

Abstract
Pyruvate dehydrogenase complex (PDHC) deficiency is a rare metabolic disorder that affects tissues with high energy demand such as the central nervous system. The clinico-radiological phenotype of Leigh's disease is one of its common presentations. We present a 9-month-old boy with rapidly progressive infantile Leigh's disease. PDHA1 gene sequencing revealed a pathological homozygous missense mutation c.131A>G or p.H44R in exon 3 consistent with PDHC deficiency. H44R is among the five mutations (H44R, R88S, G89S, R263G, and V389fs) in E1α subunit that is thiamine-responsive. The child was initiated on thiamine, riboflavin, carnitine, coenzyme Q, and sodium benzoate supplementation. Mild recovery was noted at 6 months follow up as no further episodes of encephalopathy occurred. Thereafter, the child was treated with Ketogenic diet which resulted in increased levels of activity and alertness. Despite an improving course, the child had a sudden unexpected death at the age of 21 months.
KEYWORDS: E1α subunit, Leigh's disease, pyruvate dehydrogenase complex deficiency, thiamine, X-linked
INTRODUCTION
Pyruvate dehydrogenase complex (PDHC) deficiency is a rare metabolic disorder that affects tissues with high energy demand such as the central nervous system. PDH complex comprises five major components: Pyruvate dehydrogenase (E1), dihydrolipoyl transacetylase (E2), dihydrolipoamide dehydrogenase (E3), and two regulatory enzymes, namely, pyruvate dehydrogenase phosphatase and pyruvate dehydrogenase kinase.[1] PDH serves as a link between glycolysis and the tricarboxylic acid cycle. Any defect in PDHC results in decreased acetyl-CoA synthesis and accumulation of pyruvate and lactate, causing metabolic acidosis.
The majority of PDHC deficiencies result from mutations in the X-linked pyruvate dehydrogenase (E1) α subunit gene (PDHA1).[2] The E1 subunit is known to contain a thiamine pyrophosphate (TPP) binding site. Certain mutations outside the TPP binding site such as H44R, R88S, G89S, R263G, and V389fs and within the TPP-binding site such as A1 99 T, F2 0 5L, M21 0V, W214R, and P217L are associated with good response to thiamine treatment.[3]
We report a 9-month-old infant with persistent lactic acidosis and a rapidly progressive Leigh's disease. He harbored a pathological mutation in E1α subunit of PDHC gene and showed an initial response to high dose thiamine supplementation and ketogenic diet.
CASE REPORT
A 9-month-old boy, second issue of nonconsanguineous parents, presented with recurrent regression of milestones. Birth history and the neonatal period were uneventful. At the age of 4½ months, he had an episode of lethargy, and listlessness following a minor febrile illness. During this illness, he lost his previous abilities of partial head control and social smile only to regain them back by 6 months of age. Similar episodes of regression lasting 7–10 days reoccurred at 7 months and 8½ months of age. These episodes were preceded by abnormal labored breathing and mild fever. At 8 months of age, he developed an isolated episode of apnea and cyanosis while sleeping which resolved on its own within minutes. Parents felt that the child was floppy and had difficulty in swallowing and chewing semisolid feeds. There was no history of seizures or dystonic posturing of limbs. His best-attained milestones were partial head control and social smile.
On examination, he had microcephaly (head circumference = 40.5 cm; <−3 standard deviation [SD]) and failure to thrive (Weight: 7.2 kg [at −2 SD]; Length: 67.5 cm [at −2 SD]). Cranial nerves examination, vision and hearing, were normal. Fundus was unremarkable. Both axial and appendicular tone was decreased with persistent fisting and hyporeflexia. There were no extrapyramidal signs. Serum biochemistry revealed persistent lactic acidemia on multiple occasions with arterial lactate levels ranging between 5.8 and 10.4 mmol/L (reference range <2.5 mmol/L). Cerebrospinal fluid analysis showed elevated lactate (10.5 mmol/L [reference range 1.1–2.4 mmol/L]); normal protein (14.4 mg/dl) and sugar (56 mg/dl). Pyruvate levels could not be obtained. Serum ammonia was elevated (254 mg/dl), Blood gases revealed high anion gap metabolic acidosis. Hemogram, liver function tests, renal function tests, and blood sugars were within normal limits. Endocrine workup including thyroid stimulating hormone, cortisol, parathyroid, and Vitamin D levels were normal. Tandem mass spectrometry of blood did not reveal any abnormality. Urine gas chromatography-mass spectrometry showed elevated lactate. Electrocardiograph and Echocardiogram were insignificant.
Magnetic resonance imaging (MRI) of the brain was performed twice on 1.5 tesla MRI scanner. MRI brain at 4 months of age was unremarkable except for a mild paucity of white matter. However, the second imaging performed at 9 months of age revealed bilateral T2/fluid attenuated inverse recovery hyperintense globus pallidus and substantia nigra [Figure 1] suggestive of Leigh's disease. A prominent lactate doublet with a mild decrease in NAA:Cr ratio was observed on spectroscopy.
Figure 1.
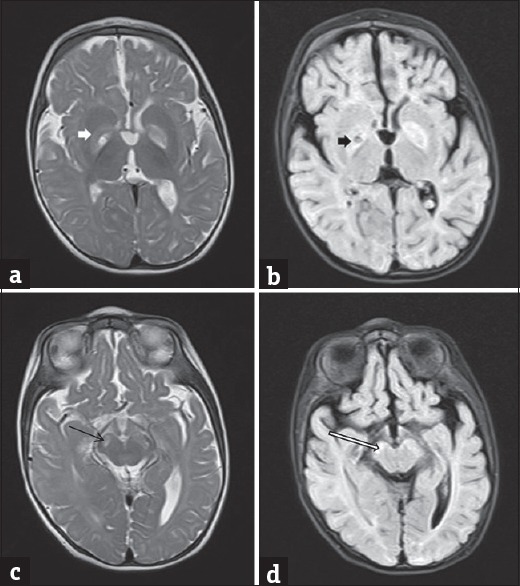
Axial T2 (a and c) and fluid attenuation inversion recovery (b and d) magnetic resonance images showing hyperintensity in the globus pallidus (white arrow in a and black arrow in b) and substantia nigra (black arrow in c and white arrow in d)
Polymerase chain reaction amplification of all coding exons and intronic flanking regions of PDHA1 gene was performed on genomic DNA of the patient. Direct sequencing of amplification products was performed in both forward and reverse directions, using automated fluorescence dideoxy sequencing method (ABI3730 platform with the software sequence scanner, and codon aligner). It revealed a homozygous missense mutation c.131A>G or p.H44R in exon 3 of PDHA1 gene. H44R is a known pathological mutation.[4] Genetic testing of parents could not be performed. Since H44R mutation has been previously described as thiamine-responsive so a high dose thiamine supplementation (500 mg/day) was initiated along with riboflavin, coenzyme Q, and carnitine.[4] At 5 months follow-up (14 months of age), no further episodes of encephalopathy occurred. There was a gain in previously lost milestones. The child achieved transient sitting with support and babbling. He was able to grasp objects, roll prone to supine, smile at a mirror image, and developed stranger anxiety. At 15 months of age, he was also started on a ketogenic diet on which he was on an improving course. However, at 21 months of age, the child had a sudden unexpected death. He was found dead at night in his crib in a pool of vomitus. An autopsy could not be performed.
DISCUSSION
PDHC deficiency is the most common biochemically proven cause of congenital lactic acidosis.[5] It is a heterogeneous disorder with broadly three clinical forms: Early neonatal form with severe lactic acidosis at birth and death in the neonatal period, an infantile form with moderate lactic acidosis but with severe psychomotor retardation and a late onset form with mild lactic acidosis, episodic ataxia, and mild developmental delay.[6] In a recent review of all published cases of PDHC deficiency between 1970 and 2010, 56.6% of cases were because of mutations in the E1α subunit.[2]
Clinico-radiological phenotype of Leigh's disease is seen in 27% of cases of PDHC deficiency.[2] Previous researchers have shown that certain mutations in the X-linked PDHA1 gene cause decreased affinity of PDHC for TPP and a consequent defective enzymatic activation. Such cases may show a good response to thiamine supplementation, especially at a high dose. H44R mutation in the PDHA1 gene in the index child is among the five mutations associated with a favorable response to thiamine.[3] Thiamine supplementation in the index child not only prevented further episodes of encephalopathy but also allowed the child to rapidly gain new milestones.
Current therapies found useful in PDH deficiency include the activation of residual PDH with dichloroacetate, the administration of cofactors such as thiamine and lipoic acid, and ketogenic diet. The clinical phenotype and choice of therapy may depend on the site and type of underlying mutation.[7] Some reports in the literature have shown a good response to thiamine supplementation in a subset of children with PDHC deficiency.[1,3,6,8] Most of these cases were assumed to have or indeed had specific mutations in the E1α subunit interfering the interaction between PDH and TPP. Naito et al. reported two cases with E1α subunit gene defect, one of which was a 10-month-old boy with Leigh's disease, who showed in vivo (clinical) response to thiamine and in vitro response to TPP, however, the degree of clinical improvement was not elaborated.[3] Similarly, most of the previously reported cases have demonstrated in vitro response but whether or not a persistent and significant clinical response is observed is unclear.[3,8]
Despite the improving course, the index child had a sudden unexpected demise. There could be many possible reasons for this. Abrupt unexplained apneic episodes secondary to brainstem dysfunction are a known feature of Leigh's disease. In addition, aspiration secondary to oropharyngeal incoordination or a cardiac arrhythmia cannot be conclusively excluded. We infer from our case that a subset of cases of Leigh's disease with underlying PDHA1 gene mutation show marked response with high dose thiamine supplementation. However, this response is mutation dependent and variable. Whether or not a sustained benefit occurs can only be deduced by a long-term follow-up of a large series of cases with a uniform treatment protocol.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Pastoris O, Savasta S, Foppa P, Catapano M, Dossena M. Pyruvate dehydrogenase deficiency in a child responsive to thiamine treatment. Acta Paediatr. 1996;85:625–8. doi: 10.1111/j.1651-2227.1996.tb14104.x. [DOI] [PubMed] [Google Scholar]
- 2.Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;105:34–43. doi: 10.1016/j.ymgme.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Ogawa Y, et al. Thiamine-responsive pyruvate dehydrogenase deficiency in two patients caused by a point mutation (F205L and L216F) within the thiamine pyrophosphate binding region. Biochim Biophys Acta. 2002;1588:79–84. doi: 10.1016/s0925-4439(02)00142-4. [DOI] [PubMed] [Google Scholar]
- 4.Jacobia SJ, Korotchkina LG, Patel MS. Characterization of a missense mutation at histidine-44 in a pyruvate dehydrogenase-deficient patient. Biochim Biophys Acta. 2002;1586:32–42. doi: 10.1016/s0925-4439(01)00083-7. [DOI] [PubMed] [Google Scholar]
- 5.Robinson BH. Scriver CR, Beaudet AL, Sly WS, Valle D. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 1995. Lactic academia (disorders of pyruvate carboxylase, pyruvate dehydrogenase) pp. 1479–99. [Google Scholar]
- 6.Di Rocco M, Lamba LD, Minniti G, Caruso U, Naito E. Outcome of thiamine treatment in a child with Leigh disease due to thiamine-responsive pyruvate dehydrogenase deficiency. Eur J Paediatr Neurol. 2000;4:115–7. doi: 10.1053/ejpn.2000.0278. [DOI] [PubMed] [Google Scholar]
- 7.Cross JH, Connelly A, Gadian DG, Kendall BE, Brown GK, Brown RM, et al. Clinical diversity of pyruvate dehydrogenase deficiency. Pediatr Neurol. 1994;10:276–83. doi: 10.1016/0887-8994(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 8.Lee EH, Ahn MS, Hwang JS, Ryu KH, Kim SJ, Kim SH. A Korean female patient with thiamine-responsive pyruvate dehydrogenase complex deficiency due to a novel point mutation (Y161C) in the PDHA1 gene. J Korean Med Sci. 2006;21:800–4. doi: 10.3346/jkms.2006.21.5.800. [DOI] [PMC free article] [PubMed] [Google Scholar]