

Abstract
Bacterial translocation (BT) has been impeccably implicated as a driving factor in the pathogenesis of a spectrum of chronic liver diseases (CLD). Scientific evidence accumulated over the last four decades has implied that the disease pathologies in CLD and BT are connected as a loop in the gut-liver axis and exacerbate each other. Pregnane X receptor (PXR) is a ligand-activated transcription factor and nuclear receptor that is expressed ubiquitously along the gut-liver-axis. PXR has been intricately associated with the regulation of various mechanisms attributed in causing BT. The importance of PXR as the mechanistic linker molecule in the gut-liver axis and its role in regulating bacterial interactions with the host in CLD has not been explored. PubMed was used to perform an extensive literature search using the keywords PXR and bacterial translocation, PXR and chronic liver disease including cirrhosis. In an adequate expression state, PXR acts as a sensor for bile acid dysregulation and bacterial derived metabolites, and in response shapes the immune profile beneficial to the host. Activation of PXR could be therapeutic in CLD as it counter-regulates endotoxin mediated inflammation and maintains the integrity of intestinal epithelium. This review mainly focuses PXR function and its regulation in BT in the context of chronic liver diseases.
Keywords: Pregnane X receptor, Bacterial translocation, Chronic liver disease, Intestinal permeability, Inflammation, Tight junctions
Core tip: Translocation of bacteria at pathological levels is a major driving factor in the progression of chronic liver diseases (CLD). However, it remains to be known whether it is the CLD condition that triggers leaky gut, or if translocation of bacteria plays an etiological role in the pathogenesis of CLD. Dysregulation of homeostasis in the gut-liver axis is considered as a crucial element that underlies the pathogenesis of BT. The nuclear receptor, pregnane X receptor (PXR) is widely expressed in gut and liver axis and is implicated in maintenance of equilibrium in the gut-liver axis. This review will summarize the various studies that have highlighted the importance of PXR as the mechanistic linker molecule in the gut-liver axis and its role in regulating bacterial translocation in the pathogenesis of cirrhosis.
INTRODUCTION
Pregnane X receptor
Pregnane X receptor (PXR) is an adopted orphan nuclear receptor (NR) that is part of a broad nuclear receptor superfamily. Specifically, PXR is encoded by NR1I2 gene and is categorized as the 2nd member of group I (nuclear receptor subfamily 1) which also comprises VDR (NR1I1) and CAR (NR1I3)[1,2]. Consistent with the majority of class 1 NR’s, PXR behaves as a transcription factor that is present in the cytosol and is activated only post ligand binding. After ligand activation, these NR’s form complexes with retinoid X receptor (RXR) and bind to DNA response elements of the genes that they regulate[3]. PXR was initially characterized as a xenobiotic receptor that senses and responds only to exogenous toxic substances and prescription drugs. Over the last two decades, PXR is widely recognized for its additional roles in sensing a range of endobiotic compounds such as bilirubin, bile acids, dietary lipids and steroid hormones, and hence is also referred to as steroid and xenobiotic receptor (SXR)[4]. PXR has been shown to be expressed in various tissues including stomach, placenta, kidney, lung, uterus and ovary, but is predominantly expressed in small intestine, colon and liver. Its increased expression in intestinal epithelium and hepatocytes also highlight its vital role in adaptive defense against xenobiotics and endobiotics exposure in intestine and liver[3,5]. A recent study has shown evidence that hepatic stellate cell (HSC) expressed PXR and its activation lead to attenuation of HSC’s differentiation and proliferation[6]. A study using mice knock out model also highlighted the expression of PXR in monocyte/macrophage cells and their role in countering inflammatory profile[7]. PXR expression has also been observed in other immune cells including T-cells and dendritic cells[8].
Identical with other NR’s, PXR contains a conserved DNA binding domain (DBD) and a flexible ligand-binding domain (LBD). Upon activation by a ligand, PXR binds to its response elements only as heterodimeric complexes that it forms with 9-cis retinoic acid receptor (RXR or NR2B) and others co-activators such as SRC-1[9]. The structural feature that makes PXR stand out from the other NR’s is its voluminous and flexible ligand-binding pocket. This enables PXR to bind and be activated by a wide range of hydrophobic ligands. Indeed, its spherical shaped ligand binding pocket has a volume greater than 1150 angstrom, making its ligand cavity one of the largest to be characterized so far[3], and on par with the NR PPAR-γ’s ligand pocket[10]. It is also important to highlight the unselective nature of the PXR LBD, enabling it to sense and respond to chemicals within a broad molecular weight range (about 250-850 kDa)[3]. An impressive range of bioactive components from herbal sources, such as Hyperforin, Paclitaxel and Guggul have also been added to the increasing list of naturally occurring PXR ligands[11].
Functions of PXR
The primary and the most conceded function of PXR is to activate genes encoding drug metabolizing and drug transporter enzymes. It acts as sensors that monitor any alteration in the levels of foreign compounds or endobiotics[3,12]. The genes that PXR activate take the responsibility of metabolizing and elimination of exogenous chemicals, and thus form the primary line of defense against toxicity challenge[12,13]. In humans, among drug metabolizing cytochrome P450 (CYP) enzymes, CYP3A is the most copiously expressed isoform in the liver and intestine[14]. Furthermore, transgenic rodent knock out models (KO)[15,16] have established beyond doubt that PXR’s are master regulators of CYP3A genes[17], which encode proteins responsible for the metabolic oxidation of more than half of the known prescription drugs[3]. Classic PXR activators such as pregnenolone 16 α-carbonitrile (PCN) and Rifampicin have also been used to validate the same. PXR also controls the expression of phase 2 conjugating enzymes such as SulT1a and UGT-1A which are primarily responsible for sulfate conjugation or glucuronidation of steroid hormones, bile acids and bilirubin[13,18-21]. Following conjugation, PXR controlled phase 3 drug transport proteins like P-glycoprotein and MRP-2 are then involved in efflux transport and elimination of the toxic metabolites[13,22].
The function of PXR extends beyond metabolism of drugs and endobiotics, which has made it a considerable area of research over the last decade. NR’s, in general, are rising as major targets for drug discovery and the identification of additional roles of PXR has given a new perspective in freshly approaching already known disease pathologies. Apart from its most researched role in inflammatory bowel disorders (IBD), PXR dysregulation has been implicated in CLDs[6,23] various cancers[24] and metabolic disorders like obesity[25]. PXR play a crucial role and aids hepatocytes in uptaking endobiotics or xenobiotics and is further involved in their metabolism and elimination[26,27]. Anti-fibrogenic activity has also been documented, wherein PXR activation by its ligand PCN prevented the transdifferentiation of hepatic stellate cells into myofibroblasts[6]. Similar observations were made when using another established PXR ligand Rifampicin, where PXR was associated with inhibition of major pro-fibrogenic factors such as transforming growth factor-β (TGF-β) and Alpha smooth muscle actin[6]. Interestingly, a significantly increased expression of PXR was observed in various tumor tissues when compared to non-neoplastic tissues, and a positive correlation was found between cell proliferation and PXR positive cells[24].
PXR has also been attributed to playing a role in energy metabolism and has been linked with diseases such as type 2 diabetes, obesity and hyperglycemia. Activation of PXR has been observed to produce a suppressive effect on hepatic gluconeogenesis. However, it has also been reported to cause hepatic steatosis by increasing lipogenesis and fatty acid uptake[25]. Increased skin inflammation was reported in PXR null mice when challenged with hapten and was associated with increased interferon gamma (INF-γ) and reduced anti-inflammatory cytokine interleukin-10 (IL-10)[28]. The numerous roles of PXR in interaction with microbial metabolites, maintaining innate immunity, epithelial integrity, countering inflammation, bile acid trafficking and detoxification are discussed in detail in the upcoming sections.
GUT MICROBIOTA AND PATHOLOGICAL TRANSLOCATION
Gut microbiome and gut-liver crosstalk
The presence of bacteria in the gut gains significant importance because of the monumental level of interaction that happens between the gut microbiome and the host. In the human body, almost 100 trillion bacteria are in constant communication with the intestinal epithelium, which spans almost 400 m2 in surface area-which is the largest in humans[29]. It does not come as a surprise that, at such a level of interaction, some of the important physiological functions of the human host including digestion, energy metabolism, maintenance of intestinal integrity and innate immune homeostasis depend largely on the balance of host-microbiome interaction[30]. These physiologic events are dependent on the extensive arsenal of microbial metabolites, which are yet to be characterized completely.
In physiological state, the commensal bacteria have influence beyond the intestine. In this context, Björkholm et al[31] have reported that more than 100 genes in the liver are differentially expressed between germ-free mice and their conventionally raised wild counterparts. However, the most gripping evidence put forth by this study was that majority of these genes that varied in expression in GF mice, were in fact related to xenobiotic metabolism. This study highlights the significance of xenobiotic sensors PXR and CAR as mechanistic links between microbes and host[31]. Thus any alterations in the gut microbiome or its sensors will be reflected in the liver functionality. Accordingly, ulcerative colitis patients have been observed to have increased susceptibility to develop primary sclerosing cholangitis[32]. Interestingly, any abnormalities in liver function are also reflected as alterations in the quality and quantity of intestinal bacteria in the gut. Patients with intrahepatic cholestasis have been shown to manifest overgrowth of bacteria in the small intestine[33]. Similar observations were made in NAFLD patients, where fat induced bile acid abnormalities were linked with bacterial dysbiosis[34]. Moreover, studies in NAFLD and chronic alcohol feeding models have observed the manifestation of intestinal inflammation, which indirectly compromises gut integrity[35,36].
Bacterial translocation
Bacterial translocation (BT) is defined as the passage of viable indigenous bacteria and bacterial products, such as endotoxin, from the intestinal lumen through the mucosa into mesenteric lymph nodes (MLNs) and other organs[13,14,37,38]. It is a common physiological event in the healthy individuals and is tightly regulated by various levels of immune and physical barriers[39]. However, BT is seen to happen physiologically at minor levels, where it is considered as a beneficial event to the host, especially in priming the host immune system[38,39]. The mucus and tightly bound intestinal epithelial lining comprise the physical barrier, while gastric acid, antimicrobial peptides (AMPs), IgA antibodies and innate immune cells form the chemical/immune barriers[38]. Physiologically dendritic cells constantly sample bacteria through their processes and help in priming the B-cells to secrete IgA. The DCs also aid in the transport of the translocating microbes to mesenteric lymph node (MLN), which serves as a central hub between the gut and rest of the body. MLN are also the location where the microbes are killed via local immune response[38]. However, when the physiologic barriers are compromised, or if the quality or quantity of bacteria is altered in the gut due to other external abnormalities such as alcohol abuse and high fructose diet, the translocation of bacteria or its products is persistent and pathologic. This phenomenon results in chronic induction of both systemic and hepatic inflammation[38,40].
Bacterial infections in patients with cirrhosis are correlated with a poor prognosis and an increased risk of mortality[40]. BT can also exacerbate the hepatic and systemic hemodynamic abnormalities of liver cirrhosis. Pathological BT is an important and emerging mechanism for the pathogenesis of CLDs. Emerging research has also shown clear evidence of increased intestinal permeability, mucosal inflammation and detection of bacteremia and endotoxemia in patients with CLD[41,42]. Indeed, spontaneous bacterial peritonitis (SBP) is considered the most evidenced clinical expression of BT, which responsible for 25%-40% of overall mortality in cirrhotic patients[43]. Pathological BT has been implicated in a range of other complications that arise in CLD, including acute-on-chronic liver failure, hepatorenal syndrome and hepatic encephalopathy (HE)[38]. The exact mechanisms of increased intestinal permeability and BT in CLD remains obscure, and only the probable pathways that might cause the phenomenon have been predicted. The cyclic cascade of events that cause BT and exacerbate CLD have been summarized in Figure 1.
Figure 1.

Cyclic cascade of bacterial translocation and associated progression of chronic liver diseases and its complications. The figure enlists the various proposed mechanisms through which chronic liver diseases conditions triggers intestinal permiability and BT. Increased intestinal permeability causes translocation of bacteria across the intestinal epithelium to the MLN and extra-intestinal sites such as liver and blood causing discrete complications in each of the systems. In the intestinal lumen, BT causes overt activation of immune cells and aggravates the pro-inflammatory cytokines in the gut. Translocation of bacteria at pathological levels to MLN is a major risk factor for systemic inflammation which leads to hyperdynamic circulation and is reflected across various organs in the form of complications such as cardiac dysfunction, hepatorenal syndrome and hepatic encephalopathy. Translocation of bacteria to the liver causes TLR4 and TLR9 mediated inflammation which further exacerbates the progression of chronic liver diseases and intestinal permeability. BT to liver also causes portal hypertension, which forms the basis of development of SBP. BT: Bacterial translocation; MLN: Mesenteric lymph node; SBP: Spontaneous bacterial peritonitis.
Possible mechanisms of BT in CLD
Compromised Bile in the intestines: Bile flow is altered in liver disease, as the organ is the major producer of bile. Bile acids have been shown to interact with NR’s such as FXR in the GI tract and keep bacteria under check. Hence a compromised bile environment may lead to overgrowth of bacteria in liver disease conditions[30,44].
Dysbiosis: It is a phenomenon inclusive of any quantitative or qualitative alterations from the symbiosis maintained between the host and microbiota[41]. Quantitative overgrowth of bacteria also called as small intestinal bacterial overgrowth is usually the first step in the phenomenon of BT. Patients with CLD, especially alcoholic hepatitis have been directly linked with developing bacterial overgrowth, low motility and increased transit time in the intestine[41]. The proportion of beneficial and less beneficial (sometimes pathogenic) bacteria is tightly maintained and is termed symbiosis. In CLD, this balance is disrupted leading to increase in pathogenic bacteria and harmful metabolites that damage the epithelium[41]. Beneficial bacteria such as lactobacillus reduce in numbers and potentially pathogenic bacteria like Enterobacteriaceae increase. Indeed, it was observed that the pathogenic bacteria are more likely to translocate across the intestinal epithelium[41].
Immune dysfunction: The mucosal immune system prevents the exposure of commensal bacteria to systemic circulation through various secretory mechanisms such as mucus and anti-microbial peptides (AMP). In cirrhotic animal models, Paneth cells, have been observed to have diminished expression of AMP’s[45].
Intestinal inflammation: Both mucosal and submucosal inflammation have been observed in patients with CLD. A pro-inflammatory cytokine profile is also actively linked with increased intestinal permeability[43,46-57]. Inflammation and its significance on intestinal permeability are discussed in more detail in the later sections of this review.
Disruption of tight junctions: A single layer of epithelial cells separate 100 trillion gut bacteria from other parts of the body. The epithelial cells are sealed tightly via tight junctions (TJ’s) that are seen to disassociate in CLD and result in the leaky gut leading to BT[48,49].
PXR AND ITS ROLE IN COUNTERACTING INFLAMMATION
Inflammation is a response mechanism to meet and overcome the challenges an organism faces from an injury or infection. However, when uncontrolled or unregulated, inflammation is often a pathological driving force in various disease conditions. In the context of this review, inflammation is the most prominent player in the pathophysiology of BT and sets the environment that causes increased intestinal permeability. Studies over the last decade have firmly established the role of PXR as a counter-regulator of inflammation[7]. Clinical studies have revealed that, in IBD patients a clear pattern of PXR downregulation was observed in inflamed tissues[50]. In this context, polymorphism of NR1I2 gene has identified to be associated with more susceptibility to IBD[51-53]. PXR has been described to interact with various components of the immune response signaling cascade to produce an immune regulating effect.
Nuclear factor kappa B (NF-κB) is a transcription factor playing a central role in regulating plethora of genes involved in innate and adaptive immunity[54]. It is well established that drug metabolism is compromised in an inflammatory environment and vice versa[55]. This phenomenon plays an important role in highlighting the counter-regulation that exists between PXR and NF-κB. PXR controls drug metabolism by binding to its response elements as heterodimers. PXR forms heterodimers with RXR-α and studies have revealed that this interaction is inhibited by the binding of p-65 subunit of NF-κB to the RXR unit of PXR heterodimer complex (Figure 2A)[56]. However, of even more interest are the observations that PXR can reciprocally inhibit NF-κB and thus making PXR an excellent target to counteract NK-κB and its associated inflammatory gene kit[57]. Zhou et al[58] demonstrated an increased NF-κB activity and inflammatory cytokine profile in PXR null mice when compared to wild type mice that constitutively express PXR. This indicates that in physiological state PXR expression keeps the NF-κB initiated inflammatory response under check. Additionally, when wild type mice were treated with PCN, a specific PXR agonist, majority of the NF-κB target genes were downregulated, an effect that was lost in PXR null mice. This suggests that PCN antagonism of NF-κB happened in a PXR dependent manner[58]. Similar findings were observed in DSS-induced colitis mice with and without PCN treatment[59]. Ultimately, PXR which is responsible for defense against chemicals, and NF-κB which is responsible for mounting an immune defense, counter-regulate each other to maintain physiological state.
Figure 2.
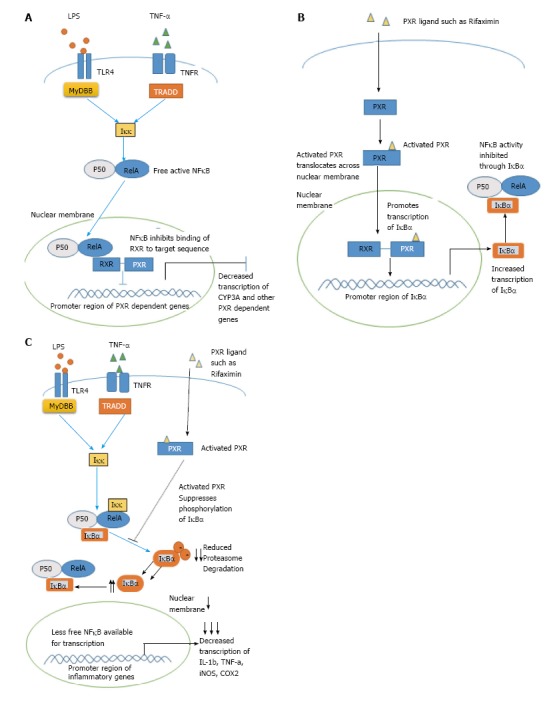
Illustration of counter-regulatory mechanisms existing between nuclear factor kappa B and pregnane X receptor. A: The p-65 subunit of NF-κB binds to the RXR unit of PXR heterodimer complex and represses its transcriptional activity; B: Activated PXR binds to the promoter region of IκBα and increases its transcription leading to indirect repression of NF-κB; C: After activation through its ligands, PXR suppresses the phosphorylation and degradation of IκBα and thus indirectly represses NF-κB activity. NF-κB: Nuclear factor kappa B; PXR: Pregnane X receptor; RXR: Retinoid X receptor; TLR: Toll like receptor.
Studies by Wallace et al[7] employed a SJL/J mice model that is characterized by increased monocyte cell infiltration into the hepatic portal tract, revealed that PXR was expressed in infiltrating monocytes. Further in SJL/J-PXR+/+ mice, activation of PXR using PCN, downregulated tumor necrosis factor alpha (TNF-α) and Interleukin-1α (IL-1α), which are cytokines controlled by NF-κB. Consistent with other studies, this effect was lost in SJL/J-PXR-/- mice[7,58]. Thus BT is also associated with considerable recruitment of monocytes cells to the lamina propria, which is accompanied by increased production of TNF-α[60,61]. Similarly, Fiorucci et al[60] observed that LPMC’s isolated from colitis mice that were treated with Rifaximin, showed complete abrogation of INFγ cytokine production. Unfortunately this study did not consider if activation of PXR by Rifaximin could have produced this effect. However, study by Cheng et al[62] using humanized PXR (hPXR) mice with DSS induced colitis showed that Rifaximin indeed works in a PXR-dependent manner and attenuated NF-κB mediated cytokines. Interestingly, TNF-α has also been associated with direct downregulation of epithelial TJ proteins[63]. In this scenario, further studies targeting activation of PXR with an aim to antagonize NF-κB-induced-TNF-α may pose as an exciting therapeutic outcome especially by controlling mononuclear cell infiltration, with an aim to attenuate intestinal epithelial damage, which is a major prequel to BT.
Even though many studies have established the mutual inhibition between PXR and NF-κB, the exact mechanism by which PXR represses NF-κB is not well understood. Indeed, Ye et al[64] showed that PXR activation by a natural PXR ligand Ginkgolide-A (GA) repressed NF-κB indirectly by enhancing the expression of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (Iκ-Bα), an inhibitory protein of NF-κB activity. When siRNA was used for silencing of PXR, GA did not increase the expression of Iκ-Bα, showing that the induction happens in a PXR dependent manner[64] (Figure 2B).
Various plant flavanols like chrysin and isorhamnetin have also been shown to inhibit NF-κB activity through a PXR dependent manner[65,66]. Studies using these flavanols in a DSS-induced colitis mouse model showed PXR mediated downregulation of NF-κB target genes including iNOS, ICAM-1 MCP-1, COX-2, TNF-α IL-2 and IL-6. Intriguingly, both studies showed that PXR activation prevented the degradation of Iκ-Bα and thus underlining the possibility that PXR might counteract NF-κB mainly through manipulation of fate of Iκ-Bα[65,66] (Figure 2C).
SUMOYLATION dependent regulation of PXR-NF-κB axis
SUMOYLATION is a post-translational modification process, in which a small ubiquitin-like modifier (SUMO) protein would be added to the ligand-binding domain of PXR protein[67]. A SUMO1 binding site was discovered in the ligand-binding domain of PXR[68]. It was revealed that post SUMOYLATION there is an increase in the transcriptional activity of PXR, marked by increased transcription of PXR target genes (Figure 3A). Also, an increase in interaction between SUMOylated PXR and NR co-repressor (NCOR1) was observed. Thus it is speculated that post-modification, PXR protein might be able to repress NF-κB indirectly by helping to keep the co-repressors (N-COR)/HDAC3 complex intact by preventing their clearance[68,69]. Vice versa, endotoxin stimulus such as LPS would signal the clearance of these repressor complex from the promotor region of pro-inflammatory genes and thus enabling NF-κB to transcribe the inflammatory profile genes[70]. A similar event has already been established to happen in another NR, PPAR-γ, which post SUMOYLATION trans-represses NF-κB by preventing recruitment of Ubc5 protein and initiates the clearance of co-repressors[71]. This mechanism opens attractive opportunities to target the PXR SUMOylation site, particularly in a scenario such as bacterial sepsis, where endotoxin stimulated NF-κB inflammatory response occurs.
Figure 3.
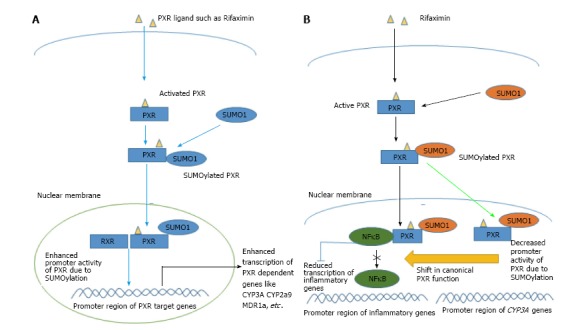
Schematic diagram illustrating the mechanism of modification of pregnane X receptor activity and function after SUMOYLATION. A: SUMO1 binding to PXR has been shown to increase its transcriptional activity; B: Sumo (3) ylation of PXR causes a shift from the canonical transcriptional function of PXR towards the transcriptional repression of NF-κB. NF-κB: Nuclear factor kappa B; PXR: Pregnane X receptor; RXR: Retinoid X receptor.
Furthermore, Hu et al[69] revealed that the PXR SUMOylation occurs as a feedback response to inflammatory stimulus such as TNF-α. They specifically identified SUMO3 chains in the post translationally modified (SUMOylated) PXR protein. An extremely interesting aspect of their discovery is that they found through their in vitro assays that SUMOylated form of PXR played a huge role in diminishing inflammation, but was hardly effective in regulating CYP3A expression. Thus, SUMOylation may shift the functional activity of PXR from a ligand-activated transcriptional inducer which upregulates xenobiotic target genes, towards a ligand-activated transcriptional repressor that brings about an immunosuppressive effect (Figure 3B).
PXR and TNF-α
Dysregulation between the intestinal epithelial cells and the innate immune system is often initiated by endotoxins such as LPS and is one of the established pathological mechanism for gut barrier disruption[73,73]. It has been observed that TNF-α acts as a central mediator of NF-κB in the initiation of mucosal inflammation[74]. Goldman et al[74] have reported that an anti-TNF approach which was effective in countering BT. Research conducted by Mencarelli et al[75] revealed that IEC on exposure to TNF-α showed significant dampening of PXR mRNA levels. However, TNF-α was completely antagonized when treated with Rifaximin, which showed significant anti-inflammatory effect through PXR activation. Further, when cells cultured from colon biopsies of IBD patients were induced with LPS and followed by Rifaximin treatment, an abrogation of the LPS induced NF-κB target genes such TNF-α, MIP-3α and IL-8 were observed. These evidence clearly indicate that effectively inducing PXR, which inhibits the effects of LPS induced TNF-α and NF-κB, may pose as a desired outcome in the treatment of BT.
PXR and MDR1 puzzle
Multi drug resistance gene (MDR) 1 is categorized under the ABC family of transporters and encodes the transmembrane protein P-glycoprotein (P-gp). MDR1 is one of the primary genes regulated by PXR and is widely expressed in intestinal epithelial cells and the liver[76]. P-gp acts as an ATP-dependent drug efflux system and is responsible for maintaining intestinal homeostasis by pushing out noxious chemicals (from drug or microbial source) from mucosa back into gut lumen[76,77]. Polymorphism in Mdr1a gene that results in a phenotype with reduced P-gp expression was observed in both Ulcerative colitis and Chron’s disease subjects[78]. Mdr1a-/- knockout mice models confirmed that, in absence of this efflux pump protein, the animals developed spontaneous colitis resembling human IBD. This effect was ameliorated by treatment with oral antibiotics indicating that reducing the bacterial burden is an effective measure to control inflammation. Reducing the toxin accumulation in the gut might be the probable mechanism for ameliorating inflammation and thus underlining the importance of a xenobiotic clearance system in the gut[77]. Langman et al[79] showed that mRNA levels of both PXR and Mdr1a were reduced in UC patients and also assumed that the dampening of PXR expression might be the probable reason for Mdr1a downregulation.
A recent study performed by Toklu et al[80] hypothesized that PXR stimulation by antibiotics rifampicin and spironolactone may cause an immunosuppressive effect through induction of Mdr1a gene (and thus P-gp protein expression). However, these observations were conflicted by Blokzijl et al[81] who showed that PXR protein levels were unchanged between inflamed and uninflamed human colons, in spite of low Mdr1a expression in the same tissues. Thus in such a scenario Mdr1a may be independent of PXR protein concentration. Ros et al[82] reported Mdr1a expression to be unaltered in liver of LPS-treated rats. Conflicting evidence regarding reduced Mdr1a levels in intestine was provided by Kalitsky-Szirtes et al[83]. Further research using PXR null mice may confirm if PXR-induced-P-gp expression is a valid mechanism in controlling BT, most probably by riding the gut of endotoxins, which may otherwise stimulate mucosal immunity.
PXR and LPS
LPS is a major component of the cell walls of gram-negative bacteria and is considered an endotoxin, which potently stimulates host innate immune response[84,85]. LPS is recognized by its specific receptor toll like receptor 4 (TLR4) and is one of the earliest inflammatory triggers to induce gut barrier disruption and BT[86]. The pro-inflammatory effects of LPS are mediated primarily through activation of transcription factors like NF-κB, which is present upstream in the inflammatory cascade[56,84]. In humans, a physiological BT state has been defined, where 5%-10% of bacteria, translocate across the intestine with minor exposures of LPS, maintaining a tightly regulated tolerance towards the gut microbes and their toxins[38]. However, when there are alterations in quantity (load) or quality (dysbiosis) of bacteria, the innate immune system is activated overtly. This is often by means of increased exposure to LPS, which stimulate immune cells such as monocytes, neutrophils and lymphocytes. These cells, in turn, produce acute response cytokines including IL-1β, IL-6 and TNF-α, creating an inflammatory profile[87-89].
LPS-induced inflammation models have highlighted the importance of PXR activity in regulating different phases of the immune response. Diminished expression of CYP3A gene was identified during infections and has been replicated in LPS-induced animal models[90,91]. Interestingly, mRNA levels of PXR were also seen to be downregulated in such models. Indeed, Moriya et al[92] have indicated that LPS treatment significantly reduced both the gene expression and activity of CYP’s, even in mice that were pre-stimulated with PXR activator PCN. LPS caused this effect by inducing cytokines, which bring about the inhibition of PXR in an NF-κB dependent manner. A similar observation was made by Gu et al[56] where the use of NF-κB suppressor SRIκBα, reversed the LPS induced downregulation of PXR, proving that “NF-κB stimulated PXR inhibition” is central in bringing out the effects of LPS. While IECs are in constant contact with bacterial products like LPS, immune cells are involved in the response to any dysregulation between IEC and microbial products. It should be noted that PXR is expressed in both these cell types and thus play a major role in regulating both the toxin challenge that is faced by IEC, as well as in response to that challenge that is brought about by immune cells. A recent study identified in the primary culture of hepatocytes (PCH) isolated from WT mice that, PCN pretreatment for 24 h alleviated LPS induced an acute response by decreasing cytokines such as IL-1β, TNF-α and IL-6. However, when PCH from PXR-null mice were treated with LPS, enhanced pro-inflammatory cytokine response was documented[93]. When PCH isolated from humanized PXR mice were pre-treated with PXR activators, it led to increased production of IL-1Ra, a natural inhibitor of IL-1β[93]. Thus PXR expression is seen to be important in both dampening endotoxin-stimulated immune response to maintain homeostasis, as well as in resolving the inflammatory state through inducing anti-inflammatory response[93,94].
Relationship between PXR, LPS and ROS
Xu et al[95,96] have revealed an interesting pathway in which LPS could suppress the expression of PXR and its associated genes. In their experiment, LPS dose-dependently suppressed PXR mRNA levels in mice and was significantly improved following antioxidants treatments[95,96]. Furthermore, inhibition of xanthine oxidase and NADPH oxidase that generates ROS, using specific inhibitors allopurinol and diphenylene iodonium respectively, led to attenuation of LPS induced PXR downregulation[95]. In this context, the antioxidant melatonin was also observed to produce a similar effect[97]. Chen et al[98] revealed that treatment with a free radical trapping agent alpha-phenyl-N-tbutylnitrone prevented LPS from downregulating PXR. Thus, it is well understood that ROS and oxidative stress have an impact in the LPS induced diminishing of PXR expression. PXR ligands such as Danshen, which have an inherent antioxidant property could be used to further understand the LPS-countering activity of PXR[11]. Research performed using PXR ligands or constitutively active PXR (VP-PXR) have revealed that LPS instigated response is regulated by PXR activation[94,99]. Hence, countering LPS induced ROS promises to be a novel opportunity to counter endotoxin-induced inflammatory response, in BT[94,99].
PXR and TLR4 crosstalk
TLR4 is a transmembrane receptor that recognizes LPS, which is a pathogen associated molecular pattern. LPS can only bind to the TLR4 complex after it has associated itself with LBP (LPS binding protein)[100]. The recognition also involves additional co-receptors such as CD-14 and MD-2 and adaptor protein MyD88[101]. Physiologically TLR4 expression and regulation is of significant importance in the intestine. Hence, TLR4 expression is tightly regulated based on the level of LPS in the gut lumen as it directly correlates with the intensity of immune response at any given time. A pathological state such as small intestinal bacterial overgrowth, may challenge this homeostasis leading to overt activation of TLR4 signaling and trigger NF-κB, which leads to barrier dysfunction and BT[101,102]. Studies have shown that the crosstalk between TLR4 and PXR could determine the homeostasis in the intestine[103]. For instance, when TLR4 was activated using its specific agonist KDO2, increased induction of mucosal TNF-α, followed by intestinal permeability was observed in both WT and PXR null mice[104]. Similarly, when PXR was activated using PCN, a clear reduction in mucosal TNF-α induction was documented. PCN activation did not have any effect in PXR null mice and thus indicating that the TLR4 inhibition was PXR-dependent. This study clearly shows that PXR and TLR4 counter-regulate each other upon their respective activation[104].
A study by Esposito et al[105] observed a similar pattern of regulation when Caco2 IECs were induced with Clostridium difficile toxin A (TcdA) to replicate ulceration and inflammation model. They found that the toxin stimulated the expression of TLR4 by 1411% in Caco2 cells. However, this phenomenon was completely reversed by Rifaximin treatment dose dependently, which down-regulated the expression of TLR4, MyD88 and NF-κB. This study also brings to light an additional pathway, where by reducing TLR4 levels following PXR activation, the NF-κB activity might be reciprocally inhibited[105]. Furthermore, PXR and TLR4 double KO mice models have shed more light into PXR’s ability to act as a mediator between the microbes and TLR4. Venkatesh et al[106] observed that PXR null mice developed leaky gut and showed increased induction of TLR’s including TLR4 (1.8 fold increase). However, in TLR4-/- and PXR-/- double KO mice model, the previously observed pathological defects in intestine disappeared. This emphasizes the impact of TLR4 expression, which was particularly high in absence of PXR, in bringing about intestinal inflammation and the gut disruption. In addition, enterocytes isolated from PXR null mice, showed similar results with TLR4 inhibitors. Thus a reciprocal relationship seems to exist between PXR and TLR4 in maintaining homeostasis. They also found that Indole-3 Propionic Acid (IPA), which is an endogenous, microbe derived ligand for PXR, activated PXR and reduced enterocyte TNF-α[106]. This is a clear example of the sensing system: PXR, and the symbiotic bacteria working in coherence to repress overt inflammation. Together, these data suggest that PXR regulates the expression of TLR4, and that PXR activation could have a therapeutic effect in BT by counteracting TLR4 mediated gut disruption.
INTESTINAL INTEGRITY AND PXR: TIGHT JUNCTIONS
PXR and maintenance of intestinal integrity
A single layer of epithelial cells serve as physical barrier that prevent the diverse contents of the gut from entering the systemic circulation and other tissues. The integrity of this barrier is governed by junctional complex proteins including TJ and adherent junctions (AJ) and are involved in sealing the gap between two adjacent cells[107]. The expression of these junctional complexes are tightly controlled and are dynamic in nature, such that allowing passage of only selected molecules across the epithelial barrier[108]. In disease state, the expression of these junctional complexes are highly compromised leading to a leaky gut, which is the major driving factor for BT and its complications[109,110].
PXR is extensively expressed by IECs and have been shown to have a direct impact on the signaling molecules that govern intestinal integrity. Accordingly, studies have shown that in PXR null mice, there is a leaky gut like pathology[106]. Indeed Venkatesh et al[106] observed reduced mRNA levels of junctional complexes such as Zonula occludens 1 (ZO1) and E-cad in PXR KO mice. However, they also found an increased expression of Claudin-2, which is associated with promoting paracellular transport of microbes and which in high expression state is linked with hyperpermeability in gut. One of the possible mechanisms through which PXR maintains expression of junctional complexes was revealed through the use of PXR-TLR4 double KO mice[106]. When both PXR and TLR4 were knocked out, the level of TJ expression was almost relatable to the levels that were found in PXR+/+ mice. Hence, PXR may preserve junctional complexes by countering TLR4 and thereby inhibiting the downstream inflammatory cytokines such as TNF-α that are stimulated by TLR4. This shows that PXR knockout state is associated with pattern of upregulation of genes that promote paracellular transport (claudin-2, TNF-α) and downregulation of genes that maintain barrier functions (ZO1) and thereby playing a vital role in maintaining intestinal integrity.
Negative regulation between PXR and MLCK
Several studies that have targeted PXR activation have attributed the preservation of the junctional complexes to PXR’s ability to interact with various intercellular signaling mediators. Myosin Light Chain Kinase (MLCK) is associated with regulation of paracellular permeability through its ability to phosphorylate myosin II regulatory light chain (MLC), which underlies the junctional complex arrangement[111]. Hence through phosphorylation of MLC, MLCK is able to stimulate actomyosin contraction and modulate TJ localization[111]. In a pathological state such as infection or inflammation TNF-α induced both the expression of MLCK, and its activity and thus influencing intestinal permeability[112]. He et al[113] observed TNF-α induced MLCK expression was increased through the stimulation of NF-κB, which acted upstream of MLCK. Hence PXR, which counter-regulates TNF-α mediated NF-κB could possibly interact with this pathway to preserve TJs. Indeed, study by Garg et al[104] reported that TNF-α exposure induced increased relocalization of ZO1 through the upregulation of MLCK expression. However, PXR activation by Rifaximin, countered the MLCK upregulation in Caco2 IEC cells, through its established function of attenuating NF-κB. The same results were reproduced in an in-vivo DSS mice model, where PCN treatment attenuated MLCK expression and protected against ZO1 mislocalization[104]. Thus by inhibiting TNF-α induced NF-κB activation, PXR is able to maintain intestinal integrity through indirectly regulating MLCK[104].
Preservation of intestinal integrity by PXR through JNK1/2 interference
PXR activation has been reported to influence on the JNK1/2 pathway. C-jun N-terminal kinase (JNKs) are kinases that are activated in response to stress stimuli including various cytokines like TNF-α and are implicated in apoptosis and inflammation[114-116]. The exact role of JNK1/2 on inflammatory disorders is still unclear. While few studies reported JNK1/2 deletion increased the severity of inflammation in DSS model[117], others showed that JNK1/2 inhibition is protective[114,118]. In this context, Mitsuyama et al[119] have reported an increased JNK1/2 expression in IECs of CD patients. Garg et al[104] have reported that TNF-α/INFγ stimulation of Caco-2 cells resulted in increased activation of JNK1/2, an effect that was associated with ZO1 mislocalization. However, this phenomenon was completely inhibited using JNK inhibitor SP600125. Most importantly, activation of PXR using Rifaximin was seen to attenuate JNK1/2 activity by inducing the transcription of growth arrest and DNA damage inducible 45β (GADD45β), a protein that is known to block JNK1/2 activity by preventing its phosphorylation. Thus PXR activation was seen to protect intestinal TJ integrity by preventing the activation/phosphorylation of JNK1/2.
PXR and CDX2: An interaction of two transcription factors
Recently a new mechanism of PXR-related immunosuppression was reported by Dou et al[120] involving the Caudal related Homeobox transcription factor, CDX2. Interestingly, CDX2 has been implicated in intestinal differentiation and in maintaining intestinal integrity[121,122]. CDX2 is a transcription factor reportedly expressed in the intestine, where it binds to the promotor region of PXR and induces PXR transcription. Further mechanistic studies may establish if CDX2 is a player involved in regulating PXR expression especially in the scenario of mucosal inflammation.
PXR AND BILE ACIDS
Bile acids play a very important physiologic role in the catabolism of cholesterol and are known to regulate bacterial overgrowth owing to their bacteriostatic properties[123,124]. Bile acids are also ligands to NR’s such as FXR and VDR[125,126]. On activation these NR’s regulate the expression of anti-microbial peptides and innate immunity genes, which keep gut microbiome outgrowth in check[123-126]. However, studies have shown that when bile acids accumulate, they can be potentially toxic, thus highlighting the importance of presence of active bile acid detoxification system to afford protection against their toxicity[127]. Lithocholic acid (LCA) is a secondary bile acid, which is considered to be toxic at higher concentrations than the basal levels and is a byproduct of gut bacterial biotransformation process[128]. Makoto Ishit et al[129] have shown that upregulation of PXR and its dependent genes happens as an adaptive response to an increase in LCA, in patients who underwent gastrectomy. They reported that gastrectomy shifted the intestinal PH towards alkaline state due to reduced gastric acid, which led to the increased thriving of LCA-producing bacteria and thus leading to increased accumulation of LCA. This study highlights that PXR is the foremost physiologic and adaptive sensor of LCA especially considering that FXR another important bile acid sensor is unresponsive to LCA[129]. More importantly, this study using the example of gastrectomy sets the precedence that other pathologic events, such as dysbiosis may also shift the balance of gut microbiome composition and ultimately influence the bile acid metabolism and the genes they control.
Bacterial dysbiosis, an established mechanism to cause BT, has been shown to affect the composition of bile acid pool[130]. Disease states such as Non-Alcoholic Fatty Liver Disease (NAFLD) are associated with both increased bile acids and alteration in bacterial gut microbiome communities[131]. Such states may shift the balance of hydrophobic and hydrophilic secondary bile acids in the overall bile acid pool. In the context of this review, this is of importance, as a linear correlation has been observed between perturbance in gut microbes, disturbance in bile metabolites and disruption of intestinal barrier homeostasis[132,133]. Increased hydrophobic bile acids such as LCA and Deoxycholic acid (DCA) have been shown to be associated with disruption of gut barrier as shown by Stentman et al[134] where high fat related concentration of hydrophobic but not hydrophilic bile acids produced barrier disruption. Hughes et al[135] made an interesting observation that at physiologic levels, LCA increased paracellular permeability in Caco2 intestinal epithelial cells, which was indicated by a decrease in trans-epithelial resistance and increase in mannitol flux. However, at the same physiologic levels, LCA was seen to increase occludin expression. It would be interesting to see if LCA induced occludin expression happens in a PXR dependent manner, as PXR is the major physiologic sensor of LCA. Also, since this study only focused on acute twelve hour effects of LCA at basal levels[135], future studies at chronic treatment times and higher doses would need to be conducted to illuminate the effect of LCA toxicity on TJ expression.
LCA-feeding has been used a standard in-vivo model to induce cholestasis in mice. Fickert et al[136], used this model to demonstrate that LCA feeding induced disruption of TJ protein ZO1 in both bile duct epithelial cells and between hepatocytes. Vu et al[137], have found the similar observations that TJ permeability was elevated followed by cholestatic dose of LCA.
Role of PXR in LCA detoxification
The role of PXR in LCA detoxification is paramount, evidenced by studies which showed that PXR KO animals were susceptible to bile toxicity and cholestasis[138]. In cholestasis, as the disease progresses the bile acid flow is impeded leading to a compromised bile environment. This is exploited by bacterial outgrowth leading to their uncontrolled translocation[139]. This highlights the fact that, PXR sensing and detoxification may serve as a prophylactic (preventive) setup to maintain homeostasis in bile metabolism and afford protection against cholestasis[140]. The role of PXR in protection against NAFLD may also be critical, as increased LCA has been associated with high-fat consuming population[141,142]. Studies by Staudinger et al[143] and Xie et al[144] have documented the various mechanisms of PXR-dependent LCA detoxification. PXR has been observed to control the expression of CYP7A1, a rate-limiting enzyme in bile acid production from cholesterol, and is seen to repress CYP7A1 following PXR activation through its ligands[143]. PXR also directly regulates the expression of Na1-independent organic anion transporter 2, a protein that is involved in uptake of bile acids by hepatocytes for metabolism[143,144]. Finally, PXR dependent CYP3A enzymes in the hepatocytes mediate hydroxylation of LCA and prime it for elimination[145]. Additional mechanisms such as sulfonation of LCA by PXR dependent SULT enzymes have also been described[146]. Transgenic mice that constitutively expressed activated PXR (VP-PXR) were seen to be resistant to the toxic effects of LCA[146]. Consequently, PXR plays a very important role prophylactically in affording protection against bile dysregulation and toxicity, as seen in closely related pathologic states such as dysbiosis and NAFLD. The activation of PXR might serve as an attractive option in preventing BT, which might result from direct or indirect effects of bile acid dysregulation.
PXR THE SENSOR OF MICROBIAL METABOLITE CUE
In a physiological state, bacteria have been shown to have an impact on host genes without direct contact[31]. Gut bacteria communicate with the host mainly through the extensive profile of microbial metabolites that they produce, which interact with a range of physiologic sensors such as NR’s in the host cells of intestine and liver[14]. The enterohepatic circulation gains major importance in this matter, as the liver and intestine interact with each other through bile and metabolite (nutrient and microbial) profile respectively. Compromised bile availability in disease states like cholestasis has been linked with bacterial dysbiosis in the intestine[147]. Similarly, metabolic disorders such as obesity and Type 1 diabetes have been implicated with alterations in the gut microbiota[148], which indicate alterations in the proportion of the microbial metabolites the microbiota produce. While some of these microbial metabolites have been characterized as essential nutrients, many other metabolites and the NR’s that they interact with are yet to be explored. PXR owing to its flexible binding domain and extensive expression pattern in gut and liver has been identified to interact with wide range of bacterial metabolites through which it is seen to maintain homeostasis along the gut-liver axis.
IPA, a bacterial product of the tryptophan metabolism is being recognized as one of the established ligands of PXR[149]. Venkatesh et al[106] highlighted the importance of IPA sensing by PXR, where PXR after activation, maintained gut barrier function by downregulating TLR4 and its downstream effector TNF-α. IPA also increased the mRNA levels of junctional proteins. They demonstrated that either loss or reduced expression of PXR (as seen in inflammatory conditions) or the loss of IPA producing bacteria (as documented through commensal depleted organism models) led to worsening of inflammation and increased intestinal permeability[106]. It was also revealed that reintroducing C. Sporogenes in the presence of their substrate L-Tryptophan in GF mice led to production of IPA and improved cell-cell junctional complex efficacy. Importantly, the same phenomenon was absent in PXR KO mice. Hence shifting the bacterial composition towards favorable metabolic profile might be an effective method to activate PXR and counteract inflammation[106]. However, it is also important to consider the level of PXR available to sense IPA, which is often compromised during inflammation.
Gut bacteria also play a vital role in the bio-transformation of various natural herbal products into forms that are beneficial to host. β-glucuronidases produced by symbiotic bacteria have been shown to convert the flavonoid baicalin into baicalein[122,150]. Interestingly, Dou et al[122] reported that both baicalein and baicalin attenuated gut inflammation induced by DSS in vivo. However, baicalin treatment did not have any effect, when the β-glucuronidase inhibitor was used to prevent the bioconversion of baicalin. The study further reported that only baicalein activated PXR to potently produce an anti-inflammatory phenotype in the colitis induced mice. Thus, PXR acts as an important mediator between bacterial derived metabolites and host, and upon activation shapes the immune profile. Table 1 summarizes the various identified natural ligands of PXR along with the effective response they produce after binding with PXR.
Table 1.
Table enlisting the documented effect of various natural occurring ligands of pregnane X Receptor
| No | Natural ligand and source | Effective response after binding of ligands with PXR | Ref. |
| 1 | Baicalein from roots of Scutellaria baicalensis Georgi | Attenuated colonic inflammation in DSS induced colitis mice model through stimulation of CDX2. ↓TNFα and IL-6 mRNA levels in intestinal mucosa | Dou et al[122] Plos One 2012 |
| 2 | Forskolin and 1,9 dideoxyforskolin from roots of Coleus forskohlii | ↑CYP3A expression in primary hepatocytes through activation of Protein Kinase A signaling pathway | Ding and Staudinger[151] J Pharmacol Exp Ther 2005 |
| 3 | Z-guggulsterone from Commiphora mukul (Guggul) | ↓CYP7A1 gene in HepG2 cells | Owsley and Chiang[152] Biochem Biophys Res Commun 2003 |
| 4 | E-guggulsterone from Commiphora mukul (Guggul) | ↑CYP3A11 and CYP3A4 mRNA levels only in cultured hepatocytes from PXR +/+ mice and not in PXR KO mice | Brobst et al[153] J Pharmacol Exp Ther 2004 |
| 5 | Hyperforin from Hypericum perforatum (St. John’s wort) | ↑CYP3A4 induction in hepatocytes. Induction of CYP2C9 gene expression was also reported in humans | Moore et al[154] Proc Natl Acad Sci USA 2000 |
| 6 | Colupulone from Humulus lupulus (Hop Extract) | ↑CYP3A4, CYP2B6 and MDR1 gene expression in primary human hepatocytes dose dependently | Teotico et al[155] Mol Pharmacol 2008 |
| 7 | Kava Kava (Piper methysticum) | ↑CYP3A4 mRNA expression in primary human hepatocytes extensively | Raucy[156] Drug Metab Dispos 2003 |
| 8 | Wu Wei Zi [Dibenzocyclooctene lignans: schisandrol B, schisandrin (A and B)] | ↑Transcription of CYP3A4, CYP2C9 and MRP2 genes in primary hepatocytes | Mu et al[157] J Pharmacol Exp Ther 2006 |
| 9 | Ginkgolide A from Gingko Biloba extract | Protection against CCL4 induced acute toxicity model in rats, ↑Iκ-Bα transcription, which in turn inhibited NF-κB | Ye et al[64] Biomol Ther (Seoul) 2016 |
| 10 | Ginkgolide B from Gingko Biloba extract | ↑Nuclear translocation of PXR, and protected HUVEC cells from drug induced apoptosis. Anti-inflammatory role by reducing VCAM-1 and E-selectin induced by TNFα | Zhou et al[158] Acta Pharmacol Sin 2016 |
↑: Upregulation; ↓: Downregulation; PXR: Pregnane X receptor.
CONCLUSION
PXR plays a pivotal role as an endobiotic and xenobiotic sensor scanning for any alterations in the environmental cues and then translates the signals into an epithelial phenotype that is protective to host. Its role in maintaining homeostasis along the gut liver axis is undisputable. The majority of studies in exploration of PXR pathways, have been conducted only in IBD and associated pathologies. However, PXR is expressed copiously along the gut liver axis and has been proved to have a huge functional impact in both liver and intestine, and poses as an excellent target in CLDs with BT. As summarized above, PXR has important functional implications in each of the major pathophysiological mechanisms attributed to causing BT in CLD states. Targeting PXR with naturally occurring herbs or other polyphenolic compounds may potentially cease BT and attenuate the progression of liver disease or the manifestation of the associated fatal complications of CLD. Moreover, even though Rifaximin is a potent agonist for PXR, it can activate only gut PXR and is associated with adverse hepatotoxic side effects. The plethora of bioactive components from natural herbs are being discovered as effective activators for both human and rodent PXR, promising a fertile research ground for future studies. Using a CCL4 induced mouse cirrhotic model, our lab is currently investigating the effect of a naturally identified PXR ligand Ginkgolide-A, to further comprehend the functional impact of PXR activation on regulating BT.
Footnotes
Conflict-of-interest statement: None declared.
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: India
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: August 22, 2017
First decision: September 19, 2017
Article in press: October 30, 2017
P- Reviewer: Higuera-de la Tijera M, Morales-Gonzalez JAA, Pan W S- Editor: Qi Y L- Editor: A E- Editor: Lu YJ
Contributor Information
Sundhar Mohandas, Liver Diseases Research Lab, Department of Biochemistry, Jawaharlal Institute of Postgraduate Medical Education and Research, Dhanvantari Nagar, Pondicherry 605006, India.
Balasubramaniyan Vairappan, Liver Diseases Research Lab, Department of Biochemistry, Jawaharlal Institute of Postgraduate Medical Education and Research, Dhanvantari Nagar, Pondicherry 605006, India.
References
- 1.Olefsky JM. Nuclear receptor minireview series. J Biol Chem. 2001;276:36863–36864. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 4.Zhou C, Verma S, Blumberg B. The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl Recept Signal. 2009;7:e001. doi: 10.1621/nrs.07001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burk O, Koch I, Raucy J, Hustert E, Eichelbaum M, Brockmöller J, Zanger UM, Wojnowski L. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR) J Biol Chem. 2004;279:38379–38385. doi: 10.1074/jbc.M404949200. [DOI] [PubMed] [Google Scholar]
- 6.Haughton EL, Tucker SJ, Marek CJ, Durward E, Leel V, Bascal Z, Monaghan T, Koruth M, Collie-Duguid E, Mann DA, et al. Pregnane X receptor activators inhibit human hepatic stellate cell transdifferentiation in vitro. Gastroenterology. 2006;131:194–209. doi: 10.1053/j.gastro.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Wallace K, Cowie DE, Konstantinou DK, Hill SJ, Tjelle TE, Axon A, Koruth M, White SA, Carlsen H, Mann DA, et al. The PXR is a drug target for chronic inflammatory liver disease. J Steroid Biochem Mol Biol. 2010;120:137–148. doi: 10.1016/j.jsbmb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schote AB, Turner JD, Schiltz J, Muller CP. Nuclear receptors in human immune cells: expression and correlations. Mol Immunol. 2007;44:1436–1445. doi: 10.1016/j.molimm.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 9.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 10.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 11.Chang TK. Activation of pregnane X receptor (PXR) and constitutive androstane receptor (CAR) by herbal medicines. AAPS J. 2009;11:590–601. doi: 10.1208/s12248-009-9135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov. 2002;1:259–266. doi: 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- 13.Timsit YE, Negishi M. CAR and PXR: the xenobiotic-sensing receptors. Steroids. 2007;72:231–246. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodwin B, Redinbo MR, Kliewer SA. Regulation of cyp3a gene transcription by the pregnane x receptor. Annu Rev Pharmacol Toxicol. 2002;42:1–23. doi: 10.1146/annurev.pharmtox.42.111901.111051. [DOI] [PubMed] [Google Scholar]
- 15.Cheng X, Klaassen CD. Regulation of mRNA expression of xenobiotic transporters by the pregnane x receptor in mouse liver, kidney, and intestine. Drug Metab Dispos. 2006;34:1863–1867. doi: 10.1124/dmd.106.010520. [DOI] [PubMed] [Google Scholar]
- 16.Li T, Chiang JY. Rifampicin induction of CYP3A4 requires pregnane X receptor cross talk with hepatocyte nuclear factor 4alpha and coactivators, and suppression of small heterodimer partner gene expression. Drug Metab Dispos. 2006;34:756–764. doi: 10.1124/dmd.105.007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 18.Vyhlidal CA, Rogan PK, Leeder JS. Development and refinement of pregnane X receptor (PXR) DNA binding site model using information theory: insights into PXR-mediated gene regulation. J Biol Chem. 2004;279:46779–46786. doi: 10.1074/jbc.M408395200. [DOI] [PubMed] [Google Scholar]
- 19.Harmsen S, Meijerman I, Febus CL, Maas-Bakker RF, Beijnen JH, Schellens JH. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother Pharmacol. 2010;66:765–771. doi: 10.1007/s00280-009-1221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strassburg CP, Kalthoff S, Ehmer U. Variability and function of family 1 uridine-5’-diphosphate glucuronosyltransferases (UGT1A) Crit Rev Clin Lab Sci. 2008;45:485–530. doi: 10.1080/10408360802374624. [DOI] [PubMed] [Google Scholar]
- 21.Hariparsad N, Chu X, Yabut J, Labhart P, Hartley DP, Dai X, Evers R. Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res. 2009;37:1160–1173. doi: 10.1093/nar/gkn1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albermann N, Schmitz-Winnenthal FH, Z’graggen K, Volk C, Hoffmann MM, Haefeli WE, Weiss J. Expression of the drug transporters MDR1/ABCB1, MRP1/ABCC1, MRP2/ABCC2, BCRP/ABCG2, and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem Pharmacol. 2005;70:949–958. doi: 10.1016/j.bcp.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Li T, Yu RT, Atkins AR, Downes M, Tukey RH, Evans RM. Targeting the pregnane X receptor in liver injury. Expert Opin Ther Targets. 2012;16:1075–1083. doi: 10.1517/14728222.2012.715634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiao E, Ji M, Wu J, Ma R, Zhang X, He Y, Zha Q, Song X, Zhu LW, Tang J. Expression of the PXR gene in various types of cancer and drug resistance. Oncol Lett. 2013;5:1093–1100. doi: 10.3892/ol.2013.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol Sci. 2012;33:552–558. doi: 10.1016/j.tips.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright MC. The impact of pregnane X receptor activation on liver fibrosis. Biochem Soc Trans. 2006;34:1119–1123. doi: 10.1042/BST0341119. [DOI] [PubMed] [Google Scholar]
- 27.Tien ES, Negishi M. Nuclear receptors CAR and PXR in the regulation of hepatic metabolism. Xenobiotica. 2006;36:1152–1163. doi: 10.1080/00498250600861827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmuth M, Moosbrugger-Martinz V, Blunder S, Dubrac S. Role of PPAR, LXR, and PXR in epidermal homeostasis and inflammation. Biochim Biophys Acta. 2014;1841:463–473. doi: 10.1016/j.bbalip.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Mani S, Boelsterli UA, Redinbo MR. Understanding and modulating mammalian-microbial communication for improved human health. Annu Rev Pharmacol Toxicol. 2014;54:559–580. doi: 10.1146/annurev-pharmtox-011613-140007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones ML, Martoni CJ, Ganopolsky JG, Labbé A, Prakash S. The human microbiome and bile acid metabolism: dysbiosis, dysmetabolism, disease and intervention. Expert Opin Biol Ther. 2014;14:467–482. doi: 10.1517/14712598.2014.880420. [DOI] [PubMed] [Google Scholar]
- 31.Björkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One. 2009;4:e6958. doi: 10.1371/journal.pone.0006958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broomé U, Chapman RW. Ulcerative colitis: sclerosing cholangitis today, cancer tomorrow? Gut. 1997;41:571–572. doi: 10.1136/gut.41.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lisowska A, Kobelska-Dubiel N, Jankowska I, Pawłowska J, Moczko J, Walkowiak J. Small intestinal bacterial overgrowth in patients with progressive familial intrahepatic cholestasis. Acta Biochim Pol. 2014;61:103–107. [PubMed] [Google Scholar]
- 34.Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L, Fung S, Fischer SE, McGilvray IG, Allard JP. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS One. 2016;11:e0151829. doi: 10.1371/journal.pone.0151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C, Lund PK. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One. 2010;5:e12191. doi: 10.1371/journal.pone.0012191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen P, Stärkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology. 2015;61:883–894. doi: 10.1002/hep.27489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berg RD, Garlington AW. Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect Immun. 1979;23:403–411. doi: 10.1128/iai.23.2.403-411.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014;60:197–209. doi: 10.1016/j.jhep.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 39.Balzan S, de Almeida Quadros C, de Cleva R, Zilberstein B, Cecconello I. Bacterial translocation: overview of mechanisms and clinical impact. J Gastroenterol Hepatol. 2007;22:464–471. doi: 10.1111/j.1440-1746.2007.04933.x. [DOI] [PubMed] [Google Scholar]
- 40.Giannelli V, Di Gregorio V, Iebba V, Giusto M, Schippa S, Merli M, Thalheimer U. Microbiota and the gut-liver axis: bacterial translocation, inflammation and infection in cirrhosis. World J Gastroenterol. 2014;20:16795–16810. doi: 10.3748/wjg.v20.i45.16795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartmann P, Seebauer CT, Schnabl B. Alcoholic liver disease: the gut microbiome and liver cross talk. Alcohol Clin Exp Res. 2015;39:763–775. doi: 10.1111/acer.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llorente C, Schnabl B. The gut microbiota and liver disease. Cell Mol Gastroenterol Hepatol. 2015;1:275–284. doi: 10.1016/j.jcmgh.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desai AP, Reau N, Reddy KG, Te HS, Mohanty S, Satoskar R, Devoss A, Jensen D. Persistent spontaneous bacterial peritonitis: a common complication in patients with spontaneous bacterial peritonitis and a high score in the model for end-stage liver disease. Therap Adv Gastroenterol. 2012;5:275–283. doi: 10.1177/1756283X11417037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schnabl B. Linking intestinal homeostasis and liver disease. Curr Opin Gastroenterol. 2013;29:264–270. doi: 10.1097/MOG.0b013e32835ff948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 46.Pendyala S, Neff LM, Suárez-Fariñas M, Holt PR. Diet-induced weight loss reduces colorectal inflammation: implications for colorectal carcinogenesis. Am J Clin Nutr. 2011;93:234–242. doi: 10.3945/ajcn.110.002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ilan Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2609–2618. doi: 10.3748/wjg.v18.i21.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee SH. Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intest Res. 2015;13:11–18. doi: 10.5217/ir.2015.13.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 50.Shakhnovich V, Vyhlidal C, Friesen C, Hildreth A, Singh V, Daniel J, Kearns GL, Leeder JS. Decreased Pregnane X Receptor Expression in Children with Active Crohn’s Disease. Drug Metab Dispos. 2016;44:1066–1069. doi: 10.1124/dmd.115.068742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dring MM, Goulding CA, Trimble VI, Keegan D, Ryan AW, Brophy KM, Smyth CM, Keeling PW, O’Donoghue D, O’Sullivan M, et al. The pregnane X receptor locus is associated with susceptibility to inflammatory bowel disease. Gastroenterology. 2006;130:341–348; quiz 592. doi: 10.1053/j.gastro.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Martínez A, Márquez A, Mendoza J, Taxonera C, Fernández-Arquero M, Díaz-Rubio M, de la Concha EG, Urcelay E. Role of the PXR gene locus in inflammatory bowel diseases. Inflamm Bowel Dis. 2007;13:1484–1487. doi: 10.1002/ibd.20252. [DOI] [PubMed] [Google Scholar]
- 53.Glas J, Seiderer J, Fischer D, Tengler B, Pfennig S, Wetzke M, Beigel F, Olszak T, Weidinger M, Göke B, et al. Pregnane X receptor (PXR/NR1I2) gene haplotypes modulate susceptibility to inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:1917–1924. doi: 10.1002/ibd.21562. [DOI] [PubMed] [Google Scholar]
- 54.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006;46:123–149. doi: 10.1146/annurev.pharmtox.46.120604.141059. [DOI] [PubMed] [Google Scholar]
- 56.Gu X, Ke S, Liu D, Sheng T, Thomas PE, Rabson AB, Gallo MA, Xie W, Tian Y. Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J Biol Chem. 2006;281:17882–17889. doi: 10.1074/jbc.M601302200. [DOI] [PubMed] [Google Scholar]
- 57.Xie W, Tian Y. Xenobiotic receptor meets NF-kappaB, a collision in the small bowel. Cell Metab. 2006;4:177–178. doi: 10.1016/j.cmet.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 58.Zhou C, Tabb MM, Nelson EL, Grün F, Verma S, Sadatrafiei A, Lin M, Mallick S, Forman BM, Thummel KE, et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116:2280–2289. doi: 10.1172/JCI26283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1114–G1122. doi: 10.1152/ajpgi.00528.2006. [DOI] [PubMed] [Google Scholar]
- 60.Fiorucci S, Distrutti E, Mencarelli A, Barbanti M, Palazzini E, Morelli A. Inhibition of intestinal bacterial translocation with rifaximin modulates lamina propria monocytic cells reactivity and protects against inflammation in a rodent model of colitis. Digestion. 2002;66:246–256. doi: 10.1159/000068362. [DOI] [PubMed] [Google Scholar]
- 61.Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. 2011;4:31–42. doi: 10.1038/mi.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng J, Shah YM, Ma X, Pang X, Tanaka T, Kodama T, Krausz KW, Gonzalez FJ. Therapeutic role of rifaximin in inflammatory bowel disease: clinical implication of human pregnane X receptor activation. J Pharmacol Exp Ther. 2010;335:32–41. doi: 10.1124/jpet.110.170225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 64.Ye N, Wang H, Hong J, Zhang T, Lin C, Meng C. PXR Mediated Protection against Liver Inflammation by Ginkgolide A in Tetrachloromethane Treated Mice. Biomol Ther (Seoul) 2016;24:40–48. doi: 10.4062/biomolther.2015.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dou W, Zhang J, Zhang E, Sun A, Ding L, Chou G, Wang Z, Mani S. Chrysin ameliorates chemically induced colitis in the mouse through modulation of a PXR/NF-κB signaling pathway. J Pharmacol Exp Ther. 2013;345:473–482. doi: 10.1124/jpet.112.201863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dou W, Zhang J, Li H, Kortagere S, Sun K, Ding L, Ren G, Wang Z, Mani S. Plant flavonol isorhamnetin attenuates chemically induced inflammatory bowel disease via a PXR-dependent pathway. J Nutr Biochem. 2014;25:923–933. doi: 10.1016/j.jnutbio.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Staudinger JL, Xu C, Biswas A, Mani S. Post-translational modification of pregnane x receptor. Pharmacol Res. 2011;64:4–10. doi: 10.1016/j.phrs.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Priyanka, Kotiya D, Rana M, Subbarao N, Puri N, Tyagi RK. Transcription regulation of nuclear receptor PXR: Role of SUMO-1 modification and NDSM in receptor function. Mol Cell Endocrinol. 2016;420:194–207. doi: 10.1016/j.mce.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 69.Hu G, Xu C, Staudinger JL. Pregnane X receptor is SUMOylated to repress the inflammatory response. J Pharmacol Exp Ther. 2010;335:342–350. doi: 10.1124/jpet.110.171744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang W, Ghisletti S, Perissi V, Rosenfeld MG, Glass CK. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol Cell. 2009;35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chakravortty D, Kumar KS. Modulation of barrier function of small intestinal epithelial cells by lamina propria fibroblasts in response to lipopolysaccharide: possible role in TNFalpha in inducing barrier dysfunction. Microbiol Immunol. 1999;43:527–533. doi: 10.1111/j.1348-0421.1999.tb02438.x. [DOI] [PubMed] [Google Scholar]
- 73.Yajima S, Morisaki H, Serita R, Suzuki T, Katori N, Asahara T, Nomoto K, Kobayashi F, Ishizaka A, Takeda J. Tumor necrosis factor-alpha mediates hyperglycemia-augmented gut barrier dysfunction in endotoxemia. Crit Care Med. 2009;37:1024–1030. doi: 10.1097/CCM.0b013e31819b53b6. [DOI] [PubMed] [Google Scholar]
- 74.Goldman G, Soffer D, Heller L, Aderka D, Lahat A, Klausner JM. Tumour necrosis factor mediates bacterial translocation after haemorrhagic shock and endotoxaemia. Eur J Surg. 2001;167:299–304. doi: 10.1080/110241501300091543. [DOI] [PubMed] [Google Scholar]
- 75.Mencarelli A, Migliorati M, Barbanti M, Cipriani S, Palladino G, Distrutti E, Renga B, Fiorucci S. Pregnane-X-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem Pharmacol. 2010;80:1700–1707. doi: 10.1016/j.bcp.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 76.Ho GT, Moodie FM, Satsangi J. Multidrug resistance 1 gene (P-glycoprotein 170): an important determinant in gastrointestinal disease? Gut. 2003;52:759–766. doi: 10.1136/gut.52.5.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 78.Schwab M, Schaeffeler E, Marx C, Fromm MF, Kaskas B, Metzler J, Stange E, Herfarth H, Schoelmerich J, Gregor M, et al. Association between the C3435T MDR1 gene polymorphism and susceptibility for ulcerative colitis. Gastroenterology. 2003;124:26–33. doi: 10.1053/gast.2003.50010. [DOI] [PubMed] [Google Scholar]
- 79.Langmann T, Moehle C, Mauerer R, Scharl M, Liebisch G, Zahn A, Stremmel W, Schmitz G. Loss of detoxification in inflammatory bowel disease: dysregulation of pregnane X receptor target genes. Gastroenterology. 2004;127:26–40. doi: 10.1053/j.gastro.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 80.Toklu HZ, Kabasakal L, Imeryuz N, Kan B, Celikel C, Cetinel S, Orun O, Yuksel M, Dulger GA. A study comparing the efficacy of antimicrobial agents versus enzyme (P-gp) inducers in the treatment of 2,4,6 trinitrobenzenesulfonic acid-induced colitis in rats. J Physiol Pharmacol. 2013;64:439–451. [PubMed] [Google Scholar]
- 81.Blokzijl H, Vander Borght S, Bok LI, Libbrecht L, Geuken M, van den Heuvel FA, Dijkstra G, Roskams TA, Moshage H, Jansen PL, et al. Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels. Inflamm Bowel Dis. 2007;13:710–720. doi: 10.1002/ibd.20088. [DOI] [PubMed] [Google Scholar]
- 82.Ros JE, Schuetz JD, Geuken M, Streetz K, Moshage H, Kuipers F, Manns MP, Jansen PL, Trautwein C, Müller M. Induction of Mdr1b expression by tumor necrosis factor-alpha in rat liver cells is independent of p53 but requires NF-kappaB signaling. Hepatology. 2001;33:1425–1431. doi: 10.1053/jhep.2001.24667. [DOI] [PubMed] [Google Scholar]
- 83.Kalitsky-Szirtes J, Shayeganpour A, Brocks DR, Piquette-Miller M. Suppression of drug-metabolizing enzymes and efflux transporters in the intestine of endotoxin-treated rats. Drug Metab Dispos. 2004;32:20–27. doi: 10.1124/dmd.32.1.20. [DOI] [PubMed] [Google Scholar]
- 84.Buer J, Balling R. Mice, microbes and models of infection. Nat Rev Genet. 2003;4:195–205. doi: 10.1038/nrg1019. [DOI] [PubMed] [Google Scholar]
- 85.Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 86.Yue C, Ma B, Zhao Y, Li Q, Li J. Lipopolysaccharide-induced bacterial translocation is intestine site-specific and associates with intestinal mucosal inflammation. Inflammation. 2012;35:1880–1888. doi: 10.1007/s10753-012-9510-1. [DOI] [PubMed] [Google Scholar]
- 87.Koj A. Initiation of acute phase response and synthesis of cytokines. Biochim Biophys Acta. 1996;1317:84–94. doi: 10.1016/s0925-4439(96)00048-8. [DOI] [PubMed] [Google Scholar]
- 88.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 89.Ruminy P, Gangneux C, Claeyssens S, Scotte M, Daveau M, Salier JP. Gene transcription in hepatocytes during the acute phase of a systemic inflammation: from transcription factors to target genes. Inflamm Res. 2001;50:383–390. doi: 10.1007/PL00000260. [DOI] [PubMed] [Google Scholar]
- 90.Li-Masters T, Morgan ET. Effects of bacterial lipopolysaccharide on phenobarbital-induced CYP2B expression in mice. Drug Metab Dispos. 2001;29:252–257. [PubMed] [Google Scholar]
- 91.Sewer MB, Barclay TB, Morgan ET. Down-regulation of cytochrome P450 mRNAs and proteins in mice lacking a functional NOS2 gene. Mol Pharmacol. 1998;54:273–279. doi: 10.1124/mol.54.2.273. [DOI] [PubMed] [Google Scholar]
- 92.Moriya N, Kataoka H, Fujino H, Nishikawa J, Kugawa F. Effect of lipopolysaccharide on the xenobiotic-induced expression and activity of hepatic cytochrome P450 in mice. Biol Pharm Bull. 2012;35:473–480. doi: 10.1248/bpb.35.473. [DOI] [PubMed] [Google Scholar]
- 93.Sun M, Cui W, Woody SK, Staudinger JL. Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes. Drug Metab Dispos. 2015;43:335–343. doi: 10.1124/dmd.114.062307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mencarelli A, Renga B, Palladino G, Claudio D, Ricci P, Distrutti E, Barbanti M, Baldelli F, Fiorucci S. Inhibition of NF-κB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur J Pharmacol. 2011;668:317–324. doi: 10.1016/j.ejphar.2011.06.058. [DOI] [PubMed] [Google Scholar]
- 95.Xu DX, Wei W, Sun MF, Wu CY, Wang JP, Wei LZ, Zhou CF. Kupffer cells and reactive oxygen species partially mediate lipopolysaccharide-induced downregulation of nuclear receptor pregnane x receptor and its target gene CYP3a in mouse liver. Free Radic Biol Med. 2004;37:10–22. doi: 10.1016/j.freeradbiomed.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 96.Xu DX, Chen YH, Wang JP, Sun MF, Wang H, Wei LZ, Wei W. Perinatal lipopolysaccharide exposure downregulates pregnane X receptor and Cyp3a11 expression in fetal mouse liver. Toxicol Sci. 2005;87:38–45. doi: 10.1093/toxsci/kfi239. [DOI] [PubMed] [Google Scholar]
- 97.Xu DX, Wei W, Sun MF, Wei LZ, Wang JP. Melatonin attenuates lipopolysaccharide-induced down-regulation of pregnane X receptor and its target gene CYP3A in mouse liver. J Pineal Res. 2005;38:27–34. doi: 10.1111/j.1600-079X.2004.00171.x. [DOI] [PubMed] [Google Scholar]
- 98.Chen YH, Wang JP, Wang H, Sun MF, Wei LZ, Wei W, Xu DX. Lipopolysaccharide treatment downregulates the expression of the pregnane X receptor, cyp3a11 and mdr1a genes in mouse placenta. Toxicology. 2005;211:242–252. doi: 10.1016/j.tox.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 99.Wang X, Fang X, Zhou J, Chen Z, Zhao B, Xiao L, Liu A, Li YS, Shyy JY, Guan Y, et al. Shear stress activation of nuclear receptor PXR in endothelial detoxification. Proc Natl Acad Sci USA. 2013;110:13174–13179. doi: 10.1073/pnas.1312065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 101.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 102.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–G1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 103.Qiu Z, Cervantes JL, Cicek BB, Mukherjee S, Venkatesh M, Maher LA, Salazar JC, Mani S, Khanna KM. Pregnane X Receptor Regulates Pathogen-Induced Inflammation and Host Defense against an Intracellular Bacterial Infection through Toll-like Receptor 4. Sci Rep. 2016;6:31936. doi: 10.1038/srep31936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garg A, Zhao A, Erickson SL, Mukherjee S, Lau AJ, Alston L, Chang TK, Mani S, Hirota SA. Pregnane X Receptor Activation Attenuates Inflammation-Associated Intestinal Epithelial Barrier Dysfunction by Inhibiting Cytokine-Induced Myosin Light-Chain Kinase Expression and c-Jun N-Terminal Kinase 1/2 Activation. J Pharmacol Exp Ther. 2016;359:91–101. doi: 10.1124/jpet.116.234096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Esposito G, Nobile N, Gigli S, Seguella L, Pesce M, d’Alessandro A, Bruzzese E, Capoccia E, Steardo L, Cuomo R, et al. Rifaximin Improves Clostridium difficile Toxin A-Induced Toxicity in Caco-2 Cells by the PXR-Dependent TLR4/MyD88/NF-κB Pathway. Front Pharmacol. 2016;7:120. doi: 10.3389/fphar.2016.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity. 2014;41:296–310. doi: 10.1016/j.immuni.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ivanov AI. Structure and regulation of intestinal epithelial tight junctions: current concepts and unanswered questions. Adv Exp Med Biol. 2012;763:132–148. doi: 10.1007/978-1-4614-4711-5_6. [DOI] [PubMed] [Google Scholar]
- 108.Weber CR. Dynamic properties of the tight junction barrier. Ann N Y Acad Sci. 2012;1257:77–84. doi: 10.1111/j.1749-6632.2012.06528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr. 2011;141:769–776. doi: 10.3945/jn.110.135657. [DOI] [PubMed] [Google Scholar]
- 110.Jiang Y, Guo C, Zhang D, Zhang J, Wang X, Geng C. The altered tight junctions: an important gateway of bacterial translocation in cachexia patients with advanced gastric cancer. J Interferon Cytokine Res. 2014;34:518–525. doi: 10.1089/jir.2013.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci. 2012;1258:34–42. doi: 10.1111/j.1749-6632.2012.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–G430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 113.He F, Peng J, Deng XL, Yang LF, Camara AD, Omran A, Wang GL, Wu LW, Zhang CL, Yin F. Mechanisms of tumor necrosis factor-alpha-induced leaks in intestine epithelial barrier. Cytokine. 2012;59:264–272. doi: 10.1016/j.cyto.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 114.Roy PK, Rashid F, Bragg J, Ibdah JA. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J Gastroenterol. 2008;14:200–202. doi: 10.3748/wjg.14.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 116.Liu G, Rondinone CM. JNK: bridging the insulin signaling and inflammatory pathway. Curr Opin Investig Drugs. 2005;6:979–987. [PubMed] [Google Scholar]
- 117.Kersting S, Reinecke K, Hilgert C, Janot MS, Haarmann E, Albrecht M, Müller AM, Herdegen T, Mittelkötter U, Uhl W, et al. Knockout of the c-Jun N-terminal Kinase 2 aggravates the development of mild chronic dextran sulfate sodium colitis independently of expression of intestinal cytokines TNFα, TGFB1, and IL-6. J Inflamm Res. 2013;6:13–23. doi: 10.2147/JIR.S36415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kersting S, Behrendt V, Kersting J, Reinecke K, Hilgert C, Stricker I, Herdegen T, Janot MS, Uhl W, Chromik AM. The impact of JNK inhibitor D-JNKI-1 in a murine model of chronic colitis induced by dextran sulfate sodium. J Inflamm Res. 2013;6:71–81. doi: 10.2147/JIR.S40092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mitsuyama K, Suzuki A, Tomiyasu N, Tsuruta O, Kitazaki S, Takeda T, Satoh Y, Bennett BL, Toyonaga A, Sata M. Pro-inflammatory signaling by Jun-N-terminal kinase in inflammatory bowel disease. Int J Mol Med. 2006;17:449–455. [PubMed] [Google Scholar]
- 120.Dou W, Mukherjee S, Li H, Venkatesh M, Wang H, Kortagere S, Peleg A, Chilimuri SS, Wang ZT, Feng Y, et al. Alleviation of gut inflammation by Cdx2/Pxr pathway in a mouse model of chemical colitis. PLoS One. 2012;7:e36075. doi: 10.1371/journal.pone.0036075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saad RS, Ghorab Z, Khalifa MA, Xu M. CDX2 as a marker for intestinal differentiation: Its utility and limitations. World J Gastrointest Surg. 2011;3:159–166. doi: 10.4240/wjgs.v3.i11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Coskun M, Troelsen JT, Nielsen OH. The role of CDX2 in intestinal homeostasis and inflammation. Biochim Biophys Acta. 2011;1812:283–289. doi: 10.1016/j.bbadis.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 123.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Claudel T, Zollner G, Wagner M, Trauner M. Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim Biophys Acta. 2011;1812:867–878. doi: 10.1016/j.bbadis.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 126.Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66:948–983. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Attili AF, Angelico M, Cantafora A, Alvaro D, Capocaccia L. Bile acid-induced liver toxicity: relation to the hydrophobic-hydrophilic balance of bile acids. Med Hypotheses. 1986;19:57–69. doi: 10.1016/0306-9877(86)90137-4. [DOI] [PubMed] [Google Scholar]
- 128.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703–722. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 129.Ishii M, Toda T, Ikarashi N, Kusunoki Y, Kon R, Ochiai W, Machida Y, Sugiyama K. Gastrectomy increases the expression of hepatic cytochrome P450 3A by increasing lithocholic acid-producing enteric bacteria in mice. Biol Pharm Bull. 2014;37:298–305. doi: 10.1248/bpb.b13-00824. [DOI] [PubMed] [Google Scholar]
- 130.Banerjee S, Sindberg G, Wang F, Meng J, Sharma U, Zhang L, Dauer P, Chen C, Dalluge J, Johnson T, et al. Opioid-induced gut microbial disruption and bile dysregulation leads to gut barrier compromise and sustained systemic inflammation. Mucosal Immunol. 2016;9:1418–1428. doi: 10.1038/mi.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stenman LK, Holma R, Korpela R. High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J Gastroenterol. 2012;18:923–929. doi: 10.3748/wjg.v18.i9.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–338. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nie YF, Hu J, Yan XH. Cross-talk between bile acids and intestinal microbiota in host metabolism and health. J Zhejiang Univ Sci B. 2015;16:436–446. doi: 10.1631/jzus.B1400327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stenman LK, Holma R, Eggert A, Korpela R. A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol. 2013;304:G227–G234. doi: 10.1152/ajpgi.00267.2012. [DOI] [PubMed] [Google Scholar]
- 135.Hughes R, Kurth MJ, McGilligan V, McGlynn H, Rowland I. Effect of colonic bacterial metabolites on Caco-2 cell paracellular permeability in vitro. Nutr Cancer. 2008;60:259–266. doi: 10.1080/01635580701649644. [DOI] [PubMed] [Google Scholar]
- 136.Fickert P, Fuchsbichler A, Marschall HU, Wagner M, Zollner G, Krause R, Zatloukal K, Jaeschke H, Denk H, Trauner M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol. 2006;168:410–422. doi: 10.2353/ajpath.2006.050404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vu DD, Tuchweber B, Raymond P, Yousef IM. Tight junction permeability and liver plasma membrane fluidity in lithocholate-induced cholestasis. Exp Mol Pathol. 1992;57:47–61. doi: 10.1016/0014-4800(92)90048-g. [DOI] [PubMed] [Google Scholar]
- 138.Uppal H, Toma D, Saini SP, Ren S, Jones TJ, Xie W. Combined loss of orphan receptors PXR and CAR heightens sensitivity to toxic bile acids in mice. Hepatology. 2005;41:168–176. doi: 10.1002/hep.20512. [DOI] [PubMed] [Google Scholar]
- 139.Alaish SM, Smith AD, Timmons J, Greenspon J, Eyvazzadeh D, Murphy E, Shea-Donahue T, Cirimotich S, Mongodin E, Zhao A, et al. Gut microbiota, tight junction protein expression, intestinal resistance, bacterial translocation and mortality following cholestasis depend on the genetic background of the host. Gut Microbes. 2013;4:292–305. doi: 10.4161/gmic.24706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kliewer SA, Willson TM. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res. 2002;43:359–364. [PubMed] [Google Scholar]
- 141.Ajouz H, Mukherji D, Shamseddine A. Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol. 2014;12:164. doi: 10.1186/1477-7819-12-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Reddy BS. Diet and excretion of bile acids. Cancer Res. 1981;41:3766–3768. [PubMed] [Google Scholar]
- 143.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA. 2001;98:3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR) Proc Natl Acad Sci USA. 2002;99:13801–13806. doi: 10.1073/pnas.212494599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ranhotra HS, Flannigan KL, Brave M, Mukherjee S, Lukin DJ, Hirota SA, Mani S. Xenobiotic Receptor-Mediated Regulation of Intestinal Barrier Function and Innate Immunity. Nucl Receptor Res. 2016;3:101199. doi: 10.11131/2016/101199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. 2012;56:1283–1292. doi: 10.1016/j.jhep.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients. 2012;4:1095–1119. doi: 10.3390/nu4081095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Brave M, Lukin DJ, Mani S. Microbial control of intestinal innate immunity. Oncotarget. 2015;6:19962–19963. doi: 10.18632/oncotarget.4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ding X, Staudinger JL. Induction of drug metabolism by forskolin: the role of the pregnane X receptor and the protein kinase a signal transduction pathway. J Pharmacol Exp Ther. 2005;312:849–856. doi: 10.1124/jpet.104.076331. [DOI] [PubMed] [Google Scholar]
- 152.Owsley E, Chiang JY. Guggulsterone antagonizes farnesoid X receptor induction of bile salt export pump but activates pregnane X receptor to inhibit cholesterol 7alpha-hydroxylase gene. Biochem Biophys Res Commun. 2003;304:191–195. doi: 10.1016/s0006-291x(03)00551-5. [DOI] [PubMed] [Google Scholar]
- 153.Brobst DE, Ding X, Creech KL, Goodwin B, Kelley B, Staudinger JL. Guggulsterone activates multiple nuclear receptors and induces CYP3A gene expression through the pregnane X receptor. J Pharmacol Exp Ther. 2004;310:528–535. doi: 10.1124/jpet.103.064329. [DOI] [PubMed] [Google Scholar]
- 154.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, Willson TM, Collins JL, Kliewer SA. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci USA. 2000;97:7500–7502. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Teotico DG, Bischof JJ, Peng L, Kliewer SA, Redinbo MR. Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol Pharmacol. 2008;74:1512–1520. doi: 10.1124/mol.108.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Raucy JL. Regulation of CYP3A4 expression in human hepatocytes by pharmaceuticals and natural products. Drug Metab Dispos. 2003;31:533–539. doi: 10.1124/dmd.31.5.533. [DOI] [PubMed] [Google Scholar]
- 157.Mu Y, Zhang J, Zhang S, Zhou HH, Toma D, Ren S, Huang L, Yaramus M, Baum A, Venkataramanan R, et al. Traditional Chinese medicines Wu Wei Zi (Schisandra chinensis Baill) and Gan Cao (Glycyrrhiza uralensis Fisch) activate pregnane X receptor and increase warfarin clearance in rats. J Pharmacol Exp Ther. 2006;316:1369–1377. doi: 10.1124/jpet.105.094342. [DOI] [PubMed] [Google Scholar]
- 158.Zhou T, You WT, Ma ZC, Liang QD, Tan HL, Xiao CR, Tang XL, Zhang BL, Wang YG, Gao Y. Ginkgolide B protects human umbilical vein endothelial cells against xenobiotic injuries via PXR activation. Acta Pharmacol Sin. 2016;37:177–186. doi: 10.1038/aps.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]