

Abstract
Reducing the levels of the androgen receptor (AR) is one of the most viable approaches to combat castration-resistant prostate cancer (CRPC). Previously, we observed that proteasomal-dependent degradation of AR in response to 2-methoxyestradiol (2-ME) depends primarily on the E3 ligase C-terminus of HSP70-interacting protein (STUB1/CHIP). Here, 2-ME stimulation activates CHIP by phosphorylation via Aurora kinase A (AURKA). Aurora A kinase inhibitors and RNAi knockdown of Aurora A transcript selectively blocked CHIP phosphorylation and AR degradation. Aurora A kinase is activated by 2-ME in S-phase as well as during mitosis, and phosphorylates CHIP at S273. Prostate cancer cells expressing a S273A mutant of CHIP have attenuated AR degradation upon 2-ME treatment compared to cells expressing wild-type CHIP, supporting the idea that CHIP phosphorylation by Aurora A activates its E3 ligase activity for the AR. These results reveal a novel 2-ME→Aurora A→CHIP→AR pathway which promotes AR degradation via the proteasome, that may offer novel therapeutic opportunities for prostate cancer.
Keywords: Aurora A, TPX2, CHIP, Androgen Receptor, 2-methoxyestradiol, Prostate cancer
Introduction
The development, treatment, and recurrence of prostate cancer all depend on the androgen receptor (AR). Tumors that develop resistance to androgen ablation therapy, called castration-resistant prostate cancer, retain AR that transmits proliferation and survival signals independent of androgens (1–3). Under these circumstances, reduced expression of the AR would be potentially beneficial. Therefore, we have undertaken studies to discover ways to increase degradation of the AR to lower its levels in prostate cancer cells.
We previously reported that treatment of human prostate cancer cell lines with 2-methoxyestradiol (2-ME) increased proteasomal degradation of the AR and that this response depended on the C-terminus of Hsp70-interacting protein (CHIP) (4). CHIP interacts with Hsp70 and Hsp90 while mediating the ubiquitination and degradation of various chaperone-associated client proteins (5, 6). Thus, CHIP acts as a link between the chaperone system and the 26S proteasome system to maintain protein homeostasis in the cytoplasm (7, 8).
CHIP has been shown to be a regulator of oncogenic pathways such as those involved in tumorigenesis, proliferation, and invasion in several malignancies, particularly breast cancer (8). CHIP has been implicated in carcinogenesis via its regulation of a number of proteins including the receptor tyrosine kinase ErbB2, hypoxia-inducible factor 1α (HIF1α), human telomerase, reverse transcriptase, Src-3, NF-κB, and c-Myc (9–12), as well as tumor suppressors such as p53, apoptosis inducing factor 1 (AIF1), and interferon regulatory factor 1 (IRF-1) (13, 14). Recent reports have established that CHIP also promotes degradation of the glucocorticoid receptor (GR) and the cystic fibrosis transmembrane conductance regulator (CFTR) (6, 15).
The activity of CHIP is regulated by interactions with chaperones and co-chaperones to shift the triage of client proteins towards either folding or degradation (10). CHIP is regulated both transcriptionally and post-transcriptionally. The mRNA levels of CHIP are up-regulated in heat-shock, overexpression of pathogenic forms of polyQ proteins, and oxidative stress conditions (16, 17). Both mRNA and protein levels of CHIP are upregulated in breast (9, 18), colorectal (14, 19), and gastric (20) cancers, and CHIP expression strongly correlates with poor prognosis. It has also been reported that miR-764-5p down-regulates CHIP in osteoblast differentiation (21).
A plethora of evidence establishes that CHIP function is regulated through post-translational modifications. One such modification is regulatory ubiquitination, which facilitates targeting of CHIP substrates for proteasomal degradation (7). A recent report has shown that ataxin-3, a deubiquitinase, associates with monoubiquitinated (at Lys 2) CHIP and provides a chain editing activity, which determines the dynamics of substrate ubiquitination by CHIP. Ataxin-3 presumably binds polyubiquitinated substrates through its ubiquitin-interacting domain and deubiquitinates CHIP to terminate the reaction (22). Furthermore, CHIP is phosphorylated on serine and threonine residues in both its N- and C-terminal regions (23). However, how phosphorylation alters CHIP function is unknown. The kinases that are responsible for CHIP phosphorylation have not been identified, except for ERK5 that associates with CHIP and increases ubiquitin ligase activity, perhaps due to conformational changes in CHIP (24). Here we show Aurora A is activated upon 2-ME treatment of prostate cancer cells, LNCaP, C4-2, 22RV1 and LAPC4. Inhibition of Aurora A kinase either by pharmacological inhibitors or by knockdown using siRNA prevented 2-ME induced CHIP phosphorylation and AR degradation. Aurora A phosphorylates CHIP at S273 and substitution of alanine for serine at this site (S273A) in CHIP attenuated 2-ME induced AR degradation in cells.
Methods and Materials
Cell culture
Androgen-dependent human prostate carcinoma, LNCaP and androgen-independent human prostate carcinoma C4-2 and 22Rv1 (freshly sourced from American Type Culture Collection, Manassas, VA, USA) were maintained in RPMI (Gibco-Life Technologies, Grand Island, NY, USA), Los Angeles prostate cancer 4, LAPC4 (a kind gift of Dr. Daniel Gioeli, Department of Microbiology, Immunology & Cancer Biology, University of Virginia, USA) were maintained in iMDM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cell lines were maintained in a 37 °C/5% CO2 humidified atmosphere.
Chemicals/Inhibitors
2-ME was from Sigma-Aldrich (St Louis, MO, USA). Kinase inhibitors were from Selleckchem.com unless otherwise mentioned.
RNA isolation and RT–PCR
Cells were treated with different doses of 2-ME for 24 h or treated with siRNA against Aurora C and total RNA was isolated using TRIzol reagent (Invitrogen-Life Technologies) according to the manufacturer’s instructions. Complementary DNA synthesis for mRNA detection was carried out using the SuperScript III first-strand synthesis system for RT–PCR (Invitrogen). mRNA was detected by qPCR using SYBR Green PCR master mix (Bio-Rad) in a Bio-Rad CFX96 cycler and quantified with Bio-Rad CFX manager software (Bio-Rad, Hercules, CA, USA).
siRNA transfection
siRNA was transfected into LNCaP or C4-2 cells using RNAiMAX (Invitrogen) as per the manufacturer’s protocol. Briefly, 2 × 105 cells were seeded in 6 cm dish in growth medium. Following day transfection complex (siRNA+RNAiMax) was added to the cells after washing once with phosphate-buffered saline, waiting for 4–6 h, then transfection complex was removed, cells washed twice with phosphate-buffered saline and growth medium restored. Twenty-four hour later drugs were added and cells harvested after 24 h unless otherwise mentioned. siRNAs used are Aurora A-5′-CAGAAGAGAAGUAGAAAUA-3′; Aurora B- 5′-GGAGAAUAGCAGUGGGACA-3′; Aurora C- 5′-GCGAGAAAUUAGAUGAACA-3′; CHIP-3′UTR- 5′-CCACUAUCUGUGUAAUAAA-3′; TPX2- 5′-CCAAAGAAGAUGAGGAAGA-3′; CDK1- 5′-GGAAUACCUAUCAGAGUAU-3′; CDK2- 5′-CCGAGAGAUCUCUCUGCUU-3′; PDK1- 5′-GACCAGAGGCCAAGAAUUU-3′ and 5′-CCGAAGAUGAGAAGAGGUU-3′ AKt- 5′-CCGAGGUGCUGGAGGACAA-3, and 5′-GGACAGAGGAGCAAGGUUU-3′; PLK1- 5′-GCACCGAAACCGAGUUAUU-3′ and 5′-GGAGGAAAGCCCUGACUGA-3′
Western blotting and antibodies
For Western blotting, cells were lysed in modified RIPA lysis buffer (50 mm Tris–HCl, 150 mm NaCl, 5 mm EDTA, 5 mm EGTA, 0.5% NP-40, 0.5% Triton X–100, 50 mm NaF, 2 mm sodium orthovanadate, 40 mm β-glycerophosphate and 1 μm microcystin) supplemented with a protease inhibitor mix (Thermo Scientific, Rockford, IL, USA). Unless otherwise described, 30 μg of total protein was resolved by SDS–polyacrylamide gel electrophoresis, transferred to filters, and immunoblotted with various antibodies (1:1000 dilution unless otherwise mentioned). The antibodies used were antiAR (sc-7305); antiTPX2; Cdk1 (17) and Cdk2 (D-12) all from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-CHIP (C386), anti-Aurora A, anti-pT288-Aurora A, anti-Aurora B and anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase) from Cell Signaling Technology (Danvers, MA, USA); cyclin B1 (CC03, Calbiochem, Billerica, MA, USA), and mouse monoclonal anti-tubulin antibody from Sigma-Aldrich.
FACS Analysis
Cells were harvested by trypsinization and fixed with 70% ethanol for 24 h at 4 °C. Fixed cells were stained in 1 ml of propidium iodide solution (0.05% NP-40, 50 mg per ml propidium iodide, and 10 mg per ml RNase A) for at least 2 h at room temperature or overnight at 4 °C. Stained cells were analyzed with a flow cytometer using CellQuest software (both from BD Biosciences, San Jose, CA, USA), cell cycle phases were analyzed by ModFit LT V3.3.11 (Mac, Verity Software House, Topshan, ME, USA).
Point Mutation
Serine to alanine or aspartic acid mutation of CHIP at S273 was carried out using Quikchange site directed mutagenesis kit as per manufacture’s instruction (Stratagene).
Immunoprecipitation and Aurora A Kinase Assays
FLAG-CHIP was immunoprecipitated from cell extracts using anti-FLAG-M2 beads (Sigma-Aldrich, St Louis, MO, USA). The beads were washed three times with lysis buffer and twice with kinase buffer (20 mM Tris-HCL, pH 7.4, 1 mM MgCl2, 25 mM KCL, 1 mM DTT and 40 ug/ml BSA). The beads were then incubated with 19 μl of kinase reaction mixture (20 mM Tris-HCL, pH 7.4, 1 mM MgCl2, 25 mM KCL, 1 mM DTT and 40ug/ml BSA, 100 μM ATP, 5 μCi of [γ-32P]ATP) and 1 ul of purified recombinant Aurora A (purchased from Millipore) at 30°C for 30 min. The reaction was stopped by the addition of 4 μl of 6X SDS sample buffer. Samples were resolved by SDS-PAGE, and the gel was stained with Coomassie Brilliant Blue and dried before autoradiography.
Mass Spectrometry analysis
LNCaP cells stably expressing FLAG-WT-CHIP were treated with or without 1 μM 2-ME for 24 h. FLAG-CHIP was affinity purified using anti-FLAG M2 beads and eluted with FLAG peptide. Elutes were separated by SDS-PAGE, stained with Coomassie Brilliant Blue and protein bands were excised. LC/MS/MS was performed using a Thermo Scientific Orbitrap Velos ETD spectrometer in the Biomedical Mass Spectrometry Laboratory, which is supported by the University of Virginia School of Medicine . Data were analyzed using Sequest algorithm.
Results
Effects of kinase inhibitors on AR degradation in prostate cancer cells
We had previously observed substantial reduction in the levels of AR protein when we treated LNCaP or C4-2 prostate cancer cells with 2-ME. This was due to proteasomal degradation that correlated with covalent modification of CHIP, based on its reduced electrophoretic mobility in SDS-PAGE (4). If cell extracts were treated with lambda phosphatase this slower migrating form of CHIP was eliminated and the faster migrating form appeared, indicating that CHIP was phosphorylated in response to 2ME treatment of the cells (data not shown). Previous reports have shown CHIP phosphorylation in response to paclitaxel (Taxol™) in different cell lines, including prostate cancer cells (PhosphoSitePlus.org). To determine which kinases are involved in the CHIP response to 2-ME, we assayed a panel of commercially available kinase inhibitors in LNCaP and C4-2 cells with or without addition of 2-ME (Supplementary Table 1). Inhibition of MEK1/2, p38 MAPK, PKA, or PKC did not prevent CHIP phosphorylation nor attenuate AR degradation upon 2-ME treatment. We observed similar results with inhibitors of PI3K family members mTOR, ATM, or DNA-PK (Supplementary Table 1). On the other hand, the PI3K inhibitors LY294002 and Wortmannin blocked both CHIP phosphorylation and AR degradation in response to 2-ME treatment. Because PI3K is upstream of PDK1 and Akt activation, we expected to mimic the effects of PI3K inhibitors by knockdown of PDK1 or Akt using specific siRNAs. To our surprise, CHIP was phosphorylated and AR degraded in response to 2-ME in LNCaP and C4-2 cells knocked down for PDK1 and Akt. In addition, inhibition of Akt using MK-2206 did not affect the responses to 2-ME. We speculate that inhibition of CHIP phosphorylation and attenuation of AR degradation by LY294002 and Wortmannin were off-target effects, or at least effects not dependent on PI3K/PDK1/Akt kinases (25, 26).
Aurora Kinase A is required for AR degradation in response to 2-ME
Cells treated with 2-ME for 24 h undergo mitotic arrest (4), therefore the effects on CHIP and AR could be dependent on kinases that are activated when cells are in mitosis. We tested whether inhibition of mitotic kinases would block responses to 2-ME. To this end, we treated LNCaP and C4-2 cells with inhibitors of Aurora A/B kinases in the presence or absence of 2-ME. Both MLN-8054 and VX-680 inhibited Aurora A activation (phosphorylation of Thr288) and CHIP phosphorylation, as well as AR degradation in response to 2-ME (Figure 1A and B). MLN-8054 and VX-680 attenuated degradation of both full length as well as truncated (AR-V7) forms of AR in 22RV1 cells (Fig 1C) upon 2-ME treatment. We observed similar results as seen Fig 1A and Fig 1B in LAPC4 cells (Fig 1D). In contrast, inhibition of Aurora B with ZM447439, or Polo-like kinase (PLK) by BI6727 or by siRNA knockdown in LNCaP and C4-2 cells, did not prevent phosphorylation of CHIP (Supplementary Table 1). We observed a slight reduction of AR degradation in presence of 2-ME when we inhibited Aurora B with ZM447439 or knocked down Aurora B or Aurora C by siRNA (see below).
Figure 1. Aurora A kinase inhibitors attenuate 2-ME induced AR degradation.
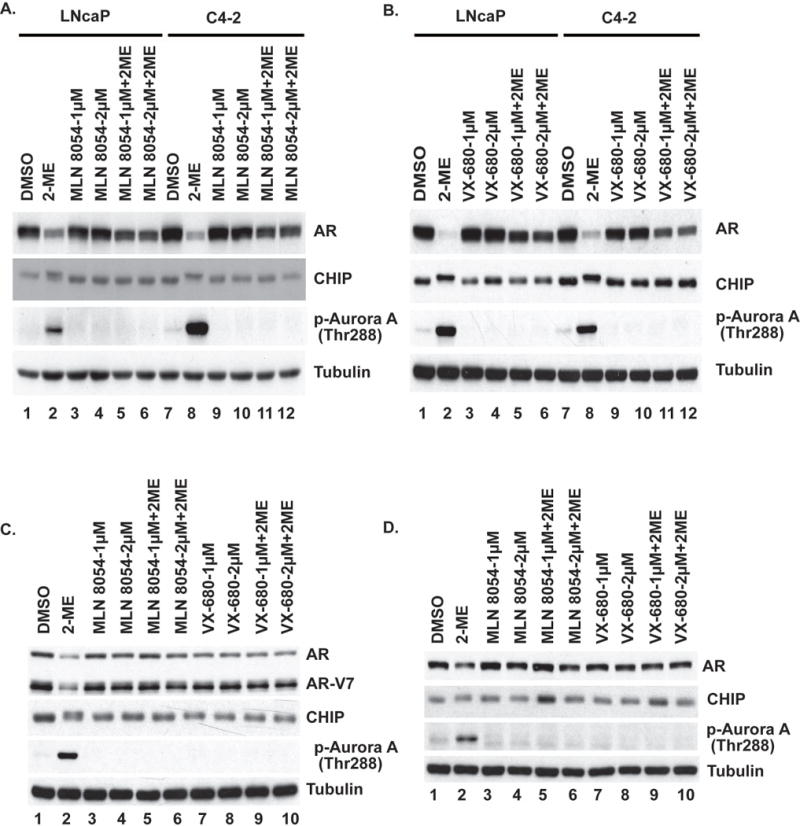
(A-B) LNCaP and C4-2, (C) 22RV1 and (D) LAPC4 cells were treated with or without 2-ME in presence or absence of Aurora A kinase inhibitor MLN8054 and/or VX-680 for 24 h. Cells were harvested, lysed and extracts were immunoblotted for indicated proteins. Tubulin served as loading control.
To compare the relative contributions of Aurora A, B, and C, we knocked them down individually or in combinations in LNCaP (Fig. 2A) and C4-2 (Fig. 2B) cells. Knockdown of Aurora A spared most of the AR from degradation upon 2-ME treatment. We observed partial protection of AR upon knockdown of Aurora B and slight preservation of AR upon knockdown of Aurora C. We did not observe any additive effects on AR levels when we knocked down combinations of Aurora A with either Aurora B or Aurora C. On the other hand, knockdown of Aurora B and Aurora C together did little to prevent AR degradation. The phosphorylation of CHIP (seen as reduced mobility in SDS-PAGE) was eliminated in cells knocked down for Aurora A alone, or combinations that included Aurora A, but not in cells knocked down for Aurora B or Aurora C (Fig. 2A and 2B). This reinforced the idea that Aurora A was phosphorylating CHIP. Furthermore, knockdown of Aurora A, but not Aurora B or Aurora C, prevented phosphorylation of CHIP and 2-ME-mediated AR degradation in cells stably expressing wild-type (WT) FLAG-tagged CHIP (Supplementary Fig. S1). We concluded that 2-ME stimulation of CHIP phosphorylation and AR degradation required Aurora A but not other kinases tested.
Figure 2. Knockdown of Aurora A attenuates 2-ME induced AR degradation.
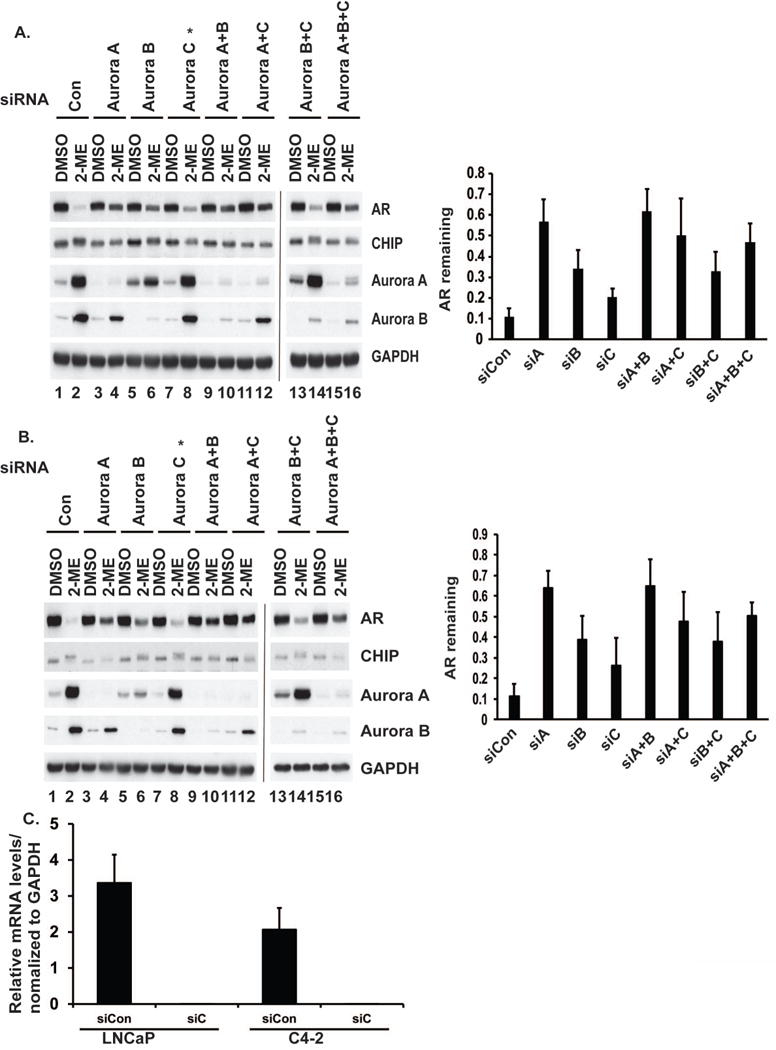
(A) LNCapP or (B) C4-2 cells were knocked down for Aurora A, B or C individually or in combination, treated with or without 2-ME, harvested and extracts were immunoblotted for AR, CHIP, Aurora A, Aurora B and GAPDH as a control. Remaining amounts of AR presented as a ratio of the band intensity of 2-ME treated vs corresponding DMSO treated cells, LNCaP (upper right) and C4-2 (lower right). Mean ± S.D. of at least three independent experiments. (C) *Due to lack of availability of an effective antibody against Aurora C, knockdown was monitored by qRT–PCR. mRNA levels are shown in LNCaP and C4-2 cells.
2-ME activates Aurora A kinase in a dose- and time-dependent manner
We treated LNCaP or C4-2 cells with increasing doses of 2-ME (Fig. 3A). At doses above 0.5 µM there was near complete loss of AR protein and an increase in CHIP phosphorylation. We also noted a large increase in the protein levels of Aurora A in both cell lines in response to 2-ME, compared to untreated cells. This increase in Aurora A protein corresponded to an increase of >10-fold in mRNA levels of Aurora A in response to 2-ME treatments (Fig. 3B). The phosphorylation of Thr288 (indicative of Aurora A kinase activation) increased in parallel to the increase in Aurora A protein level. The phosphorylation of CHIP and degradation of AR correlated closely with Aurora A up-regulation and activation.
Figure 3. 2-ME activates Aurora A kinase in dose and time dependent manner.
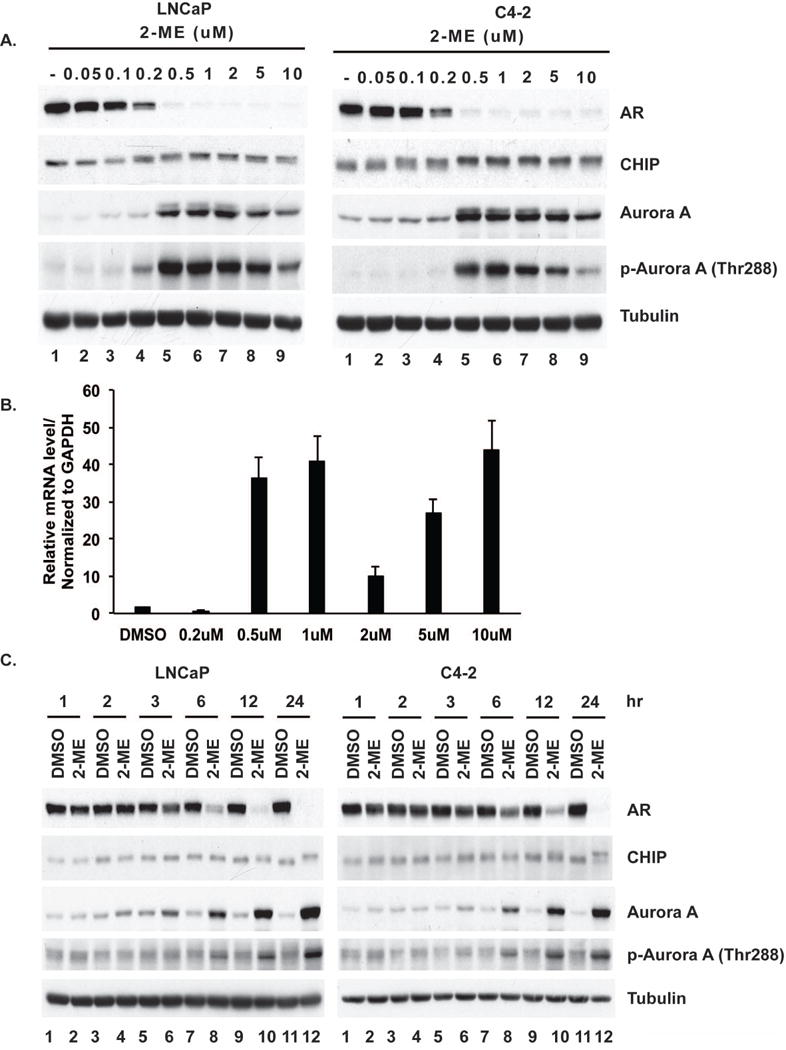
(A) LNCaP or C4-2 cells were treated with increasing concentrations of 2-ME for 24 h. (B) mRNA levels of LNCaP cells from (A) and (C) with 2μM of 2-ME were harvested at indicated times. Extracts were immunoblotted for AR, CHIP, Aurora A or pThr288-Aurora A and tubulin as control.
We next examined the kinetics of the response to 2-ME (Fig. 3C). LNCaP and C4-2 cells were treated with 2 µM 2-ME or DMSO as vehicle control and harvested at different time points for Western blotting. AR degradation and CHIP phosphorylation were obvious at 6, 12, and 24 h in both cell lines treated with 2-ME (lanes 8, 10, 12). Aurora A protein up-regulation and Thr288 phosphorylation in 2-ME-treated cells increased progressively from 6 through 24 h. Aurora A up-regulation could be detected as early as 3 h in C4-2 cells (lane 6) and even earlier (lane 4) in LNCaP cells. These results showed that 2-ME elicited up-regulation and activation of Aurora A that corresponded to CHIP phosphorylation and AR degradation.
2-ME activates Aurora A kinase in S phase of the cell cycle
Prolonged treatment with higher doses of 2-ME induces mitotic arrest in LNCaP and C4-2 prostate cancer cells (4). These M phase cells also have higher levels of cyclin B1 compared to untreated cells (Fig. 4A). To determine whether Aurora A kinase is activated by 2-ME without mitotic arrest, we treated prostate cancer cells for 24 h with aphidicholin, an inhibitor of the replicative DNA polymerase. Cells were then treated with or without 2-ME for an additional 24 h in the presence of aphidicholin. In the presence or absence of 2-ME, aphidicholin arrested cells at G1-S, as determined by DNA content using FACS analysis (Fig. 4B, left). The Aurora A protein levels were not increased in these cells, nor were the levels of cyclin B1, consistent with the cells not being in mitosis. However, Aurora A was activated by 2-ME in S-phase cells, based on phosphorylation of Thr288. Activation of the Aurora A by 2-ME corresponded with severe reduction in the levels of AR protein. These results show that Aurora A can be activated by 2-ME in cells that are not in M phase. Perhaps more importantly, AR degradation is stimulated by 2-ME outside of mitosis, reducing the possibility that mitotic kinases other than Aurora A are involved in this response.
Figure 4. 2-ME activates Aurora A Kinase in cells arrested in S phase.
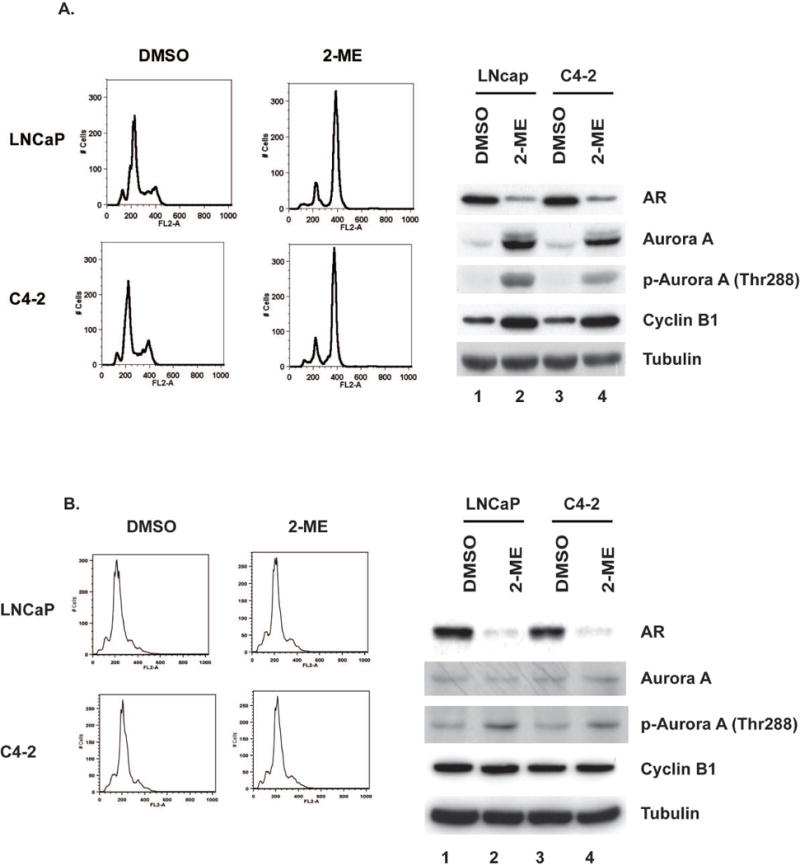
(A) Flow cytometry analysis of unsynchronized LNCaP and C4-2 treated with 2-ME for 24h (upper left). Immunoblots for indicated proteins and tubulin as a control (upper right). (B) LNCaP or C4-2 cells were held in G1/S by adding aphidicholin for 24 h. Cells were treated with or without 2-ME (2 μM) for additional 24 h in continued presence of aphidicholin. DNA content of LNCaP and C4-2 (Lower left) and immunoblotted for indicated proteins and tubulin serves as control (lower right)
Phosphorylation of CHIP by Aurora A requires TPX2
TPX2 is a protein that binds to and activates Aurora A (27). We knocked down TPX2 in LNCaP and C4-2 cells using siRNA and treated the cells with and without 2-ME for 24 h. Pre-knockdown, TPX2 protein levels were increased robustly in response to 2-ME treatment, and this corresponded to a large increase in activation (i.e., phosphorylation of Thr288) of Aurora A (Fig. 5A, lanes 2 and 6). Knockdown of TPX2 essentially eliminated phosphorylation of Aurora A in response to 2-ME. In addition, without TPX2, CHIP was not phosphorylated and AR escaped degradation. Thus, TPX2 and Aurora A were both required for these responses. Cells arrested in S-phase expressed about the same levels of TPX2 as unsynchronized cells (Fig. 5B). Under these conditions, activation of Aurora A by 2-ME did not involve TPX2 up-regulation. Presumably there was a sufficient level of TPX2 without up-regulation to support activation of Aurora A. These results further support the hypothesis that 2-ME activates Aurora A/TPX2, which in turn phosphorylates CHIP, leading to enhanced AR degradation.
Fig 5. Knock down of TPX2 attenuates 2-ME induced AR degradation.
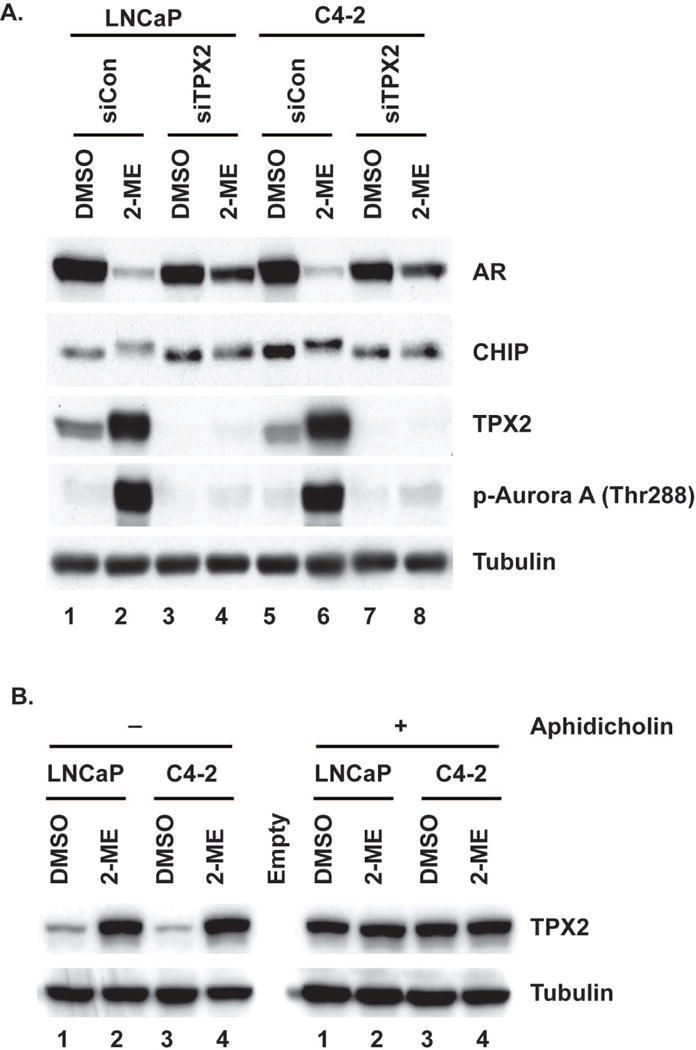
(A) LNCaP and C4-2 cells were depleted of TPX2 and treated with or without 2-ME for 24 h. Cells were harvested and extracts were immunoblotted for indicated proteins. Tubulin serves as control. (B) Extracts from Fig 4 were immunoblotted for TPX2, tubulin serves as control.
CHIP phosphorylation and AR degradation are independent of CDKs
We produced LNCaP cells stably expressing FLAG-CHIP and treated them with or without 2-ME for 24 h. FLAG-CHIP was recovered from cell extracts on anti-FLAG beads and eluted with FLAG peptide, and the tryptic peptides were analyzed for phosphorylation by LC-MS/MS. Analysis of the relative recovery of the phospho- and dephospho- peptides revealed that the peptide with pSer273 was enriched 12- to 15-fold in 2-ME-treated cells compared to untreated cells (Table 1). In addition, there was phosphorylation of Ser19 in both treated and untreated cells, and 2-fold induction of the doubly phosphorylated peptide with pSer19 and pSer23, with a corresponding decrease in the amount of singly phosphorylated peptide. Thus, addition of 2-ME to living cells increased phosphorylation of CHIP predominantly at Ser273.
Table 1.
Analysis of CHIP phosphorylation upon 2-ME treatment of prostate cancer cell. Fold change of CHIP phosphorylation upon 2-ME treatment of LNCaP cells stably expressing FLAG-WT-CHIP.
| Sequence | Phosphosite | 2ME/DMSO | |
|---|---|---|---|
|
|
S19 | −2X | |
|
|
S19 and S23 | +4X | |
|
|
S273 | +12X to +15X |
Phosphorylation of CHIP at Ser19, Ser23, and Ser273 seems incompatible with direct phosphorylation by Aurora A because these serine residues all are adjacent to prolines, and Aurora A does not phosphorylate Ser-Pro in peptides (28). In fact, phosphorylation of Ser-Pro has not been observed among known Aurora A substrates in cells (29). Instead, Ser-Pro is known to be a consensus site for phosphorylation by cyclin-dependent kinases (CDKs). A recent article reported that Cdk5 phosphorylates CHIP on Ser19, and this inhibits apoptosis (30). We therefore treated LNCaP and C4-2 cells with or without 2-ME in the presence or absence of the broad-specificity CDK inhibitor roscovitine (Supplementary Fig. S2A). Roscovitine did not prevent CHIP phosphorylation or AR degradation in response to 2-ME treatment. To provide additional evidence, we knocked down Cdk1 and Cdk2 individually or in combination (Supplementary Fig. S2B). These knockdowns caused no noticeable reduction in CHIP phosphorylation or AR degradation in 2-ME-treated cells. These results appear to rule out the involvement of CDKs in 2-ME-induced CHIP phosphorylation and AR degradation.
Aurora A phosphorylates CHIP at S273 in living cells
We next tested whether Aurora A directly phosphorylates CHIP utilizing a “back-phosphorylation” strategy (31) and by substituting Ser273 in CHIP with Ala to prevent phosphorylation at this site. We treated cells expressing FLAG-tagged WT CHIP with or without 1 μM 2-ME for 24 h, conditions where the Aurora A phosphorylation site(s) would become occupied in the WT protein. This was indeed the case, based on anti-FLAG immunoblotting that showed reduced mobility of FLAG-CHIP in 2-ME treated vs. untreated cells (Fig. 6A, lanes 3 vs. 2). We expressed FLAG-tagged WT and S273A CHIP, recovered the proteins using anti-FLAG beads and performed an in vitro kinase assay with purified recombinant Aurora A and [32P]γ-ATP. The WT FLAG-CHIP recovered from untreated cells was 32P-phosphorylated by Aurora A (Fig. 6B, lane 2), but there was not phosphorylation of WT FLAG-CHIP recovered from 2-ME-treated cells (Fig. 6B, lane 3). This demonstrated that the Aurora A phosphorylation site in FLAG-CHIP was occupied in 2-ME treated cells. Purified Aurora A also failed to phosphorylate FLAG-CHIP S273A (Fig. 6B, lane 4), consistent with our mapping of this as the primary phosphosite in 2-ME treated cells. These results demonstrated that Ser273 was the site of Aurora A phosphorylation in CHIP in cells treated with 2-ME.
Figure 6. Aurora A phosphorylates CHIP at S273.
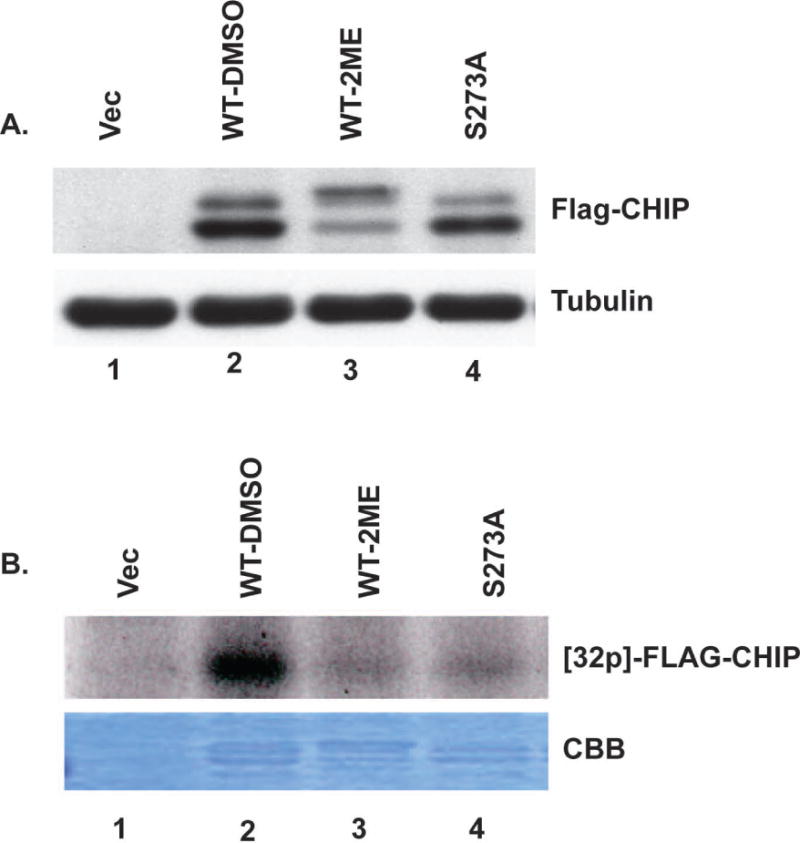
(A) Cells stably expressing FLAG-WT-CHIP were treated with or without 2-ME for 24 hrs, harvested and immunoblotted using anti-FLAG antibody (upper panel). Immunoblotting for tubulin serves as loading control (lower panel). (B) FLAG-WT-CHIP and FLAG-CHIP S273A were pulled down with M2 beads and used as substrates in kinase assays performed using bacterially expressed recombinant purified Aurora A and radiolabeled γ-ATP. Upper panel shows autoradiogram of 32P incorporated into FLAG-CHIP. Coomassie Brilliant Blue (CBB) staining of gel shows protein bands.
CHIP S273A mutation attenuates 2-ME-induced AR degradation
To address the role of CHIP Ser273 phosphorylation in AR degradation, we depleted endogenous CHIP using siRNA against the 3′-UTR of CHIP in cells stably expressing empty vector or untagged WT, S273A, or S273D versions of CHIP and treated these cells with or without 2-ME for 24 h. As shown in Fig. 7A knockdown of endogenous CHIP reduced the extent of AR degradation induced by 2-ME (lane 4 vs. 2). This degradation was restored in cells expressing WT CHIP (lane 6), whereas cells expressing CHIP S273A exhibited less AR degradation in response to 2-ME compared to cells expressing WT CHIP (lane 8 vs. 6). CHIP Ser273 was substituted with Asp as a phosphomimetic residue, and this S273D version was compared to WT and S273A. The quantitative amount or AR remaining after 2ME stimulation (even numbered lanes in panel A) from at least three independent experiments are shown in Fig. 7B, revealing statistically significant less AR degradation with knockdown of CHIP or substitution of Ser273. These data support the conclusion that CHIP phosphorylation at Ser273 enhances AR degradation.
Figure 7. S273A substitution in CHIP attenuates 2-ME induced AR degradation.
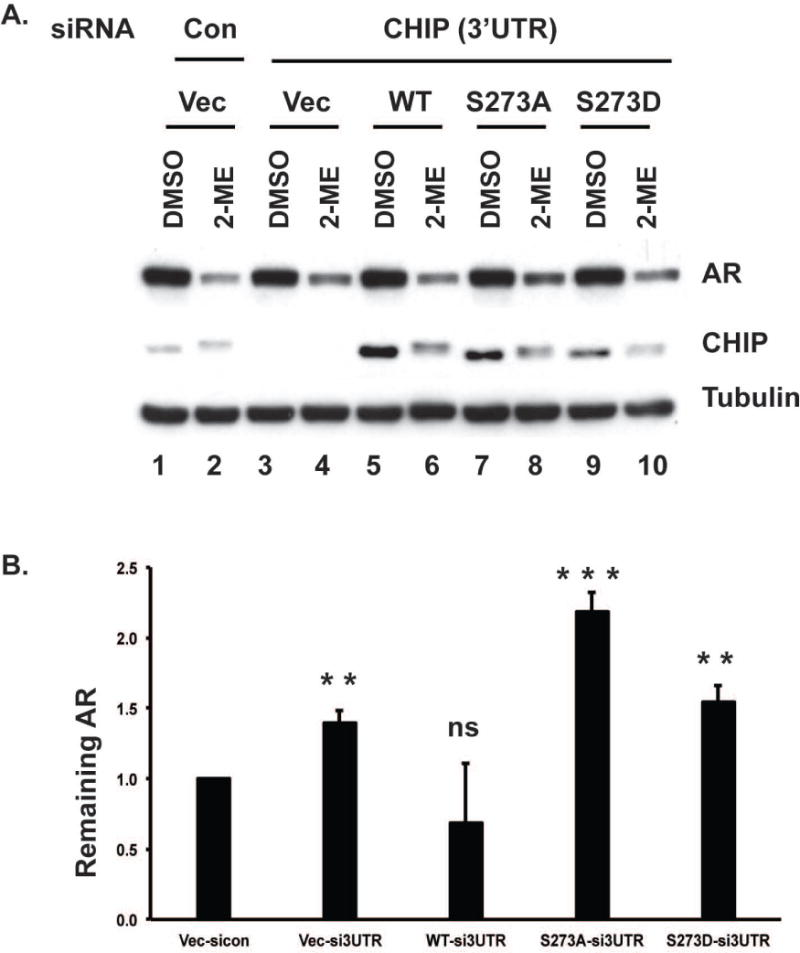
(A). C4-2 cells stably expressing empty vector, WT CHIP or CHIP S273 mutants were depleted of endogenous CHIP using siRNA against 3′-UTR, and treated with or without 2-ME for 24 h, then harvested and immunoblotted for AR, CHIP and tubulin as a control. (B) Quantification of remaining AR expressed as a ratio of the band intensity of 2-ME treated vs corresponding DMSO treated cells. The fold change was calculated relative to AR level in 2ME-treated control cells stably expressing empty vector.. Mean ± S.D. of at least three independent experiments. Statistical significance presented as- ns- not significant, *p<0.01, **p<0.001 and ***p<0.0001.
Discussion
AR signaling is critical for the progression of prostate cancer, so understanding the pathways that promote AR degradation is one step toward combating castration-resistant prostate cancer. We report here that Aurora kinase A phosphorylates CHIP and promotes AR degradation in LNCaP, C4-2, 22RV1 and LAPC4 prostate cancer cells upon 2-ME treatment. We suggest this novel 2-ME→Aurora A→CHIP→AR pathway might be exploited in developing new therapeutics. It is important to note that 22RV1 cells express alternately spliced forms of the AR that contribute to castration resistance and are degraded in response to this novel pathway.
Aurora A kinase is known to regulate mitotic entry, spindle formation, and centrosome maturation. Experimental overexpression of Aurora A overrides the mitotic spindle checkpoint and induces resistance to paclitaxel (32). Several early studies have shown that Aurora A is overexpressed in various malignancies, including prostate cancer (33, 34). These facts in part prompted development of Aurora A kinase inhibitors as anti-cancer drugs. Aurora A is activated at mitotic entry and this suggests AR degradation might depend on 2-ME induction of mitosis. However we found that 2-ME activates Aurora A and AR degradation in both S and G2-M phases of the cell cycle, so the actions of 2-ME cannot simply be attributed to cells being arrested in mitosis.. A non-canonical role for Aurora A in DNA replication has been reported (35, 36), consistent with our observation of Aurora A activation in aphidicholin arrested cells. The activation of Aurora A in response to 2-ME can be explained in part by the increase of Aurora A mRNA levels. Binding of TPX2 to Aurora A stabilizes an active kinase conformation and prevents Thr288 dephosphorylation (37). We discovered Aurora A activation and CHIP-mediated AR degradation are both TPX2-dependent in mitosis. TPX2 is also present in non-mitotic cells, but it is unknown whether interphase activation of Aurora A is TPX2 dependent. Inducing TPX2 synthesis or accumulation would be predicted to activate Aurora A and cause degradation of AR.
In mammals, Aurora A, B, and C possess distinctive roles, Aurora A phosphorylates multiple substrates and promotes mitotic entry by activation of cyclin B1/Cdk1 (38, 39) and activation of PLK1 (40, 41). Aurora B provides the catalytic activity to the chromosome passenger complex (CPC) (42), and Aurora C is required for spermatogenesis and oocyte development (43). We observed an attenuation of 2-ME-induced AR degradation when we knocked down Aurora B, even though CHIP was still phosphorylated. However, simultaneous knockdown of Aurora B and C was unable to restore 2-ME-induced AR degradation, suggesting that 2-ME promoted CHIP phosphorylation and AR degradation primarily, if not exclusively, via activated Aurora A.
Our mass spectrometry results revealed that 2-ME treatment increased the phosphorylation of CHIP at Ser273 by 12- to 15-fold. Ser273 is followed by proline in a consensus sequence (S/T*]PX[K/R) for CDKs (44). However, inhibitors and knockdowns indicated that CDKs are not responsible for CHIP Ser273 phosphorylation or AR degradation. Because the S273A mutation prevented in vitro phosphorylation of CHIP by Aurora A and attenuated 2-ME-induced AR degradation in cells, we conclude that phosphorylation of Ser273 in CHIP stimulates AR degradation. The most straightforward possibility is that Aurora A directly phosphorylates Ser273 in CHIP. However, Aurora A has a well-defined consensus sequence which does not match the Ser273 site with an adjacent Pro residue. It is conceivable that an unknown Aurora A-dependent kinase phosphorylates Ser273 in CHIP in 2-ME treated cells. Such a kinase would have to co-precipitate with CHIP or contaminate purified recombinant Aurora A that were used in kinase assays. Then again, Aurora A itself may be responsible. CHIP functions in a multiprotein complex with chaperones that might render the conformation of the S273 site reactive with Aurora A. We cannot rule out the possibility that Aurora A also enhances AR degradation through direct phosphorylation of AR. Aurora A was reported to phosphorylate AR at T282 and S293 and activate transcription, but no decrease in AR levels in response to Aurora A phosphorylation was noted. Complicating the issue, that article was subsequently retracted (PMID 27825092). Thus, we cannot exclude the possibility that Aurora A might activate AR degradation by phosphorylation of both enzyme and substrate, CHIP and AR respectively.
A recent report has shown that a complex of PC-1, an androgen-responsive transcription factor, and CHIP degrades the AR in mitosis (45). Whether 2-ME-mediated AR degradation involves PC-1 is unknown. However, the interphase activation of Aurora would be expected to be PC-1-independent. AR function is cell cycle-dependent, and S308 phosphorylation by Cdk1 regulates its localization and transcriptional activity (46). Additionally, phosphorylation of CHIP at Ser20 by Cdk5 promotes tAIF-mediated neuronal death (30). These observations raise the question of whether phosphorylation of CHIP or AR by CDKs may contribute to AR degradation. However, our results eliminate CDK involvement in 2-ME-induced CHIP phosphorylation, for both treatment with roscovitine, which inhibits Cdk1, Cdk2, and Cdk5, and simultaneous knockdown of Cdk1 and Cdk2 failed to prevent 2-ME-induced CHIP phosphorylation and AR degradation.
We raise three potentially important therapeutic implications regarding activation of Aurora in promotion of AR degradation. First, any chemotherapeutic regimen, including microtubule-disrupting agents, should promote AR degradation by arresting cells in mitosis when Aurora A is activated. Doxetaxel is highly active in castrate resistant prostate cancer and we suggest some of its activity could be explained by Aurora A mediated AR degradation. Second, if Aurora A levels are rate-limiting in promoting AR degradation then Aurora A levels could be a potential biomarker for response to Aurora-activating agents. Third, agents that inhibit Aurora activity would be expected to increase AR levels and signaling, thereby promoting prostate cancer growth. Aurora A inhibitors have been tested in clinical trials since 2005. At least seventy trials of such inhibitors have been initiated in different cancers. We suggest these agents (33, 47-50) including MLN8054 and VX-680 might have unintended adverse effects in prostate cancer due to protection of AR from degradation.
Supplementary Material
Acknowledgments
We thank Dr. Cam Patterson, New York Presbyterian Hospital/Weill Cornell Medical Center, New York, USA, for providing us wild type CHIP construct. This work was supported by grant NIH-CA192669 and the generous support of Charles Burnett III.
Funding: NIH-CA192669 to JML
Footnotes
Conflict of Interest: Authors have no financial interest in relation to this work
References
- 1.Coutinho I, Day TK, Tilley WD, Selth LA. Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocrine-related cancer. 2016;23(12):T179–T97. doi: 10.1530/ERC-16-0422. [DOI] [PubMed] [Google Scholar]
- 2.Devlin HL, Mudryj M. Progression of prostate cancer: multiple pathways to androgen independence. Cancer letters. 2009;274(2):177–86. doi: 10.1016/j.canlet.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD. Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocrine-related cancer. 2004;11(3):459–76. doi: 10.1677/erc.1.00525. [DOI] [PubMed] [Google Scholar]
- 4.Sarkar S, Brautigan DL, Parsons SJ, Larner JM. Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene. 33(1):26–33. doi: 10.1038/onc.2012.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19(6):4535–45. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3(1):93–6. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 7.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8(4):303–8. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun C, Li HL, Shi ML, Liu QH, Bai J, Zheng JN. Diverse roles of C-terminal Hsp70-interacting protein (CHIP) in tumorigenesis. J Cancer Res Clin Oncol. 140(2):189–97. doi: 10.1007/s00432-013-1571-5. [DOI] [PubMed] [Google Scholar]
- 9.Kajiro M, Hirota R, Nakajima Y, Kawanowa K, So-ma K, Ito I, et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol. 2009;11(3):312–9. doi: 10.1038/ncb1839. [DOI] [PubMed] [Google Scholar]
- 10.Paul I, Ghosh MK. The E3 ligase CHIP: insights into its structure and regulation. Biomed Res Int. 2014:918183. doi: 10.1155/2014/918183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sisoula C, Gonos ES. CHIP E3 ligase regulates mammalian senescence by modulating the levels of oxidized proteins. Mech Ageing Dev. 132(5):269–72. doi: 10.1016/j.mad.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99(20):12847–52. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J Biol Chem. 285(6):3651–63. doi: 10.1074/jbc.M109.068577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Ren F, Feng Y, Wang D, Jia B, Qiu Y, et al. CHIP/Stub1 functions as a tumor suppressor and represses NF-kappaB-mediated signaling in colorectal cancer. Carcinogenesis. 35(5):983–91. doi: 10.1093/carcin/bgt393. [DOI] [PubMed] [Google Scholar]
- 15.Esser C, Scheffner M, Hohfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J Biol Chem. 2005;280(29):27443–8. doi: 10.1074/jbc.M501574200. [DOI] [PubMed] [Google Scholar]
- 16.Dikshit P, Jana NR. The co-chaperone CHIP is induced in various stresses and confers protection to cells. Biochem Biophys Res Commun. 2007;357(3):761–5. doi: 10.1016/j.bbrc.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Stankowski JN, Zeiger SL, Cohen EL, DeFranco DB, Cai J, McLaughlin B. C-terminus of heat shock cognate 70 interacting protein increases following stroke and impairs survival against acute oxidative stress. Antioxid Redox Signal. 14(10):1787–801. doi: 10.1089/ars.2010.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patani N, Jiang W, Newbold R, Mokbel K. Prognostic implications of carboxyl-terminus of Hsc70 interacting protein and lysyl-oxidase expression in human breast cancer. J Carcinog. 9:9. doi: 10.4103/1477-3163.72505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruckova E, Muller P, Nenutil R, Vojtesek B. Alterations of the Hsp70/Hsp90 chaperone and the HOP/CHIP co-chaperone system in cancer. Cell Mol Biol Lett. 17(3):446–58. doi: 10.2478/s11658-012-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan L, Liu DB, Lu HF, Long GX, Mei Q, Hu GY, et al. Decreased expression of the carboxyl terminus of heat shock cognate 70 interacting protein in human gastric cancer and its clinical significance. Oncol Rep. 28(4):1392–8. doi: 10.3892/or.2012.1957. [DOI] [PubMed] [Google Scholar]
- 21.Guo J, Ren F, Wang Y, Li S, Gao Z, Wang X, et al. miR-764-5p promotes osteoblast differentiation through inhibition of CHIP/STUB1 expression. J Bone Miner Res. 27(7):1607–18. doi: 10.1002/jbmr.1597. [DOI] [PubMed] [Google Scholar]
- 22.Scaglione KM, Zavodszky E, Todi SV, Patury S, Xu P, Rodriguez-Lebron E, et al. Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol Cell. 43(4):599–612. doi: 10.1016/j.molcel.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105(31):10762–7. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo CH, Le NT, Shishido T, Chang E, Lee H, Heo KS, et al. Novel role of C terminus of Hsc70-interacting protein (CHIP) ubiquitin ligase on inhibiting cardiac apoptosis and dysfunction via regulating ERK5-mediated degradation of inducible cAMP early repressor. FASEB J. 24(12):4917–28. doi: 10.1096/fj.10-162636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrido G, Vernos I. Non-centrosomal TPX2-Dependent Regulation of the Aurora A Kinase: Functional Implications for Healthy and Pathological Cell Division. Frontiers in oncology. 2016;6:88. doi: 10.3389/fonc.2016.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrari S, Marin O, Pagano MA, Meggio F, Hess D, El-Shemerly M, et al. Aurora-A site specificity: a study with synthetic peptide substrates. Biochem J. 2005;390(Pt 1):293–302. doi: 10.1042/BJ20050343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Science signaling. 2011;4(179):rs5. doi: 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim C, Yun N, Lee J, Youdim MB, Ju C, Kim WK, et al. Phosphorylation of CHIP at Ser20 by Cdk5 promotes tAIF-mediated neuronal death. Cell Death Differ. 2016;23(2):333–46. doi: 10.1038/cdd.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matejovicova M, Kaplan P, Mubagwa K, Raeymaekers L, Pongo E, Flameng W. Phosphorylation by protein kinases A and C of myofibrillar proteins in rabbit stunned and non-stunned myocardium. Journal of molecular and cellular cardiology. 1997;29(12):3189–202. doi: 10.1006/jmcc.1997.0534. [DOI] [PubMed] [Google Scholar]
- 32.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer cell. 2003;3(1):51–62. doi: 10.1016/s1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- 33.Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015;34(5):537–45. doi: 10.1038/onc.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee EC, Frolov A, Li R, Ayala G, Greenberg NM. Targeting Aurora kinases for the treatment of prostate cancer. Cancer research. 2006;66(10):4996–5002. doi: 10.1158/0008-5472.CAN-05-2796. [DOI] [PubMed] [Google Scholar]
- 35.Tsunematsu T, Arakaki R, Yamada A, Ishimaru N, Kudo Y. The Non-Canonical Role of Aurora-A in DNA Replication. Frontiers in oncology. 2015;5:187. doi: 10.3389/fonc.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsunematsu T, Takihara Y, Ishimaru N, Pagano M, Takata T, Kudo Y. Aurora-A controls pre-replicative complex assembly and DNA replication by stabilizing geminin in mitosis. Nature communications. 2013;4:1885. doi: 10.1038/ncomms2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12(4):851–62. doi: 10.1016/s1097-2765(03)00392-7. [DOI] [PubMed] [Google Scholar]
- 38.Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114(5):585–98. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 39.Satinover DL, Brautigan DL, Stukenberg PT. Aurora-A kinase and inhibitor-2 regulate the cyclin threshold for mitotic entry in Xenopus early embryonic cell cycles. Cell cycle. 2006;5(19):2268–74. doi: 10.4161/cc.5.19.3316. [DOI] [PubMed] [Google Scholar]
- 40.Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455(7209):119–23. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 41.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320(5883):1655–8. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terada Y. Role of chromosomal passenger complex in chromosome segregation and cytokinesis. Cell structure and function. 2001;26(6):653–7. doi: 10.1247/csf.26.653. [DOI] [PubMed] [Google Scholar]
- 43.Kimmins S, Crosio C, Kotaja N, Hirayama J, Monaco L, Hoog C, et al. Differential functions of the Aurora-B and Aurora-C kinases in mammalian spermatogenesis. Molecular endocrinology. 2007;21(3):726–39. doi: 10.1210/me.2006-0332. [DOI] [PubMed] [Google Scholar]
- 44.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15(24):7060–9. [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Zhang H, Zhang X, Wang P, Wang H, Huang F, et al. PC-1 works in conjunction with E3 ligase CHIP to regulate androgen receptor stability and activity. Oncotarget. 2016 doi: 10.18632/oncotarget.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koryakina Y, Knudsen KE, Gioeli D. Cell-cycle-dependent regulation of androgen receptor function. Endocrine-related cancer. 2015;22(2):249–64. doi: 10.1530/ERC-14-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dees EC, Infante JR, Cohen RB, O’Neil BH, Jones S, von Mehren M, et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer chemotherapy and pharmacology. 2011;67(4):945–54. doi: 10.1007/s00280-010-1377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Investigational new drugs. 2012;30(6):2411–32. doi: 10.1007/s10637-012-9798-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macarulla T, Cervantes A, Elez E, Rodriguez-Braun E, Baselga J, Rosello S, et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Molecular cancer therapeutics. 2010;9(10):2844–52. doi: 10.1158/1535-7163.MCT-10-0299. [DOI] [PubMed] [Google Scholar]
- 50.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104(10):4106–11. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.