

Abstract
The plasma zymogens factor XI (fXI) and prekallikrein (PK) are activated by factor XIIa (fXIIa) during contact activation. Polyanions such as DNA and RNA may contribute to thrombosis and inflammation partly by enhancing PK and fXI activation. We examined PK and fXI activation in the presence of nucleic acids, and determine the effects of the cofactor high molecular weight kininogen (HK) on the reactions. In the absence of HK, DNA and RNA induced of fXI autoactivation. Proteases known to activate fXI (fXIIa and thrombin) did not enhance this process appreciably. Nucleic acids had little effect on PK activation by fXIIa in the absence of HK. HK had significant but opposite effects on PK and fXI activation. HK enhanced fXIIa activation of PK in the presence of nucleic acids, but blocked fXI autoactivation. Thrombin and fXIIa could overcome the HK inhibitory effect on autoactivation, indicating these proteases are necessary for nucleic acid-induced fXI activation in an HK-rich environment such as plasma. In contrast to PK, which requires HK for optimal activation, fXI activation in the presence of nucleic acids depends on anion binding sites on the fXI molecule. The corresponding sites on PK are not necessary for PK activation. Our results indicate that HK functions as a cofactor for PK activation in the presence of nucleic acids in a manner consistent with classic models of contact activation. However, HK has, on balance, an inhibitory effect on nucleic acid-supported fXI activation and may function as a negative regulator of fXI activation.
INTRODUCTION
Polymers such as polyphosphates (Poly-P) and nucleic acids have procoagulant effects when added to blood in vitro, and are thought to contribute to thrombosis and inflammation in animal models and in human disease (1-7). These polyanions induce activation of the plasma kallikrein-kinin system by a process called contact activation (1-4,8-10). Exposure of blood to artificial surfaces triggers reciprocal conversion of the zymogens factor XII (fXII) and prekallikrein (PK) to the proteases factor XIIa (fXIIa) and α-kallikrein by limited proteolysis (8-10). The reactions are supported by high molecular weight kininogen (HK), a protein that facilitates PK binding to the surface (8-12). Contact activation, or a process similar to it, appears to occur at a basal level in healthy mice (13), suggesting the presence of naturally occurring counterparts to the artificial surfaces that induce the process in vitro. Poly-P and nucleic acids are considered strong candidates for such cofactors.
Plasma components of the kallikrein-kinin system (fXII, PK and HK) contribute to thrombosis in a variety of animal models (1,13-19) and may play a role in human thrombosis, particularly when blood is exposed to foreign surfaces such as during extracorporeal circulation (20-22). The kallikrein-kinin system may influence thrombus formation by several mechanisms, with fXIIa-mediated conversion of factor XI (fXI) to the protease factor XIa (fXIa) playing a significant part (23). FXI is a homolog of PK (24,25) that is also activated by thrombin (26-30). FXI activation by thrombin is enhanced by many of the same polyanions that support contact activation in plasma (31-33).
PK and fXI circulate in plasma largely as complexes with HK (12). During contact activation on non-biologic surfaces such as kaolin, HK may facilitate fXI binding to the surface in the same manner that it supports PK binding (11). However, there is little data on the effects of HK on fXI or PK activation in the presence of biologic polyanions that may support the processes in vivo. Here we use nucleic acid-supported protease activation as a model system to study PK and fXI activation, and to assess the effects of HK on activation. We show that HK enhances PK activation in the presence of nucleic acids in a manner consistent with models of contact activation on non-biologic surfaces. However, FXI appears to interact with nucleic acids in a different manner, relying on specific anion binding sites, with HK serving an inhibitory or modulatory role in fXI activation.
MATERIALS AND METHODS
Materials
Human plasma; Precision Biologicals. α-Thrombin, fXI and fXIa; Hematologic Technologies. Corn trypsin inhibitor (CTI), PK, fXIIa, HK, and HKa; Enzyme Research Lab. RecombiPlasTin human tissue factor; Instrumentation Laboratory. Diisopropylfluorophosphate (DIP), bovine serum albumin (BSA), bovine pancrease deoxyribonuclease I (DNase), and ribonuclease I-AS (RNase); Sigma-Aldrich. Phosphatidylcholine:phosphatidylserine (PC/PS) vesicles, Avanti Polar Lipids. Argatroban, GlaxoSmith Kline. S-2302 (H-D-prolyl-L-phenyl-alanyl-L-arginine-p-nitroaniline dihydrochloride), S-2366 (L-pyro-Glu-L-Pro-L-Arg-p- nitroaniline dihydrochloride); DiaPharma. Z-Gly-Gly-Arg-AMC, Bachem. Genomic DNA was prepared from human blood leukocytes by phenol-chloroform extraction. 32 and 64 base pair species of double stranded DNA were prepared using oligonucleotides generated in the Vanderbilt University Medical Center DNA core facility. RNA was isolated from C57Bl/6 mouse liver in Trizol. DNA and RNA were dissolved in water or TBE buffer, respectively, and stored at -80 0C. Anti-fXI IgG O1A6 (34) and anti-FXIIa IgG D06 (35) have been described.
Recombinant fXI
Human fXI was expressed in HEK293 fibroblasts and purified from media as described (36). Wild type fXI (fXI-WT), and variants with alanine replacing the active site serine (fXI-Ala557) (30); Arg250, Lys252, Lys253 and Lys255 which form an anion binding site (ABS) on the A3 domain (fXI-ABS1) (37,38); and Lys529, Arg530 and Arg532 which form an ABS on the catalytic domain (fXI-ABS2) (37,39) were prepared. FXI was incubated with 500 μM DIP on ice for 1 hr to inhibit fXIa, followed by dialysis into 4 mM Sodium Acetate-HCl, 150 mM NaCl, pH 5.3.
Recombinant PK
Human wild type PK (PK-WT) and PK with alanine replacing Lys531, Arg532 and Lys537 (PK-ABS2) were expressed in HEK293 cells under serum free conditions (Dulbecco’s Modified Eagle Medium supplemented with insulin, transferring and selenium) using the expression system used for fXI (36). Sodium acetate was added to conditioned media to 50 mM to adjust pH to 5.2, followed by application to an SP Sepharose column and elution with a 100-1000 mM NaCl gradient. PK-containing fractions were pooled, dialyzed against 25 mM Tris-HCl 50 mM NaCl pH 8.0 and reapplied to the SP Sepharose column. Elution was with a 50-1000 mM NaCl gradient. PK containing fractions were dialyzed into 4 mM sodium acetate, 150 mM NaCl, pH 5.2 (Supplemental Fig.1A).
Chromogenic assays for fXI and PK activation
FXI (30 nM) or PK (60 nM) activation was carried out in standard buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 10 μM ZnCl2, 0.1% PEG-8000) at 37 °C. Some reactions contained DNA or RNA (0.05 to 100 μg/ml), HK or HKa (30 to 60 nM), fXIIa (2.5 nM for fXI, 50 pM for PK) or thrombin (2.5 nM). For reactions with fXI, 5 μl samples were removed at various times into 25 μl Polybrene (320 μg/ml) containing 8 μM Argatroban (for reactions with thrombin) or 1.6 μM CTI (for reactions with fXIIa). For reactions with PK 5 μl samples were removed into 75 μl Polybrene (130 μg/ml) containing 13 nM IgG D06. Then 10 μl of 1 mM S-2366 (for fXIa) or S-S2302 (for α-kallikrein) was added and ∆OD 405 nm was followed on a microplate reader. Results were compared to standard curves constructed with fXIa or α-kallikrein.
FXI-Ala557 cleavage
FXI-Ala557 (200 nM) was incubated in standard buffer at 37 °C with fXIIa or thrombin (15 nM), in the absence or presence of DNA (30 μg/ml) or RNA (5 μg/ml), and in the absence or presence of HK (200 nM). At various times, aliquots were removed into reducing SDS-sample buffer, followed by size fractionation on 10% acrylamide-SDS gels. Gels were stained with GelCode Blue (Thermo) and analyzed by densitometry.
Thrombin generation
Thrombin generation in plasma was measured at 37 °C on a Fluoroskan Ascent analyzer as described (29,30). Briefly, assays were performed in 96-well round bottom polypropylene plates. Plasma was supplemented with 415 μM Z-Gly-Gly-Arg-AMC, 5 μM PC:PS vesicles, 1.6 μM CTI or vehicle, 50 μg/ml of the anti-fXI IgG O1A6 or vehicle, tissue factor (0.5 pM) or vehicle, and DNA (25 μg/ml). Supplemented plasma (100 μl) was mixed with 20 μl of 20 mM HEPES pH 7.4, 100 mM CaCl2, 6% BSA and fluorescence (excitation λ 390 nm, emission λ 460 nm) was monitored. Each set of conditions was tested in duplicate. Peak thrombin generation and area under the curve (Endogenous Thrombin Potential, ETP) were calculated using Thrombinoscope Analysis software, version 3.0.
Thrombin generation in plasma supplemented with human neutrophils
Human neutrophils were isolated from acid-citrate dextrose anticoagulated blood by centrifugation through a percol gradient, and suspended in RPMI-1640 medium at 1×103/μL. Cells (100 μl) were placed in 96 well polypropylene platelets and allowed to adhere for one hour in 5% CO2 humidified air at 37 °C, followed by incubation with vehicle or phorbol-myristate acetate (100 ng/ml) for 4 hrs with or without DNase (100 U/ml). For fluorescent microscopy, media was replaced with fresh media containing 10 μM Hoechst 33342 and 1 μM SytoxGreen. For thrombin generation assays, media was replaced with 100 μl of normal plasma supplemented with 415 μM Z-Gly-Gly-Arg-AMC, 5 μM PC:PS vesicles, 1.6 μM CTI or vehicle, 100 U/ml DNase or vehicle, and 50 μg/ml of the anti-fXI IgG O1A6 or vehicle. Thrombin generation was initiated by addition of 20 μl of 20 mM HEPES pH 7.4, 100 mM CaCl2, 6% BSA.
RESULTS
Nucleic acids induce fXI autoactivation
Polyanions such as dextran sulfate (27), heparin (40) and Poly-P (31,37) induce fXI autoactivation through by a template mechanism in which fXI and fXIa molecules bind in proximity to each other on the polymer. The template mechanism is characterized by a bell-shaped distribution for polyanion-dependence. DNA and RNA support fXI autoactivation, with nucleic acid-dependence distributions characteristic of this mechanism (Fig.1A). The template mechanism requires polymers greater than a certain length to support binding of protease and substrate in proximity to each other. Consistent with this, small species of double stranded DNA (32 and 64 nucleotides in length) did not support fXI autoactivation (Supplemental Fig.2). PK did not undergo autoactivation in the presence of DNA or RNA (Fig.1B).
Figure 1. Nucleic acids induce autoactivation of fXI but not PK.
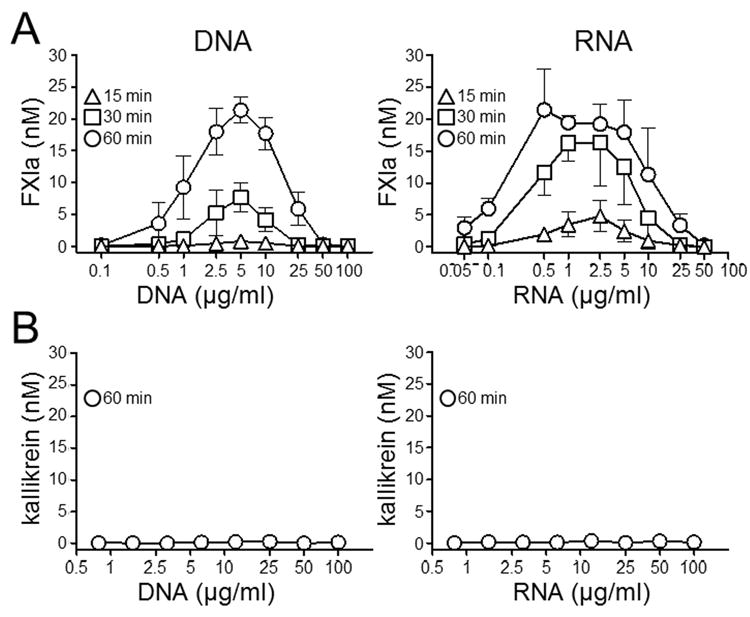
(A) Plasma fXI (30 nM) or (B) PK (60 nM) was incubated with varying concentrations of genomic DNA or total cellular RNA for 15 (∆), 30 (□) or 60 (○) minutes. At designated times fXIa or kallikrein activity where measured by chromogenic assay. Error bars are +/− one SD.
FXI and PK activation by thrombin or fXIIa in the presence of nucleic acids
Fig.2A shows progress curves demonstrating nucleic acid support for fXI autoactivation (No Protease), and nucleic acid enhancement of fXI activation by fXIIa or thrombin. The effect of DNA or RNA is negated by including DNAse (Fig.2B) or RNAse (Fig.2C) in the respective reactions. FXIIa or thrombin have small effects on the rate of fXI activation relative to autoactivation alone, suggesting nucleic acids do not enhance fXI activation by thrombin or fXIIa significantly. To examine this, we used a fXI-Ala557, a variant that does not autoactivate because its active site serine is replaced with alanine (Fig.3A). Rates of cleavage of fXI-Ala557 by thrombin were increased a modest 3-fold and 2-fold in the presence of DNA and RNA, respectively (Fig.3B). Interestingly, nucleic acids inhibited fXI-Ala557 cleavage by fXIIa (Fig.3C), suggesting that nucleic acids modulate fXIIa activity resulting in loss of capacity to activate fXI, or perhaps that fXI is in a conformation after binding to nucleic acids that renders the activation cleavage site inaccessible to fXIIa. As discussed, nucleic acid polymers do not induce PK autoactivation nor do they enhance PK activation by fXIIa appreciably (Fig.2D). Thrombin does not activate PK (30).
Figure 2. FXI and PK activation by fXIIa or thrombin in the presence of nucleic acids.
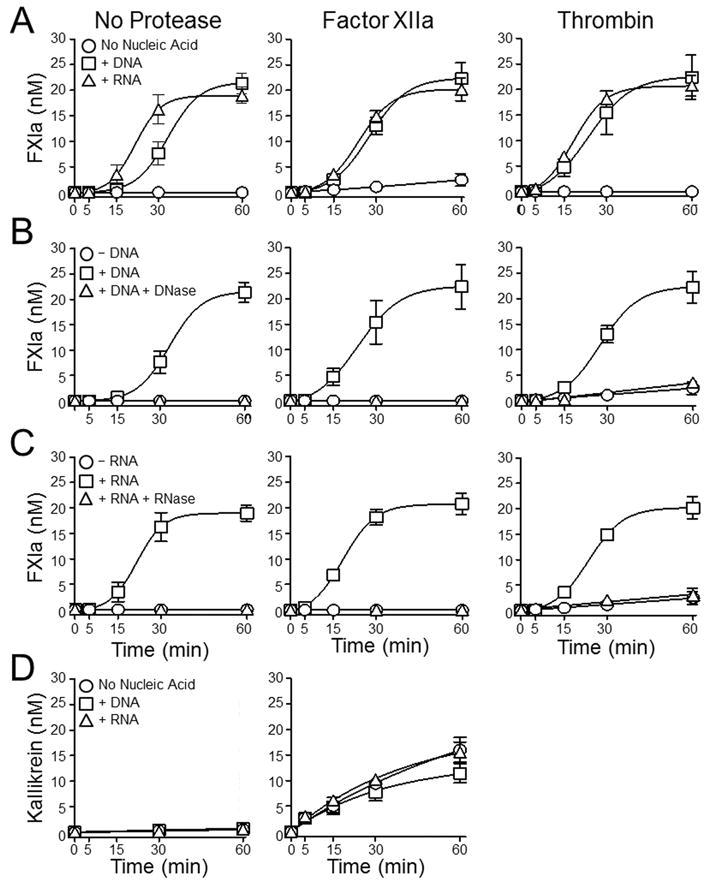
(A) Plasma fXI (30 nM) was incubated without an activating protease (No Protease) or with 2.5 nM fXIIa, or 2.5 nM thrombin, in the absence of nucleic acid (○), in the presence of 5 μg/ml DNA (□), or in the presence of 1 μg/ml RNA (∆). (B and C) Plasma fXI (30 nM) was incubated without activating protease (No Protease) or with 2.5 nM fXIIa, or 2.5 nM thrombin. Reactions in panel B were run in the absence of nucleic acid (○), in the presence of 5 μg/ml DNA (□), or in the presence of 5 μg/ml DNA treated with DNAse (∆). Reactions in panel C were run in the absence of nucleic acid (○), in the presence of 1 μg/ml RNA (□), or in the presence of 1 μg/ml RNA treated with RNAse (∆). For panels A-C, at indicated time points, aliquots were tested for fXIa activity by chromogenic assay. (D) Plasma PK (60 nM) was incubated without an activating protease (No Protease) or with 50 pM fXIIa, in the absence of nucleic acid (○), in the presence of 5 μg/ml DNA (□), or in the presence of 1 μg/ml RNA (∆). At indicated time points, aliquots were tested for kallikrein activity by chromogenic assay. For all panels error bars are +/−one SD.
Figure 3. Cleavage of fXI-Ala557 by fXIIa or thrombin in the presence of nucleic acids.
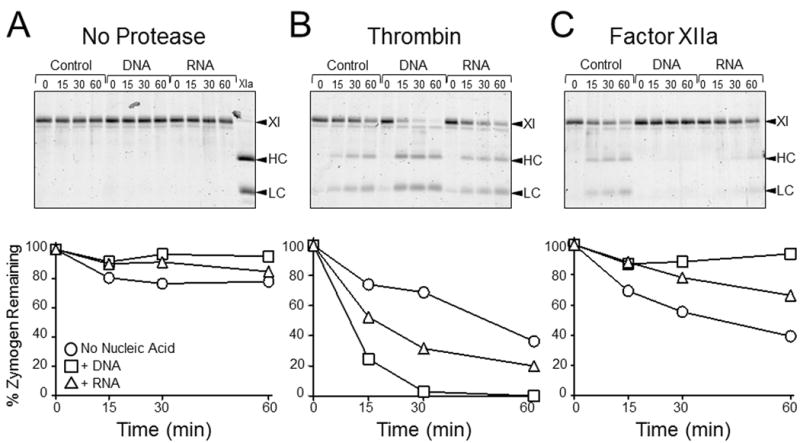
(Top Row) SDS-PAGE of fXI-Ala557 (200 nM) incubated with vehicle (control), 30 μg/ml DNA, or 5 μg/ml RNA (A) without a protease, (B) with 15 nM thrombin, or (C) with 15 nM fXIIa. Incubation times in minutes are shown at the tops of gels. Abbreviations: XI – zymogen fXI, HC – fXIa heavy chain, LC – fXIa light chain. (Bottom Row) Reduction of the 80 kDa fXI-Ala557 zymogen band on the gels immediately above each graph, as determined by densitometry. Results are for reactions without nucleic acid (○), with DNA (□), or with RNA (∆).
HK enhances PK activation in the presence of nucleic acids
We postulated that the modest effects of nucleic acids on fXI activation by thrombin or fXIIa, and the lack of an effect on PK activation, reflected the absence of HK in the system. In plasma nearly all fXI and most PK is bound to HK (11,41). PK activation by fXIIa was enhanced modestly by HK in the absence of nucleic acids, and increased severa-lfold when genomic DNA or total RNA was present (Fig.4A). Small DNA oligonucleotides had relatively little effect (Supplemental Fig.3). HK did not induce PK autoactivation on nucleic acids (Fig.4B). During PK activation, HK was converted to a form migrating with a standard for HKa, a form of HK with the bradykinin sequence removed (Fig.4C, Supplemental Fig.1B). With time HKa under-went additional cleavage. HKa supports PK activation by fXIIa in the presence of nucleic acids (Fig.4D), indicating the HK does not have to be in its single chain (uncleaved) form to express cofactor activity. As with HK, HKa does not support PK activation on nucleic acids if fXIIa is not present (Fig.4E).
Figure 4. Effects of HK and HKa on PK activation in the presence of nucleic acids.
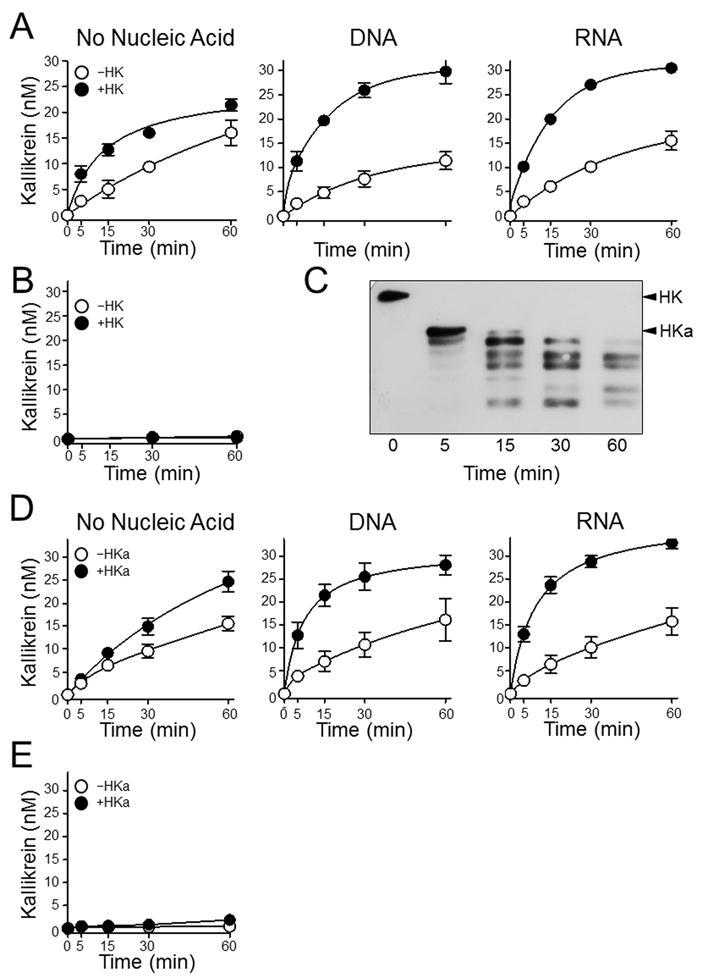
(A) Plasma PK (60 nM) was incubated with 50 pM fXIIa in the absence (○) or presence (●) of 60 nM HK; in the absence of nucleic acids (No Nucleic Acids), in the presence of 5 μg/ml DNA, or in the presence of 1 μg/ml RNA. (B) Plasma PK (60 nM) incubated in the absence (○) or presence (●) of 60 nM HK, without fXIIa or nucleic acids. For panels A and B, at the indicated time points, aliquots were tested for kallikrein activity by chromogenic assay. (C) Western blot for HK using samples from the reaction containing HK in the left-hand image in panel A. Positions of standard for HK and HKa are indicated at the right of the image. (D and E) Same for panels A and B, respectively except that HK was replaced with 60 nM HKa. For panels A, B, D and E, error bars are +/− one SD.
HK inhibits fXI activation in the presence of nucleic acids
HK had a strong inhibitory effect on fXI autoactivation (Fig.5A). However, with HK, thrombin largely restored fXI activation with DNA, and partly restored it with RNA (Fig.5A). Recall that fXIIa did not contribute appreciably to fXI activation in the presence of nucleic acids when HK was not present (Fig.2A), and that nucleic acids inhibit fXIIa activation of fXI-Ala557 (Fig.3C). Despite this, in reactions with HK, fXIIa restored fXI activation on DNA, and partially restored activation with RNA (Fig.5A). We examined the effects of HK on fXI cleavage by thrombin and fXIIa using fXI-Ala557. With nucleic acids, thrombin cleaves fXI-Ala557 in the presence (Fig.6B) or absence (Fig.3B) of HK at roughly similar rates. While nucleic acids inhibited fXIIa activation of fXI in the absence of HK (Fig.3C), in its presence fXI activation by fXIIa occurs at rates similar to those in reactions performed without nucleic acids (Fig.6C). This suggests that HK overcomes the inhibitory effects of nucleic acids on fXI activation by fXIIa. The sigmoidal progress curves generated during fXI activation by thrombin or fXIIa in the presence of HK and nucleic acids (Fig.5A) strongly suggest that fXI autoactivation contributes to fXIa generation. FXIa generated initially by α-thrombin or fXIIa may be sufficient to overcome the effect of HK on autoactivation.
Figure 5. Effects of HK and HKa on fXI activation in the presence of nucleic acids.

(A) Plasma fXI (30 nM) was incubated with vehicle (○,●), 5 μg/ml DNA (□,■), or 1 μg/ml RNA (∆,▲) in the absence (○,□,∆) or presence (●,■,▲) of 30 nM HK. Reactions included no activating protease (No Protease), 2.5 nM fXIIa or 2.5 nM thrombin. At indicated time points, aliquots were tested for fXIa activity by chromogenic assay. (B) Western blot for HK using samples from the reactions containing HK and DNA shown in panel A. Positions of standard for HK and HKa are indicated at the right of the images. (C) Same for panel A except that HK was replaced with 30 nM HKa. For panels A and C error bars are +/−one SD.
Figure 6. Cleavage of fXI-Ala557 by fXIIa or thrombin in the presence of nucleic acids and HK.
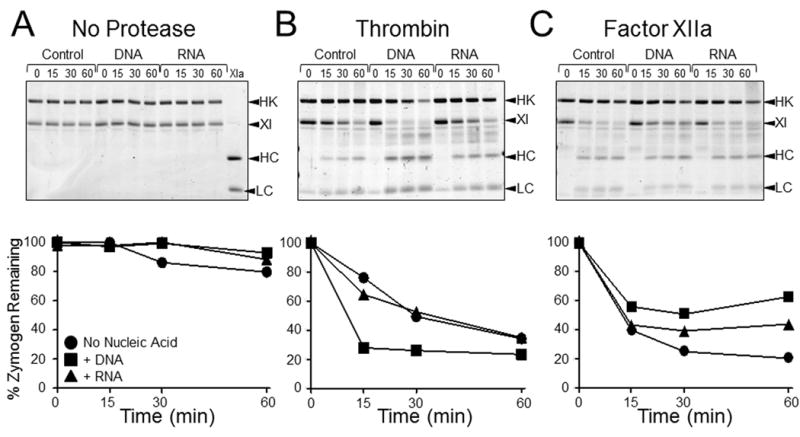
(Top Row) SDS-PAGE of recombinant fXI-Ala557 (200 nM) and plasma HK (200 nM) incubated with vehicle (control), 30 μg/ml DNA, or 5 μg/ml RNA (A) without a protease, (B) with 15 nM thrombin, or (C) with 15 nM fXIIa. Incubation times in minutes are shown at the tops of gels. Abbreviations: HK − high molecular weigh-kininogen, XI − zymogen fXI, HC − fXIa heavy chain, LC − fXIa light chain. (Bottom Row) Reduction of the 80 kDa fXI-Ala557 zymogen band on the gels immediately above each graph, as determined by densitometry. Results are for reactions without nucleic acid (●), with DNA (■), or with RNA (▲).
HK is cleaved during fXI activation in the presence of nucleic acids
In reactions in which HK inhibits fXI autoactivation in the presence of DNA, HK is cleaved to a form that co-migrates with an HKa standard (Fig.5B). As fXIa has been reported to cleave HK to release bradykinin (42), the result indicates that some fXIa must be formed in the reaction. Addition of fXIIa or thrombin enhanced HK cleavage (Fig.5B), consistent with an increased rate of fXIa generation (Fig.5A). HK inhibition of fXI auto-activation may, then, be partly due to HK acting as a competitive substrate for fXIa. However, this may not completely explain HK’s inhibitory properties, as HKa also inhibits fXI autoactivation (Fig.5C).
ABSs contribute to fXI activation on nucleic acids
Each fXI subunit has an ABS on the A3 domain (ABS1) and another on the catalytic domain 170-loop (ABS2) (37-39). Previously, using fXI species with alanine replacements for residues in the ABSs, we noted that ABS1 and ABS2 contribute comparably to fXI activation with Poly-P (37). Both ABSs contributed to fXI activation by thrombin and fXIIa in the presence of DNA (Fig.7A) or RNA (Fig.7B), with an absolute requirement for ABS2. The observation that HK supports fXI-Ala557 cleavage by fXIIa in the presence of nucleic acids raises the possibility that HK facilitates fXI binding to nucleic acids, obviating the need for ABS-mediated interactions. However, in the presence of DNA, HK did not reverse the defect in fXI-ABS2 activation with thrombin or fXIIa (Fig.7C). PK lacks basic residues on the A3 domain corresponding to fXI ABS1, but has residues in the 170-loop corresponding to fXI ABS2 (24). However, in contrast to fXI-ABS2, elimination of PK ABS2 did not affect the ability of HK to enhance PK activation by fXIIa in the presence of DNA (Fig.7D).
Figure 7. Activation of fXI and PK anion-binding site (ABS) variants in the presence of nucleic acids.
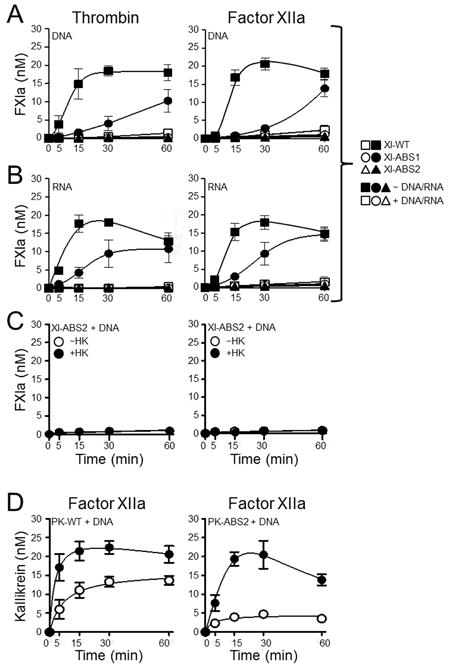
Recombinant fXI species (30 nM) were incubated without (□,○,∆) or with (■,●,▲) (A) 5 μg/ml DNA or (B) 1 μg/ml RNA, and with 2.5 nM thrombin (left) or 2.5 nM fXIIa (right). Symbols: fXI-WT (□,■), fXI-ABS1 (○,●), or fXI-ABS2 (∆,▲). (C) FXI-ABS2 (30 nM) was incubated with 5 μg/ml DNA and 2.5 nM thrombin (left) or 2.5 nM fXIIa (right) without (○) or with (●) 30 nM HK. (D) 60 nM PK-WT (left) or PK-ABS2 (right) were incubated with 50 pM fXIIa in the presence of 5 μg/ml DNA without (○) or with (●) 60 nM HK. For all reactions, at indicated time points, aliquots were tested for fXIa or kallikrein activity by chromogenic assay. Error bars are +/−one SD.
DNA-induced thrombin generation in plasma
The data suggest that nucleic acid-induced fXI activation in plasma would require fXIIa or thrombin, because spontaneous fXI autoactivation would be inhibited by HK. Adding DNA to plasma induces thrombin generation that is blocked by the anti-fXI IgG O1A6 (Fig.8A). In the absence of tissue factor, nucleic acids appear to trigger thrombin generation through contact activation, as the fXIIa inhibitor CTI also blocks the process. The absence of thrombin generation in reactions with CTI supports the premise that HK limits fXI autoactivation. To examine DNA-dependent thrombin-mediated fXI activation, we induced clotting with a combination of DNA (to induce contact activation) and tissue factor (to initiate thrombin generation through factor VIIa). A burst of thrombin generation is produced (Fig.8A), a portion of which is inhibited by CTI and reflects the contribution of fXIIa. FXIIa-independent thrombin generation, which reflects the tissue factor contribution, was reduced by O1A6, consistent with thrombin-mediated fXI activation. These data support the premise that fXIIa or thrombin are required for fXI activation in plasma in the presence of HK, and that fXI will not undergo autoactivation in an HK-rich environment without one of these enzymes.
Figure 8. Effect of DNA on fXI-dependent thrombin generation in plasma.
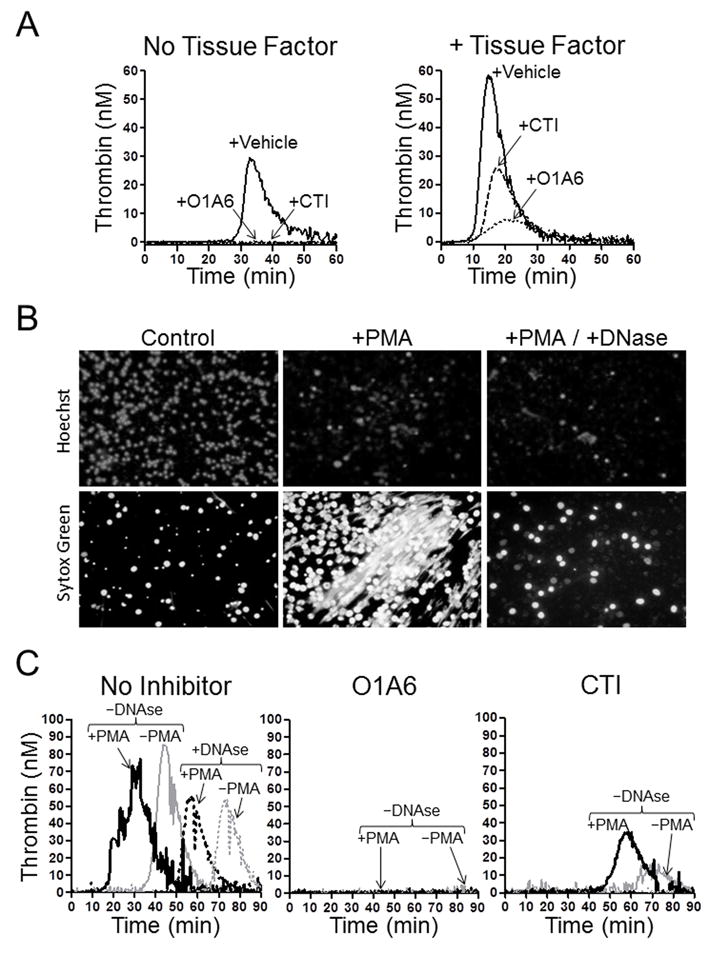
(A) Thrombin generation measured in normal plasma after addition of 25 μg/ml DNA without (left hand panel) or with (right hand panel) 0.5 pM TF. Assays were run in the presence of vehicle (solid line), the anti-fXI IgG O1A6 (dotted line) or the fXIIa inhibitor CTI (dashed line). (B) Images of neutrophils stimulated with vehicle (control) or 100 ng/ml phorbol myristate acetate (PMA) with or without 100 U/mL DNAse. Cell permeable Hoechst stain (top row) indicates DNA within intact cell nuclei. Sytox Green staining (bottom row) demonstrates extracellular DNA or DNA within dying cells that have lost the ability to exclude the dye. PMA treated cells form neutrophil extracellular traps (NETs, middle column), which are not observed in the presence of DNase (right column). Images were made on an Inverted Fluorescence Microscope (89404-464) using a V10MP camera (VWR), and Motic Images Plus 2.0 software. (C) Thrombin generation in normal plasma supplemented with neutrophils treated with vehicle (gray lines, –PMA) or PMA (black lines, + PMA), without (solid lines) or with (dashed lines) DNAse in the absence of an inhibitor (No inhibitor), or in the presence of O1A6 or CTI. For reactions containing O1A6, no thrombin generation was detected with or without DNAse treatment. For reactions with CTI, some activity was detected in samples not treated with DNAse.
Stimulated neutrophils may extrude their chromatin in the form of NETs (42,43), which have been implicated in thrombus formation in humans and mice (4-7). PMA-stimulated neutrophils produce NETs that are degraded by DNAse (Fig.8B). Adding unstimulated neutrophils to plasma leads to thrombin generation (Fig.8C, gray solid line left panel), perhaps due to nucleic acids released from dead cells, as the effect is reduced and delayed by DNase (Fig.8C, gray dashed line). PMA stimulated neutrophils promote earlier thrombin generation than unstimulated cells (Fig.8C, black solid line) that is also DNase sensitive (Fig.8C, black dashed line). Thrombin generation in neutrophil-supplemented plasma is blocked by O1A6 (Fig.8C middle panel), demonstrating the importance of fXI, and is significantly reduced by CTI (Fig.8C right panel), consistent with a requirement for fXIIa in fXI activation.
DISCUSSION
During contact activation, fXIIa catalyzes conversion of PK and fXI to the proteases α-kallikrein and fXIa, respectively (8-10,23). As, fXI arose through a duplication of the PK gene (25), the proteins are homologs and share structural and functional properties (23,24). In addition to being substrates for fXIIa, PK and FXI circulate as complexes with HK (12,41), and their active forms cleave HK (8-10,42). However, fXI has evolved novel properties that allow it to contribute to thrombin generation independently of contact activation. As examples, fXIa contains a unique exosite that facilitates factor IX activation (36), and changes to the fXI activation site permit cleavage by thrombin (30). The evidence presented here indicates that PK and fXI also differ in their manner of interaction with nucleic acids.
Our data are consistent with those from earlier studies showing that DNA or RNA support activation of contact factors through a template mechanism in which polynucleotides of a certain length are required to permit substrate and activating protease to bind in proximity to each other. (3,33,43,44). RNA also induces autoactivation of the serine protease factor VII activating protease by this mechanism (45). In this regard, genomic DNA and total cellular RNA appear to behave similarly to other polyanions that support FXI activation in a size-dependent manner, such as polyphosphate and dextran sulfate (27,31,32,46). While this suggests that the negatively charged phosphate-containing backbone of nucleic acid polymers form the points of interaction with positively charged regions on contact factors, this may not completely describe the situation. Gansler et al. demonstrated that hairpin-forming RNA and DNA oligomers are better at inducing plasma contact activation than linear oligomers, largely due to their ability to bind HK (44). Nucleic acid secondary structure, therefore, may be an important determinant of activity, and the procoagulant effects of genomic DNA and total RNA may be due to subfractions of the total material with appropriate secondary structure. Alternatively, nucleic acids may enhance contact factor activation through more than one mechanism, with a template mechanism based on polymer length and charge interactions, and secondary structure contributing to HK-mediated processes involving PK activation and kallikrein activity.
In our study, thrombin and fXIIa had small effects on fXI and PK activation in the presence of nucleic acids beyond what could be accounted for by autoactivation. This differs from experience with other polyanions (27,31,32,40), As both thrombin and fXIIa bind DNA and RNA (43), we postulated that their weak effects were due to the absence of HK. During contact activation on non-biologic surfaces, HK serves as a cofactor for fXIIa-mediated PK activation, and kallikrein-mediated fXII activation, by facilitating PK binding to the surface (11,12). HK probably plays a similar role in fXI activation on such surfaces, although the effect may be smaller than with PK. In the activated partial thromboplastin time (aPTT) assay, coagulation is initiated by adding a non-biologic activator of contact activation such as silica to plasma. The aPTT of plasma lacking HK is longer than those of plasmas lacking PK or fXI, supporting the impression that HK enhances activation of both zymogens. The goal of our study was to determine if HK produced similar effects with physiologically relevant cofactors such as nucleic acids
HK enhances PK activation by fXIIa in the presence of DNA or RNA, consistent with proposed mechanisms for PK activation on non-biologic surfaces. However, HK had the opposite effect on fXI activation. FXI is converted to fXIa by fXIIa, thrombin, or fXIa (26,27,31,32,37). Activation by fXIIa or thrombin can occur in the absence of a surface, but activation by fXIa (autoactivation) is surface-dependent (27,32,37). HK and its cleaved form HKa inhibit nucleic acid-dependent fXI autoactivation. Interestingly, while thrombin or fXIIa had little effect on fXI activation when HK was not present, both restored (at least partly) fXI activation in reactions with HK. This effect does not appear to be due to HK enhancement of thrombin-mediated fXI activation, and while HK overcame the inhibitory effect of nucleic acids on fXIIa-mediated fXI activation, the effect was small. Rather, the sigmoidal progress curves indicate fXI autoactivation occurs despite the presence of HK, implying that thrombin or fXIIa generates a threshold amount of fXIa that overcomes HK inhibition of autoactivation. As fXIa cleaves HK during activation, HK and fXI may compete for fXIa, and generation of fXIa independently of autoactivation may overcome the competitive inhibition by increasing the amount of fXIa in the reaction.
FXI binds avidly to DNA (3,43), while PK does not appear to do so (3). Previously, we reported that Poly-P enhancement of fXI activation requires ABSs on the fXI A3 (ABS1) and catalytic (ABS2) domains (37). While fXI variants lacking either binding site perform normally in aPTT assays (38,39), they are defective in Poly-P-initiated clotting assays (37), suggesting an important difference between the two types of activators. In the present study, ABS2 was required to observe the nucleic acid effect on fXI activation. Its contribution was HK-independent, as HK did not compensate for loss of ABS2. Removal of the corresponding ABS2 site on PK, in contrast, did not impair HK support of PK activation. These results support the conclusion that HK is required for PK interactions with surfaces including nucleic acids (3,44), while fXI interacts directly with polyanions through its positively charged ABSs. This highlights a major difference between these homologs.
Our results support a model for nucleic acid-induced plasma contact activation in which HK functions as a cofactor to enhance fXIIa activation of PK, and reciprocal activation of fXII and PK, by facilitating PK/kallikrein interaction with the nucleic acid. HK also may permit FXI activation by FXIIa in the presence of DNA or RNA, without producing the enhancement seen with PK activation. HK is not required for FXI activation by thrombin. Furthermore, FXI must interact directly with nucleic acids through its ABSs for optimal activation to occur. Indeed, with FXI, HK may serve a negative regulatory role by preventing autoactivation in the absence of fXIIa or thrombin. This may serve to limit the thrombotic potential of FXI. FXI activation by thrombin is thought to have a role in hemostasis (47), and the observation that HK does not have an enhancing effect on this reaction is consistent with the observation that HK-deficient individuals do not have a hemostatic defect, while fXI-deficient patients may experience abnormal bleeding.
Supplementary Material
Acknowledgments
The authors wish to acknowledge support from awards HL81326 and HL58837 from the National Heart, Lung and Blood Institute.
References
- 1.Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renné T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrissey JH, Choi SH, Smith SA. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood. 2012;119:5972–5979. doi: 10.1182/blood-2012-03-306605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kannemeier C, Shibamiya A, Nakazawa F, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA. 2007;104:6388–6393. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischer S, Preissner KT. Extracellular nucleic acids as novel alarm signals in the vascular system. Mediators of defense and disease Hamostaseologie. 2013;33:37–42. doi: 10.5482/HAMO-13-01-0001. [DOI] [PubMed] [Google Scholar]
- 6.Pinegin B, Vorobjeva N, Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun Rev. 2015;14:633–640. doi: 10.1016/j.autrev.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136–144. doi: 10.1111/j.1538-7836.2011.04544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long AT, Kenne E, Jung R, Fuchs TA, Renné T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. 2016;14:427–437. doi: 10.1111/jth.13235. [DOI] [PubMed] [Google Scholar]
- 9.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28–39. doi: 10.1111/jth.13194. [DOI] [PubMed] [Google Scholar]
- 10.Danese E, Montagnana M, Lippi G. Factor XII in hemostasis and thrombosis: active player or (innocent) bystander? Semin Thromb Hemost. 2016;42:682–688. doi: 10.1055/s-0036-1571338. [DOI] [PubMed] [Google Scholar]
- 11.Wiggins RC, Bouma BN, Cochrane CG, Griffin JH. Role of high-molecular-weight kininogen in surface-binding and activation of coagulation Factor XI and prekallikrein. Proc Natl Acad Sci USA. 1977;74:4636–4640. doi: 10.1073/pnas.74.10.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson RE, Mandle R, Jr, Kaplan AP. Studies of binding of prekallikrein and Factor XI to high molecular weight kininogen and its light chain. Proc Natl Acad Sci USA. 1979;76:4862–4866. doi: 10.1073/pnas.76.10.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Revenko AS, Gao D, Crosby JR, Bhattacharjee G, Zhao C, May C, Gailani D, Monia BP, MacLeod AR. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. 2011;118:5302–5311. doi: 10.1182/blood-2011-05-355248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renné T, Pozgajová M, Grüner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Q, Tucker EI, Pine MS, Sisler I, Matafonov A, Sun MF, White-Adams TC, Smith SA, Hanson SR, McCarty OJ, Renné T, Gruber A, Gailani D. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–3989. doi: 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matafonov A, Leung PY, Gailani AE, Grach SL, Puy C, Cheng Q, Sun MF, McCarty OJ, Tucker EI, Kataoka H, Renné T, Morrissey JH, Gruber A, Gailani D. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. 2014;123:1739–1746. doi: 10.1182/blood-2013-04-499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bird JE, Smith PL, Wang X, Schumacher WA, Barbera F, Revelli JP, Seiffert D. Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait. Thromb Haemost. 2012;107:1141–1150. doi: 10.1160/th-11-10-0682. [DOI] [PubMed] [Google Scholar]
- 18.Stavrou EX, Fang C, Merkulova A, Alhalabi O, Grobe N, Antoniak S, Mackman N, Schmaier AH. Reduced thrombosis in Klkb1-/- mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood. 2015;125:710–719. doi: 10.1182/blood-2014-01-550285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merkulov S, Zhang WM, Komar AA, Schmaier AH, Barnes E, Zhou Y, Lu X, Iwaki T, Castellino FJ, Luo G, McCrae KR. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111:1274–1281. doi: 10.1182/blood-2007-06-092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wendel HP, Jones DW, Gallimore MJ. FXII levels, FXIIa-like activities and kallikrein activities in normal subjects and patients undergoing cardiac surgery. Immunopharmacology. 1999;45:141–144. doi: 10.1016/s0162-3109(99)00067-3. [DOI] [PubMed] [Google Scholar]
- 21.Plötz FB, van Oeveren W, Bartlett RH, Wildevuur CR. Blood activation during neonatal extracorporeal life support. J Thorac Cardiovasc Surg. 1993;105:823–832. [PubMed] [Google Scholar]
- 22.Jaffer IH, Fredenburgh JC, Hirsh J, Weitz JI. Medical device-induced thrombosis: what causes it and how can we prevent it? J Thromb Haemost. 2015;13(Suppl 1):S72–81. doi: 10.1111/jth.12961. [DOI] [PubMed] [Google Scholar]
- 23.Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015;13:1383–1395. doi: 10.1111/jth.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujikawa K, Chung DW, Hendrickson LE, Davie EW. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–2424. doi: 10.1021/bi00357a018. [DOI] [PubMed] [Google Scholar]
- 25.Ponczek MB, Gailani D, Doolittle RF. Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost. 2008;6:1876–1883. doi: 10.1111/j.1538-7836.2008.03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–7358. [PubMed] [Google Scholar]
- 27.Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 28.von dem Borne PA, Mosnier LO, Tans G, Meijers JC, Bouma BN. Factor XI activation by meizothrombin: stimulation by phospholipid vesicles containing both phosphatidylserine and phosphatidylethanolamine. Thromb Haemost. 1997;78:834–839. [PubMed] [Google Scholar]
- 29.Kravtsov DV, Matafonov A, Tucker EI, Sun MF, Walsh PN, Gruber A, Gailani D. Factor XI contributes to thrombin generation in the absence of factor XII. Blood. 2009;114:452–458. doi: 10.1182/blood-2009-02-203604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matafonov A, Sarilla S, Sun MF, Sheehan JP, Serebrov V, Verhamme IM, Gailani D. Activation of factor XI by products of prothrombin activation. Blood. 2011;118:437–445. doi: 10.1182/blood-2010-10-312983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118:6963–6970. doi: 10.1182/blood-2011-07-368811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geng Y, Verhamme IM, Smith SB, Sun MF, Matafonov A, Cheng Q, Smith SA, Morrissey JH, Gailani D. The dimeric structure of factor XI and zymogen activation. Blood. 2013;121:3962–3969. doi: 10.1182/blood-2012-12-473629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, Liaw PC. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34:1977–1984. doi: 10.1161/ATVBAHA.114.304114. [DOI] [PubMed] [Google Scholar]
- 34.Tucker EI, Marzec UM, White TC, Hurst S, Rugonyi S, McCarty OJ, Gailani D, Gruber A, Hanson SR. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–944. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kokoye Y, Ivanov I, Cheng Q, Matafonov A, Dickeson SK, Mason M, Sexton DJ, Renné T, McCrae K, Feener EP, Gailani D. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb Res. 2016;140:118–124. doi: 10.1016/j.thromres.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng Y, Verhamme IM, Messer A, Sun MF, Smith SB, Bajaj SP, Gailani D. A sequential mechanism for exosite-mediated factor IX activation by factor XIa. J Biol Chem. 2012;287:38200–38209. doi: 10.1074/jbc.M112.376343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geng Y, Verhamme IM, Smith SA, Cheng Q, Sun M, Sheehan JP, Morrissey JH, Gailani D. Factor XI anion-binding sites are required for productive interactions with polyphosphate. J Thromb Haemost. 2013;11:2020–2028. doi: 10.1111/jth.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao M, Abdel-Razek T, Sun MF, Gailani D. Characterization of a heparin binding site on the heavy chain of factor XI. J Biol Chem. 1998;273:31153–31159. doi: 10.1074/jbc.273.47.31153. [DOI] [PubMed] [Google Scholar]
- 39.Yang L, Sun MF, Gailani D, Rezaie AR. Characterization of a heparin-binding site on the catalytic domain of factor XIa: mechanism of heparin acceleration of factor XIa inhibition by the serpins antithrombin and C1-inhibitor. Biochemistry. 2009;48:1517–1524. doi: 10.1021/bi802298r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gailani D, Broze GJ., Jr Effects of glycosaminoglycans on factor XI activation by thrombin. Blood Coagul Fibrinolysis. 1993;4:15–20. [PubMed] [Google Scholar]
- 41.Thompson RE, Mandle R, Jr, Kaplan AP. Association of factor XI and high molecular weight kininogen in human plasma. J Clin Invest. 1977;60:1376–1380. doi: 10.1172/JCI108898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott CF, Silver LD, Purdon AD, Colman RW. Cleavage of human high molecular weight kininogen by factor XIa in vitro. Effect on structure and function. J Biol Chem. 1985;260:10856–10863. [PubMed] [Google Scholar]
- 43.Vu TT, Leslie BA, Stafford AR, Zhou J, Fredenburgh JC, Weitz JI. Histidine-rich glycoprotein binds DNA and RNA and attenuates their capacity to activate the intrinsic coagulation pathway. Thromb Haemost. 2016;115:89–98. doi: 10.1160/TH15-04-0336. [DOI] [PubMed] [Google Scholar]
- 44.Gansler J, Jaax M, Leiting S, Appel B, Greinacher A, Fischer S, Preissner KT. Structural requirements for the procoagulant activity of nucleic acids. PLoS One. 2012;7:e50399. doi: 10.1371/journal.pone.0050399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakazawa F, Kannemeier C, Shibamiya A, Song Y, Tzima E, Schubert U, Koyama T, Niepmann M, Trusheim H, Engelmann B, Preissner KT. Extracellular RNA is a natural cofactor for the auto-activation of Factor VII-activating protease (FSAP) Biochem J. 2005;385(Pt 3):831–838. doi: 10.1042/BJ20041021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gailani D, Broze GJ., Jr Factor XI activation by thrombin and factor XIa. Semin Thromb Hemost. 1993;19:396–404. doi: 10.1055/s-2007-993291. [DOI] [PubMed] [Google Scholar]
- 47.Wheeler AP, Gailani D. Why factor XI is a clinical concern. Expert Rev Hematol. 2016;9:629–637. doi: 10.1080/17474086.2016.1191944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.