

Abstract
While the peripheral nervous system has the capacity to regenerate following a nerve injury, it is often at a slow rate and results in unsatisfactory recovery, leaving patients with reduced function. Many regeneration associated genes have been identified over the years, which may shed some insight into how we can manipulate this intrinsic regenerative ability to enhance repair following peripheral nerve injuries. Our lab has identified the membrane bound protease beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1), or beta secretase, as a potential negative regulator of peripheral nerve regeneration. When beta secretase activity levels are abolished via a null mutation in mice, peripheral regeneration is enhanced following a sciatic nerve crush injury. Conversely, when activity levels are greatly increased by overexpressing beta secretase in mice, nerve regeneration and functional recovery are impaired after a sciatic nerve crush injury. In addition to our work, many substrates of beta secretase have been found to be involved in regulating neurite outgrowth and some have even been identified as regeneration associated genes. In this review, we set out to discuss BACE1 and its substrates with respect to axonal regeneration and speculate on the possibility of utilizing BACE1 inhibitors to enhance regeneration following acute nerve injury and potential uses in peripheral neuropathies.
Keywords: peripheral nerve, axonal regeneration, beta-site amyloid precursor protein-cleaving enzyme 1
Introduction
One of the many unsolved mysteries in neuroscience is the unknown reasons why the peripheral nervous system (PNS) is able to regenerate its axons following injury while the central nervous system (CNS) cannot. The search for understanding this fundamental difference between the peripheral and central nervous systems has led to many discoveries of the cellular changes, genes, and pathways that regulate how the PNS is able to regenerate. There is now a good understanding of the steps required in order for the peripheral nerve to begin to regenerate and how these steps differ from the CNS. The first step is the clearance of axonal and myelin debris in order to make space for regenerating axons to grow into. In the PNS, macrophages, and to some extent Schwann cells, begin to phagocytose axonal and myelin debris (Stoll et al., 1993; Brück, 1997; Martini et al., 2008; Niemi et al., 2013; Mietto et al., 2015). In the CNS, resident microglia, and an influx of systemic macrophages, are responsible for debris clearance, however, there is some evidence that the CNS response to debris clearance is very slow and contributes to the limited axonal regeneration (George and Griffin, 1994; Ferguson et al., 2008; Kigerl et al., 2009). Next, the supporting cells need to provide aid to the regenerating axons by guiding them towards the correct end target. The Schwann cells of the PNS carry this out by undergoing dedifferentiation and sending out many processes to create tubes, called Bünger bands, for which the regenerating axons can enter and grow through before reaching their target and getting remyelinated (Fawcett and Keynes, 1990; Griffin et al., 2010; Toy and Namgung, 2013). In the CNS, astrocytes become activated and begin to proliferate and secrete inhibitory proteoglycans, which make up a majority of the glial scar (Toy and Namgung, 2013). This process is detrimental to axonal regeneration and provides a significant barrier for repair following CNS trauma. Another step that must occur for successful regeneration is when neurons and supporting cells alter their gene expression profiles to a pro-regenerative state. The cells of the PNS upregulate various growth factors, transcription factors, and adhesion molecules, collectively termed regeneration associated genes (RAGs), which all work together to enhance neurite outgrowth (Seijffers et al., 2007; Huebner and Strittmatter, 2009; Painter et al., 2014; Gordon and English, 2016). On the other hand, CNS cells generally do not express high levels of RAGs. In fact, oligodendrocytes express various myelin-associated inhibitors, such as Nogo-A and myelin-associated glycoprotein, while neurons express axon regeneration inhibitor molecules, like repulsive guidance molecule and Semaphorin A (Huebner and Strittmatter, 2009).
Our lab has discovered a novel role of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1), or β-secretase, in regulating peripheral axonal regeneration. BACE1 is a transmembrane protease that is involved in the cleavage and processing of a wide variety of membrane bound proteins (Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Hu et al., 2006; Willem et al., 2006; Kuhn et al., 2007; Hemming et al., 2009; Zhou et al., 2012; Pigoni et al., 2016). What is interesting with regards to peripheral nerve regeneration is that many of BACE1's substrates have been implicated in the regulation of axonal regeneration and neurite outgrowth. Some have even been identified as genes which are upregulated following nerve injury. We have demonstrated, using genetically modified mouse models, that β-secretase levels negatively regulate peripheral axonal regeneration following axonal injury (Figure 1). When β-secretase was genetically knocked out, we observed an increased number of regenerating axons (Farah et al., 2011; Farah, 2012; Liu et al., 2016). Mice which transgenically overexpressed human β-secretase in their neurons had impaired regeneration following a sciatic nerve crush (Tallon et al., 2017).
Figure 1.

Beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) activity levels inversely regulate peripheral axonal repair.
With low levels of BACE1 activity, peripheral nerve repair is accelerated. When BACE1 activity levels are high, axonal repair is reduced. We hypothesize that this relationship is on a sliding scale where slight changes in BACE1 expression have a small effect on how effective the repair process is.
Despite the fact that peripheral nerves are able to regenerate, the outcome of this regeneration is often poor and insufficient (Seddon, 1942; Sunderland, 1951; Fawcett and Keynes, 1990; Griffin et al., 2010). Peripheral nerve regeneration must overcome many obstacles, including slow regeneration rates, inefficient axonal guidance, and the degeneration of end targets. Currently, the only available treatment option for patients is the surgical reconnection of the nerve (Palispis and Gupta, 2017). Various novel therapeutic options are being explored to improve outcomes such as enhanced nerve conduits, stem cell treatments, and small molecule drugs to enhance regeneration (Gordon and English, 2016; Palispis and Gupta, 2017). Our recent data, together with the greater body of research on BACE1's substrates’ impact on axonal growth, points towards a possible novel application for BACE1 inhibitors currently being investigated.
β-Secretase and Its Many Substrates
BACE1's role in amyloid precursor protein processing
β-secretase is a transmembrane aspartyl protease which cleaves mostly type I membrane bound proteins, generating both a membrane bound and a soluble fragment. This enzyme has been well studied in the context of Alzheimer's disease (AD) as it is one of the main enzymes responsible for generating amyloid beta plaques (Aβ), a pathological hallmark of the disease (Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001). In order to generate Aβ, BACE1 cleaves amyloid precursor protein (APP) into a membrane bound C-terminal fragment (CTF) and a soluble APPβ fragment. The CTF is then further cleaved by γ-secretase to generate the Aβ fragment that then goes on to aggregate and form plaques. A reduction in β-secretase activity, either genetically or pharmacologically, leads to a decrease in the production of Aβ (Vassar et al., 1999; Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001; Kennedy et al., 2016). The fact that BACE1 is one of two important enzymes involved in generating Aβ plaques has led many drug companies to investigate BACE1 inhibitors as potential therapeutics for AD (Sankaranarayanan et al., 2008; Chang et al., 2011; May et al., 2015; Kennedy et al., 2016; Cebers et al., 2017). This large interest in inhibiting BACE1 has led many to ask the question, what other substrates, if any, does BACE1 cleave?
BACE1 impacts peripheral myelination via neuregulin-1 type III
As it turns out, BACE1 is a rather promiscuous enzyme and over 60 putative substrates have been identified (Kitazume et al., 2001; Lichtenthaler et al., 2003; Hemming et al., 2009; Gersbacher et al., 2010; Zhang et al., 2011; Zhou et al., 2012). Fortunately, despite the large number of substrates, BACE1 knock out (KO) mice are viable with some moderate behavioral phenotypes (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001; Laird et al., 2005; Savonenko et al., 2008). One of the more striking phenotypes observed was the reduction in the myelination of the peripheral nerves in these mice. The thickness of the myelin is markedly decreased, but not completely absent, and the presence of an increase in unmyelinated groups of axons, called Remak bundles, can be seen in nerve bundles in the periphery (Hu et al., 2006, 2008; Willem et al., 2006; Velanac et al., 2012). While reduced peripheral myelination is cause for concern, this phenotype appears to be a developmental issue. When adult mice are given a BACE1 inhibitor, others (Sankaranarayanan et al., 2008) and our lab (unpublished observation) have observed that the myelination of uninjured peripheral axons is not changed. This observation of altered myelination gave a clue as to what other substrates BACE1 may be cleaving. As it turns out, BACE1 is involved in the cleavage of neuregulin-1 (NRG1) type III (Willem et al., 2006; Hu et al., 2008). NRG1 type III is an important player in myelinating the peripheral nervous system (Michailov et al., 2004; Taveggia et al., 2005). The cleavage and activation of NRG1 type III is not solely dependent on BACE1, as it is also cleaved by a disintegrin and metalloprotease 17 (ADAM17) (La Marca et al., 2011). This parallel pathway allows for some myelination to occur and may be the reason for the presence of some myelination in total KO mice.
BACE1 activity influences axonal guidance via multiple adhesion molecules
Another phenotype that has caused some concern for BACE1 inhibitor use is the potential issues with axonal guidance. In BACE1 KO mice, there appears to be some defects in axonal connections in the central nervous system, most notably in the olfactory bulb and mossy fiber projections (Hitt et al., 2012). This finding has led to the speculation that BACE1 inhibitors may negatively impact memory and learning by impairing synaptic plasticity (Laird et al., 2005; Savonenko et al., 2008). Since some of the potential BACE1 substrates are involved in cell-cell adhesion, such as neural cell adhesion molecule 1 (NCAM1) (Hemming et al., 2009), it likely follows that this may be the reason behind the axonal guidance issues. Indeed, two of BACE1's identified substrates, L1 and close homolog of L1 (CHL1) (Kuhn et al., 2012), are thought to be involved in axonal guidance (Zhou et al., 2012). These molecules are members of the immunoglobulin superfamily and have been identified as being important in proper neurogenesis. A loss of L1 or CHL1 leads to behavioral abnormalities and decreased cognitive function in mice (Montag-Sallaz et al., 2002; Pratte et al., 2003). There are also known mutations in humans which lead to mental retardation as well as schizophrenia (Kurumaji et al., 2001; Weller and Gärtner, 2001; Sakurai et al., 2002; Chen et al., 2005).
Another protein that was identified as a BACE1 substrate is contactin-2, also known as Axonin-1 or transiently expressed axonal surface glycoprotein-1 (TAG-1) (Kuhn et al., 2012). This protein is also a member of the immunoglobulin superfamily and is expressed on the surface of axons as well as the Schwann cells of the PNS (Yamamoto et al., 1986; Traka et al., 2002). A loss of contactin-2 led to a reduced axonal growth speed and impaired guidance in zebrafish (Wolman et al., 2008). Following a sciatic nerve crush, contactin-2 was found to be upregulated in Schwann cells near the lesion (Soares et al., 2005). BACE1 cleavage of contactin-2 also appears to have a negative impact on the level of contactin-2 found on the cell surface of primary mouse neurons (Gautam et al., 2014).
One more BACE1 substrate that has been identified as being an important regulator for neurite outgrowth is Seizure-related gene 6 (Sez-6) (Kuhn et al., 2012). It has been implicated as having an important role in regulating neurite outgrowth and connectivity in the developing neocortex. Cultured cortical neurons from Sez-6 null mice showed more extensive neurite branching, however, they also had a reduction in neurite length (Gunnersen et al., 2007). Another group observed a decrease in neurite length when administering a short hairpin RNA (shRNA) against Sez-6 in PC12 cells treated with nerve growth factor (NGF) (Zhang et al., 2011).
As many of BACE1's substrates appear to be important in regulating axonal outgrowth and neurite branching, regulating BACE1 activity levels may have implications going beyond AD. Our lab has been studying the effects of BACE1 activity levels on the efficacy of peripheral nerve regeneration following acute nerve injury and hypothesize that BACE1 activity levels have an inverse relationship with regenerative efficacy (Farah et al., 2011; Tallon et al., 2017).
How do Peripheral Nerves Regenerate?
Stages of peripheral axonal regeneration following nerve injury
The stages of degeneration and regeneration an axon undergoes following peripheral nerve injury are well studied. First, the distal portion of the injured axons undergo Wallerian degeneration, where both the axoplasm and myelin sheath begin to break up. Next, the axonal and myelin debris are cleared away in order for optimal regeneration to occur (Fawcett and Keynes, 1990; Brück, 1997; Griffin et al., 2010). This is carried out mainly by the phagocytic action of infiltrating macrophages as well as the Schwann cells (Stoll et al., 1989; Martini et al., 2008). Once the debris is cleared, the proximal portion of the nerve is able to extend a tiny axonal sprout out towards the area of denervation. In less severe injuries, such as a nerve crush, the axonal sprouts may be able to navigate their way back into denervated Schwann cell tubes. This is advantageous as the neural tube acts as a highway for the axon to be properly guided towards its appropriate target. In more severe injuries, such as a partial or complete nerve transection, the Schwann cell tube is no longer connected and the proximal axonal sprout can have a difficult time correctly navigating towards the open end of the tube. The final stage comes when the axon reaches its target and is then able to mature and increase its size, as well as becoming remyelinated by the Schwann cells. Axonal sprouts that are unable to reach the vacated Schwann cell tubes and reinnervate the target muscle are generally pruned back to the main axon branch. In some cases, the unguided sprout can become tangled up and form a neuroma.
Axonal sprouting from neighboring intact axons contributes to reinnervation
When the distal portion of the axon is unable to regenerate, or the area of denervation is too far away from the distal site, neighboring intact axons can also send out axonal sprouts from its own axon to reinnervate nearby denervated neuromuscular junctions (Gordon and Borschel, 2017). The terminal Schwann cell sitting on the denervated neuromuscular junction (NMJ) will become activated and send out numerous processes (Reynolds and Woolf, 1992; Woolf et al., 1992; Kang et al., 2003, 2014). Eventually, a process will connect with a nearby intact axonal sprout and create a bridge between the intact axonal sprout and the denervated NMJ. This then allows the axonal sprout to be guided towards the denervated NMJ where it can then reinnervate the NMJ. These axonal sprouts can form from multiple regions along intact axons (Brown et al., 1981). Axonal sprouts, termed nodal sprouts, can emerge along the main axon branch at the nodes of Ranvier where the myelin sheath does not cover the axon. The sprouts can also emerge closer to the NMJs, called pre-terminal sprouts, where the myelin sheath ends just before the NMJ to allow for the terminal bouton to interact with the NMJ. Finally, there can also be terminal axonal sprouts which come directly from the terminal bouton innervating the NMJ. Terminal axonal sprouts are easiest to identify as axonal sprouts as the normal morphology of the terminal bouton at the NMJ only has one axonal connection (Brown et al., 1981). In cases of disease or injury, these terminal to terminal connections can be seen and scored as an axonal sprout. While axonal sprouting to neighboring targets is an important process to maintain connections under conditions of neuronal cell loss, it is a limited process. The size of any particular motor unit is bound by the surviving neuron's ability to metabolically sustain such a large number of connections (Gordon et al., 2004; Hegedus et al., 2008). Eventually, the motor unit becomes too large and the neuron cannot afford to continue to sprout axons.
Regeneration associated genes
In order to initiate axonal sprouting, the gene expression profiles change to a pro-regenerative state. With the recent advances of microarrays, and now RNA Seq, the number of genes that are differentially expressed following acute peripheral nerve injury is ever growing (Seijffers et al., 2007; Ma et al., 2011; Painter et al., 2014). Many of these regeneration associated genes (RAGs) are anti-apoptotic, transcription factors, or are involved in cytoskeleton remodeling, cell growth, and cell-cell adhesion. In order to accommodate the rapid increase in transcription, many transcription factors become upregulated following injury and have been observed to enhance regeneration, with c-Jun, signal transducer and activator of transcription 3 (STAT3) and activating transcription factor 3 (ATF3) being notable (Schwaiger et al., 2000; Raivich et al., 2004; Seijffers et al., 2007; Lindå et al., 2011). Many kinase cascades, such as the mitogen-activated protein kinase (MAPK) pathway, have been identified as RAGs, however, there is some debate over whether these pathways are involved in adult peripheral regeneration (Liu and Snider, 2001). Neurotrophic factors, such as insulin-like growth factor (IGF) 1, IGF2 and brain derived neurotrophic factor (BDNF), are also increased following injury and have been shown to positively impact axonal regeneration (Kanje et al., 1989; Glazner et al., 1993; Hammarberg et al., 2000). There is also a large upregulation of the expression of cell adhesion molecules following nerve injuries, such as CHL1 (Zhang et al., 2000). Proteins that aid in the interaction between the cell surface and the cytoskeletal components of the cell, such as growth associated protein 43 (GAP43), are upregulated following injury in order to prepare for the large cytoskeleton remodeling involved in axonal outgrowth (Skene and Willard, 1981; Frey et al., 2000a; Bomze et al., 2001). The genes listed here are just a snapshot of the large number of genes whose expression levels are altered in order for neurons to undergo the extensive and complicated process of regeneration.
Problems in Peripheral Nerve Regeneration
Peripheral nerves must overcome many obstacles for proper regeneration
The ability for regeneration in the PNS is especially important since peripheral nerves are less protected than those in the CNS and are injured much more frequently. While the peripheral nervous system contains the capacity for regeneration, it is often slow and incomplete, leading to less than favorable outcomes following severe nerve trauma (Figure 2) (Seddon, 1942; Sunderland, 1951; Griffin et al., 2010). This is especially true in humans where the average rate of regeneration is around 1 mm per day. Depending on the site of injury and how far away it is from the target muscle, the regeneration rate may be too slow for the axon to reach the muscle before other changes begin to happen that are detrimental to regeneration (Höke, 2006; Gordon et al., 2011). When a muscle is denervated for too long the NMJs begin to breakdown into fragments, which can greatly hinder the regenerating axon's ability to reinnervate its target muscle (Wu et al., 2014). In addition to the breakdown of NMJs, the receptiveness of the supporting Schwann cells for the regenerating axon also becomes reduced following prolonged denervation (Höke, 2011). All of these changes contribute to significantly limit the effectiveness of peripheral nerve regeneration and can lead to suboptimal clinical outcomes. Additionally, axonal regeneration is very inefficient and many of the axonal sprouts that are sent out never end up reaching their target. Another major setback is that even when the axon is able to reach its target, the myelination and efficiency of the nerve remains impaired, even long after the connection has stabilized (Sanders and Whitteridge, 1946; Fawcett and Keynes, 1990).
Figure 2.
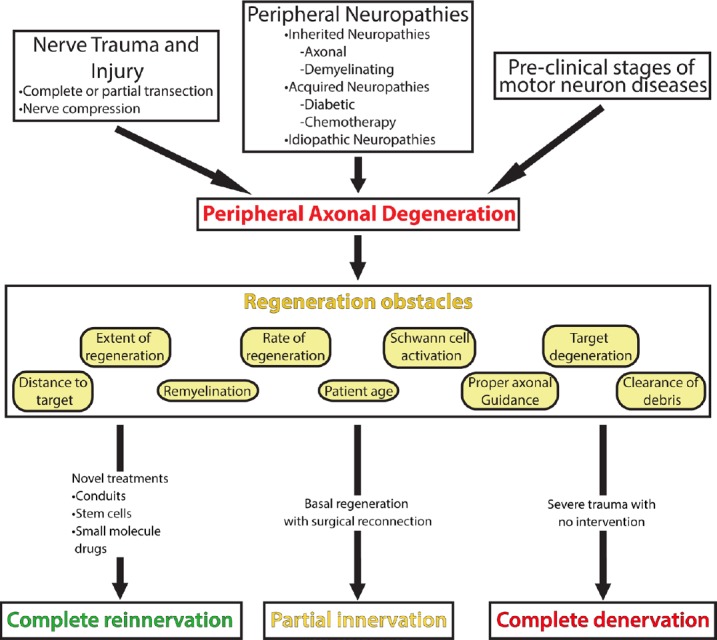
Problems in peripheral nerve regeneration.
There are many ways axons of the peripheral nervous system can get injured by nerve trauma, various peripheral neuropathies, or early stages of motor neuron diseases. The injuries that arise from these events lead to axonal damage and denervation. In order for regeneration to occur, many obstacles, which are detailed in the regeneration obstacles box, need to be overcome. With the current intervention of surgical repair and the basal state of regeneration, the extent of reinnervation is typically only partially complete. With the development of novel treatments, we hope that complete reinnervation can be achieved. In cases of severe trauma with no surgical intervention, there is a risk of no reinnervation occurring, leaving the target organ completely denervated.
Peripheral nerve regeneration is further impaired by age and disease
These issues are even greater when the repair process itself becomes reduced either due to aging or neurodegenerative disorders (Verdú et al., 2000). In terms of aging, the rate and number of regenerating axons in elderly patients and animals are significantly reduced following a peripheral nerve injury (Verdú et al., 2000). All of the key steps in peripheral nerve regeneration are reduced in aged rats. The peripheral nerves do not sprout as extensively, myelin debris clearance is delayed and impaired, and Schwann cell activation is inhibited (Verdú et al., 2000; Painter et al., 2014). In cases of peripheral neuropathies or motor neuron diseases with distal axonal degeneration, the rate of axonal regeneration may be overwhelmed by the rate of axonal degeneration occurring via the disease process (Gordon et al., 2004; Hegedus et al., 2007, 2008). This is further complicated by subsequent neuronal cell loss which reduces the number of available axons for regenerating. This then increases the burden on the surviving axons to generate axonal sprouts and increase their motor unit size.
Therapeutic options for enhancing regeneration are absent
Despite insufficient repair following axonal insult to the peripheral nervous system, either due to trauma or disease, there are currently no therapeutics available to enhance regeneration. As it stands right now, the best available treatment is surgical reattachment of the distal portion of the nerve to the proximal stump in hopes that the proximal stump can sprout into the vacated Schwann cell tubes to accelerate repair (Palispis and Gupta, 2017). Even with the best surgical techniques however, this still leaves many without full recovery. Current research to enhance regeneration focuses on developing nerve conduits which improve the connection between the proximal and distal stumps (Lin et al., 2013), this approach does not aid those who suffer from severely pinched nerves or those suffering from neurodegenerative diseases, nor does it address the slow rate of axonal growth. Other potential therapeutics have looked into electrically stimulating the nerves to enhance regeneration. Some research has shown that when the proximal portion of an injured nerve is stimulated, axonal growth is enhanced (Xu et al., 2014). However, this strategy has the potential to be an invasive and painful experience for the patient. What is needed is a minimally invasive therapy that can be administered following successful reattachment, most likely together with a nerve conduit, which can accelerate the growth rate.
β-Secretase as a Novel Regulator of Peripheral Nerve Regeneration
BACE1 activity levels inversely control peripheral nerve regeneration
Recently, our lab has identified a novel role for BACE1 in modulating peripheral nerve regeneration following injury. Based on our data, we have determined that BACE1 activity levels have an inverse effect on the efficacy of peripheral nerve regeneration. We observed a significant improvement in peripheral nerve regeneration following a sciatic nerve crush in BACE1 KO mice when compared with their wild type (WT) littermates (Farah et al., 2011). The nerves in the KO mice were able to grow further on top of having a greater number of regenerated axons compared with WT mice. There was also an improvement in muscle reinnervation in the BACE1 KO mice. These experiments provided interesting data highlighting a novel role for BACE1 in regulating peripheral nerve regeneration. However, these experiments did not narrow down where BACE1 was exerting its effects. The enhanced regeneration could be due to increasing the outgrowth of the axons themselves, enhanced macrophage recruitment and activity, or even enhanced Schwann cell activation and axonal support.
In order to explore where BACE1 was acting and to further understand its role in regeneration, we performed similar experiments in transgenic mice overexpressing human BACE1, solely in neurons (Tallon et al., 2017). Following a sciatic nerve crush, we observed a marked decrease in peripheral nerve regeneration in mice overexpressing BACE1. These mice had reduced axonal outgrowth early on and a decrease in the number of regenerated axons. We also observed a significant reduction in the reinnervation of the gastrocnemius muscle in overexpressing mice. This impaired reinnervation was also reflected in electrophysiological experiments where transgenic mice had a significant delay in the recovery of the compound muscle action potential in the plantar muscles. Based on our own data, we have concluded that neuronal BACE1 activity levels inversely influence axonal regeneration in the periphery following injury.
While BACE1 activity's influence over the neuronal component of peripheral nerve regeneration is an interesting piece of the puzzle, we also need to investigate the role that macrophage and Schwann cell BACE1 activity levels play in enhancing regeneration as well. Our original study using global BACE1 KO mice indicated that macrophages are not only recruited in greater numbers, but also engulf more myelin debris following a peripheral nerve injury (Farah et al., 2011). We also observed an increase in a macrophage signaling receptor, tumor necrosis factor receptor 1 (TNFR1), expression in the distal stump of the injured nerve in both BACE1 KO mice and WT mice treated with a BACE1 inhibitor (Liu et al., 2016). Studies investigating how BACE1 activity levels influence the initial activation of Schwann cells following peripheral nerve injuries has not yet been studied. However, there is evidence which indicates that BACE1 activity is involved in peripheral nerve remyelination following axonal outgrowth and is an important for proper remyelination to occur (Hu et al., 2008, 2015). In order to truly elucidate how BACE1 activity levels influence each component of the peripheral nerve regeneration machinery our lab is currently conducting experiments utilizing conditional KO mice for each of these components.
BACE1 substrates themselves influence axonal regeneration
In addition to our own data, there has been some evidence that some of the substrates of BACE1 are also involved in regulating peripheral nerve regeneration. A few studies have found that the processing of APP itself has an effect on neurite outgrowth. Knocking out APP in cultured neurons has led to enhanced neurite outgrowth (Perez et al., 1997; Young-Pearse et al., 2008), while soluble APPβ has been shown to have an inhibiting effect on neurite outgrowth (Li et al., 1997). While interesting, the effects APP processing has on peripheral nerve regeneration has been inconclusive. The processing of another BACE1 substrate, NCAM, has also been shown to have an impact on neurite outgrowth and branching. Cleavage of NCAM to produce ectodomain shedding reduces neurite outgrowth, which can be rescued by preventing the release of the ectodomain (Hinkle et al., 2006). Additionally, the cleavage of CHL1 by BACE1 also impacts neurite outgrowth. When the release of CHL1's cleavage product is prevented, the growth cone no longer collapses and promotes neurite extension (Barão et al., 2015). Taken together with our own data, neuronal BACE1 activity on its various substrates may play an important role in regulating peripheral nerve regeneration by modulating cell adhesion molecules (Figure 3).
Figure 3.
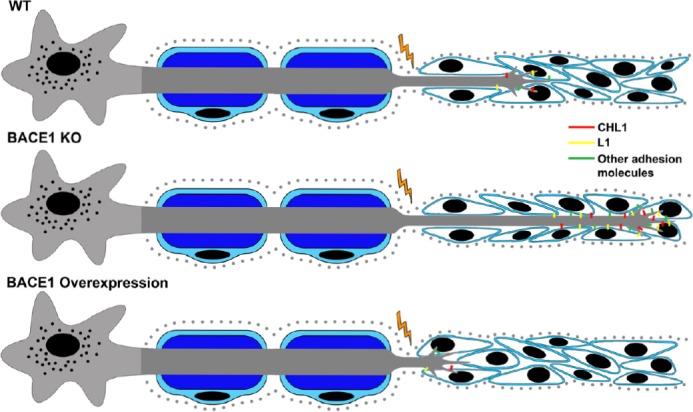
Potential mechanism for how neuronal beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) activity levels regulate peripheral axon growth.
Since adhesion molecules are essential for proper axonal outgrowth and interaction with Schwann cells (depicted in blue), we hypothesize that BACE1 regulates peripheral axonal outgrowth by modulating the cell surface expression levels of various adhesion molecules, such as close homolog of L1 (CHL1) and L1. In wild type (WT) mice, adhesion molecules are moderately expressed on the cell surface leading to basal regeneration rates. With reduced BACE1 expression, as in knock out (KO) mice, adhesion molecules are not cleaved and therefore remain at the cell surface in greater numbers, enhancing interactions with Schwann cells and leading to increased growth. When BACE1 is overexpressed, there is more cleavage causing fewer adhesion molecules to be expressed on the cell surface, impairing Schwann cell interactions and reducing axonal growth.
β-Secretase Inhibitors as a Potential Therapeutic Strategy for Enhancing Peripheral Nerve Regeneration
BACE1 inhibitors developed for AD have good safety profiles
As we mentioned earlier in this review, because BACE1 has a large role in generating Aβ plaques in AD, many pharmaceutical companies have made great efforts to develop small molecule BACE1 inhibitors. Early efforts were held back due to the liver toxicity seen with the Eli Lilly BACE1 inhibitor LY2886721 during their phase II trials. This led many to speculate whether BACE1 inhibitors were a feasible target for AD. However, recent advances in the development of these drugs has shown more promise. Recently, Merck announced that their latest BACE1 inhibitor, verubecestat (MK-8931), was well tolerated at various doses in patients enrolled in their phase I clinical trial (Kennedy et al., 2016). Verubecestat also showed a robust decrease in CSF Aβ production, indicative of proper target engagement. In addition to verubecestat, AstraZeneca/Lilly also have a promising small molecule BACE1 inhibitor, AZD3293, currently in phase II/III clinical trials. This drug was also well tolerated in phase I trials and showed a marked decrease in CSF Aβ production (Cebers et al., 2017).
Unfortunately, Merck recently stopped their mild/moderate Phase III trial for verubecestat due to a lack of significant clinical changes observed. Despite being a disappointing setback, this result was unsurprising to those in the field as Aβ levels tend to plateau and neuronal death is widespread by the time clinical symptoms begin to appear. While this is bad news for AD, all may not be lost for those who have sunk billions into developing BACE1 inhibitors. Since high BACE1 activity appears to have a negative impact on peripheral nerve regeneration, we hypothesize that BACE1 inhibitors may be a potential drug to fill the therapeutic void of compounds for enhancing peripheral nerve regeneration. Indeed, when we administered an older BACE1 inhibitor for 7 days following a sciatic nerve crush, we observed an apparent increase in regenerated axons as well as enhanced axonal debris clearance (Farah et al., 2011). This is a promising start for determining whether BACE1 inhibitors may find a new life as peripheral nerve regeneration enhancers.
In addition to the generation of safe, well-tolerated BACE1 inhibitors, treating peripheral nerve injuries with small molecule inhibitors may not have to worry about the blood-brain barrier issue that hinders CNS drug development. Unlike the blood-brain barrier, the nerve-blood barrier is rather leaky and would most likely allow larger molecules through at lower concentrations (Mellick and Cavanagh, 1968). This may be beneficial and could potentially allow for the development of more potent BACE1 inhibitors that while too large to cross the blood-brain barrier, may be able to cross the nerve-blood barrier. Being able to get a high efficiency with a lower dose would also lower the risk of toxicity issues that has been seen with some BACE1 inhibitors at higher doses and would give a therapeutic advantage to treating peripheral nerve injuries with BACE1 inhibitors.
Potential issues surrounding the use of BACE1 inhibitors following acute peripheral nerve injuries
While utilizing BACE1 inhibitors as a means to enhance regeneration following a peripheral nerve injury has a lot of promise, due to BACE1's promiscuous nature, potential issues of inhibition need to be closely monitored. Firstly, because BACE1's cleavage of NRG1 type III is involved in the myelination of peripheral nerves it is important to think about how this may negatively impact peripheral nerve remyelination as well as the existing myelin on uninjured axons. In the case of uninjured nerves, their myelination does not appear to be altered with BACE1 inhibition as we have seen in our own lab (unpublished data) as well as by other groups (Sankaranarayanan et al., 2008). For regenerating axons, remyelination would most likely be reduced for as long as BACE1 activity is being reduced. This hypomyelination would impact the ability of the axons to fire in a coherent manner and may have a negative impact on the effectiveness of the regenerated axons to engage with its targets correctly. How large this deficit would be is a factor that needs to be studied with regards to administering BACE1 inhibitors following a peripheral axon injury. While this deficit could be similar to the hypo-remyelination observed in BACE1 KO mice (Hu et al., 2008, 2015), it may be ameliorated by only administering the BACE1 inhibitors for a short time period, a couple of weeks at most, immediately following the injury. This dosage schedule would induce increased axonal outgrowth during the early stages of repair and once the inhibition of BACE1 is abolished, remyelination would be allowed to occur in a normal capacity and would hopefully allow for proper axonal firing to occur. Additionally, there is some evidence that BACE1 cleavage of NRG1 type III is not essential for the induction of myelination (Velanac et al., 2012). Therefore, the amount of remyelination that occurs with BACE1 inhibition may be sufficient for proper nerve repair.
Another issue that could potentially arise when using BACE1 inhibitors is that there may be possible defects with the muscle spindles. In the absence of BACE1, mice have defects in muscle spindle formation and maturation (Cheret et al., 2013). Additionally, when BACE1 was inhibited in adult mice, Cheret et al. (2013) found that muscle spindle maintenance was impaired as was motor coordination. While this finding may be troubling for the use of BACE1 inhibitors, phase I clinical trial reports about adverse responses to BACE1 administration in human patients did not report any issues with motor coordination even at the highest doses administered (Kennedy et al., 2016). The lack of issues seen in human patients following BACE1 inhibitor treatment leads us to believe that many of the issues surrounding possible pitfalls associated with BACE1 inhibition may not be as severe as originally thought.
BACE1 inhibitors may be useful in early stage peripheral neurodegenerative diseases
We also hypothesize that these inhibitors may be helpful in the early stages of peripheral neurodegenerative disorders. In the earliest stages of motor neuron disease, axonal degeneration typically precedes neuronal cell body death (Azzouz et al., 1997; Frey et al., 2000b; Fischer et al., 2004; Schaefer et al., 2005; Pun et al., 2006; Hegedus et al., 2007). These neurons also die back at different rates depending on their susceptibility to degeneration based on multiple factors such as fiber type, size, and length (Frey et al., 2000b; Pun et al., 2006; Hegedus et al., 2007; Saxena and Caroni, 2007; Saxena et al., 2009, 2013; Tallon et al., 2016). We theorize that motor function, and therefore quality of life, may be prolonged in the early stages of these diseases if the surviving motor neurons could be encouraged to sprout and regrow in an attempt to reinnervate denervated muscle. It is true that even with a disease background, peripheral nerves do have the capacity to sprout, however limited it may be (Brown et al., 1981; Schäfers et al., 2002; Gordon et al., 2004; Tallon et al., 2016). If the rate of axonal sprouting and outgrowth can be enhanced using a BACE1 inhibitor, it may be able to keep up with the rate of degeneration for a time, especially in slowly progressing disorders.
BACE1 inhibitor treatment may avoid axonal guidance issues
BACE1 inhibitors being used to enhance peripheral nerve regeneration may also be able to overcome many of the axonal guidance issues that have been identified. Many of these studies were performed in null mice, either of BACE1 or its substrates, and using an inhibitor that would not completely abolish BACE1 activity levels may lead to a less severe phenotype with regards to axonal guidance. Additionally, these studies were also focusing on how a loss of BACE1 or its substrates affects axonal guidance in the CNS. Due to the wide variety of BACE1 substrates, it may be possible that the substrates involved in impaired axonal guidance in the CNS may not be as important for similar functions in the peripheral nervous system. An alternative hypothesis is that these substrates may interact with different molecules and cells in the periphery which lead to a different phenotype. This makes sense, as axons in the CNS interact with oligodendrocytes and astrocytes while the PNS axons interact with Schwann cells. Indeed, our lab has not noticed any morphological differences in how NMJs are innervated in uninjured BACE1 KO mice. Another issue to note is that many of the null studies may be related to developmental defects, as the mice are lacking BACE1 and its substrates from the beginning. Using BACE1 inhibitors for the treatment of peripheral neurodegeneration would occur after the bulk of the synaptic connections had been formed, and depending on the type of neurodegeneration, would not need to be a very prolonged treatment and would allow proper guidance to occur in normal pruning.
Conclusion
Our current understanding of how BACE1 is involved in regulating axonal regeneration is still in its infancy. However, we have been able to identify an interesting relationship between BACE1 activity levels and how effective the axons are at regenerating following an acute nerve injury. Our own data has determined that BACE1 activity levels inversely regulate axonal regeneration and this finding has interesting clinical implications. If the administration of BACE1 inhibitors following an acute nerve injury can help to increase the rate and efficacy of peripheral nerve regeneration, it would fill a much-needed therapeutic gap for acute nerve injuries. Additionally, BACE1 inhibitors may also be useful in the early stages of peripheral neuropathies as a means for enhancing the rate of axonal regeneration, which may be able to compensate for the progressive loss of axons and leading to prolonged function.
Footnotes
Conflicts of interest: None declared.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review reports:
Reviewer 1: Mohammad Reza Farahpour, Islamic Azad University Urmia Branch, Iran.
Reviewer 2: Ben Christensen, University of Utah, USA.
Comments to authors: The authors’ present a literature review of BACE1, specifically related to its role in peripheral nerve regeneration, as well as a brief synopsis of degenerative and regenerative process in the PNS following injury. Overall, the authors do a good job of reviewing this topic and incorporating relevant literature. A few topics in relation to BACE1 and peripheral regeneration could be included to enhance this manuscript's impact.
Funding: This work was supported by the Muscular Dystrophy Association and R01NS079339 from the National Institutes of Neurological Disease and Stroke of the National Institutes of Health.
References
- Azzouz M, Leclerc N, Gurney M, Warter JM, Poindron P, Borg J. Progressive motor neuron impairment in an animal model of familial amyotrophic lateral sclerosis. Muscle Nerve. 1997;20:45–51. doi: 10.1002/(sici)1097-4598(199701)20:1<45::aid-mus6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Barão S, Gärtner A, Leyva-Díaz E, Demyanenko G, Munck S, Vanhoutvin T, Zhou L, Schachner M, López-Bendito G, Maness PF, De Strooper B. Antagonistic effects of BACE1 and APH1B-γ-secretase control axonal guidance by regulating growth cone collapse. Cell Rep. 2015;12:1367–1376. doi: 10.1016/j.celrep.2015.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomze HM, Bulsara KR, Iskandar BJ, Caroni P, Skene JH. Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci. 2001;4:38–43. doi: 10.1038/82881. [DOI] [PubMed] [Google Scholar]
- Brück W. The role of macrophages in Wallerian degeneration. Brain Pathol. 1997;7:741–752. doi: 10.1111/j.1750-3639.1997.tb01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Holland RL, Hopkins WG. Motor nerve sprouting. Annu Rev Neurosci. 1981;4:17–42. doi: 10.1146/annurev.ne.04.030181.000313. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Cebers G, Alexander RC, Haeberlein SB, Han D, Goldwater R, Ereshefsky L, Olsson T, Ye N, Rosen L, Russell M, Maltby J, Eketjall S, Kugler AR. AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer's disease. J Alzheimers Dis. 2017;55:1039–1053. doi: 10.3233/JAD-160701. [DOI] [PubMed] [Google Scholar]
- Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, Tang J. Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011;25:775–784. doi: 10.1096/fj.10-167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QY, Chen Q, Feng GY, Lindpaintner K, Chen Y, Sun X, Chen Z, Gao Z, Tang J, He L. Case-control association study of the close homologue of L1 (CHL1) gene and schizophrenia in the Chinese population. Schizophr Res. 2005;73:269–274. doi: 10.1016/j.schres.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Cheret C, Willem M, Fricker FR, Wende H, Wulf-Goldenberg A, Tahirovic S, Nave KA, Saftig P, Haass C, Garratt AN, Bennett DL, Birchmeier C. Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles. EMBO J. 2013;32:2015–2028. doi: 10.1038/emboj.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah MH. BACE1 influences debris clearance and axonal regeneration in injured peripheral nerve. J Peripher Nerv Syst. 2012;17(Suppl 3):30–33. doi: 10.1111/j.1529-8027.2012.00428.x. [DOI] [PubMed] [Google Scholar]
- Farah MH, Pan BH, Hoffman PN, Ferraris D, Tsukamoto T, Nguyen T, Wong PC, Price DL, Slusher BS, Griffin JW. Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J Neurosci. 2011;31:5744–5754. doi: 10.1523/JNEUROSCI.6810-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Keynes RJ. Peripheral nerve regeneration. Annu Rev Neurosci. 1990;13:43–60. doi: 10.1146/annurev.ne.13.030190.000355. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Laux T, Xu L, Schneider C, Caroni P. Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J Cell Biol. 2000a;149:1443–1454. doi: 10.1083/jcb.149.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000b;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam V, D’Avanzo C, Hebisch M, Kovacs DM, Kim DY. BACE1 activity regulates cell surface contactin-2 levels. Mol Neurodegener. 2014;9:4. doi: 10.1186/1750-1326-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George R, Griffin JW. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Exp Neurol. 1994;129:225–236. doi: 10.1006/exnr.1994.1164. [DOI] [PubMed] [Google Scholar]
- Gersbacher MT, Kim DY, Bhattacharyya R, Kovacs DM. Identification of BACE1 cleavage sites in human voltage-gated sodium channel beta 2 subunit. Mol Neurodegener. 2010;5:61. doi: 10.1186/1750-1326-5-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazner GW, Lupien S, Miller JA, Ishii DN. Insulin-like growth factor II increases the rate of sciatic nerve regeneration in rats. Neuroscience. 1993;54:791–797. doi: 10.1016/0306-4522(93)90248-e. [DOI] [PubMed] [Google Scholar]
- Gordon T, English AW. Strategies to promote peripheral nerve regeneration: electrical stimulation and/or exercise. Eur J Neurosci. 2016;43:336–350. doi: 10.1111/ejn.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, Borschel GH. The use of the rat as a model for studying peripheral nerve regeneration and sprouting after complete and partial nerve injuries. Exp Neurol. 2017;287:331–347. doi: 10.1016/j.expneurol.2016.01.014. [DOI] [PubMed] [Google Scholar]
- Gordon T, Hegedus J, Tam SL. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease. Neurol Res. 2004;26:174–185. doi: 10.1179/016164104225013806. [DOI] [PubMed] [Google Scholar]
- Gordon T, Tyreman N, Raji MA. The basis for diminished functional recovery after delayed peripheral nerve repair. J Neurosci. 2011;31:5325–5334. doi: 10.1523/JNEUROSCI.6156-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, Pan B, Polley MA, Hoffman PN, Farah MH. Measuring nerve regeneration in the mouse. Exp Neurol. 2010;223:60–71. doi: 10.1016/j.expneurol.2009.12.033. [DOI] [PubMed] [Google Scholar]
- Gunnersen JM, Kim MH, Fuller SJ, De Silva M, Britto JM, Hammond VE, Davies PJ, Petrou S, Faber ES, Sah P, Tan SS. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56:621–639. doi: 10.1016/j.neuron.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Höke A. Mechanisms of Disease: what factors limit the success of peripheral nerve regeneration in humans? Nat Clin Pract Neurol. 2006;2:448–454. doi: 10.1038/ncpneuro0262. [DOI] [PubMed] [Google Scholar]
- Höke A. A (heat) shock to the system promotes peripheral nerve regeneration. J Clin Invest. 2011;121:4231–4234. doi: 10.1172/JCI59320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarberg H, Piehl F, Risling M, Cullheim S. Differential regulation of trophic factor receptor mRNAs in spinal motoneurons after sciatic nerve transection and ventral root avulsion in the rat. J Comp Neurol. 2000;426:587–601. doi: 10.1002/1096-9861(20001030)426:4<587::aid-cne7>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol. 2008;586:3337–3351. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS One. 2009;4:e8477. doi: 10.1371/journal.pone.0008477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle CL, Diestel S, Lieberman J, Maness PF. Metalloprotease-induced ectodomain shedding of neural cell adhesion molecule (NCAM) J Neurobiol. 2006;66:1378–1395. doi: 10.1002/neu.20257. [DOI] [PubMed] [Google Scholar]
- Hitt B, Riordan SM, Kukreja L, Eimer WA, Rajapaksha TW, Vassar R. β-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J Biol Chem. 2012;287:38408–38425. doi: 10.1074/jbc.M112.415505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hu J, Dai L, Trapp B, Yan R. Axonal and Schwann cell BACE1 is equally required for remyelination of peripheral nerves. J Neurosci. 2015;35:3806–3814. doi: 10.1523/JNEUROSCI.5207-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Hu X, He W, Diaconu C, Tang X, Kidd GJ, Macklin WB, Trapp BD, Yan R. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 2008;22:2970–2980. doi: 10.1096/fj.08-106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner EA, Strittmatter SM. Axon regeneration in the peripheral and central nervous systems. Results Probl Cell Differ. 2009;48:339–351. doi: 10.1007/400_2009_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Tian L, Thompson W. Terminal Schwann cells guide the reinnervation of muscle after nerve injury. J Neurocytol. 2003;32:975–985. doi: 10.1023/B:NEUR.0000020636.27222.2d. [DOI] [PubMed] [Google Scholar]
- Kang H, Tian L, Mikesh M, Lichtman JW, Thompson WJ. Terminal Schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J Neurosci. 2014;34:6323–6333. doi: 10.1523/JNEUROSCI.4673-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanje M, Skottner A, Sjoberg J, Lundborg G. Insulin-like growth factor I (IGF-I) stimulates regeneration of the rat sciatic nerve. Brain Res. 1989;486:396–398. doi: 10.1016/0006-8993(89)90531-3. [DOI] [PubMed] [Google Scholar]
- Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, Egan M, Ereshefsky L, Hodgson RA, Hyde LA, Jhee S, Kleijn HJ, Kuvelkar R, Li W, Mattson BA, Mei H, Palcza J, Scott JD, Tanen M, Troyer MD, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer's disease patients. Sci Transl Med. 2016;8:363ra150. doi: 10.1126/scitranslmed.aad9704. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y. Alzheimer's beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci U S A. 2001;98:13554–13559. doi: 10.1073/pnas.241509198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn PH, Marjaux E, Imhof A, De Strooper B, Haass C, Lichtenthaler SF. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J Biol Chem. 2007;282:11982–11995. doi: 10.1074/jbc.M700356200. [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A, Haass C, Roßner S, Bräse S, Lichtenthaler SF. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31:3157–3168. doi: 10.1038/emboj.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurumaji A, Nomoto H, Okano T, Toru M. An association study between polymorphism of L1CAM gene and schizophrenia in a Japanese sample. Am J Med Genet. 2001;105:99–104. [PubMed] [Google Scholar]
- La Marca R, Cerri F, Horiuchi K, Bachi A, Feltri ML, Wrabetz L, Blobel CP, Quattrini A, Salzer JL, Taveggia C. TACE (ADAM17) inhibits Schwann cell myelination. Nat Neurosci. 2011;14:857–865. doi: 10.1038/nn.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Roch JM, Sundsmo M, Otero D, Sisodia S, Thomas R, Saitoh T. Defective neurite extension is caused by a mutation in amyloid beta/A4 (A beta) protein precursor found in familial Alzheimer's disease. J Neurobiol. 1997;32:469–480. [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, De Strooper B, Seed B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem. 2003;278:48713–48719. doi: 10.1074/jbc.M303861200. [DOI] [PubMed] [Google Scholar]
- Lin MY, Manzano G, Gupta R. Nerve allografts and conduits in peripheral nerve repair. Hand Clin. 2013;29:331–348. doi: 10.1016/j.hcl.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Lindå H, Sköld MK, Ochsmann T. Activating transcription factor 3, a useful marker for regenerative response after nerve root injury. Front Neurol. 2011;2:30. doi: 10.3389/fneur.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Fissel JA, Tasnim A, Borzan J, Gocke A, Calabresi PA, Farah MH. Increased TNFR1 expression and signaling in injured peripheral nerves of mice with reduced BACE1 activity. Neurobiol Dis. 2016;93:21–27. doi: 10.1016/j.nbd.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Liu RY, Snider WD. Different signaling pathways mediate regenerative versus developmental sensory axon growth. J Neurosci. 2001;21:RC164. doi: 10.1523/JNEUROSCI.21-17-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Ma CH, Omura T, Cobos EJ, Latremoliere A, Ghasemlou N, Brenner GJ, van Veen E, Barrett L, Sawada T, Gao F, Coppola G, Gertler F, Costigan M, Geschwind D, Woolf CJ. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest. 2011;121:4332–4347. doi: 10.1172/JCI58675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Fischer S, López-Vales R, David S. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia. 2008;56:1566–1577. doi: 10.1002/glia.20766. [DOI] [PubMed] [Google Scholar]
- May PC, Willis BA, Lowe SL, Dean RA, Monk SA, Cocke PJ, Audia JE, Boggs LN, Borders AR, Brier RA, Calligaro DO, Day TA, Ereshefsky L, Erickson JA, Gevorkyan H, Gonzales CR, James DE, Jhee SS, Komjathy SF, Li L, et al. The potent BACE1 inhibitor LY2886721 elicits robust central Abeta pharmacodynamic responses in mice, dogs, and humans. J Neurosci. 2015;35:1199–1210. doi: 10.1523/JNEUROSCI.4129-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellick R, Cavanagh JB. The function of the perineurium and its relation to the flow phenomenon within the endoneurial spaces. Proc Aust Assoc Neurol. 1968;5:521–525. [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Mietto BS, Mostacada K, Martinez AM. Neurotrauma and inflammation: CNS and PNS responses. Mediators Inflamm. 2015;2015:251204. doi: 10.1155/2015/251204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag-Sallaz M, Schachner M, Montag D. Misguided axonal projections, neural cell adhesion molecule 180 mRNA upregulation, and altered behavior in mice deficient for the close homolog of L1. Mol Cell Biol. 2002;22:7967–7981. doi: 10.1128/MCB.22.22.7967-7981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernández L, Lindborg JA, Mandell D, Zigmond RE. A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J Neurosci. 2013;33:16236–16248. doi: 10.1523/JNEUROSCI.3319-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MW, Brosius Lutz A, Cheng YC, Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX, Wagers AJ, Havton LA, Barres B, Omura T, Woolf CJ. Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron. 2014;83:331–343. doi: 10.1016/j.neuron.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palispis WA, Gupta R. Surgical repair in humans after traumatic nerve injury provides limited functional neural regeneration in adults. Exp Neurol. 2017;290:106–114. doi: 10.1016/j.expneurol.2017.01.009. [DOI] [PubMed] [Google Scholar]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH. The beta-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17:9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigoni M, Wanngren J, Kuhn PH, Munro KM, Gunnersen JM, Takeshima H, Feederle R, Voytyuk I, De Strooper B, Levasseur MD, Hrupka BJ, Muller SA, Lichtenthaler SF. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol Neurodegener. 2016;11:67. doi: 10.1186/s13024-016-0134-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratte M, Rougon G, Schachner M, Jamon M. Mice deficient for the close homologue of the neural adhesion cell L1 (CHL1) display alterations in emotional reactivity and motor coordination. Behav Brain Res. 2003;147:31–39. doi: 10.1016/s0166-4328(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Reynolds ML, Woolf CJ. Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J Neurocytol. 1992;21:50–66. doi: 10.1007/BF01206897. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K, Johnson-Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nichols NF, Power M, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- Sakurai K, Migita O, Toru M, Arinami T. An association between a missense polymorphism in the close homologue of L1 (CHL1, CALL) gene and schizophrenia. Mol Psychiatry. 2002;7:412–415. doi: 10.1038/sj.mp.4000973. [DOI] [PubMed] [Google Scholar]
- Sanders FK, Whitteridge D. Conduction velocity and myelin thickness in regenerating nerve fibres. J Physiol. 1946;105:152–174. [PubMed] [Google Scholar]
- Sankaranarayanan S, Price EA, Wu G, Crouthamel MC, Shi XP, Tugusheva K, Tyler KX, Kahana J, Ellis J, Jin L, Steele T, Stachel S, Coburn C, Simon AJ. In vivo beta-secretase 1 inhibition leads to brain Abeta lowering and increased alpha-secretase processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther. 2008;324:957–969. doi: 10.1124/jpet.107.130039. [DOI] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Mechanisms of axon degeneration: from development to disease. Prog Neurobiol. 2007;83:174–191. doi: 10.1016/j.pneurobio.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron. 2013;80:80–96. doi: 10.1016/j.neuron.2013.07.027. [DOI] [PubMed] [Google Scholar]
- Schäfers M, Schmidt C, Vogel C, Toyka KV, Sommer C. Tumor necrosis factor-alpha (TNF) regulates the expression of ICAM-1 predominantly through TNF receptor 1 after chronic constriction injury of mouse sciatic nerve. Acta Neuropathol. 2002;104:197–205. doi: 10.1007/s00401-002-0541-9. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Schwaiger FW, Hager G, Schmitt AB, Horvat A, Hager G, Streif R, Spitzer C, Gamal S, Breuer S, Brook GA, Nacimiento W, Kreutzberg GW. Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT) Eur J Neurosci. 2000;12:1165–1176. doi: 10.1046/j.1460-9568.2000.00005.x. [DOI] [PubMed] [Google Scholar]
- Seddon HJ. A classification of nerve injuries. Br Med J. 1942;2:237–239. doi: 10.1136/bmj.2.4260.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–7920. doi: 10.1523/JNEUROSCI.5313-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Skene JH, Willard M. Characteristics of growth-associated polypeptides in regenerating toad retinal ganglion cell axons. J Neurosci. 1981;1:419–426. doi: 10.1523/JNEUROSCI.01-04-00419.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares S, Traka M, von Boxberg Y, Bouquet C, Karagogeos D, Nothias F. Neuronal and glial expression of the adhesion molecule TAG-1 is regulated after peripheral nerve lesion or central neurodegeneration of adult nervous system. Eur J Neurosci. 2005;21:1169–1180. doi: 10.1111/j.1460-9568.2005.03961.x. [DOI] [PubMed] [Google Scholar]
- Stoll G, Griffin JW, Li CY, Trapp BD. Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J Neurocytol. 1989;18:671–683. doi: 10.1007/BF01187086. [DOI] [PubMed] [Google Scholar]
- Stoll G, Li CY, Trapp BD, Griffin JW. Expression of NGF-receptors during immune-mediated and lysolecithin-induced demyelination of the peripheral nervous system. J Neurocytol. 1993;22:1022–1029. doi: 10.1007/BF01235746. [DOI] [PubMed] [Google Scholar]
- Sunderland S. A classification of peripheral nerve injuries producing loss of function. Brain. 1951;74:491–516. doi: 10.1093/brain/74.4.491. [DOI] [PubMed] [Google Scholar]
- Tallon C, Rockenstein E, Masliah E, Farah MH. Increased BACE1 activity inhibits peripheral nerve regeneration after injury. Neurobiol Dis. 2017;106:147–157. doi: 10.1016/j.nbd.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Tallon C, Russell KA, Sakhalkar S, Andrapallayal N, Farah MH. Length-dependent axo-terminal degeneration at the neuromuscular synapses of type II muscle in SOD1 mice. Neuroscience. 2016;312:179–189. doi: 10.1016/j.neuroscience.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy D, Namgung U. Role of glial cells in axonal regeneration. Exp Neurobiol. 2013;22:68–76. doi: 10.5607/en.2013.22.2.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Dupree JL, Popko B, Karagogeos D. The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J Neurosci. 2002;22:3016–3024. doi: 10.1523/JNEUROSCI.22-08-03016.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Velanac V, Unterbarnscheidt T, Hinrichs W, Gummert MN, Fischer TM, Rossner MJ, Trimarco A, Brivio V, Taveggia C, Willem M, Haass C, Mobius W, Nave KA, Schwab MH. Bace1 processing of NRG1 type III produces a myelin-inducing signal but is not essential for the stimulation of myelination. Glia. 2012;60:203–217. doi: 10.1002/glia.21255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdú E, Ceballos D, Vilches JJ, Navarro X. Influence of aging on peripheral nerve function and regeneration. J Peripher Nerv Syst. 2000;5:191–208. doi: 10.1046/j.1529-8027.2000.00026.x. [DOI] [PubMed] [Google Scholar]
- Weller S, Gärtner J. Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): Mutations in the L1CAM gene. Hum Mutat. 2001;18:1–12. doi: 10.1002/humu.1144. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Wolman MA, Sittaramane VK, Essner JJ, Yost HJ, Chandrasekhar A, Halloran MC. Transient axonal glycoprotein-1 (TAG-1) and laminin-alpha1 regulate dynamic growth cone behaviors and initial axon direction in vivo. Neural Dev. 2008;3:6. doi: 10.1186/1749-8104-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Reynolds ML, Chong MS, Emson P, Irwin N, Benowitz LI. Denervation of the motor endplate results in the rapid expression by terminal Schwann cells of the growth-associated protein GAP-43. J Neurosci. 1992;12:3999–4010. doi: 10.1523/JNEUROSCI.12-10-03999.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Chawla A, Spinner RJ, Yu C, Yaszemski MJ, Windebank AJ, Wang H. Key changes in denervated muscles and their impact on regeneration and reinnervation. Neural Regen Res. 2014;9:1796–1809. doi: 10.4103/1673-5374.143424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Kou Y, Zhang P, Han N, Yin X, Deng J, Chen B, Jiang B. Electrical stimulation promotes regeneration of defective peripheral nerves after delayed repair intervals lasting under one month. PLoS One. 2014;9:e105045. doi: 10.1371/journal.pone.0105045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Boyer AM, Crandall JE, Edwards M, Tanaka H. Distribution of stage-specific neurite-associated proteins in the developing murine nervous system recognized by a monoclonal antibody. J Neurosci. 1986;6:3576–3594. doi: 10.1523/JNEUROSCI.06-12-03576.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Young-Pearse TL, Chen AC, Chang R, Marquez C, Selkoe DJ. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 2008;3:15. doi: 10.1186/1749-8104-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang J, Wu Y, Zhou Y, Shao Z, Kang X, Ma L, Li M, Liu L, Shi H. Sez-6 may play an important role in neurite outgrowth through the PKCgamma signaling pathways. Z Naturforsch C. 2011;66:614–620. doi: 10.1515/znc-2011-11-1211. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Roslan R, Lang D, Schachner M, Lieberman AR, Anderson PN. Expression of CHL1 and L1 by neurons and glia following sciatic nerve and dorsal root injury. Mol Cell Neurosci. 2000;16:71–86. doi: 10.1006/mcne.2000.0852. [DOI] [PubMed] [Google Scholar]
- Zhou L, Barão S, Laga M, Bockstael K, Borgers M, Gijsen H, Annaert W, Moechars D, Mercken M, Gevaert K, De Strooper B. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem. 2012;287:25927–25940. doi: 10.1074/jbc.M112.377465. [DOI] [PMC free article] [PubMed] [Google Scholar]