I. , Rota‐Stabelli, O. , & Anfora, G. (2013). Draft genome sequence of the Wolbachia endosymbiont of Drosophila suzukii . Genome Announcements, 1, e00032–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouthamer, R. , Breeuwert, J. A. , Luck, R. F. , & Werren, J. H. (1993). Molecular identification of microorganisms associated with parthenogenesis. Nature, 361, 66–68. [DOI] [PubMed] [Google Scholar]
- Sutton, E. R. , Harris, S. R. , Parkhill, J. , & Sinkins, S. P. (2014). Comparative genome analysis of Wolbachia strain wAu. BMC Genomics, 15, 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori, H. , Watabe, H. , Fuyama, Y. , Zhang, Y. , & Aotsuka, T. (2006). Drosophila subpulchrella, a new species of the Drosophila suzukii species subgroup from Japan and China (Diptera: Drosophilidae). Entomological Science, 9, 121–128. [Google Scholar]
- Teixeira, L. , Ferreira, Á. , & Ashburner, M. (2008). The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster . PLoS Biology, 6, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli, M. (1994). Evolution of incompatibility‐inducing microbes and their hosts. Evolution, 48, 1500–1513. [DOI] [PubMed] [Google Scholar]
- Turelli, M. , & Hoffmann, A. A. (1995). Cytoplasmic incompatibility in Drosophila simulans: Dynamics and parameter estimates from natural populations. Genetics, 140, 1319–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli, M. , Lipkowitz, J. R. , & Brandvain, Y. (2014). On the Coyne and Orr‐igin of species: Effects of intrinsic postzygotic isolation, ecological differentiation, X‐chromosome size, and sympatry on Drosophila speciation. Evolution, 68, 1176–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks, A. R. , Turelli, M. , Harcombe, W. R. , Reynolds, K. T. , & Hoffmann, A. A. (2007). From parasite to mutualist: Rapid evolution of Wolbachia in natural populations of Drosophila . PLoS Biology, 5, 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, L. A. , Araujo‐Jnr, E. V. , Ahmed, M. Z. , & Welch, J. J. (2015). The incidence of bacterial endosymbionts in terrestrial arthropods. Proceedings of the Royal Society of London B, 282, 20150249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, M. L. , Sun, V. L. , Vamathevan, L. , Riegler, M. , Deboy, R. , Brownlie, J. C. , … Eisen, J. A. (2004). Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: A streamlined genome overrun with mobile genetic elements. PLoS Biology, 2, 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Hou, Z. H. , Qian, Y. H. , Kang, H. , & Zeng, Q.‐T. (2012). Increasing the data size to accurately reconstruct the phylogenetic relationships between nine subgroups of the Drosophila melanogaster species group (Drosophilidae, Diptera). Molecular Phylogenetics and Evolution, 62, 214–223. [DOI] [PubMed] [Google Scholar]
- Zabalou, S. , Charlat, S. , Nirgianaki, A. , Lachaise, D. , Mercot, H. , & Bourtzis, K. (2004). Natural Wolbachia infections in the Drosophila yakuba species complex do not induce cytoplasmic incompatibility but fully rescue the wRi modification. Genetics, 167, 827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our nuclear data from D. subpulchrella are available from GenBank under accession numbers MF908506–MF909523. For wSpc, the Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession NTHL00000000. The version described in this paper is version NTHL01000000.