

Abstract
In the past decade, a shift toward targeted therapies in non-small-cell lung cancer following molecular profiling has dramatically changed the way advanced adenocarcinoma is treated. However, tumor cells inevitably acquire resistance to such therapies, circumventing any sustained clinical benefit. As the genomic classification of lung cancer continues to evolve and as the mechanisms of acquired resistance to targeted therapies become elucidated and more improved target-specific drugs come into sight, the future will see more promising results from the clinic through the development of new therapeutic strategies to overcome, or prevent the development of, resistance for lung cancer patients.
Keywords: Non-small-cell lung cancer, Oncogenic drivers, TKIs, RTKs, Acquired resistance
Introduction
Lung cancer has been the leading cause of cancer-related mortality in the past decade among men and women worldwide, with an estimated 160,000 deaths due to lung cancer predicted in 2014 for the USA alone [1–3]. Lung cancer has a 5-year overall survival rate of just 16 % for all stages, and survival has only minimally improved in the last three decades despite advances in surgical techniques, radiation, and systemic treatment [4]. Lung cancer is classified based on histology as either small-cell lung cancer (SCLC, 15 % of cases) or non-small-cell lung cancer (NSCLC, 85 % of cases), with NSCLC further subtyped into squamous cell carcinoma, large-cell carcinoma, and adenocarcinoma [3, 5]. Surgery is the mainstay treatment in operable patients with NSCLC; however, only 15 % of patients are diagnosed at an early stage [4]. For SCLC, chemotherapy remains the cornerstone for therapy of this disease [6, 7].
In metastatic NSCLC, chemotherapy has been the standard treatment for a long time, with different platinum-based chemotherapy regimens only offering modest clinical benefit, with response rates (RR) between 17 and 32 % and median overall survival between 7.4 and 9.9 months [8–11]. These results have only been marginally improved by the addition of bevacizumab, an antivascular endothelial growth factor antibody, to chemotherapy in patients with nonsquamous histological subtypes [12].
In the past decade, a deeper understanding of the molecular abnormalities present in NSCLC that define disease subsets has been achieved, particularly in adenocarcinoma. This evolving genomic classification of NSCLC has helped to guide therapeutic strategies. The discovery of oncogenic drivers led to the design of therapies targeting tumors harboring specific gene alterations that cause aberrant signaling and proliferation [13]. The majority of these oncogenic drivers are tyrosine kinases, proteins that are key regulators of cellular proliferation, growth, and survival. Constitutive activation of these kinases through mutations can be targeted using small-molecule tyrosine kinase inhibitors (TKIs) or antibodies directed at receptor tyrosine kinases (RTKs), which have provided greater clinical benefit toward tumors that are oncogene-dependent [14–16].
Currently, most patients with NSCLC, particularly adenocarcinomas, routinely undergo a biopsy for molecular profiling of their tumor. The molecular analysis of the biopsy helps to provide insight into the molecular and histological characteristics of the disease to guide treatment decisions. Testing for EGFR mutations and anaplastic lymphoma kinase (ALK) fusions, regardless of sex, race, smoking history, or other risk factors, are prioritized over other molecular predictive tests [17–22]. This paradigm for targeted therapies in oncology allows the right patient to receive the most active therapy, while sparing those that are unlikely to benefit from the cost and potential morbidity associated with unresponsive therapeutic interventions. However, despite the clinical success of targeted therapies in NSCLC and other indications, responses typically last less than a year as tumors inevitably acquire resistance to such targeted therapies [5, 16]. Here, we review some of the known oncogenic drivers (EGFR, ALK, ROS1, RET, etc.) in NSCLC, its targeted therapies, and the acquired resistance to these therapeutic interventions that are observed in patients (Table 1).
Table 1.
Molecular targets and therapies in lung adenocarcinoma
| Molecular targets | Prevalence (%) | More commonly associated patient characteristics | Targeted therapies |
|---|---|---|---|
| RAS | 30 | Former/current smokers | None |
| EGFR | 10–18 Caucasian; 40–55 Asian | East Asian, female, never smokers | Erlotinib, gefitinib, afatinib |
| ALK | 3–7 | Young, never smokers | Crizotinib, LDK378 |
| ROS | 1–2 | Young, never smokers | Crizotinib |
| RET | 1–2 | Never smokers | Vandetanib, cabozantinib |
| PIK3CA | 2 | Concurrent with other oncogenic drivers | GDC-0941, BKM120 |
| BRAF | 3–5 | Former/current smokers | Debrafenib |
| HER2 | 1–4 | Female, never smokers | Trastuzumab, pertuzumab, afatinib |
| RIT | 2b | Not available | GDC-0941 |
| NTRK1 | 3b | Female, never smokers | Crizotinib |
| MET | 11 | Mutually exclusive with EGFR mutations | Crizotinib, onartuzumab, tivantinib |
| FGFR1 | 1–3 | Male, smokersa | AZD4547 |
EGFR
The epidermal growth factor receptor (EGFR) belongs to a family of RTKs that include ERBB2, ERBB3, and ERBB4 (also known as HER2, HER3, and HER4). Activating mutations in EGFR occur within the kinase domain in exons 18 to 21 and are mutually exclusive to KRAS mutations, which are the most prevalent oncogenic driver within NSCLC (Fig. 1). EGFR mutations are almost exclusively found in adenocarcinoma histology. Particular patient characteristics are more commonly associated with EGFR mutations: East Asian origin (40–55 %), female gender (22 %), and never-smokers (42 %) [26–29]. In Caucasians, the overall incidence of EGFR mutations is 10–15 %. Initially, the discovery that EGFR was overexpressed in many epithelial tumors galvanized the design for anti-EGFR inhibitors. Gefitinib, erlotinib, and afatinib are small-molecule TKIs that can penetrate the cell membrane and bind to the ATP pocket of the EGFR kinase domain, whereas cetuximab is an anti-EGFR antibody that prevents ligand-induced receptor dimerization [13].
Fig. 1.
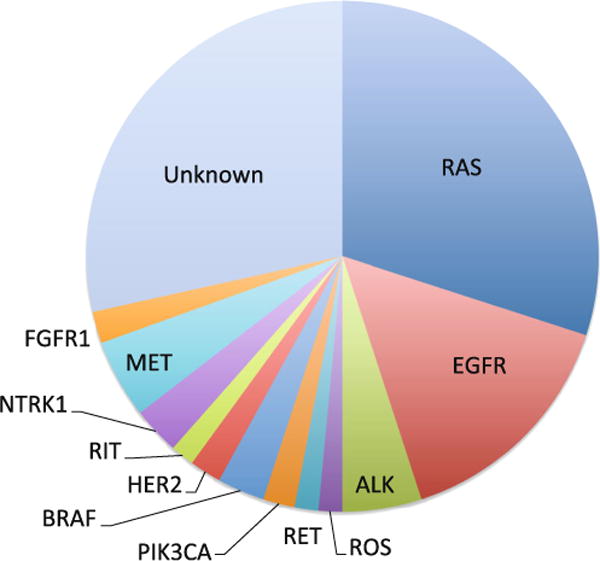
The pie chart summarizes the prevalence of genetic mutations in adenocarcinoma of the lung
In 2004, it was discovered that certain mutations in the kinase domain of EGFR lead to increased growth factor signaling and are associated with increased response rate of NSCLC patients to EGFR TKI treatment [30, 31]. After a large number of studies and investigations of copy number and expression of EGFR as potential biomarkers of activity of EGFR inhibitors, it became clear that the presence of activating mutations in EGFR (in particular exon 19 deletions and exon 21 substitutions such as L858R, which represent over 90 % of these activating mutations) was the best predictor of response [30, 31]. These findings eventually led to phase III studies that determined that NSCLC patients harboring EGFR-activating mutations achieved superior overall response rates (ORR) and progression-free survival (PFS) with EGFR TKIs compared to chemotherapy alone in the first line treatment of advanced NSCLC [32, 33]. The IPASS study, which compared gefitinib to standard chemotherapy with carboplatin and paclitaxel in East Asian patients with adenocarcinoma and a light or never smoking history, concluded the superiority of gefitinib as initial treatment [32]. The subsequent studies investigated the same question in patients harboring EGFR-activating mutations (Table 2). Most of these studies were conducted in Asia, where the incidence of EGFR mutations is 3–4-fold higher than in the USA or Europe [29]; the EURTAC study, conducted mainly in Spain and a few other European countries, was able to replicate the results obtained in Asia [38]. All of these studies provided strong evidence for an increased response rate and increased PFS but not overall survival. The crossover to EGFR TKIs that occurred in patients who received chemotherapy first possibly explains the lack of increased overall survival. Also, of interest is that the overall survival in studies selecting for EGFR-positive mutation status is superior to that of studies with EGFR-negative populations (about 22 vs 12 months) indicating that the presence of EGFR mutations also has prognostic significance [34]. In part, this increased survival may be also due to the reduced incidence of comorbidities in this mainly never-smoker population.
Table 2.
Phase III trials comparing first-line treatment of EGFR TKIs to chemotherapy on patients with EGFR-positive mutation status
| Study (reference) | Treatments | PFS (months) | OS (months) |
|---|---|---|---|
| IPASS [32, 34]a | Gefitinib vs carbo/paclitaxel | 9.5 vs 6.3 | 21.6 vs 21.9 |
| First-SIGNAL [35] | Gefitinib vs cis/gem | 8 vs 6.3 | 27.2 vs 25.6 |
| WJTOG3405 [36] | Gefitinib vs cis/doce | 9.2 vs 6.3 | 36 vs 39 |
| NEJSG [37] | Gefitinib vs carbo/paclitaxel | 10.8 vs 5.4 | 30.5 vs 23.6 |
| EURTAC [38] | Erlotinib vs chemotherapy | 9.7 vs 5.2 | 19.3 vs 19.5 |
| OPTIMAL [39] | Erlotinib vs carbo/gem | 13.1 vs 4.6 | 22.7 vs 28.9 |
| LUX-Lung 3 [33]** | Afatinib vs cis/pemetrexed | 11.1 vs 6.9 | 16.6 vs 14.8b |
PFS progression-free survival, OS overall survival
IPASS study included East Asians with advanced pulmonary adenocarcinoma who were light or never smokers; the data above represents the EGFR-positive subgroup of the randomized study
Preliminary OS data (25th percentile), not statistically significant
Despite the impressive high response rates to EGFR TKIs in EGFR-mutant NSCLC, none of the patients with advanced NSCLC can be cured with these treatments, and prolonged clinical benefit is limited due to acquired drug resistance. Acquired resistance can be defined as progression of the disease after responding in a patient harboring an EGFR-activating mutation while on continuous treatment with single-agent EGFR TKI [40].
The increasing use of repeat biopsies at the time of progression has offered the opportunity to study the mechanisms underlying resistance to EGFR TKIs as well as other drugs. Knowing the mechanism of resistance may have implication for the next treatment and potential participation in clinical trials of novel agents that are targeting the ensuing genetic abnormality [17]. The most common resistance mechanism to erlotinib and gefitinib is the development of the secondary mutation, T790Min EGFR exon 20, which occurs in approximately 50–60 % of patients [41–43]. This threonine-to-methionine mutation abrogates the inhibitory activity of the drug by inducing steric hindrance in the ATP-binding pocket of EGFR and also increases the binding affinity of ATP [44]. Resistance may arise from an undetectable minor T790M clone that is present in a small fraction of tumor cells prior to EGFR TKI treatment, which then gets propagated upon erlotinib or gefitinib therapy [45]. Other secondary mutations in EGFR have been reported in erlotinib- and gefitinib-relapsed patients. D761Y mutation in EGFR was found in a metastatic brain lesion of a patient who acquired resistance to gefitinib [46]; the EGFR secondary mutation T854A was reported in another case [47], and one patient who was on gefitinib treatment for 40 months acquired a L747S secondary mutation in EGFR [48]. These secondary mutations, however, occur at much less frequency than the T790M mutation.
Of interest, second (afatinib and dacomitinib) and third generation EGFR TKIs (CO-1686 and AZD9291) have been developed to increase efficacy and cope with acquired secondary mutations which first generation TKIs confer; these inhibitors appear active on the T790M mutations while sparing wild-type EGFR in preclinical models [33, 49–51]. Afatinib gained FDA approval in mid-2013 based on the LUX-Lung 3 trial [33]. The activity of second generation inhibitors against the T790M mutation in clinical studies has been disappointing, primarily due to the subtherapeutic dose administered because of the dose-limiting toxicities by on-target wild-type EGFR inhibition, which commonly leads to skin rash and diarrhea [52]. On the contrary, CO-1686 and AZD9291 have already demonstrated significant activity in patients with EGFR-mutant advanced NSCLC that failed prior EGFR TKIs and do not appear to have the typical side effects related to inhibition of wild-type EGFR, which are the most common side effects of the first generation EGFR TKIs (gefitinib and erlotinib) which have a weak effect on wild-type EGFR [50, 51]. However promising, the data on third-generation EGFR inhibitors CO-1686 and AZD9291 are preliminary, and we are still awaiting the full results of the phase I clinical trials [51, 52].
In addition to secondary EGFR mutations, activation of bypass pathways is a frequently occurring theme in the mechanisms of resistance to RTK-targeted inhibition. Amplification of the MET oncogene was initially observed in around 20 % of EGFR-mutant NSCLC patients who acquire resistance to EGFR TKIs, but its real incidence is probably a more modest 5 % [5]. This amplification occasionally coexists with the T790M secondary mutation [53, 54]. MET oncogene encodes a RTK, which activates the phosphoinositide-3-kinase (PI3K)/Akt signal transduction pathway independent of EGFR through phosphorylation of ERBB3, creating a bypass mechanism that promotes resistance to EGFR TKIs. Activation of MET can also be driven by increased production of its ligand, hepatic growth factor (HGF), which is sufficient to activate downstream signaling and to promote resistance to EGFR TKIs [55, 56].
HER2 amplification, by fluorescence in situ hybridization (FISH), is also observed in 12 % of patient tumor samples that acquired resistance to EGFR TKIs, compared to 1 % of EGFR TKI naive tumor samples. HER2 amplification was mutually exclusive of T790M secondary mutation in this study [57]. AXL kinase, another RTK, has also been shown to drive resistance to EGFR TKIs either through overexpression of AXL or upregulation of its ligand GAS6 [58]. AXL activation confers drug resistance through the mitogen-activated protein kinase (MAPK), PI3K/Akt, and NFκB signaling pathways. Twenty percent of tumor samples from patients that acquired resistance to EGFR TKIs showed increased expression of AXL, and this overexpression was mutually exclusive with MET amplification [58]. Mutations in the PIK3CA gene encoding the p110α catalytic subunit of PI3K were observed in 5 % of tumor samples that acquired resistance to EGFR TKIs [43]. However, PI3K-activating mutations usually coexist with other oncogenic drivers, including EGFR, KRAS, or ALK [59]. The effect of concurrent mutations on acquired resistance to EGFR TKIs remains to be explored.
Histological transformation of NSCLC to SCLC occurs in 14 % of patients; none of whom demonstrated EGFR T790M mutation or MET amplification; however, one patient developed a PIK3CA mutation [43]. These patients were sensitive to standard SCLC chemotherapy. In another study of tumor specimens at the time of acquired resistance, only 3 % of patients had transformed to SCLC histology [60].
The notion that MET amplification, AXL kinase activation, and PIK3CA mutations are involved in the mechanisms of acquired resistance offers the rationale of using combinatorial therapies targeting EGFR and MET, or EGFR and AXL kinase, or EGFR and PIK3CA to extend the survival of EGFR-mutant NSCLC patients [52, 53, 58]. Various clinical trials are currently designed to target the moment of occurrence of resistance with a new agent in combination with EGFR TKI (NCT01887886 and NCT01487265). However, limitations involving overlapping toxicities in combination therapy can lead to suboptimal dosing of each agent [52].
ALK
Anaplastic lymphoma kinase (ALK) is a RTK that belongs to the insulin receptor family on the basis of their similarities in the kinase domain [61]. In 3–7 % of NSCLC, ALK fusion proteins occur, causing constitutive activation of ALK and mediating oncogenesis through the MAPK, JAK/STAT, and PI3K/Akt signal transduction pathways [62–64]. ALK gene rearrangements can occur through different fusion partners, including KIF5B-, TFG-, KLC1-, PTPN3-, STRN-, and the most common, EML4-ALK [61]. At least seven variants of EML4-ALK have been identified based on the number of exons of EML4 fused to ALK kinase domain [65]. The EML4-ALK fusions generated by a small inversion within the short arm of chromosome 2 typically occur in young never-smokers, are mutually exclusive of EGFR and KRAS mutations and have brought forth new treatment options for this subset of NSCLC [66]. Patients are screened for the presence of ALK-positive NSCLC using an FDA-approved break-apart FISH method to detect for the presence of ALK rearrangement [67]. Crizotinib, an ALK, ROS1, and MET inhibitor, is a first-in-class FDA-approved targeted therapy for NSCLC patients harboring ALK fusion protein. It was shown to be clinically efficacious in ALK-positive NSCLC with a response rate close to 60 % [68], and when compared to chemotherapy in a phase 3 open-label trial in patients who had failed one prior platinum-based regimen, the median PFS and RR in the crizotinib group was 7.7 months and 65 %, respectively, versus 3.0 months and 20 % [69]. However, in this phase 3 trial, there was no improvement in overall survival probably due to the crossover of patients from chemotherapy to crizotinib [69]. Crizotinib therapy was shown to be associated with improved overall survival compared with crizotinib-naive patients in a retrospective analysis [70].
Similarly to EGFR TKIs, acquired resistance to crizotinib can occur via secondary mutations in ALK protein, which are observed in 22–36 % of ALK-positive NSCLC patients [71, 72]. The most common secondary mutation in ALK is the gatekeeper L1196M mutation, analogous to the T790M gatekeeper mutation in EGFR [73]. The leucine-to-methionine substitution causes steric hindrance in the binding pocket of ALK, thus preventing crizotinib from binding effectively. Additionally, other secondary mutations in ALK have been observed in crizotinib-resistant patient samples, including T1151ins, G1202R, S1206Y, L1152R, and G1269A among others [72–75]. Although the mechanism in which specific secondary mutations confer resistance to crizotinib is unknown, it is likely that the mutations either reduce the binding affinity of crizotinib to ALK or increase the ATP affinity for the mutant ALK, or both.
Second generation ALK inhibitors that are more potent than crizotinib, such as ceritinib (LDK378) and alectinib (CH5424802), are currently under clinical investigation (Table 3). In a subset of 80 crizotinib-resistant ALK-positive patients, ceritinib treatment resulted in a 56 % ORR [77]. Alectinib shows antitumor activity against the gatekeeper L1196M mutation in ALK that frequently arises in acquired resistance to crizotinib [83]. In a single-arm, open-label phase II study, alectinib was given to ALK-inhibitor-naive patients, of whom 93.5 % achieved an objective response [78]. Alectinib achieved a 54 % response rate in 37 evaluable crizotinib-resistant patients (preliminary data) [79]. These studies validate the necessity for second and third generation TKIs to overcome acquired resistance or to delay the development of resistance. However, multiple minor resistant populations within a tumor mediated by different oncogenic drivers may exist; thus, the efficacy of a potent single-agent therapy may be limited due to the intratumor heterogeneity.
Table 3.
ALK TKIs in the clinic
| ALK inhibitors (reference) | ROS1 activity | Status | NCT identifier |
|---|---|---|---|
| Crizotinib [68, 69, 76] | Yes | Approved for ALK-positive NSCLC; investigational for ROS1 | NCT00585195 |
| Ceritinib (LDK378) [77] | Yes | Phase II/III | NCT01828099, NCT01828112 |
| Alectinib (CH5424802) [78, 79] | No | Phase I/II | NCT01588028 |
| AP26113 [80] | Yes | Phase I/II | NCT01449461 |
| ASP3026 [81] | Yes | Phase I | NCT01284192 |
Modified from Brahmer et al. [82]
Furthermore, mechanisms of crizotinib resistance can also occur through copy number gain of the ALK gene, either alone or together with secondary mutations in ALK [71, 72]. Other mechanisms that have been observed in patients include activation of bypass signaling involving KIT amplification and increased phospho-EGFR levels due to elevated ligand levels [71]. One patient had both KIT amplification and a secondary G1202R ALK mutation, further confirming the notion that multiple mechanisms of resistance can be activated in the same patient [71]. EGFR-activating mutations have also been observed in patients who acquired resistance to crizotinib, as well as mutations in the KRAS oncogene [72]; however, in another study of 25 criztonib-resistant ALK-positive patients, these mutations were not evident [66].
ROS1
ROS1, remotely related to the ALK and insulin receptor family, is another RTK that can form oncogenic fusion proteins to define a new molecular class of NSCLC. ROS1 gene fusions are observed in around 1–2 % of NSCLC patients; all of whom have adenocarcinoma and are typically young never-smokers [84, 85]. ROS1 translocations does not overlap with other oncogenic drivers and can rearrange with several different partners, including SDC4, CD74, and GOPC, while still harboring the kinase domain that is constitutively active [84, 86]. CD74-ROS1, observed in 30 % of ROS translocations, was found to mediate invasiveness through phosphorylation of extended synaptotagmin-like protein, E-Syt1 [87]. Given that the ROS1 and ALK kinases share high sequence homology with 77 % at the ATP-binding site [88], crizotinib has shown to be efficacious in patients harboring ROS1 translocations [76, 85], with a 56 % RR [89]. However, acquired resistance to crizotinib inevitably occurs. The glycine-to-arginine substitution at codon 2032 in the ROS1 kinase domain has been observed in a crizotinib-treated CD74-ROS1-positive NSCLC patient [90]. In another case, a patient with SDC4-ROS1 NSCLC experienced disease progression after successful treatment with crizotinib, and the resistant biopsy suggested an oncogenic switch mediated by EGFR activation as the potential mechanism of resistance [91]. Ongoing clinical trials are testing second- and third-generation ALK inhibitors ceritinib (LDK378), AP26113, and ASP3026 in ROS1-positive tumors (NCT01964157, NCT01449461, and NCT01284192).
RET
Another oncogenic driver in NSCLC is the fusion of the RTK RET, and similar to ROS1 gene rearrangements, RET gene fusions are mutually exclusive of other driver mutations and are found in around 1–2 % of NSCLC patients, typically never-smokers [84, 92, 93]. RET fusions were originally found in papillary thyroid cancers and could fuse with several genes including KIF5B and lead to constitutive activation of RET kinase [86, 92]. RET-rearranged NSCLC all show moderate to well differentiated tumor cells, and histopathological features are common between patient biopsies including positive staining for thyroid transcription factor 1 (TTF-1) and napsin A and negative staining for thyroglobulin [92]. Tumor cell nuclei also displayed prominent intranuclear inclusions [86]. RET can be targeted with TKIs such as vandetanib and cabozantinib [3]. In the preliminary data of a phase II trial of cabozantinib (NCT01639508), two of three NSCLC patients with RET fusion-positive confirmed partial responses (one with a novel TRIM33-RET fusion), while the third patient had prolonged disease stabilization [94].
MET
In addition to the MET amplifications in EGFR TKI-treated EGFR-mutant NSCLC patients, high copy number of MET gene has been observed in up to 11 % of NSCLC patients (resected from primary tumor site in lung) and correlated with shorter survival time [95]. However, different techniques are used to determine MET gene amplification and overexpression, including FISH and immunohistochemistry (IHC) analysis, which may lead to discordant results. For example, using a FISH assay to determine MET gene amplification will not clearly differentiate between real MET gene amplification and polysomy. Thus, it remains critical to standardize and optimize for real MET gene amplification or MET overexpression to better select and define the patient population that will benefit from anti-MET therapies [95, 96].
Various therapies targeting the HGF/MET pathway are in the clinic, including crizotinib, onartuzumab, and tivantinib [3]. However, multiple studies have cast doubt on tivantinib’s ability to selectively inhibit MET to induce cell death, including tivantinib’s potential to inhibit microtubule dissociation as one of its off-target effects [97, 98]. In one case, an ALK-negative patient with de novo MET-amplified NSCLC achieved a rapid and durable response to crizotinib therapy, exemplifying crizotinib’s ability to effectively inhibit MET and validating METas an oncogenic driver [99]. Onartuzumab is a humanized monovalent monoclonal antibody directed at MET to prevent HGF ligand binding [100]. In a randomized phase II trial comparing onartuzumab and erlotinib versus placebo and erlotinib in MET-positive NSCLC patients (confirmed by IHC), patients treated with onartuzumab showed improved PFS and OS. Clinical outcomes were worse in MET-negative patients treated with onartuzumab plus erlotinib than control, highlighting the importance of companion diagnostic testing [100]. Preclinical models suggest that MET and KRAS gene amplifications along with activation of EGFR pathway serve as mechanisms of acquired resistance to MET TKIs [101, 102].
HER2
The initial discovery of EGFR mutations in NSCLC galvanized subsequent interest in finding other oncogenic drivers. HER2 kinase domain mutations were present in around 1–4 % of primary lung tumors [103–105]. Mutations in exon 20 of HER2 are mutually exclusive with KRAS (except for one patient) and EGFR mutations, observed predominantly in female never-smokers with adenocarcinoma subtype [105]. Several HER2-targeted therapies have been developed, including antibodies directed at HER2 like trastuzumab and pertuzumab and small-molecule TKIs like afatinib and lapatinib [105]. Recently, trastuzumab has been shown to control disease at a rate of 96 % (n=15) and is currently being tested in clinical trials for patients with HER2 mutation, amplification, or IHC-positive NSCLC (NCT00004883 and NCT00758134) [105]. Lapatinib, however, has been unsuccessful in demonstrating clinical benefit to NSCLC patients with HER2 gene amplification or kinase mutations [106].
BRAF
BRAF is a serine/threonine kinase downstream of KRAS in the MAP kinase signaling pathway, and mutational activation of BRAF occurs in approximately 60 % of human melanoma [107]. In particular, the V600E BRAF mutation accounts for over 90 % of BRAF mutations in melanoma, and targeted therapies, such as vemurafenib, have shown over 50 % overall response rate for V600EBRAF-driven advanced melanomas [108, 109]. In lung adenocarcinomas, 3–5 % of patients have missense mutations in the BRAF gene with a little over 50 % of the BRAF mutations represented by V600E; other BRAF mutations included G469A and D594G [108]. These patients were typically former or current smokers [108, 110]. Debrafenib is a selective inhibitor of mutant BRAF that has demonstrated clinical efficacy in a phase II study in previously treated BRAF V600E-mutated NSCLC patients. Preliminary results show a RR of 54 %, validating BRAF as an actionable target in NSCLC [111]. In one case, a BRAF-mutant NSCLC patient treated with debrafenib acquired resistance through an oncogenic mutation in KRAS [112]. NSCLC patients with non-BRAF V600E mutations are unlikely to benefit from V600E specific BRAF inhibitors such as debrafenib; thus, inhibiting downstream targets such as MEK may offer clinical benefit [113].
PI3K
Activation of phosphatidylinositol 3-kinase (PI3K), an intracellular membrane-bound kinase, regulates cell growth, proliferation, and survival. Activating mutations in PIK3CA gene, which encodes the catalytic subunit of PI3K, are observed in 1–4 % of NSCLC patients of primarily squamous cell carcinoma histology [113]. PIK3CA mutations cause aberrant signaling through the PI3K/Akt/mTOR pathway; however, they often coexist with other oncogenic drivers such as EGFR, KRAS, or ALK in about 2 % of lung adenocarcinomas [59]. Ongoing phase II trials are being conducted in NSCLC patients to examine combinations of PI3K inhibitors with chemotherapy or other targeted agents (NCT01297491, NCT01493843, and NCT01487265).
Other oncogenic drivers
RAS oncogenes (KRAS and NRAS) are oncogenic drivers observed in about 30 % of lung cancer, mainly adenocarcinomas; however, there have been a lack of clinically efficacious drugs targeting RAS in the clinic [3, 25]. Clinical trials of targeted therapies inhibiting proteins downstream of RAS (i.e., MEK inhibitors) are ongoing for NSCLC patients with a KRAS-positive mutation (NCT01362296 and NCT01229150). Recently, gene fusions that include the kinase domain of NTRK1 have been observed in 3.3 % of lung cancer patients with otherwise unknown oncogenic alterations, and this gene rearrangement can transform cells in vivo [24]. One patient with MPRIP-NTRK1 fusion gene consented to treatment with crizotinib and achieved minor radiographic response before the disease progressed after 3 months [24]. Another study has observed activating mutations in the small GTPase gene RIT1 in 2 % of lung adenocarcinoma patients with otherwise unknown oncogenic mutations, and mutated RIT1 induces transformation of cells into the malignant phenotype in vitro and in vivo [25]. Mutations in DDR2 kinase gene occur in around 4 % of lung squamous cell carcinomas, and potentially could be sensitive to the multi-targeted TKI dasatinib in patients harboring this mutation [114]. Gene amplification of FGFR1, a membrane-bound RTK, occurs in 21 % of squamous cell carcinoma and 1–3 % of adenocarcinomas [113, 115]. FGFR1 amplification in lung adenocarcinomas tends to be more common in male heavy smokers; however, this analysis of patient characteristics was not statistically significant [23]. Several FGFR TKIs are under clinical investigation, such as AZD4547, a selective inhibitor of FGFR1/2/3 that is in a proof-of-concept phase II clinical trial (NCT01795768) in patients with FGFR1-amplified squamous cell carcinoma of the lung [3].
Targeted immunotherapy and future direction
Immunotherapies are achieving promising results in the clinic and have emerged not only as an effective way to inhibit oncogenic proteins or survival factors but also to enrich the power of the immune system [116]. The interaction between ligand PD-L1 and PD-1 on activated T cells plays a crucial role for tumor evasion [3]. High levels of PD-L1 on tumor cells have been reported in NSCLC patients and targeting PD-1 and PD-L1 with nivolumab and MPDL3280A, respectively, has demonstrated antitumor activity in clinical trials (NCT01673867, NCT01721759, NCT01846416, NCT01903993, and NCT01375842) [3, 82, 116, 117]. Immunotherapies may be especially beneficial to heavy smokers who accumulate many somatic mutations, where one single driver mutation may not exist such as squamous cell carcinomas and SCLCs. In addition, acquired mutations to TKIs may lead to the upregulation of PD-L1 in tumor cells. In this regard, immunotherapies may help to address the challenge of acquired resistance to TKIs. The re-biopsy of NSCLC tumors at recurrence is becoming increasingly critical to understand the underlying molecular changes causing acquired resistance to treatment. Molecular profiling of the tumor provides clinicians a rational therapeutic strategy in the next line of treatment to overcome resistance. However, the feasibility of obtaining quality biopsies in lung tumors remains a great challenge due to tumor location, potential harm to the patient, and disease status among others [3].
Technological advances in sequencing may help to overcome this challenge. Next generation and exome sequencing of tumors without known oncogenic drivers helped to discover new oncogenic drivers such as NTRK1 and RIT1, respectively [24, 25]. In addition, advances in next-generation deep sequencing (NGS) may potentially detect mechanisms of resistance through noninvasive means before the clinical diagnosis of progressive disease is observed [52]. The noninvasive analysis of acquired resistance through exome-wide sequencing of circulating tumor DNA has yielded comparable results, for example, the detection of T790M mutation in EGFR of a gefitinib-resistant NSCLC patient [118]. In addition, with NGS, one can also identify minor populations of resistance clones in a heterogeneous tumor. Genomic testing of NSCLCs alongside histological testing will allow for candidate patients to be identified for optimal therapeutic intervention, ultimately reducing the risk of clinical trial failure and increasing patient benefit through personalized medicine.
Acknowledgments
This work was supported by the Lombardi Comprehensive Cancer Center at Georgetown University.
Footnotes
Conflict of interest: The authors declare that they have no conflict of interests.
Contributor Information
Arjan Gower, Georgetown University, Washington, DC, USA.
Yisong Wang, Georgetown University, Washington, DC, USA.
Giuseppe Giaccone, Georgetown University, Washington, DC, USA.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Shames D, Wistuba I. The evolving genomic classification of lung cancer. J Pathol. 2014;232:121–133. doi: 10.1002/path.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Cancer Society. Cancer facts and figures 2012. 2012 [Accessed January 13, 2014]. Available from: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-031941.pdf.
- 5.Hrustanovic G, Lee B, Bivona T. Mechanisms of resistance to EGFR targeted therapies. Cancer Biol Ther. 2013;14:304–314. doi: 10.4161/cbt.23627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spiro S, Silvestri G. One hundred years of lung cancer. Am J Respir Crit Care Med. 2005;172:523–529. doi: 10.1164/rccm.200504-531OE. [DOI] [PubMed] [Google Scholar]
- 7.Metro G, Cappuzzo F. Emerging drugs for small-cell lung cancer. Expert Opin Emerg Drugs. 2009;14:591–606. doi: 10.1517/14728210903206983. [DOI] [PubMed] [Google Scholar]
- 8.Kelly K, Crowley J, Bunn P, Jr, Presant C, Grevstad P, Moinpour C, Ramsey S, Wozniak A, Weiss G, Moore D, et al. Randomized phase III trial of paclitaxel plus carboplatin versus vinorelbine plus cisplatin in the treatment of patients with advanced non-small-cell lung cancer: a Southwest Oncology Group trial. J Clin Oncol. 2001;19:3210–3218. doi: 10.1200/JCO.2001.19.13.3210. [DOI] [PubMed] [Google Scholar]
- 9.Schiller J, Harrington D, Belani C, Langer C, Sandler A, Krook J, Zhu J, Johnson D. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 10.Scagliotti G, De Marinis F, Rinaldi M, Crino L, Gridelli C, Ricci S, Matano E, Boni C, Marangolo M, Failla G, et al. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J Clin Oncol. 2002;20:4285–4291. doi: 10.1200/JCO.2002.02.068. [DOI] [PubMed] [Google Scholar]
- 11.Bonanno L, Favaretto A, Rosell R. Platinum drugs and DNA repair mechanisms in lung cancer. Anticancer Res. 2014;34:493–502. [PubMed] [Google Scholar]
- 12.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson D. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 13.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;11:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 15.Weinstein I. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 16.Niederst M, Engelman J. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal. 2013;6:re6. doi: 10.1126/scisignal.2004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas A, Rajan A, Lopez-Chavez A, Wang Y, Giaccone G. From targets to targeted therapies and molecular profiling in non-small cell lung carcinoma. Ann Oncol. 2013;24:577–585. doi: 10.1093/annonc/mds478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim E, Herbst R, Wistuba I, Lee J, Blumenschein G, Jr, Tsao A, Stewart D, Hicks M, Erasmus J, Jr, Gupta S, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardarella S, Ortiz T, Joshi V, Butaney M, Jackman D, Kwiakowski D, Yeap B, Jänne P, Linderman N, Johnson B. The introduction of systematic genomic testing for patients with non-small-cell lung cancer. J Thorac Oncol. 2012;12:1767–1774. doi: 10.1097/JTO.0b013e3182745bcb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linderman N, Cagle P, Beasley M, Chitale D, Dacic S, Giaccone G, Jenkins R, Kwiatkowski D, Saldivar J, Squire J, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn. 2013;15:415–453. doi: 10.1016/j.jmoldx.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Linderman N, Cagle P, Beasley M, Chitale D, Dacic S, Giaccone G, Jenkins R, Kwiatkowski D, Saldivar J, Squire J, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8:823–859. doi: 10.1097/JTO.0b013e318290868f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linderman N, Cagle P, Beasley M, Chitale D, Dacic S, Giaccone G, Jenkins R, Kwiatkowski D, Saldivar J, Squire J, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch Pathol Lab Med. 2013;137:828–860. doi: 10.5858/arpa.2012-0720-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadgeel S, Chen W, Cote ML, Bollig-Fischer A, Land S, Schwartz AG, Bepler G. Fibroblast growth factor receptor 1 amplification in non-small cell lung cancer by quantitative real-time PCR. PLoS One. 2013;8:e79820. doi: 10.1371/journal.pone.0079820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, Mahale S, Davies K, Aisner D, Pilling A, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19:1469–1472. doi: 10.1038/nm.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berger A, Imielinski M, Duke F, Wala J, Kaplan N, Shi G, Andres D, Meyerson M. Oncogenic RIT1 mutations in lung adenocarcinoma. Oncogenesis. 2014 doi: 10.1038/onc.2013.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weir B, Woo M, Getz G, Perner S, Ding L, Beroukhim R, Lin W, Province M, Kraja A, Johnson L, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber B, Hager H, Sorensen B, McCulloch T, Mellemgaard A, Khalil A, Nexo E, Meldgaard P. EGFR mutation frequency and effectiveness of erlotinib: a prospective observational study in Danish patients with non-small cell lung cancer. Lung Cancer. 2014;83:224–230. doi: 10.1016/j.lungcan.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 28.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, Brzostowski EB, Riely GJ, Kris MG, Zakowski MF, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res. 2012;18:6169–6177. doi: 10.1158/1078-0432.CCR-11-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch T, Bell D, Sordella R, Gurubhagavatula S, Okimoto R, Brannigan B, Harris P, Haserlat S, Supko J, Haluska F, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 31.Paez J, Jänne P, Lee J, Tracy S, Greulich H, Gabriel S, Herman P, Kaye F, Lindeman N, Boggon T, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 32.Mok T, Wu Y, Thongprasert S, Yang C, Chu D, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 33.Sequist L, Yang J, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater S, Orlov S, Tsai C, Boyer M, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 34.Fukuoka M, Wu Y, Thongprasert S, Sunpaweravong P, Leong S, Sriuranpong V, Chao T, Nakagawa K, Chu D, Saijo N, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS) J Clin Oncol. 2011;29:2866– 2874. doi: 10.1200/JCO.2010.33.4235. [DOI] [PubMed] [Google Scholar]
- 35.Han JY, Park K, Kim SW, Lee DH, Kim HY, Kim HT, Ahn MJ, Yun T, Ahn JS, Suh C, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trail in never-smokers with adenocarcinoma of the lung. J Clin Oncol. 2012;10:1122–1128. doi: 10.1200/JCO.2011.36.8456. [DOI] [PubMed] [Google Scholar]
- 36.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of epidermal growth factor receptor (WJTOG3405): an open label, randomized phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 37.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 38.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 39.Zhou C, Wu Y, Chen G, Feng J, Liu X, Wang C, Zhang S, Wang J, Zhou S, Ren S, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 40.Jackman D, Pao W, Riely G, Engelman J, Kris M, Jänne P, Lynch T, Johnson B, Miller V. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–360. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobayashi S, Boggon T, Dayaram T, Jänne P, Kocher O, Meyerson M, Johnson B, Eck M, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 42.Pao W, Miller V, Politi K, Riely G, Somwar R, Zakowski M, Kris M, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sequist L, Waltman B, Dias-Santagata D, Digumarthy S, Turke A, Fidias P, Bergethon K, Shaw A, Gettinger S, Cosper A, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;75:ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yun C, Mengwasser K, Toms A, Woo M, Greulich H, Wong K, Meyerson M, Eck M. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, Suehisa H, Ouchida M, Aoe K, Aoe M, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66:7854–7858. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 46.Balak M, Gong Y, Riely G, Somwar R, Li A, Zakowski M, Chiang A, Yang G, Ouerfelli O, Kris M, et al. NovelD761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–6501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 47.Bean J, Riely G, Balak M, Marks J, Ladanyi M, Miller V, Pao W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–7525. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costa D, Schumer S, Tenen D, Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol. 2008;26:1182– 1184. doi: 10.1200/JCO.2007.14.9039. [DOI] [PubMed] [Google Scholar]
- 49.Engelman J, Zejnullahu K, Gale C, Lifshits E, Gonzales A, Shimamura T, Zhao F, Vincent P, Naumov G, Bradner J, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 50.Water A, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ranson M, Pao W, Kim D, Kim S, Ohe Y, Felip E, Planchard D, Ghiorghiu S, Cantarini M, Jänne P. LATE BREAKING ABSTRACT: preliminary results from a Phase I study with AZD9291: an irreversible inhibitor of epidermal growth factor receptor (EGFR) activating and resistance mutations in non-small-cell lung cancer (NSCLC) Eur Cancer Congr Abstr. 2013;33 [Google Scholar]
- 52.Chong C, Jänne P. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engelman J, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park J, Lindeman N, Gale C, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 54.Bean J, Brennan C, Shih J, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turke B, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, et al. Hepatocyte growth factor induces gefitinib resistance of lung ade-nocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 57.Takezawa K, Pirazzoli V, Arcila M, Nebhan C, Song X, de Stanchina E, Ohashi K, Janjigian Y, Spitzler P, Melnick M, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaft J, Arcila M, Paik P, Lau C, Riely G, Pietanza M, Zakowski M, Rusch V, Sima C, Ladanyi M, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther. 2012;11:485–491. doi: 10.1158/1535-7163.MCT-11-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu H, Arcila M, Rekhtman N, Sima C, Zakowski M, Pao W, Kris M, Miller V, Ladanyi M, Riely G. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hallberg B, Palmer R. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13:685–700. doi: 10.1038/nrc3580. [DOI] [PubMed] [Google Scholar]
- 62.Soda M, Choi Y, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 63.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 64.Li Y, Ye X, Liu J, Zha J, Pei L. Evaluation of EML4-ALK fusion proteins in non-small cell lung cancer using small molecule inhibitors. Neoplasia. 2011;13:1–11. doi: 10.1593/neo.101120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong D, Leung E, So K, Tam I, Sihoe A, Cheng L, Ho K, Au J, Chung L, Pik Wong M. The EML4-ALKfusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115:1723–1733. doi: 10.1002/cncr.24181. [DOI] [PubMed] [Google Scholar]
- 66.Gainor J, Varghese A, Ou S, Kabraji S, Awad M, Katayama R, Pawlak A, Mino-Kenudson M, Yeap B, Riely G, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19:4273–4281. doi: 10.1158/1078-0432.CCR-13-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang N, Liu Y, Ma L, Wang L, Hao X, Yuan Z, Lin D, Li D, Zhou Y, Lin H, et al. The molecular detection and clinical significance of ALK rearrangement in selected advanced non-small cell lung cancer: ALK expression provides insights into ALK targeted therapy. PLoS One. 2014;9:e84501. doi: 10.1371/journal.pone.0084501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kwak E, Bang Y, Camidge D, Shaw A, Solomon B, Maki R, Ou S, Dezube B, Jänne P, Costa D, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaw A, Kim D, Nakagawa K, Seto T, Crinó L, Ahn M, De Pas T, Besse B, Solomon B, Blackhall F, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 70.Shaw T, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou SH, Butaney M, et al. Effect of crizotinib on overall survival in patients with advanced non-small- cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;120:ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doebele R, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choi Y, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 74.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–6060. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tartarone A, Lazzari C, Lerose R, Conteduca V, Improta G, Zupa A, Bulotta A, Aieta M, Gregorc V. Mechanisms of resistance to EGFR tyrosine kinase inhibitors gefitinib/erlotinib and to ALK inhibitor crizotinib. Lung Cancer. 2013;81:328–336. doi: 10.1016/j.lungcan.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 76.Bos M, Gardizi M, Schildhaus HU, Heukamp LC, Geist T, Kaminsky B, Zander T, Nogova L, Scheffler M, Dietlein M, Kobe C, Holstein A, Maintz D, Büttner R, Wolf J. Complete metabolic response in a patient with repeatedly relapsed non-small cell lung cancer harboring ROS1 gene rearrangement after treatment with crizotinib. Lung Cancer. 2013;81:142–143. doi: 10.1016/j.lungcan.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 77.Shaw A, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189– 1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013;14:590–598. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 79.Ou S, Gadgeel S, Chiappori A, Riely G, Lee R, Garcia L, Tatsuno M, Tanaka T, Gandhi L. LATE BREAKING ABSTRACT: safety and efficacy analysis of RO5424802/CH5424802 in anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) patients who have failed crizotinib in a dose-finding phase I study (AF-002JG, NCT01588028) Eur Cancer Congr Abstr. 2013;44 [Google Scholar]
- 80.Camidge D, Bazhenova L, Salgia R, Weiss G, Langer C, Shaw A, Narasimhan N, Dorer D, Rivera V, Zhang J, et al. First-inhuman dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: updated results. J Clin Oncol Abstr. 2013;31:8031. [Google Scholar]
- 81.Patnaik A, LoRusso P, Ball H, Bahceci E, Yuen G, Papadopoulos K, Kittaneh M, Tolcher A. Pharmacokinetics and safety of an oral ALK inhibitor, ASP3026, observed in a phase I dose escalation trial. J Clin Oncol Abstr. 2013;31:2606. [Google Scholar]
- 82.Brahmer J, Tykodi S, Chow L, Hwu W, Topalian S, Hwu P, Drake C, Camacho L, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–690. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 84.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 85.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang R, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suehara Y, Arcila M, Wang L, Hasanovic A, Ang D, Ito T, Kimura Y, Drilon A, Guha U, Rusch V, et al. Identification of KIF5B-RET and GOPC-ROS1 fusions in lung adenocarcinomas through a comprehensive mRNA-based screen for tyrosine kinase fusions. Clin Cancer Res. 2012;18:6599–6608. doi: 10.1158/1078-0432.CCR-12-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jun H, Johnson H, Bronson RT, de Feraudy S, White F, Charest A. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012;72:3764–3767. doi: 10.1158/0008-5472.CAN-11-3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chin L, Soo R, Soong R, Ou S. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol. 2012;7:1625–1630. doi: 10.1097/JTO.0b013e31826baf83. [DOI] [PubMed] [Google Scholar]
- 89.Ou S, Bang Y, Camidge D, Riely G, Salgia R, Shapiro G, Solomon B, Engelman J, Kwak E, Clark J, et al. Efficacy and safety of crizotinib in patients with advanced ROS1-rearranged non-small cell lung cancer (NSCLC) J Clin Oncol Abstr. 2013;31:8032. [Google Scholar]
- 90.Awad M, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, Friboulet L, Huang D, Falk M, Timofeevski S, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368:2395–2401. doi: 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davies K, Mahale S, Astling D, Aisner D, Le A, Hinz T, Vaishnavi A, Bunn P, Jr, Heasley L, Tan A, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One. 2013;8:e82236. doi: 10.1371/journal.pone.0082236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pao W, Hutchinson K. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 94.Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, Ross J, Miller V, Ginsberg M, Zakowski M, et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013;3:630–635. doi: 10.1158/2159-8290.CD-13-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta M, Buttitta F, Incarbone M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27:1667–1674. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sadiq A, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31:1089–1096. doi: 10.1200/JCO.2012.43.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Basilico C, Pennacchietti S, Vigna E, Chiriaco C, Arena S, Bardelli A, Valdembri D, Serini G, Michieli P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res. 2013;19:2381–2392. doi: 10.1158/1078-0432.CCR-12-3459. [DOI] [PubMed] [Google Scholar]
- 98.Katayama R, Aoyama A, Yamori T, Qi J, Oh-hara T, Song Y, Engelman JA, Fujita N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73:3087–3096. doi: 10.1158/0008-5472.CAN-12-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ou S, Kwak E, Siwak-Tapp C, Dy J, Bergethon K, Clark J, Camidge D, Solomon B, Maki R, Bang Y, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6:942–946. doi: 10.1097/JTO.0b013e31821528d3. [DOI] [PubMed] [Google Scholar]
- 100.Spigel D, Ervin T, Ramlau R, Daniel D, Goldschmidt J, Jr, Blumenschein G, Jr, Krzakowski M, Robinet G, Godbert B, Barlesi F, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31:4105–4114. doi: 10.1200/JCO.2012.47.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cepero V, Sierra J, Corso S, Ghiso E, Casorzo L, Perera T, Comoglio P, Giordano S. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res. 2010;70:7580–7590. doi: 10.1158/0008-5472.CAN-10-0436. [DOI] [PubMed] [Google Scholar]
- 102.McDermott U, Pusapati R, Christensen J, Gray N, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010;70:1625–1634. doi: 10.1158/0008-5472.CAN-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, Wistuba I, Fong K, Toyooka S, Shimizu N, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 104.Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’Meara S, Smith R, Parker A, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525– 526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 105.Mazières J, Peters S, Lepage B, Cortot A, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31:1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 106.Ross H, Blumenschein G, Jr, Aisner J, Damjanov N, Dowlati A, Garst J, Rigas J, Smylie M, Hassani H, Allen K, et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin Cancer Res. 2010;16:1938– 1949. doi: 10.1158/1078-0432.CCR-08-3328. [DOI] [PubMed] [Google Scholar]
- 107.Davies H, Bignell G, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett M, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 108.Paik P, Arcila M, Fara M, Sima C, Miller V, Kris M, Ladanyi M, Riely G. Clinical characteristics of patients with lung adeno-carcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046– 2051. doi: 10.1200/JCO.2010.33.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sosman J, Kim K, Schuchter L, Gonzalez R, Pavlick A, Weber J, McArthur G, Hutson T, Moschos S, Flaherty K, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, Viola P, Pullara C, Mucilli F, Buttitta F. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29:3574–3579. doi: 10.1200/JCO.2011.35.9638. [DOI] [PubMed] [Google Scholar]
- 111.Planchard D, Mazieres J, Riely G, Rudin C, Barlesi F, Quoix E, Souquet P, Socinski M, Switzky J, Ma B, et al. Interim results of phase II study BRF113928 of dabrafenib in BRAF V600E mutation–positive non-small cell lung cancer (NSCLC) patients. ASCO Meet Abstr. 2013;31:8009. [Google Scholar]
- 112.Rudin C, Hong K, Streit M. Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. J Thorac Oncol. 2013;8:e41–e42. doi: 10.1097/JTO.0b013e31828bb1b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oxnard G, Binder A, Jänne P. New targetable oncogenes in non-small-cell lung cancer. J Clin Oncol. 2013;31:1097–1104. doi: 10.1200/JCO.2012.42.9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hammerman P, Sos M, Ramos A, Xu C, Dutt A, Zhou W, Brace L, Woods B, Lin W, Zhang J, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011;1:78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dutt A, Ramos A, Hammerman P, Mermel C, Cho J, Sharifnia T, Chande A, Tanaka KE, Stransky N, Greulich H, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sliwkowski M, Mellman I. Antibody therapeutics in cancer. Science. 2013;341:1192–1198. doi: 10.1126/science.1241145. [DOI] [PubMed] [Google Scholar]
- 117.Topalian S, Hodi F, Brahmer J, Gettinger S, Smith D, McDermott D, Powderly J, Carvajal R, Sosman J, Atkins M, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, et al. Noninvasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]