

Abstract
Desipramine is a secondary tricyclic amine, which is primarily metabolized by cytochrome 2D6. It shows a high volume of distribution (Vss) (10–50 L/kg) due to its high lipophilicity, unspecific phospholipid binding, and lysosomal trapping. The objective of this study was to develop and qualify a physiologically based pharmacokinetic (PBPK) model for desipramine, which accounts for the high Vss of the drug following intravenous and oral administration of doses up to 100 mg. The model also accounts for the extended time to reach maximum concentration after oral dosing due to enterocyte trapping. Once developed and qualified in adults, we characterized the dynamic changes in metabolism and pharmacokinetics of desipramine after birth by scaling the system‐specific parameters of the model from adults to pediatrics. The developed modeling strategy provides a prototypical workflow that can also be applied to other drugs with similar properties and a high volume of distribution.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Desipramine is a secondary tricyclic amine, which exerts a high volume of distribution (10–50 L/kg) due to its high lipophilicity, unspecific phospholipid binding, and lysosomal trapping.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The objective of this study was to develop and qualify a physiologically based pharmacokinetic model for desipramine, which accounts for the high volume of distribution of the drug following intravenous and oral administration of doses up to 100 mg.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ We developed an age‐ranging PBPK model for desipramine that is able to account for the drug's atypical distribution behavior due to phospholipid binding and lysosomal trapping as well as changes in drug clearance due to growth and maturation.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The developed modeling strategy provides a prototypical workflow that can also be applied to other drugs with similar properties and a high volume of distribution.
Desipramine is a secondary tricyclic amine, which is indicated for the treatment of depression.1, 2 Desipramine is primarily metabolized by cytochrome 2D6 (CYP2D6) to its 2‐hydroxy metabolite,3 which might itself possess some clinical antidepressant activity.4 The US Food and Drug Administration (FDA) issued a black‐box warning for desipramine because of the increased risk of suicidal thinking and behavior in children, adolescents, and young adults. Desipramine is also used off‐label in adults and pediatrics for the treatment of enuresis anxiety and attention‐deficit hyperactivity disorder (ADHD).5, 6, 7, 8 However, there is also concern among clinicians about the use of desipramine in pediatrics due to QT prolongation9, 10 and cases of death following administration and accumulation of imipramine and its active metabolite desipramine.11 Given its use in patients across a wide age range, it is important to understand desipramine's dose concentration–response relationship in adults and changes therein with age when attempting to establish safe and effective dosing regimen for patients of different age groups. This is likely to become even more important in the next few years due to a recent recommendation of the US Preventative Services Task Force (USPSTF) indicating the need of screening the general adult population, including pregnant and postpartum women, for depression.12
Dosing regimens for special patient populations are typically obtained by scaling adult dosing regimen by either size (e.g., by body weight (BW)) or function (e.g., organ function in renally or hepatically impaired patients). In particular, dosing regimens for children are routinely obtained by allometrically scaling clearance (CLchild = CLadult (BWchild/BWadult)0.75) and volume of distribution (Vdchild = Vdadult · BWchild/BWadult) using body weight based functions.13 However, in the case of desipramine, the situation is more complex due to the fact that the drug exerts unique physicochemical properties, which result in phospholipid binding and lysosomal trapping14, 15 and, thus, a high volume of distribution (Vss) (10–50 L/kg). The high volume of distribution indicates that most of the drug is located in the tissue, which poses a challenge for the treatment of desipramine overdosing due to ineffective hemoperfusion or hemodialysis.1 The appropriate characterization of these physicochemical properties is consequently important for characterizing desipramine's pharmacokinetics and, thus, for selecting safe and effective dosing regimen. The use of physiologically based pharmacokinetic (PBPK) modeling and simulation approaches is one way of achieving this objective, as it allows for a clear distinction between system‐specific parameters, drug‐specific parameters, and trial design parameters16 and, thus, for characterizing the dynamic interplay between the drug and the biological system on a physiological basis. While system‐specific parameters characterize the function of the underlying biological system, drug‐specific parameters account for the physicochemical properties of a given drug and for how the drug interacts with the biological system. Trial design parameters specify the conditions that pertain to the setup of the clinical trial population, such as dosing regimen and frequency, age, and gender of the trial population. Given the high level of physiological detail and the use of drug‐specific properties, PBPK modeling shows great promise for appropriately characterizing desipramine's pharmacokinetics, in particular its atypical disposition kinetics. In addition, information on growth and maturation can be used to scale system‐specific parameters from e.g., adults to pediatrics.13 PBPK approaches can consequently be used to obtain dosing regimen in children.
The objective of our research was to develop and qualify a PBPK model for desipramine that integrates information on the physicochemical (drug‐specific) properties of the drug and its specific interactions with the biological system, particularly with subcellular components, in a stepwise fashion in GastroPlus (Figure 1): First, we developed and qualified a PBPK model for desipramine in adults that is able to characterize the drug's concentration–time profiles following intravenous (i.v.) administration giving the increased volume of distribution due phospholipid binding and/or lysosomal trapping a special consideration. Second, the adult i.v. model was then further expanded to an oral absorption model taking the presystemic binding in enterocytes into consideration. Once developed and qualified, the adult model was finally expanded to children by accounting for growth and maturation from neonates to adults.
Figure 1.
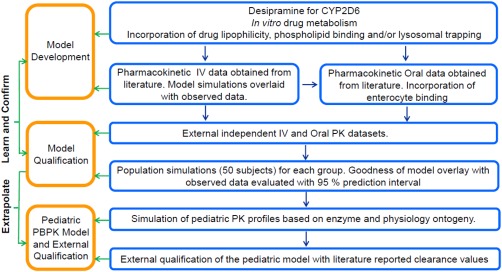
Model building and qualification workflow for the establishment of an age‐ranging desipramine PBPK model.
METHODS
Development of the adult desipramine PBPK model
GastroPlus v. 8.6 (Simulation Plus, Lancaster, CA) was used to develop an age‐ranging PBPK model for desipramine. The software provides a built‐in detailed whole‐body PBPK modeling framework, which incorporates a wide range of anatomical, physiological, and drug disposition parameters and characteristics.17 This information is based on age, gender, and genetic differences between populations and represents a curated summary of available literature data. In addition, the split between drug‐specific, system‐specific, and trial design parameters allows for a physiologically based description of the dynamic interplay between the drug, i.e., desipramine, and the biological system, i.e., the human body under various conditions, such as fed vs. fasted, adult vs. child, or normal organ function vs. organ impaired.
Drug‐specific parameters for desipramine were obtained from the literature and in silico predictions (ADMET Predictor v. 6.5, Simulations Plus; Table 1). The Advanced Compartmental Absorption and Transit (ACAT) model was used to simulate desipramine dissolution, absorption, and intestinal metabolism. The ACAT model combines the compartmental absorption and transit (CAT) model18 with finite dissolution, pH dependence, absorption from stomach or colon, in addition to the seven compartments of the small intestine, carrier‐mediated transport, and transporter densities. The built‐in intestinal physiology for human fasted state with the default absorption scale factor (ASF) model (Opt logD SA/V6.1) was used. ASF is a multiplier used to scale the effectively permeability (Peff) to account for variations in pH effects, absorptive surface areas, and other passive absorption rate‐determining effects that differ from one compartment to another. Literature reported Peff for desipramine19 was used in the model. The systemic disposition was modeled assuming that all tissues are perfusion‐limited. Partition coeffcients (Kp) were calculated in GastroPlus using the Lukacova (Rodgers–Single) method.20 The Lukacova method used for calculating Kp is given in the Supplementary Material.
Table 1.
Drug‐specific parameters for desipramine obtained from a literature search or in silico predictions (ADMET Predictor v. 6.5)
The Population Estimates for Age Related (PEAR) Physiology module was used to match the trial design specific parameter input in GastroPlus to the corresponding observations of the literature‐reported clinical trials. The built‐in GastroPlus algorithm was utilized to account for sex, age, and body weight‐dependent changes in the physiological and anatomical parameters, such as blood flows, cardiac output, organ/tissue volumes. In vitro Vmax and Km values for CYP2D6 (68 pmol/min/mg and 6.1 μM, respectively21) were used to define in vivo unbound hepatic intrinsic clearance (CLint). Since CYP2D6 is also expressed in the gut, these Vmax and Km values were also used to compute the CYP2D6‐mediated clearance in the gut. CYP2D6‐mediated clearance in the gut was lower than that in the liver due to the reduced CYP2D6 expression in the gut vs. the liver. Given that desipramine is only filtered in the kidneys, renal clearance was estimated as fu*GFR, where fu represents the fraction of unbound drug in plasma and GFR the glomerular filtration rate.
Qualification of the adult desipramine PBPK model
The PBPK model for desipramine was developed and qualified in a stepwise fashion (Figure 1) in order to reduce the uncertainty associated with its structural setup and associated parameter values. First, desipramine's pharmacokinetics were characterized following i.v. administration in order to characterize systemic clearance and to avoid convolution by first‐pass and presystemic absorption effects. Tissue partition coefficients, i.e., Kp values, were adjusted using optimized B/P ratios (2.4 in males and 2.7 in females). These higher B/P ratios were used to calculate desipramine's Kps and volume of distribution (Vss) in light of its phospholipid binding and lysosomal trapping. The equations for calculating Kps and Vss are complex because they consider physiological fluid volumes and binding to plasma proteins as well as cellular components and are provided as reference material for the reader in the Supplementary Material. The B/P ratio for both males and females was reverted back to the predicted value of 1.03 once the higher volume of distribution was captured. The model was originally developed using observed PK data following i.v. bolus administration of 12.5 mg desipramine22 and externally qualified by overlaying model‐based predictions with observed data following the i.v. administration of 50 mg desipramine,23 which further increased our confidence in the model given that it was able to predict plasma concentration–time profiles across an almost 4‐fold dose‐range.
Once developed and qualified for i.v. administration, the PBPK model was expanded to account for presystemic metabolism or nonspecific binding/lysosomal trapping in enterocytes, if any, following oral administration. To that end, the fraction unbound in enterocytes was optimized to 0.55% in order to appropriately characterize the observed concentration–time profile. The PBPK model for oral desipramine administration was developed for a 50 mg dose24 and was externally qualified with a 100 mg dose.25
In addition, the model's performance in adults was evaluated by calculating AUC0‐t and Cmax ratios for observations vs. predictions as well as the use of visual predictive checks. The structural model was deemed acceptable if the ratio of observed/predicted AUC0‐t and Cmax was contained within the 0.8–1.25 range of model‐based predictions, which is more stringent than the 2‐fold range that is typically applied for evaluating the predictive performance of PBPK models.26, 27 Visual predictive checks were performed to qualitatively and quantitatively evaluate the predictive performance of the model on a population basis.26 To that end, a virtual population of 50 subjects was generated via Monte Carlo simulations and model‐based population predictions were superimposed with observed plasma concentrations. In addition, 2.5th and 97.5th percentiles were generated for the model‐based population predictions and overlaid with the respective percentiles of the observed plasma concentrations. The population simulations incorporated 10–30% variability on various system and drug‐dependent parameters based on GastroPlus built‐in values as well as literature reported variability values.
Development of the pediatric desipramine PBPK model
The GastroPlus PEAR Physiology module was used to build the pediatric PBPK model for children of different age groups ranging from newborns to 15‐year‐olds. Age groups (0, 0.5, 1, 2.5, 5, 10, and 15 years) were selected to represent the classification given in the ICH guideline for neonates (birth to 1 month), infants (1 month to 2 years), children (2–12 years), and adolescents (12–16 years). The built‐in GastroPlus ontogeny patterns for CYP2D6 were used to characterize changes in clearance after birth. The enzyme maturation patterns are provided by the expression levels of CYP2D6 in the liver and the gut. Renal clearance was scaled in GastroPlus using the PEAR Physiology module, which accounts for maturational changes in plasma protein levels (resulting in age‐dependent changes in fu) and GFR. The B/P ratio was scaled from adults to children in GastroPlus by accounting for age‐dependent changes in hematocrit.17 Changes in other physiological parameters, such as organ weight and composition or blood flows, with age were also considered during the development of the pediatric desipramine PBPK model.
Due to the lack of reported experimental concentration–time in children, the model was externally qualified by comparing model‐predicted clearance values (calculated by noncompartmental analysis of simulated profiles) with reported clearance values from the literature.23, 24, 25, 26, 27, 28
RESULTS
Model building and qualification results in adults
Our PBPK model for desipramine describes the drug's observed exposure following both i.v. (Figure 2 a,d) and Oral (Figure 3 a,d) administration reasonably well. The observed/predicted ratios for i.v. AUC0‐t (0.97 (model development) and 0.87 (external model qualification)), oral AUC0‐t (0.88 (model development) and 0.98 (external model qualification)), and Cmax (0.90 (method development) and 1.04 (external qualification)) were well within the 0.8–1.25‐fold limits. This indicates that the model predicted values are in good agreement with the respective observed values for doses from 12.5–100 mg. The PBPK model predicted clearance (Figure 4) was also similar to that of the observed clearance for adults from the published literature.22, 23
Figure 2.
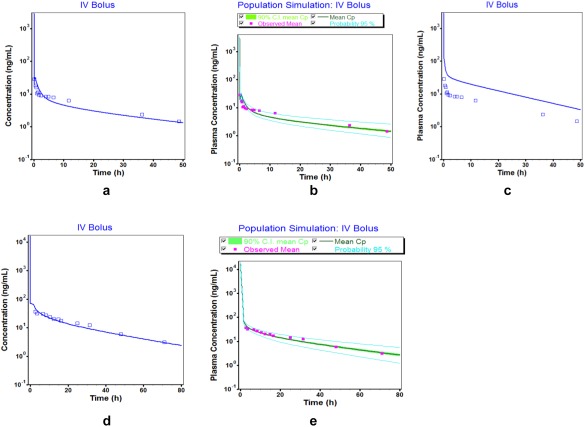
Comparison of simulated (lines) and observed (points) plasma concentration–time profiles of desipramine following i.v. administration: (a) mean simulated Cp‐time profile after 12.5 mg i.v. bolus administration used for model building; (b) population simulation (mean, 2.5–97.5th percentiles) for 12.5 mg i.v. bolus administration used for model building; (c) mean simulated Cp‐time profile after 12.5 mg i.v. bolus administration without increased volume of distribution; (d) mean predicted Cp‐time profile for 50 mg i.v. bolus administration used for external model qualification; (e) population simulation (mean, 2.5–97.5th percentiles) for 50 mg i.v. bolus administration used for external model qualification. Observed data are obtained from refs. 22, 23.
Figure 3.
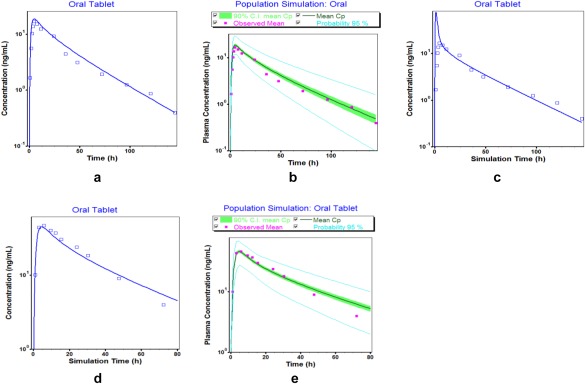
Comparison of simulated (lines) and observed (points) plasma concentration–time profiles of desipramine following oral administration: (a) mean simulated Cp‐time profile after 50 mg oral administration used for model building; (b) population simulation (mean, 2.5–97.5th percentiles) for 50 mg oral administration used for model building; (c) mean simulated Cp‐time profile after 50 mg oral administration with no phospholipid binding or lysosomal trapping in enterocytes; (d) mean predicted Cp‐time profile for 100 mg oral administration used for external model qualification; (e) population simulation (mean, 2.5–97.5th percentiles) for 100 mg oral administration used for external model qualification. Observed data are obtained from refs. 24, 25.
Figure 4.
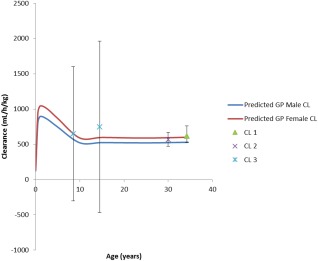
Changes in body weight normalized clearance as a function of age. Model‐based predictions for males (blue solid line) and females (red solid line) were overlaid with respective mean (±SD) literature data (CL 1,22 CL 2,23 CL 328).
The Kp values in all simulations were calculated using an increased B/P ratio that was used to adjust the Vss to respective literature values. This is an important optimization to obtain a good estimate of Vss, which when not included in the model led to a poor predictive performance of the pharmacokinetic profile and Vss lying outside the clinically observed values and pharmacokinetic parameters outside of the predefined 0.8–1.25 limits (Figure 2 c) for the i.v. dosing. For the oral desipramine model, the percent unbound drug in the enterocytes was optimized to 0.55% in order to account for the effect of desipramine binding to phospholipids and/or accumulation in lysosomes in enterocytes on the rate at which drug appears in plasma. This optimization was used in the oral desipramine model development to achieve a good overlay of the observed and simulated concentration–time profiles at a dose of 50 mg (Figure 3 a). We gained confidence in this optimized value of 0.55% by predicting plasma concentration–time profiles in the presence (Figure 3 a) and absence (Figure 3 c) of enterocyte binding following oral administration at the current dose level (50 mg). Our results clearly indicate a mismatch between model‐based predictions and observations (Figure 3 c) if enterocyte binding was not considered. This value of 0.55% was qualified with an external dataset for oral route at a higher dose (100 mg) which was not used for model building (Figure 3 d). This optimization consequently represents an important step in the development of the desipramine PBPK model. The 2.5–97.5th percentiles of the population simulation in 50 virtual subjects also captured the observed clinical data suggesting good model predictions as compared to the observed concentrations with the visual predictive checks (Figure 2 b,e, Figure 3 b,e).
Once developed and qualified, the adult model was scaled to pediatrics by accounting for growth and maturation after birth. In the absence of actual concentration–time data in pediatrics, we used the model‐based predictions (Figure 5) to compute key pharmacokinetic parameters, particularly clearance, and compared them to reported literature values. Respective computed clearance values normalized by body weight are in good agreement with those reported in the literature, as shown in Figure 4. We expect this normalized clearance values to increase in younger children given that both organ, i.e., liver, size, and CYP2D6 enzyme ontogeny play a role. Such nonmonotonic changes in clearance have been previously reported for other enzyme‐substrate combinations in children for midazolam (CYP3A4), theophylline (CYP1A2),29 and 27 other substrates.30 The changes in computed desipramine clearance and Vss are provided in the Supplementary Table.
Figure 5.
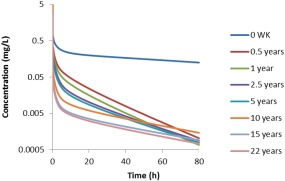
Simulated concentration–time curves of desipramine in males of different ages following the i.v. administration of 12.5 mg desipramine.
DISCUSSION
Most of the current PBPK applications focus on the characterization of changes in clearance due to genetic polymorphisms, drug–drug interactions, or growth and maturation, while atypical distribution patterns due to, e.g., phospholipid binding or lysosomal trapping, remain largely understudied for small molecules. The primary objective of our study was to address this limitation by developing a respective case study using desipramine, a tricyclic antidepressant that shows atypical tissue distribution, as an example. Desipramine's atypical tissue distribution is due to phospholipid binding as well as lysosomal trapping and cannot be captured by any of the typically employed Kp calculation methods, such as Poulin and Theil,31, 32 Berezhkovskiy,33 and Rodgers et al.34, 35
To overcome this limitation, we calculated desipramine's Kp values using a higher B/P ratio, which mimics a stronger interaction with acidic phospholipids than estimated based on in silico B/P ratios.34, 35, 36 However, a clear distinction between phospholipid binding and lysosomal trapping is difficult because the phospholipid content is highest in tissues with high lysosomal volumes.34 As a consequence, the increased B/P ratio served as a hybrid surrogate for both effects in our PBPK model. Once this information was incorporated into the adult desipramine PBPK model, model‐based predictions in adults were in good agreement with respective clinical observations following 12.5 and 50 mg i.v. dosing regimens (Figure 2).
In addition, desipramine's oral bioavailability is reported to be around 40%22 with a time to reach maximum concentration (Tmax) of around 4–6 h. This is somewhat unusual for a high‐permeability compound, such as desipramine, where one would expect a faster Tmax. This discrepancy between high permeability and prolonged Tmax might be due to desipramine's phospholipid binding and lysosomal trapping in the gut wall because it explains the fast disappearance of drug from the gut lumen and relatively slow appearance in plasma. This phenomenon of fraction of unbound drug in the enterocytes was captured by using an optimized value of 0.55%. We also considered presystemic CYP2D6 metabolism in the gut, but given that CYP2D6 is expressed at less than 1% of the corresponding liver microsome levels, its impact on intestinal desipramine metabolism was found negligible. This is in line with the corresponding literature37 and resulting model predictions are in good agreement with observed plasma concentrations following administration of 50 mg and 100 mg desipramine, respectively.
The secondary objective of our study was to expand the PBPK model developed in adults to children given its off‐label use in this patient population. This expansion is in line with recent advances and applications of PBPK models in pediatric drug development and regulatory decision‐making.13, 38 In particular, it was noted in a 2014 FDA workshop that PBPK modeling provides a viable option for dose selection in children “since dose‐response information for efficacy and safety in special populations is often lacking.”39 Numerous case examples for extrapolating adult pharmacokinetics to children using PBPK modeling and simulation approaches have become available in recent years in the literature.40, 41, 42 Furthermore, the FDA Advisory Committee has voted in favor of the routine use of PBPK modeling in pediatric drug development for identifying dose, optimizing clinical trial design, and evaluating the impact of organ impairment.43
We expanded our adult PBPK for desipramine to children by accounting for growth and maturation after birth, which allowed for the prediction of nonmonotonic changes in drug clearance for i.v. from neonates to adults (Figure 4) (Supplementary Table). These nonmonotonic changes in clearance are the combined result of changes in body and organ size as well as changes in metabolizing enzyme expression and activity levels.13 Even though an external qualification of the developed pediatric PBPK model for desipramine was not possible due to the lack of available concentration–time data in children, our confidence in the predictive performance of the pediatric model increased stepwise during the model building process. This is particularly due to the fact that: 1) pediatric clearance values that were computed from the PBPK model‐predicted concentration–time profiles were in good agreement with respective literature‐reported values down to the age of 8.5 years (Figure 5); 2) physiological changes in plasma protein levels (resulting in age‐dependent changes in fu), GFR, hematocrit (resulting in age‐dependent changes in the B/P ratio), organ size, and blood flow were taken into consideration; and 3) nonmonotonic changes in clearance in young children are also reported for a plethora of other drugs that are cleared via different CYP pathways.
CONCLUSION
In summary, we developed a PBPK model for desipramine that accounts for desipramine's atypical distribution behavior due to phospholipid binding and lysosomal trapping. Once developed and qualified in adults, we expanded the PBPK model to children by accounting for growth and maturation after birth. Although physiologically based, the model would benefit from a prospective qualification in children prior to its application for efficacy and safety predictions in this special patient population.
Supporting information
Supplementary table: Simulated changes in clearance (CL) and Volume of Distribution (Vss) from neonates to adults
Computation of Tissue distribution Coefficients and Volume of Distribution in GastroPlus
GastroPlus source code
Conflict of Interest
The authors declare no conflicts of interest.
Author Contributions
T.S.S., V.L., and S.S. wrote the article; T.S.S., V.L., and S.S. designed the research; T.S.S. performed the research. T.S.S. analyzed the data.
References
- 1. Sallee, F. & Pollock, B. Clinical pharmacokinetics of imipramine and desipramine. Clin. Pharmacokinet. 18, 346–364 (1990). [DOI] [PubMed] [Google Scholar]
- 2. Rudorfer, M.V. & Potter, W.Z. Metabolism of tricyclic antidepressants. Cell. Mol. Neurobiol. 19, 373–409 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Potter, W.Z. et al Active metabolites of imipramine and desipramine in man. Clin. Pharmacol. Ther. 31, 393–401 (1982). [DOI] [PubMed] [Google Scholar]
- 4. DeVane, C. , Savett, M. & Jusko, W. Desipramine and 2‐hydroxy‐desipramine pharmacokinetics in normal volunteers. Eur. Clin. Pharmacol. 19, 61–64 (1981). [DOI] [PubMed] [Google Scholar]
- 5. Donnelly, M. et al Treatment of childhood hyperactivity with desipramine: plasma drug concentration, cardiovascular effects, plasma and urinary catecholamine levels, and clinical response. Clin. Pharmacol. Ther. 39, 72–81 (1986). [DOI] [PubMed] [Google Scholar]
- 6. Oates, J.A. , Wood, A.J. , Potter, W.Z. , Rudorfer, M.V. & Manji, H. The pharmacologic treatment of depression. N. Engl. J. Med. 325, 633–642 (1991). [DOI] [PubMed] [Google Scholar]
- 7. Wilens, T.E. et al Six‐week, double‐blind, placebo‐controlled study of desipramine for adult attention deficit hyperactivity disorder. Am . J. Psychiatry. 153, 1147–1153 (1996). [DOI] [PubMed] [Google Scholar]
- 8. Biederman, J. , Baldessarini, R.J. , Wright, V. , Knee, D. & Harmatz, J.S. A double‐blind placebo controlled study of desipramine in the treatment of ADD: I. Efficacy. J. Am. Acad. Child Adolesc. Psychiatry 28, 777–784 (1989). [DOI] [PubMed] [Google Scholar]
- 9. Scahill, L. , Chappell, P.B. , Kim, Y.S. , Schultz, R.T. , Katsovich, L. , Shepherd, E. , Arnsten, A.F. , Cohen, D.J. , Leckman, J.F . A placebo‐controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. 158, 1067–1074 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Riddle, M.A. , Geller, B. & Ryan, N. Another sudden death in a child treated with desipramine. J. Am. Acad. Child Adolesc. Psychiatry 32, 792–797 (1993). [DOI] [PubMed] [Google Scholar]
- 11. Swanson, J.R. , Jones, G.R. , Krasselt, W. , Denmark, & L.N. Ratti, F. Death of two subjects due to imipramine and desipramine metabolite accumulation during chronic therapy: a review of the literature and possible mechanisms. J. For. Sci. 42, 335–339 (1997). [PubMed] [Google Scholar]
- 12.<http://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/depression-in-adults-screening1>
- 13. Samant, T.S. , Mangal, N. , Lukacova, V. & Schmidt, S. Quantitative clinical pharmacology for size and age scaling in pediatric drug development: A systematic review. J. Clin. Pharmacol. 55, 1207–1217 (2015). [DOI] [PubMed] [Google Scholar]
- 14. Daniel, W. , Bickel, M. & Honegger, U. The contribution of lysosomal trapping in the uptake of desipramine and chloroquine by different tissues. Pharmacol. Toxicol. 77, 402–406 (1995). [DOI] [PubMed] [Google Scholar]
- 15. Daniel, W.A. & Wöjcikowski, J. Contribution of lysosomal trapping to the total tissue uptake of psychotropic drugs. Pharmacol. Toxicol. 80, 62–68 (1997). [DOI] [PubMed] [Google Scholar]
- 16. Johnson, T.N. & Rostami‐Hodjegan, A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr. Anaesth. 21, 291–301 (2011). [DOI] [PubMed] [Google Scholar]
- 17. GastroPlus Manual, version 8.5, April 2013; Simulations Plus, Inc, Lancaster, CA. [Google Scholar]
- 18. Lawrence, X.Y. , Lipka, E. , Crison, J.R. & Amidon, GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption . Advanced Drug Deliv. Rev. 19, 359–376 (1996). [DOI] [PubMed] [Google Scholar]
- 19. Lennernas H. Human in vivo regional intestinal permeability: importance for pharmaceutical drug development. Mol. Pharm. 11, 12–23 (2014). [DOI] [PubMed] [Google Scholar]
- 20. Lukacova, P.N. , Lave, T. , Fraczkiewicz, G. , Bolger, M.B. & Woltosz, W.S. General approach to calculation of tissue:plasma partition coefficients for physiologically based pharmacokinetic (PBPK) modeling, Poster Session presented at: 2008 AAPS National Annual Meeting and Exposition, November 16‐20, 2008, Atlanta, GA.
- 21. Ball, S.E. , Ahern, D. , Scatina, J. & Kao, J. Venlafaxine: in vitro inhibition of CYP2D6 dependent imipramine and desipramine metabolism; comparative studies with selected SSRIs, and effects on human hepatic CYP3A4, CYP2C9 and CYP1A2. Br. J. Clin. Pharmacol. 43, 619–626 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ciraulo, D.A. , Barnhill, J.G. & Jaffe, J.H. Clinical pharmacokinetics of imipramine and desipramine in alcoholics and normal volunteers. Clin. Pharmacol. Ther. 43, 509–518 (1988). [DOI] [PubMed] [Google Scholar]
- 23. Brøsen, K. & Gram, L.F. First‐pass metabolism of imipramine and desipramine: impact of the sparteine oxidation phenotype. Clin. Pharmacol. Ther. 43, 400–406 (1988). [DOI] [PubMed] [Google Scholar]
- 24. Kurtz, D.L. , Bergstrom, R.F. , Goldberg, M.J. & Cerimele, BJ. The effect of sertraline on the pharmacokinetics of desipramine and imipramine. Clin. Pharmacol. Ther. 62, 145–156 (1997). [DOI] [PubMed] [Google Scholar]
- 25. Brøsen, K. , Otton, S.V. & Gram, L.F. Imipramine demethylation and hydroxylation: impact of the sparteine oxidation phenotype. Clin. Pharmacol. Ther. 40, 543–559 (1986). [DOI] [PubMed] [Google Scholar]
- 26. Maharaj, A. & Edginton, A. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacometrics Syst. Pharmacol. 3, 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khalil, F. & Läer, S. Physiologically based pharmacokinetic models in the prediction of oral drug exposure over the entire pediatric age range—sotalol as a model drug. AAPS J. 16, 226–239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen, L.G. et al Absence of effect of stimulants on the pharmacokinetics of desipramine in children. Pharmacotherapy 19, 746–752 (1999). [DOI] [PubMed] [Google Scholar]
- 29. Björkman, S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br. J. Clin. Pharmacol. 59, 691–704 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ginsberg, G. et al Evaluation of child/adult pharmacokinetic differences from a database derived from the therapeutic drug literature. Toxicol. Sci. 66, 185–200 (2002). [DOI] [PubMed] [Google Scholar]
- 31. Poulin, P. & Theil, FP. A priori prediction of tissue: plasma partition coefficients of drugs to facilitate the use of physiologically‐based pharmacokinetic models in drug discovery. J. Pharm Sci. 89, 16–35 (2000). [DOI] [PubMed] [Google Scholar]
- 32. Poulin, P. , Schoenlein, K. & Theil, F.P. Prediction of adipose tissue: plasma partition coefficients for structurally unrelated drugs. J. Pharm. Sci. 90, 436–447 (2001). [DOI] [PubMed] [Google Scholar]
- 33. Berezhkovskiy, L,M. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J. Pharm. Sci. 93, 1628–1640 (2004). [DOI] [PubMed] [Google Scholar]
- 34. Rodgers, T. , Leahy, D. & Rowland, M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J. Pharm. Sci. 94, 1259–1276 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 36. Rodgers, T. & Rowland, M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 24, 918–933 (2007). [DOI] [PubMed] [Google Scholar]
- 37. Thelen, K. & Dressman, JB. Cytochrome P450‐mediated metabolism in the human gut wall. J. Pharm. Pharmacol. 61, 541–558 (2009). [DOI] [PubMed] [Google Scholar]
- 38. Rostami‐Hodjegan, A. , Tamai, I. & Pang, K.S. Physiologically based pharmacokinetic (PBPK) modeling: It is here to stay! Biopharm. Drug Dispos. 33, 47–50 (2012). [DOI] [PubMed] [Google Scholar]
- 39. Wagner, C. et al Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA Public Workshop on PBPK. CPT Pharmacometrics Syst. Pharmacol. 4, 226–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ginsberg, G. , Hattis, D. , Russ, A. & Sonawane, B. Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: implications for assessing children's risks from environmental agents. J. Toxicol. Environ. Health Part A 67, 297–329 (2004). [DOI] [PubMed] [Google Scholar]
- 41. Jiang, X. , Zhao, P. , Barrett, J. , Lesko, L. & Schmidt, S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacometrics Syst. Pharmacol. 2, e80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parrott, M.N. et al Development of a physiologically based model for oseltamivir and simulation of pharmacokinetics in neonates and infants. Clin. Pharmacokinet. 50, 613–623 (2011). [DOI] [PubMed] [Google Scholar]
- 43. Summary Minutes of the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology March 14, 2012; Food and Drug Administration Center for Drug Evaluation and Research. <http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceutical%20SciencesandClinicalPharmacology/UCM306989.pdf>. Accessed 12 May 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table: Simulated changes in clearance (CL) and Volume of Distribution (Vss) from neonates to adults
Computation of Tissue distribution Coefficients and Volume of Distribution in GastroPlus
GastroPlus source code