

Abstract
Human inducible nitric oxide synthase (iNOS) is active as a dimer of two identical subunits. Each subunit has an amino-terminal oxygenase domain that binds the substrate l-Arg and the cofactors heme and tetrahydrobiopterin and a carboxyl-terminal reductase domain that binds FMN, FAD, and NADPH. We previously demonstrated that a subdomain in the oxygenase domain encoded by exons 8 and 9 is important for dimer formation and NO synthesis. Further, we identified Trp-260, Asn-261, Tyr-267, and Asp-280 as key residues in that subdomain. In this study, using an Escherichia coli expression system, we produced, purified, and characterized wild-type iNOS and iNOS-Ala mutants. Using H2O2-supported oxidation of Nω-hydroxy-l-Arg, we demonstrate that the iNOS mutants' inabilities to synthesize NO are due to selective defects in the oxygenase domain activity. Detailed characterization of the Asp-280–Ala mutant revealed that it retains a functional reductase domain, as measured by its ability to reduce cytochrome c. Gel permeation chromatography confirmed that the Asp-280–Ala mutant exists as a dimer, but, in contrast to wild-type iNOS, urea-generated monomers of the mutant fail to reassociate into dimers when incubated with l-Arg and tetrahydrobiopterin, suggesting inadequate subunit interaction. Spectral analysis reveals that the Asp-280–Ala mutant does not bind l-Arg. This indicates that, in addition to dimerization, proper subunit interaction is required for substrate binding. These data, by defining a critical role for Asp-280 in substrate binding and subunit interactions, give insights into the mechanisms of regulation of iNOS activity.
Nitric oxide (NO), an important signaling and cytotoxic molecule, is synthesized from l-Arg by isoforms of NO synthase (NOS), the constitutive endothelial and neuronal and the high-output inducible enzymes (1, 2). The latter form, termed iNOS (inducible NOS), is widely expressed in diverse cell types under transcriptional regulation by inflammatory mediators (2, 3).
The human iNOS gene contains 26 exons and encodes a protein of 131 kDa (4–6). Human iNOS, like all NOSs, has three domains: (i) an amino-terminal oxygenase domain (residues 1–504) that binds heme, tetrahydrobiopterin (H4B), and l-Arg; (ii) a carboxyl-terminal reductase domain (residues 537-1153) that binds FMN, FAD, and NADPH; and (iii) an intervening calmodulin (CaM)-binding domain (residues 505–536) that regulates electron transfer between the oxygenase and reductase domains (7–9). Whereas CaM binds to the constitutive isoforms in response to an increase in calcium, it is tightly coupled to iNOS at basal calcium levels (10). Therefore, iNOS is notably distinguished from the constitutive isoforms by its sustained production of a relatively large amount of NO. The high-output production of NO, although suited for iNOS function as a host-defense agent, could cause inflammation and tissue injury. iNOS has been implicated in the pathogenesis of many diseases, including Alzheimer's, asthma, lung cancer, transplant rejection, cerebral infarct, glaucoma, inflammatory bowel disease, arthritis, and septic shock (11, 12). Understanding the regulation of NO synthesis by iNOS will assist in efforts to modulate its levels.
For the synthesis of NO, iNOS is active only as a homodimer in which the subunits align in a head-to-head manner, with the oxygenase domains forming a dimer and the reductase domains existing as independent monomeric extensions (7). Posttranslational assembly into active form, including subunit dimerization and substrate and cofactor binding, represents an important critical locus for therapeutic interventions for the regulation of NO synthesis. We previously demonstrated that in the oxygenase domain of human iNOS, a subdomain encoded by exons 8 and 9 is important for dimer formation and NO synthesis (13). Further, by using alanine-scanning mutagenesis, we identified Trp-260, Asn-261, Tyr-267, and Asp-280 as key residues in that subdomain (14). The identified residues are strictly conserved among all NOS isoforms and across species. In the present study, we used purified recombinant human iNOS and iNOS mutants to characterize the specific key residues in the subdomain encoded by exons 8 and 9. This approach together with crystallographic interpretation of the data allowed us to assess the contribution of these residues on iNOS structure and function. Our work defines a critical role for Asp-280 in substrate binding and subunit interactions.
Materials and Methods
Mutagenesis.
Site-specific, oligonucleotide-directed mutagenesis was performed by using the QuikChange mutagenesis system (Stratagene) (13). All mutations were confirmed by DNA sequence analysis.
Cell Culture, Transfection, and Cell Lysis.
Human embryonic kidney (HEK) 293 cells were cultured at 37°C in 5% CO2 in improved minimum essential medium (Biofluids, Rockville, MD) supplemented with 10% heat-inactivated FBS (HyClone). cDNA of human iNOS or iNOS mutants was inserted into the expression vector pRc/CMV (Invitrogen). Cationic lipid-mediated transient transfection was performed by using the desired DNA, Lipofectamine, and a transfection-enhancing Plus reagent (Life Technologies, Gaithersburg, MD) according to the manufacturer's instructions. After 23 h, medium was collected for nitrite measurements. After gentle rinsing with PBS, the cell layer was lysed on ice for 45 min in 40 mM Bis-Tris propane buffer, pH 7.7/150 mM NaCl/10% glycerol/25 mM sodium taurocholate and protease inhibitors. Lysates were centrifuged (16,000 × g, 5 min, 4°C), and supernatants were stored at −80°C. Total protein concentrations were determined by bicinchoninic acid reagent (Pierce) by using BSA as a standard.
Bacterial Expression Vectors.
Human iNOS cDNA was subcloned into the Escherichia coli expression vector pCWori+ (15, 16). To minimize N-terminal proteolysis during purification, the N-terminal 70 aa of human iNOS were deleted, as described for the generation of Δ 65 mouse iNOS oxygenase domain (17). A sequence coding for a His6 tag was inserted at the N-terminal end of the protein. Expression of active human iNOS in E. coli depends on coexpression of CaM (16). CaM cDNA was cloned from human brain mRNA (CLONTECH) by reverse transcription–PCR and inserted into pACYC184 (18). A pCWori+ E. coli expression vector for human iNOS oxygenase domain (iNOSox) was also generated. Using human iNOS cDNA as a template, PCR was used to amplify cDNA encoding residues 71–504 and to add a COOH-terminal His6 tag followed by a stop codon.
Recombinant Protein Production.
A vector expressing human iNOS or iNOS mutant was cotransformed with a vector expressing human CaM into proteinase-deficient E. coli strain BL21DE3 (Novagen). Cells were grown in Terrific Broth (Sigma) containing 125 μg/ml ampicillin and 40 μg/ml chloramphenicol. Cultures were shaken at 250 rpm at 37°C until an OD of 1.0 at 600 nm was reached. Protein expression was induced by adding 0.5 mM isopropyl β-d-thiogalactoside plus 0.4 mM of the heme precursor δ-aminolevulinic acid, and shaking was continued for 42 h at 25°C (15, 16). The production of iNOSox followed a similar procedure except CaM coexpression was not required and, thus, chloramphenicol was omitted from culture medium (17).
Recombinant Protein Purification.
Cells were harvested by centrifugation at 4,000 × g for 20 min at 4°C. The cell pellet was resuspended in a minimal volume of lysis buffer [40 mM N-(2-hydroxyethyl)piperazine-N-(3-propane sulfonic acid) (EPPS), pH 7.6, containing 1 mg/ml lysozyme, 10% glycerol, 150 mM NaCl, 0.5 mM l-Arg, 4 μM H4B, 2 μM FAD, 2 μM FMN, 0.5 μg/ml of the protease inhibitors leupeptin hemisulfate and pepstatin A, and 1 mM of PMSF]. Bacterial suspensions were incubated with mild agitation at 4°C for 30 min to allow for lysozyme action. Cells were broken by sonication for three 20-s pulses with a 2-min rest on ice between pulses, followed by three cycles of freezing and thawing (15, 16). Cell debris was removed by centrifugation at 30,000 × g for 30 min at 4°C. The supernatant was applied to a column containing Ni+2-NTA Sepharose 4B metal chelate affinity resin (Novagen) preequilibrated with 40 mM EPPS, pH 7.6, containing 150 mM NaCl, 10% glycerol, and 0.5 mM l-Arg (buffer A). The column was washed with 5 bed volumes of buffer A and then with buffer A plus 25 mM imidazole. Bound protein was eluted by buffer A containing 150 mM imidazole. Heme-containing fractions were pooled and concentrated by using centriprep-30 (Millipore) (15). The concentrated protein was dialyzed at 4°C against three changes of buffer A containing 4 μM H4B and either 1 mM DTT or 2 mM 2-mercaptoethanol. The protein was purified further by using a column containing 2′,5′-ADP-Sepharose equilibrated with 40 mM Bis-Tris buffer, pH 7.6, containing 1 mM l-Arg, 3 mM DTT, 4 μM H4B, 4 μM FAD, 10% glycerol, and 150 mM NaCl (buffer B). The column was washed with buffer B plus 0.5 M NaCl to eliminate nonspecific binding, and iNOS was eluted with buffer B containing 5 mM 2′-AMP. The heme-containing fractions were pooled, concentrated, and dialyzed at 4°C against buffer B containing 0.5 mM l-Arg, 1 mM DTT, 4 μM H4B, 4 μM FAD, and 10% glycerol and stored in aliquots at −80°C until further analysis (16, 17). For purification of iNOSox, one-step Ni+2 affinity purification was done as above except both cell lysis and purification were done in the absence of l-Arg, H4B, FMN, or FAD (19).
Generation of iNOS Monomers by Urea.
Monomers of iNOSox and iNOSoxD280A mutant were prepared as described (20). Briefly, the proteins were incubated at 4°C for 90 min in 40 mM EPPS buffer, pH 7.6, containing 5 M urea and 2 mM DTT. Urea concentration then was reduced by sequential dialysis for 2 h against 500 ml of buffer containing 2 M urea followed by overnight dialysis in 500 ml of buffer containing 0.1 M urea.
Gel-Permeation Chromatography.
Size-exclusion chromatography was done at 4°C by using a Superdex column and an FPLC system (Amersham Pharmacia). The column was equilibrated with 40 mM EPPS, pH 7.6/5% glycerol/0.5 mM DTT/150 mM NaCl. iNOS proteins (100 μg) were injected in 125 μl of sample volume, and the protein in the column effluent was monitored at 280 nm by using a flow-through detector. The column was calibrated with the following Mr standards: bovine thyroglobulin (67,000), bovine γ-globulin (158,000), ovalbumin (43,000), horse myoglobulin (17,000), and vitamin B12 (1,350) (13, 20).
NOS Activity.
NOS activity, in transfected HEK293 cells, was determined by measuring nitrite accumulation in culture medium. A 400-μl sample of culture medium was mixed with 400 μl of Griess reagent for 10 min, and absorbance at 543 nm was recorded (13, 21). For recombinant iNOS protein, NO synthesis was quantitated at 37°C by using the HbO2 assay (9, 15). Briefly, iNOS (0.5–1 μg) was added to a cuvette containing 40 mM EPPS buffer, pH 7.6, supplemented with 10 μM oxyhemoglobin, 0.3 mM DTT, 1 mM l-Arg, 0.1 mM NADPH, 4 μM FAD, 4 μM FMN, 4 μM H4B, 100 units/ml catalase, 10 units/ml superoxide dismutase, and 0.1 mg/ml BSA in a final volume of 1.0 ml. The NO-mediated conversion of HbO2 to methemoglobin was monitored over time as the absorbance increase at 401 nm and quantitated by using an extinction coefficient of 38 mM−1⋅cm−1 (22).
Oxygenase Domain Activity.
Oxygenase domain activity was measured by following H2O2-supported oxidation of Nω-hydroxy-l-Arg (l-NOHA) at 37°C for 10 min (19, 23). The reaction was carried out in 100 μl of total volume containing 40 mM EPPS (pH 7.6), 150–500 nM oxygenase domain protein, 1 mM NOHA, 0.5 mM DTT, 30 mM H2O2, 10 units/ml superoxide dismutase, 0.5 mg/ml BSA, and variable concentrations of H4B (0–1,000 μM). Reactions were initiated by 30 mM H2O2 and stopped by 1,500 units of catalase. Nitrite production was quantitated as above.
Cytochrome c Reductase Activity.
Cytochrome c reductase activity was measured at 37°C by following NADPH-dependent increase in absorbance at 550 nm and quantitated by using an extinction coefficient of 21 mM−1⋅cm−1 (7, 15).
UV-Visible Spectroscopy.
Binding of l-Arg, H4B, and imidazole was monitored by spectral perturbation analysis. Spectra were obtained on proteins in 40 mM EPPS, pH 7.6, containing 5% glycerol and 2 mM 2-mercaptoethanol. The iNOS-ferrous-CO adduct absorbing at 444 nm was used to quantitate heme protein content by using an extinction coefficient of 74 mM−1⋅cm−1 (17, 24, 25).
Results and Discussion
Mutational Analysis of Key Residues in the Subdomain Encoded by Exons 8 and 9 of Human iNOS.
The subdomain encoded by exons 8 and 9 of the oxygenase domain of human iNOS, containing residues 242–335, is critical for dimer formation and NO synthesis (13). In a preliminary study, 16 conserved residues of that subdomain were selected for alanine-scanning mutagenesis. This analysis identified Trp-260, Asn-261, Tyr-267, and Asp-280 as key residues for iNOS activity. Replacing any of these four residues with Ala abolished NO synthesis capacity (14). To characterize these residues further, we produced iNOS mutants with conserved mutations for each of the four residues. The iNOS mutants were characterized after their expression in HEK293 epithelial cells. This cell line does not express any NOS genes (13, 26). iNOS mutants with Phe replacing Trp-260 (W260F), Gln replacing Asn-261 (N261Q), and Phe replacing Tyr-267 (Y267F) had reduced iNOS activity compared with wild-type iNOS (Fig. 1A). Asp-280 was subjected to a more detailed analysis by mutating it to Glu (conservative mutation), Ser (nonconservative mutation), or Asn (charge-altering mutation). After expression of these mutants in HEK293 cells, even the conservative mutation Asp to Glu markedly reduced NO production. The Ser and the Asn substitution abolished the bulk of NO production. The NO production, however, was completely lost with the Ala replacement (Fig. 1B). These results confirmed that the residues Trp-260, Asn-261, Tyr-267, and Asp-280 of the subdomain encoded by exons 8 and 9 are key residues for NO synthesis by iNOS.
Figure 1.

NO production of wild-type human iNOS and iNOS mutants. HEK 293 cells were transfected with plasmids containing vector only, iNOS, or iNOS mutant. (A) iNOS mutants with Phe replacing Trp-260 (W260F), Gln replacing Asn-261 (N261Q), or Phe replacing Tyr-267 (Y267F). (B) iNOS mutants with Glu (E), Asn (N), Ser (S), or Ala (A) replacing Asp-280 (D280). Nitrite accumulation in the culture medium was assessed 72 h (A) or 36 h (B) after transfection. Data are means ± SD of three experiments. Invisible error bars are too small to be drawn.
Effects of Mutations of Key Residues in the Subdomain Encoded by Exons 8 and 9 on Oxygenase Domain Activity.
iNOS activity, measured by the enzyme ability to produce NO, reflects the presence of functional reductase and oxygenase domains. We hypothesized that the inability for the iNOS-Ala mutants to produce NO reflects a defect in the oxygenase domain activity. The functional integrity of the oxygenase domain can be tested selectively by measuring the H2O2-supported oxidation of l-NOHA (17, 23). We used an E. coli expression system to produce and purify wild-type iNOSox (residues 71–504) and iNOSox mutants with Ala replacing one of the key residues (Trp-260, Asn-261, Tyr-267, or Asp-280). The first 70 amino-terminal residues were deleted to minimize N-terminal proteolysis; this does not affect the activity or dimerization (17). SDS/PAGE analysis confirmed the size and the purity of the recombinant proteins (Fig. 2A). The ability of human iNOSox and each human iNOSox-Ala mutant to catalyze the H2O2-supported nitrite formation from l-NOHA at different concentrations of H4B (0–1,000 μM) was analyzed (Fig. 2B). As expected, human iNOSox (wild type) exhibited an oxygenase domain activity that increased with increasing concentration of H4B. In contrast, the iNOSoxD280A mutant had no detectable activity even at the highest H4B concentration. The remaining three iNOSox-Ala mutants, W260A, N261A, and Y267A, displayed minimal residual activity that was detectable only at very high concentrations of H4B. The activity of wild-type human iNOSox was similar to that of mouse iNOSox (data not shown).
Figure 2.

Characterization of oxygenase domains of human iNOS and iNOS mutants. Using E. coli, the oxygenase domains (residues 71–504 plus a COOH-terminal His6 tag) of wild-type human iNOS (iNOSox) and iNOS mutants with Ala replacing Asp-280 (D280A), Tyr-267 (Y267A), Asn-261 (N261A), or Trp-260 (W260A) were generated and purified. (A) SDS/PAGE analysis confirmed the size and the purity of the recombinant proteins. (B) Oxygenase domain activity was assessed by measuring H2O2-supported oxidation of Nω-hydroxy-l-Arg. Protein samples were preincubated for 30 min with the indicated concentrations of H4B before initiating the reaction with H2O2. Nitrite values are the amount formed in 10 min at 37°C. Data are means ± SD of three experiments.
Evaluation of Dimer Formation by iNOS Mutants by Using Gel-Permeation Chromatography.
Preliminary observations with partially denaturing SDS/PAGE analysis of transfected HEK293 cell lysates suggested that replacing Trp-260, Asn-261, or Tyr-267 with Ala resulted in inactive iNOS monomers, whereas replacing Asp-280 with Ala resulted in loss of NO synthesis without altering dimerization (14). This suggested that Ala substitution of Asp-280 might inactivate iNOS by mechanisms independent of the ability of the oxygenase domains to form the dimer interface. To test this hypothesis, we analyzed purified recombinant iNOSoxD280A by gel-permeation chromatography. Parallel experiments using wild-type iNOSox (representing an active dimer form of iNOSox) and iNOSoxY267A (representing an inactive monomer form of iNOSox) were done under similar conditions. After its purification in the absence of l-Arg and H4B, wild-type iNOSox eluted as an equilibrium mixture that was primarily dimeric with a minor monomer content (Fig. 3A, trace 1). Upon addition of 5 mM l-Arg and 20 μM H4B, the equilibrium was shifted toward 100% dimer content (Fig. 3A, trace 3). These results are similar to data for mouse iNOSox (17). Interestingly, in contrast to wild-type iNOSox, the catalytically inactive mutant iNOSoxD280A was totally dimeric even in the absence of l-Arg and H4B (Fig. 3A, trace 2) and remained dimeric after incubation with l-Arg and H4B (Fig. 3A, trace 4). Thus, despite its fully dimeric hydrodynamic behavior, iNOSoxD280A was completely inactive catalytically. The absence of monomeric iNOSoxD280A indicated that in contrast to wild-type iNOSox that seems to exist in an equilibrium mixture of monomers and dimers, iNOSoxD280A is in a “locked” dimer position that does not revert to monomer even in the absence of l-Arg and H4B. These data suggest that replacing Asp-280 with Ala brings a unique, incorrigible defect in subunit interaction that results in an inactive dimer.
Figure 3.

Gel-permeation chromatography of oxygenase domains of wild-type human iNOS and iNOS mutants. (A) Human iNOSox and iNOSoxD280A were analyzed in the absence (traces 1 and 3, respectively) and in the presence (traces 2 and 4, respectively) of 5 mM l-Arg and 20 μM H4B. (B) Elution profiles of urea-generated monomers of iNOSox and iNOSoxD280A (traces 1 and 3, respectively) and of the urea monomers after incubation of iNOSox with 5 mM l-Arg and 20 μM H4B (trace 2) or of iNOSoxD280A with 50 mM l-Arg and 1 mM H4B (trace 4). (C) iNOSoxY267A was analyzed in the absence or the presence of 50 mM l-Arg and 1 mM H4B (traces 1 and 2, respectively). (D) Calibration with Mr standards. The estimated relative retention of the oxygenase domain dimer and monomer is indicated. Letters D and M denote dimer and monomer, respectively. Data represent two experiments.
To investigate the role of Asp-280 in subunit interaction, we examined the ability of urea-generated monomers of wild-type iNOSox and iNOSoxD280A to redimerize in the presence of l-Arg and H4B. Both wild-type iNOSox and iNOSoxD280A became monomeric after exposure to 5 mM urea (Fig. 3B, traces 1 and 3, respectively). Urea-generated monomers of wild-type iNOSox reverted into dimers after their incubation with 5 mM l-Arg and 20 μM H4B (Fig. 3B, trace 2). In contrast, urea-generated monomers of iNOSoxD280A failed to dimerize even after incubation with 50 mM l-Arg and 1 mM H4B. These data indicate that iNOSoxD280A can exist either as a dimer or as a urea-generated monomer but fail to spontaneously convert back and forth between the dimer and the monomer forms. Thus, the inactivity of the iNOSoxD280A dimer could be the result, in part, of an inadequate subunit interaction as evidenced by the inability of reassociation of the urea-generated monomers in the presence of l-Arg and H4B.
In contrast to both wild-type iNOSox and iNOSoxD280A, iNOSoxY267A was primarily monomeric when purified in the absence of l-Arg and H4B (Fig. 3C, trace 1), and it remained mostly monomeric even after incubation with up to 50 mM l-Arg and 1 mM H4B (Fig. 3C, trace 2). These concentrations of l-Arg and H4B are considerably higher than those used to achieve 100% dimerization of wild-type iNOSox (5 mM l-Arg and 20 μM H4B; Fig. 3A, trace 3). These experiments indicate that both Asp-280 and Tyr-267 are critical residues for iNOS activity. However, the molecular mechanisms by which they exert their effects are quite distinct. Replacing Asp-280 with Ala results in an inactive dimer, whereas replacing Trp-267 with Ala leads to a failure of dimerization.
Characterization of the Effects of Asp-280-to-Ala Mutation on Dimerization and Activity of Full-Length iNOS.
Replacing Asp-280 with Ala resulted in the formation of an inactive oxygenase domain dimer. However, NOS activity requires both a functional oxygenase domain and a functional reductase domain that shuttles electrons from NADPH to the heme iron of the oxygenase domain (2, 7). Therefore, we investigated the functional role of Asp-280 on dimerization and activity of full-length iNOS. We used E. coli to produce and purify recombinant wild-type iNOS and an iNOS mutant with Ala replacing Asp-280. Gel-permeation chromatography done in the presence of 10 mM l-Arg and 20 μM H4B revealed that full-length iNOSD280A migrates as a dimer, similar to wild-type iNOS (data not shown). This confirms that the Asp-280-to-Ala mutation in the oxygenase domain has no effect on the hydrodynamic properties of full-length iNOS.
We next characterized the effect of the Asp-280-to-Ala mutation on the activity of full-length iNOS. The mutant iNOSD280A had no detectable NOS activity, measured by HbO2 capture assay, as compared with wild type, which had a specific activity of 800–1,000 nmol/min per mg (Fig. 4A). Similarly, the oxygenase domain-mediated catalytic activity, measured by H2O2-supported l-NOHA oxidation, was deficient in iNOSD280A compared with wild-type iNOS (Fig. 4B). In contrast, the mutant iNOSD280A had a functional reductase domain, as determined by measuring the NADPH-dependent cytochrome c reduction (Fig. 4C). These results suggest that the loss of NOS ability of iNOSD280A is a result of a selective defect in the oxygenase domain activity. They also indicate that the Asp-280-to-Ala mutation has no abrogative effect on reductase domain folding or electron transfer within the reductase domain.
Figure 4.
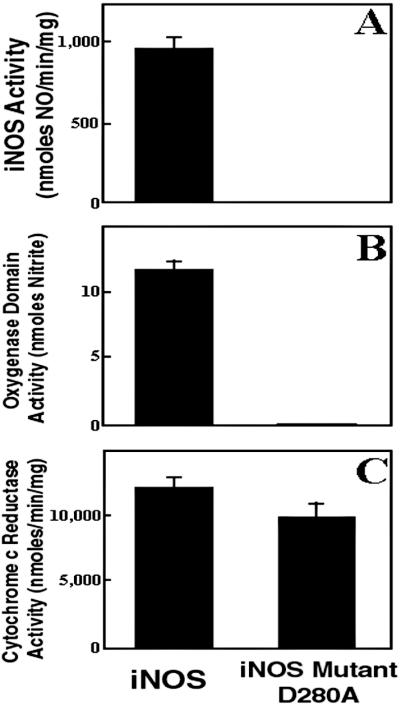
Determination of iNOS activity (A), oxygenase domain activity (B), and cytochrome c reductase activity (C) of human iNOS and iNOS D280A mutant. NO production was evaluated by using HbO2 capture assay. Oxygenase domain activity was assessed by measuring the H2O2-supported oxidation of Nω-hydroxy-l-Arg. Data are mean ± SD of three experiments.
Spectral Analyses.
To determine the underlying defect responsible for loss of catalytic activity induced by the Asp-280-to-Ala mutation, spectral perturbation analysis was used to determine heme-, l-Arg-, and H4B-binding capacity for both iNOSoxD280A and full-length iNOSD280A (17, 24, 25). Parallel experiments were done by using wild-type iNOSox and full-length iNOS under similar conditions. Spectra of 7 μM wild-type iNOSox, in 40 mM EPPS, pH 7.6, containing 5% glycerol (vol/vol) and 5 mM 2-mercaptoethanol, were recorded at 28°C. Maximum absorbance was at 417 nm, typical of low-spin hexa-coordinate heme protein (Fig. 5A). After incubation with 1 mM l-Arg and 20 μM H4B for 10 min at 28°C, the wild-type iNOSox spectrum shifted toward high-spin penta-coordinate heme species, with maximum absorbance at 395 nm. The addition of 2 mM imidazole converted heme to the low-spin state, with maximum absorbance at 427 nm. The addition of 5 mM l-Arg to the imidazole-bound heme reverted its absorbance to 395 nm. Carbon monoxide (CO) binding to reduced-heme shifted its absorbance to 444 nm, indicating that its heme iron is bound to cysteine thiolate. Similar to that of wild type, spectra of iNOSoxD280A showed the initial low-spin heme at 417 nm (Fig. 5B). However, the addition of l-Arg and H4B did not convert the heme of iNOSoxD280A to a high-spin form. Although iNOSox280A could bind imidazole, converting its heme absorbance to 427 nm, the addition of excess l-Arg could not convert heme back to its high-spin state as seen in wild-type iNOSox. CO binding followed by dithionite reduction of iNOSox280A produced spectra with predominantly a peak at 420 nm, with very little absorbance at 444 nm. This indicates that in spite of the preserved ability of iNOSox280A to bind heme, the coordination geometry of its heme was markedly different from that of wild type. Similar findings were observed when full-length proteins were used for wild-type iNOS and iNOSD28A (Fig. 5 C and D, respectively). These results indicate that the Asp-280-to-Ala mutation abolishes the substrate-binding ability of iNOS. They further suggest that iNOS dimerization per se is not sufficient to confer substrate-binding capacity or activity and that proper subunit interaction in the dimer is required.
Figure 5.

Light absorbance spectral analysis of iNOSox (A), iNOSoxD280A (B), iNOS (C), and iNOSD280A (D). Inset represents line designation of each spectral trace for A–D, except that spectra with “buffer only” and after CO reduction pertain to A and B only. (A) Spectra of 7 μM iNOSox, recorded in 40 mM EPPS, pH 7.6, containing 5% glycerol (vol/vol) and 5 mM 2-mercaptoethanol at 28°C (−), had maximum absorbance at 417 nm, typical of low-spin hexa-coordinate heme protein. After incubation for 10 min at 28°C with 1 mM l-Arg and 20 μM H4B (… ), iNOSox spectra shifted toward high-spin penta-coordinate heme species, with maximum absorbance at 395 nm. Addition of 2 mM imidazole (—) converted heme to the low-spin state, with maximum absorbance at 427 nm. Addition of 5 mM l-Arg (-.-.-) to the imidazole-bound heme reverted its absorbance to 395 nm. CO binding reduced heme, shifting its absorbance to 444 nm. (B) Spectra of iNOSoxD280A showed the initial low-spin heme at 417 nm but did not convert to high spin with the addition of l-Arg and H4B. Although iNOSox280A could bind imidazole converting its heme absorbance to 427 nm but addition of excess l-Arg could not convert heme back to its high-spin state as seen in iNOSox. (C) Spectra of iNOS exhibiting behavior similar to iNOSox. (D) Spectra of iNOS D280A exhibiting behavior similar to iNOSoxD280A. Spectra are representative of two experiments.
Structural and Functional Implications.
Human iNOS residues 242–335 (encoded by exons 8 and 9), corresponding to mouse iNOS residues 236–329, form a subdomain in the iNOS subunit (8, 13, 27). The crystal structures of the overall fold of residues 236–329 of mouse iNOS are shown in Fig. 6A. The subdomain encoded by exons 8 and 9 forms a wing of the “winged β-sheet” that structures the heme pocket, and it stabilizes the iNOS oxygenase-domain subunit fold. The α-β-α sandwich motif of this subdomain is peripheral to the dimer interface but structures part of the active center channel and stabilizes the substrate-binding helix α7a (27). The mouse iNOS residues Trp-254, Asn-256, Tyr-260, and Asp-274 (which correspond to Trp-260, Asn-261, Tyr-267, and Asp-280 in human iNOS) are critical for stabilizing the structural integrity of this subdomain and the active center. Mouse Asp-274 forms a hydrogen-bonding network with Arg-382 and residues Asp-376 and Glu-371 on helix α7a, which participate in substrate binding and the dimer interface. Asp-376 and Glu-371 interact directly with the substrate l-Arg and the intermediate Nω-hydroxy-l-Arg (Fig. 6B). Asp-376 hydrogen-bonds directly to the substrate carboxylate group, whereas Glu-371 interacts with the guanidium group.
Figure 6.

(A) The subdomain encoded by exons 8 and 9 (residues 236–329 of mouse iNOS, corresponding to residues 242–335 of human iNOS) in the crystal structure of the H4B-bound mouse Δ 65 iNOSox dimer (PDB ID code 1NOD). Mouse iNOS residues corresponding to the human residues mutated in this study are labeled. iNOSox dimer (blue and brown subunits), heme (gray), H4B, and zinc (Zn) are shown as an α-carbon trace. The subdomain encoded by exons 8 and 9 (ribbons of darker color, purple and maroon) forms a wing of the “winged β-sheet” that structures the heme pocket and stabilizes the subunit fold. W254 (W260 in human iNOS), N255 (N261, human), Y261 (Y267, human), and D274 (D280, human) are critical for stabilizing the structural integrity of the subdomain and active center (yellow side chains). (B) Specific interaction of D274 (human D280) in the active-center channel and its role in stabilizing the substrate-binding site. Residue D274 forms a hydrogen-bonding network with Arg (R) 382 and residues D376 and E371 on the substrate-binding helix α7a that interact directly with substrate l-Arg and intermediate l-NOHA (shown in gray). Thus, mutation of D274 (human D280) removes important buttressing hydrogen bonds to conserved residues that stabilize substrate binding and catalytic activity.
Thus, mutation of human Asp-280 removes important buttressing hydrogen bonds for conserved residues that stabilize substrate binding. This mechanism of “remote control” destabilization results in impairment of substrate binding. Moreover, Asp-280 mutation to Ala destabilizes α7a and thereby adversely affects oxygenase-subunit association. The latter, by causing improper subunit interaction, led to the assembly of a “defective” dimer. Human residues Trp-260, Asn-261, and Tyr-267 are not close to the active site, yet they are important structural residues in the subdomain encoded by exons 8 and 9 (Fig. 6A). Mouse residue Tyr-261 (human Tyr-267) packs with other hydrophobic residues and hydrogen-bonds to Glu-322 side chain and Tyr-341 main chain. Asn-255 (human Asn-261) stabilizes a wall of the active-center channel and forms a buried hydrogen bond with Leu-301 within the subdomain. Trp-254 (human Trp-260) packs in a hydrophobic core of the subdomain. Thus, mutation of any of human residues Trp-260, Asn-261, or Tyr-267 likely will destabilize the iNOS oxygenase domain subunit and its ability to dimerize.
Because of the diverse biological function of NO and its involvement in many physiological and pathological processes, understanding how cells control the levels of NO production is fundamental to efforts aimed at its regulation. In this context, in vivo regulation of NOSs, both transcriptional and posttranslational, has been the subject of intensive investigation. The molecular diversity of iNOS mRNA generated by alternative splicing may represent one such regulation. We have reported previously that there are several human iNOS mRNA isoforms produced by alternative splicing (28). Cloning and characterization of one of these splice variants, a variant with skipping of exons 8 and 9, led to the identification of a subdomain encoded by exons 8 and 9 as critical for iNOS activity and dimerization (13). A similar pattern of alternative splicing of mRNA has been reported for human and mouse neuronal NOS (29, 30) as well as for NOS cloned from Drosophila (31). The presence of a conserved pattern of alternative splicing in vertebrates and flies and among various NOSs suggests a common function for the resulting isoforms. Because these isoforms retain reductase domain activity, their function could be to produce superoxide or to transfer electrons derived from NADPH to a yet unidentified electron acceptor.
The in-frame deletion of 105 aa (exons 9 and 10) reported for a neuronal NOS splice variant includes the putative substrate-binding pocket and the residues Glu-592 and Asp-597 (corresponding to mouse iNOS Glu-371 and Asp-376) as well as the conserved pterin-binding residue Arg-596 (30). Thus, as expected, this deletion results in a protein with no NOS catalytic activity. In contrast, deletion of exons 8 and 9 in human iNOS does not include substrate/cofactor binding, but it does include structural elements important for the global iNOS oxygenase fold. Deletions or point mutations within this subdomain can modulate the dimerization and heme function from a distal site.
The data provided in this study outline important structural elements in iNOS and define functional mechanisms by which they affect NO synthesis. This adds to our understanding of the regulation of NO synthesis by iNOS and lays the groundwork for further studies aimed at modulating NO levels.
Acknowledgments
We thank Drs. M. Zouhair Atassi and C. S. Raman for critical review of the manuscript. The study was supported by The American Lung Association, Caroline Wiess Law Fund for Molecular Medicine, The Methodist Foundation, a T. T. Chao Scholar Award, Veterans Administration Medical Research, and awards from the National Institutes of Health (5T32HL07747-08 and AR39162).
Abbreviations
- iNOS
inducible nitric oxide synthase
- iNOSox
iNOS oxygenase domain
- EPPS
N-(2-hydroxyethyl)piperazine-N-(3-propane sulfonic acid)
- iNOSoxD280A
iNOS oxygenase domain with Ala replacing Asp-280
- CaM
calmodulin
- H4B
tetrahydrobiopterin
- HEK
human embryonic kidney
- l-NOHA
Nω-hydroxy-l-Arg
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Palmer R M, Ferrige A G, Moncada S. Nature (London) 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 2.Marletta M A. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 3.Xie Q-W, Cho H J, Calaycay J, Mumford R A, Swiderek K M, Lee T D, Ding A, Troso T, Nathan C. Science. 1992;256:225–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 4.Geller D A, Lowenstein C J, Shapiro R A, Nussler A K, Di Silvio M, Wang S C, Nakayama D K, Simmons R L, Snyder S H, Billiar T R. Proc Natl Acad Sci USA. 1993;90:3491–3495. doi: 10.1073/pnas.90.8.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charles I G, Palmer R M J, Hickery M S, Bayliss M T, Chubb A P, Hall V S, Moss D W, Moncada S. Proc Natl Acad Sci USA. 1993;90:11419–11423. doi: 10.1073/pnas.90.23.11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chartrain N A, Geller D A, Koty P P, Sitrin N F, Nussler A K, Hoffman E P, Billiar T R, Hutchinson N I, Mudgett J S. J Biol Chem. 1994;269:6765–6772. [PubMed] [Google Scholar]
- 7.Ghosh D K, Stuehr D J. Biochemistry. 1995;34:801–807. doi: 10.1021/bi00003a013. [DOI] [PubMed] [Google Scholar]
- 8.Crane B R, Arvai A S, Gachhui R, Wu C, Ghosh D K, Getzoff E D, Stuehr D J, Tainer J A. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 9.Abu-Soud H M, Stuehr D J. Proc Natl Acad Sci USA. 1993;90:10769–10772. doi: 10.1073/pnas.90.22.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho H J, Xie Q-W, Calaycay J, Mumford R A, Swiderek K M, Lee T D, Nathan C. J Exp Med. 1992;176:599–604. doi: 10.1084/jem.176.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan C. J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg J B. Mol Med. 1998;9:557–591. doi: 10.1007/BF03401758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eissa N T, Yuan J, Haggerty C M, Choo E K, Moss J. Proc Natl Acad Sci USA. 1998;95:7625–7630. doi: 10.1073/pnas.95.13.7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eissa N T, Haggerty C M, Palmer C D, Patton W, Moss J. Am J Respir Cell Mol Biol. 2001;24:616–620. doi: 10.1165/ajrcmb.24.5.4324. [DOI] [PubMed] [Google Scholar]
- 15.Roman L J, Sheta E A, Martasek P, Gross S S, Liu Q, Masters B S S. Proc Natl Acad Sci USA. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu C, Zhang J, Abu-Soud H, Ghosh D K, Stuehr D J. Biophys Res Commun. 1996;222:439–444. doi: 10.1006/bbrc.1996.0763. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh D K, Wu C, Pitters E, Moloney M, Werner E R, Mayer B, Stuehr D J. Biochemistry. 1997;36:10609–10619. doi: 10.1021/bi9702290. [DOI] [PubMed] [Google Scholar]
- 18.Chang A C Y, Cohen S N. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh D K, Crane B R, Ghosh S, Wolan D, Gachhui R, Crooks C, Presta A, Tainer J A, Getzoff E D, Stuehr D J. EMBO J. 1999;18:6260–6270. doi: 10.1093/emboj/18.22.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh D K, Abu-Soud H M, Stuehr D J. Biochemistry. 1996;35:1444–1449. doi: 10.1021/bi9521295. [DOI] [PubMed] [Google Scholar]
- 21.Green L C, Wagner D A, Glogowski J, Skipper P L, Wishnok J S, Tannenbaum S R. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 22.Kelm M, Schrader J. Circ Res. 1990;66:1561–1575. doi: 10.1161/01.res.66.6.1561. [DOI] [PubMed] [Google Scholar]
- 23.Pufahl R A, Wishnok J S, Marletta M A. Biochemistry. 1995;34:1930–1941. doi: 10.1021/bi00006a014. [DOI] [PubMed] [Google Scholar]
- 24.Stuehr D J, Ikeda-Saito M. J Biol Chem. 1992;267:20547–20550. [PubMed] [Google Scholar]
- 25.McMillan K, Masters B S. Biochemistry. 1993;32:9875–9880. doi: 10.1021/bi00089a001. [DOI] [PubMed] [Google Scholar]
- 26.Xie Q-W, Leung M, Fuortes M, Sassa S, Nathan C. Proc Natl Acad Sci USA. 1996;93:4891–4896. doi: 10.1073/pnas.93.10.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crane B R, Arvai A S, Ghosh D K, Wu C, Getzoff E D, Stuehr D J, Tainer J A. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 28.Eissa N T, Strauss A J, Haggerty C M, Choo E K, Chu S C, Moss J. J Biol Chem. 1996;271:27184–27187. doi: 10.1074/jbc.271.43.27184. [DOI] [PubMed] [Google Scholar]
- 29.Hall A V, Antoniou H, Wang Y, Cheung A H, Arbus A M, Olson S L, Lu W C, Kau C-L, Marsden P A. J Biol Chem. 1994;269:33082–33090. [PubMed] [Google Scholar]
- 30.Iwasaki T, Hori H, Hayashi Y, Nishino T, Tamura K, Oue S, Iizuka T, Ogura T, Esumi H. J Biol Chem. 1999;274:17559–17566. doi: 10.1074/jbc.274.25.17559. [DOI] [PubMed] [Google Scholar]
- 31.Regulski M, Tully T. Proc Natl Acad Sci USA. 1995;92:9072–9076. doi: 10.1073/pnas.92.20.9072. [DOI] [PMC free article] [PubMed] [Google Scholar]