

Abstract
The metabolism of testosterone to 6β-hydroxytestosterone (6β-OH-T) is a commonly used assay to evaluate human CYP3A enzyme activities. However, previous reports have indicated that CYP3A7 also produces 2α-hydroxytestosterone (2α-OH-T) and that a 2α-OH-T/6β-OH-T ratio may be a unique endogenous biomarker of the activity of the enzyme. Until now, the full metabolite and kinetic profile for testosterone hydroxylation by CYP3A7 has not been fully examined. To this end, we performed a complete kinetic analysis of the 6β-OH-T, 2α-OH-T, and 2β-hydroxytestosterone metabolites for recombinant Supersome CYP3A4, CYP3A5, and CYP3A7 enzymes and monitored metabolism in fetal and adult human liver microsomes for comparison. In general, a decrease in the velocity of the reaction was observed between CYP3A4 and the two other enzymes, with CYP3A7 showing the lowest metabolic capacity. Interestingly, we found that the 2α-OH-T/6β-OH-T ratio varied with substrate concentration when testosterone was incubated with CYP3A7, suggesting that this ratio would likely not function well as a biomarker for CYP3A7 activity. In silico docking studies revealed at least two different binding modes for testosterone between CYP3A4 and CYP3A7. In CYP3A4, the most energetically favorable docking mode places testosterone in a position with the methyl groups directed toward the heme iron, which is more favorable for oxidation at C6β, whereas for CYP3A7 the testosterone methyl groups are positioned away from the heme, which is more favorable for an oxidation event at C2α. In conclusion, our data indicate an alternative binding mode for testosterone in CYP3A7 that favors the 2α-hydroxylation, suggesting significant structural differences in its active site compared with CYP3A4/5.
Introduction
The human cytochrome P450 CYP3A enzymes CYP3A4, CYP3A5, and CYP3A7 are well-known for their role in the metabolism of many drugs (Zanger and Schwab, 2013; Ortiz de Montellano, 2015), toxicants (Fæste et al., 2011; Kittler et al., 2014), and steroids, including cholesterol (Bodin et al., 2002; Honda et al., 2011), estradiol (Yamazaki et al., 1998), testosterone (Waxman et al., 1988; Williams et al., 2002; Krauser et al., 2004; Leeder et al., 2005), dehydroepiandrosterone (DHEA) (Miller et al., 2004) and its sulfate derivative (DHEA-S) (Kitada et al., 1987a). CYP3A4 represents the major cytochrome P450 monooxygenase expressed in the adult liver and small intestine (Shimada et al., 1994; Paine et al., 2006; Michaels and Wang, 2014) and together with CYP3A5 has been estimated to metabolize at least 50% of the currently prescribed drugs (Rendic and Guengerich, 2015). Conversely, CYP3A7 is the major cytochrome P450 enzyme expressed in the fetal and neonatal liver (Schuetz et al., 1993; Stevens et al., 2003) with only limited expression observed in the adult liver (Sim et al., 2005). During the fetal period, CYP3A7 is thought to have a critical role in placental estriol synthesis by controlling the 16α-hydroxylation of DHEA-S (Kitada et al., 1987a,b). Despite a high degree of identity in their protein sequences (>82%) (Shen et al., 2004), the CYP3A4, CYP3A5, and CYP3A7 enzymes have demonstrated significant differences in their substrate specificity (Ohmori et al., 1998), catalytic efficiency (Williams et al., 2002; Shen et al., 2004), and/or regioselectivity (Gorski et al., 1994; Ohmori et al., 1998; Wang et al., 1998). Over the last 2 decades, numerous studies have been conducted to characterize these CYP3A enzymes using mutagenic (He et al., 2003; Lee et al., 2003; Kumar et al., 2006; Torimoto et al., 2007; Zhao et al., 2012), kinetic (Nakamura et al., 2003; Dai et al., 2004; Huang et al., 2004; Isin and Guengerich, 2006; Shuster et al., 2014), or crystallographic approaches (Williams et al., 2004; Yano et al., 2004; Ekroos and Sjogren, 2006), but many questions remain concerning the structural determinants controlling substrate binding, specificity, and regioselectivity, particularly with regard to the less well studied CYP3A7. This is important because, in many instances, CYP3A7 can be the main CYP3A drug elimination pathway in neonates and infants.
Testosterone is a probe substrate metabolized by the CYP3A enzymes in a regioselective and stereoselective manner (Mäenpää et al., 1993; Williams et al., 2002; Krauser et al., 2004; Krauser and Guengerich, 2005; Leeder et al., 2005). Three major hydroxylated metabolites, 6β-hydroxytestosterone (6β-OH-T), 2β-hydroxytestosterone (2β-OH-T), and 2α-hydroxytestosterone (2α-OH-T), have been identified from reactions involving recombinant CYP3A enzyme (Williams et al., 2002; Leeder et al., 2005) and human liver microsome (HLM) incubations (Mäenpää et al., 1993; Leeder et al., 2005). Indeed, testosterone 6β-hydroxylation is a commonly used assay to characterize the CYP3A enzyme activity in a variety of test systems (Sohl et al., 2009). Williams et al. (2002) determined the catalytic parameters of recombinant CYP3A4, CYP3A5, and CYP3A7 for 10 different substrates, including the conversion of testosterone to 6β-OH-T. They observed equal or reduced catalytic activity for CYP3A5 compared with CYP3A4 and considerably decreased activity for CYP3A7, particularly in regard to the production of 6β-OH-T. Williams et al. (2002) also reported that CYP3A7 produced a substantial amount of the 2α-OH-T metabolite. Afterward, Leeder et al. (2005) demonstrated that 2α-OH-T is a predominant testosterone metabolite produced by CYP3A7, both in recombinant enzyme systems and fetal HLMs (fHLMs). Additionally, they found that the production of 2α-OH-T was proportional to the human fetal hepatic microsomal CYP3A content and that the 2α-hydroxylation and 6β-hydroxylation activities were highly correlated in the fHLMs, raising the possibility that a ratio of the concentration of 2α-OH-T to that of 6β-OH-T might serve as an endogenous biomarker for CYP3A7 activity. If this were to be the case, it could prove extremely useful for dosing calculations and pharmacokinetic modeling in infants, as there currently is no known endogenous biomarker for CYP3A7 and it is not ethical to dose infants with probe substrate cocktails to determine enzymatic activity (Strougo et al., 2014).
Therefore, we set out to examine the kinetics, regioselectivity, and stereoselectivity of testosterone oxidation in the CYP3A family and assess the suitability of the 2α-OH-T/6β-OH-T metabolite ratio as an endogenous biomarker for CYP3A7 activity. In particular, our study had the following three goals: 1) to determine and compare the kinetic parameters of the 6β-OH-T, 2β-OH-T, and 2α-OH-T metabolite formation for the three human CYP3A enzymes under steady-state conditions; 2) to determine the metabolite ratios as a function of testosterone substrate concentration; and 3) to compare the correlation of the 2α-OH-T/6β-OH-T ratio between adult HLMs and fHLMs. Our results indicate that whereas 2α-OH-T is a predominant metabolite formed by CYP3A7 in both Supersomes and fHLMs, the 2α-OH-T/6β-OH-T ratio varies as a function of testosterone concentration, indicating that the regiospecificity of substrate oxidation is controlled by the concentration of the parent substrate, suggesting that for CYP3A7 multiple ligand binding may favor production of the 6β-OH-T metabolite over the 2α-OH-T metabolite at higher concentrations of substrate. Furthermore, this result may have implications for the patterns of oxidation of other endogenous steroids that are also CYP3A7 substrates, such as DHEA and DHEA-S.
Materials and Methods
Materials.
The metabolite standards, 6β-, 2β-, and 2α-hydroxytestosterone, were purchased from Steraloids (Newport, RI). The internal standard, 6β-hydroxytestosterone-d3, was purchased from Cerilliant (Round Rock, TX). Testosterone, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, and NADP were obtained from Sigma-Aldrich (St. Louis, MO). All other chemicals and solvents used were obtained from standard suppliers and were of reagent or analytical grade. Supersomes of CYP3A4, CYP3A5, and CYP3A7, coexpressed with human cytochrome P450 reductase and human cytochrome b5, in Sf9 insect cells using a baculovirus expression system were purchased from Corning (Corning, NY).
HLMs.
Individual male HLMs, lot numbers DDV and RGS, were purchased from BioreclamationIVT (Hicksville, NY). Individual male fHLMs, lot numbers 5 (120 days estimated gestational age) and 18 (100 days estimated gestational age), were prepared as previously described (Shuster et al., 2014). Quantification of the CYP3A protein content and protein concentration determination for the fHLMs were also performed as previously described (Shuster et al., 2014).
Recombinant CYP3A Enzyme and HLM In Vitro Activity Assays.
The recombinant CYP3A incubation reactions (250 µl) contained various concentrations of testosterone (2.5–500 µM) that were dissolved in methanol (1% v/v); CYP3A4 (5 pmol/ml), CYP3A5 (20 pmol/ml), or CYP3A7 (50 pmol/ml) Supersomes; 100 mM potassium phosphate buffer (pH 7.4); and 3 mM MgCl2. After preincubation at 37°C for 3 minutes, the reactions prepared in triplicate were initiated by the addition of an NADPH-generating system consisting of NADP+ (1 mM), glucose-6-phosphate (10 mM), and glucose-6-phosphate dehydrogenase (2 IU/ml). The reactions were incubated for 10 minutes at 37°C under agitation and were stopped by the addition of ice-cold methanol (250 µl) containing 400 ng/ml 6β-hydrotestosterone-d3 (internal standard). Incubations without the NADPH-generating system served as negative controls. Protein was collected by centrifugation at 1800g for 10 minutes at 4°C. Supernatants were transferred to high-performance liquid chromatography vials, and aliquots of 5 µl were analyzed by LC-MS/MS. The HLM incubation reactions (100 µl) were prepared similarly except that the concentration of testosterone was 50, 100, or 250 µM with 0.25 mg/ml microsomal protein, and the reactions were stopped after 20 minutes by the addition of ice-cold methanol (100 µl) containing 400 ng/ml 6β-hydrotestosterone-d3 (internal standard). Duplicate determinations were performed for each HLM lot.
Analytical Methods.
Samples were analyzed by LC-MS/MS with a Waters Acquity Ultra-Performance Liquid Chromatography system interfaced by electrospray ionization with a Waters Quattro Premier XE Triple Quadrupole Mass Spectrometer (Waters, Milford, MA) in positive mode and with multiple reaction monitoring (MRM) scan type. The following source conditions were applied: 1.5 kV for the capillary voltage; 30 V for the cone voltage; 120°C for the source temperature; 350°C for the desolvation temperature; 50 l/h for the source gas flow; and 650 l/h for the desolvation gas flow. The following mass transitions and collision energies (CEs) were used to detect the respective analytes as follows: 305 > 269 (CE = 15 V) for the hydroxyl testosterone metabolites; 308.5 > 272.5 (CE = 15 V) for the internal standard 6β-hydroxytestosterone-d3; and 289 > 253 (CE = 15 V) for testosterone. Testosterone and its hydroxylated metabolites were separated on a Waters BEH C18 column (1.7 µm, 2.1 × 100 mm) with 0.5 mM ammonium acetate in water or methanol (0.4 ml/min) using the following gradient: 50% organic (MeOH) held for 1 minute, increased to 57% over 2.5 minutes followed by an increase to 98% over 1.5 minutes and held at 98% for 0.9 minute. Metabolites were quantified by back-calculation of a weighted (1/×), linear least-squares regression. The regression fit used the analyte/internal standard peak area ratios calculated from the calibration standards (ranging from 0.025 to 20 µM for 6β-OH-T and 0.0125 to 20 µM for 2α-OH-T and 2β-OH-T). The MS peaks were integrated using QuanLynx software (version 4.1; Waters). The limit of detection for the LC-MS/MS method was 0.0125 µM for the 6β-OH-T metabolite and 0.00625 µM for the 2α-OH-T and 2β-OH-T metabolites. For each testosterone concentration, mean metabolite formation rate values obtained from triplicate determinations for the three recombinant CYP3A enzymes were fit to the Michaelis-Menten (hyperbolic) or the Hill (sigmoidal) equation using GraphPad Prism software (version 4.03; GraphPad Software, LaJolla, CA).
Docking of Testosterone in the CYP3A4-Midazolam Crystal Structure and CYP3A7 Homology Model.
To identify the potential binding modes of testosterone to CYP3A4 and CYP3A7, an in silico molecular docking strategy was employed using the University of California, San Francisco (UCSF) DOCK 6.7 software suite, under academic license (UCSF; http://dock.compbio.ucsf.edu/DOCK_6/index.htm) (Huang et al., 2006; Fan et al., 2009; Allen et al., 2015). The 3D conformer of testosterone was downloaded directly from the PubChem web site (https://pubchem.ncbi.nlm.nih.gov/compound/testosterone#section=3D-Conformer), parameterized for partial charges, and used for all docking studies described below. In the case of CYP3A4, testosterone was docked into the active site of the ligand-free CYP3A4-midazolam (MDZ) crystal structure, as originally obtained by Sevrioukova and Poulos (2017) (Protein Data Bank ID code, 5TE8). For CYP3A7, a homology-model generated by Swiss-Model from the CYP3A4-MDZ crystal structure was used as a receptor template (available online at https://swissmodel.expasy.org/repository/uniprot//P24462) (Arnold et al., 2006; Benkert et al., 2011; Biasini et al., 2014). This particular crystal structure and homology model were chosen for docking templates due to the fact that this structure, unlike other CYP3A4 structures, captures a conformational rearrangement in the F–G loop region which is transmitted to the adjacent D, E, H, and I helices, ultimately resulting in a collapse of the active site cavity and positioning of the substrate in an orientation favorable for oxidation (Sevrioukova and Poulos, 2017). CYP3A7 shares 88% amino acid sequence identity with CYP3A4 (Shen et al., 2004), indicating that the overall structural fold should be similar between the two enzymes, which gave us a high degree of confidence in our docking results. Prior to conducting the docking simulations with CYP3A4, the MDZ ligand was removed from the active site. Extraneous information not required for the docking simulation, including the headers, connect records, and waters, were deleted from the Protein Data Bank text file. Additionally, all hydrogens were removed from the receptor using the SELECT function from the UCSF Chimera 1.11.2 software suite (Pettersen et al., 2004). The CYP3A4 receptor was then prepared for docking by using the UCSF Chimera 1.11.2 DOCK PREP function and assigning the heme prosthetic group to the A polypeptide chain. A net charge of +2 was assigned to the heme (oxyferrous) iron, and a net charge of −2 was assigned to the heme pyrrole nitrogen. Once preparation of the receptor had been completed, it was saved in the MOL2 format. Sphere generation was accomplished by inputting a dot molecular surface parameterization of the receptor surface into the SPHGEN module of the DOCK software suite. The dot molecular surface parameterization is accomplished by rolling a ball the size of a water molecule over the Van der Waals surface of the receptor. The surface normal vector at each surface point is computed and used to calculate the size of each sphere generated. After initial sphere generation, the total number of sphere clusters was pruned to only the clusters representing the active site of the protein using the DOCK SHOWSPHERE module. This resulted in the largest sphere cluster being contained within the active site. This sphere cluster was further pruned by eliminating all spheres that crossed the plane of the active site heme. Next, a molecular docking grid was generated by enclosing the spheres in a rectangular box and using the GRID utility to obtain files representing both the electrostatic and Van der Waals forces according to the following equation:
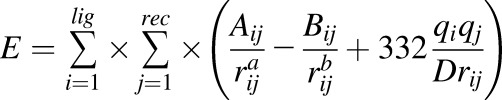 |
where each term is a double sum over ligand atoms i and receptor atoms j (Kuntz et al., 1982). The GRID utility then generates files representing the contact score and the energy score, which are used in the docking routine. The final size of the docking grid box was 16.7 × 25.9 × 16.7 Å. As a control to determine whether the initial docking parameters functioned appropriately, MDZ was redocked to the empty CYP3A4 receptor template (data not shown). The docking parameters were as follows: maximum number of orientations was set to between 50 and 1000, the Van der Waals energy component from between 20 and 25, and the maximum number of iterations to 500. For docking simulations conducted on the CYP3A7 receptor template with a single testosterone molecule already bound in the active site, the docking pose of the initial testosterone molecule was in the position most favorable for the 2α-OH-T attack. The VIEWDOCK utility of UCSF Chimera 1.11.2 was used for visualization of the docking poses and measuring distances and angles between atoms of interest.
Results
Testosterone Hydroxylation by Recombinant CYP3A4, CYP3A5, and CYP3A7.
The kinetic parameters for testosterone 6β-hydroxylation have been previously determined for the recombinant CYP3A4, CYP3A5, and CYP3A7 enzymes (Williams et al., 2002), but the kinetic characteristics for the 2β-hydroxylation and 2α-hydroxylation, other predominant CYP3A metabolic oxidations, have not been thoroughly investigated. In the present work, we used recombinant CYP3A Supersomes and an LC-MS/MS–based assay to determine the CYP3A-specific kinetic parameters for testosterone 6β-, 2β-, and 2α-hydroxylation. Figure 1 presents representative MRM chromatograms for the hydroxylated metabolites formed in testosterone incubations with the recombinant CYP3A4, CYP3A5, and CYP3A7 enzymes in the presence and absence of the NADPH-regenerating system. Under steady-state kinetics, the 6β-OH-T and 2β-OH-T metabolites were detected for all three CYP3A enzymes, whereas the 2α-OH-T metabolite was quantifiable only with the CYP3A7 enzyme and was not observed under steady-state conditions for CYP3A4 and CYP3A5. The formation of 2α-OH-T was observed only in CYP3A4 and CYP3A5 incubations when higher enzyme concentrations or significantly longer incubation times were used, leading to conditions outside of the parameters defined by steady-state kinetics (Segel, 1975). Minor amounts of other testosterone metabolites were also present but were not quantified for the purposes of this study. The metabolite formation data and the best-fit kinetic model for testosterone 6β- and 2β-hydroxylation by CYP3A4 and CYP3A5 are presented in Figs. 2 and 3, respectively. Figure 4 illustrates the metabolite formation data and the associated kinetic fit for testosterone 6β-, 2β-, and 2α-hydroxylation by CYP3A7. The R2 coefficient of determination for the 6β-OH-T and 2β-OH-T formation by CYP3A4 was 0.9839 and 0.9866, respectively, for the fit using the Hill equation kinetic model compared with 0.9838 and 0.9862 for the fit using the Michaelis-Menten equation kinetic model. The R2 coefficient of determination for the 6β- and 2β-OH-T formation by CYP3A5 was 0.9924 and 0.9905, respectively, for the fit using the Hill equation kinetic model compared with 0.9805 and 0.9666 for the fit using the Michaelis-Menten equation kinetic model. The R2 coefficient of determination values for 6β-OH-T, 2β-OH-T, and 2α-OH-T formation by CYP3A7 were 0.9925, 0.9890, and 0.9722, respectively, for the fit using the Hill equation kinetic model compared with 0.9802, 0.9855, and 0.9459 for the fit using the Michaelis-Menten equation kinetic model. In general, the data for 6β-OH-T and 2β-OH-T formation fit best to a Hill equation kinetic model, although a model based on the Michaelis-Menten equation also resulted in a similar fit based on the criteria used. CYP3A7 2α-OH-T formation fit best to the Hill equation with a Hill coefficient of 1.5. Table 1 summarizes the kinetic parameters determined for testosterone 6β-, 2β-, and 2α-hydroxylation with respect to the various CYP3A enzymes considered. The S50 and Vmax values determined for testosterone 6β-hydroxylation are largely in agreement with previous results obtained by Williams et al. (2002). Although the maximal reaction velocity is generally lower for CYP3A7 compared with CYP3A4 and CYP3A5, the Vmax values between testosterone 6β- and 2β-hydroxylation varied 18-fold, 4-fold, and 5-fold for CYP3A4, CYP3A5 and CYP3A7, respectively, and only 1-fold between 6β- and 2α-hydroxylation for CYP3A7. Furthermore, the affinity for testosterone appeared to be greater for the CYP3A7 enzyme, with S50 values of 11.8, 12.2, and 6.6 µM for 6β-, 2β-, and 2α-hydroxylation, respectively. The 2β-OH-T/6β-OH-T and 2α-OH-T/6β-OH-T metabolite ratios were calculated as a function of testosterone concentration (Fig. 5). Interestingly, the 2α-OH-T/6β-OH-T metabolite ratio calculated for CYP3A7 was ≥1 at low concentrations of testosterone; however, it decreased to <1 as the concentration of testosterone increased, while the 2β-OH-T/6β-OH-T metabolite ratio for the three CYP3A enzymes remained constant throughout the entire substrate concentration range tested.
Fig. 1.

Testosterone hydroxylation by recombinant CYP3A4, CYP3A5, and CYP3A7 enzymes. Representative MRM chromatograms for hydroxylated testosterone metabolites (MRM transitions: 305 > 269 atomic mass unit) formed in incubations of testosterone (50 µM) with CYP3A4 (A), CYP3A5 (B), and CYP3A7 (C) Supersomes for 10 minutes in the presence (continuous black trace) or absence (dotted gray trace) of the NADPH-generating system.&
Fig. 2.

Kinetics of testosterone hydroxylation by the recombinant CYP3A4 enzyme. Testosterone 6β-hydroxylation (A) and 2β-hydroxylation (B) by CYP3A4 Supersomes fitted using the Hill equation. Each data point represents the average of assays performed in triplicate, with error bars representing the S.E.
Fig. 3.

Kinetics of testosterone hydroxylation by the recombinant CYP3A5 enzyme. Testosterone 6β-hydroxylation (A) and 2β-hydroxylation (B) by CYP3A5 Supersomes fitted using the Hill equation. Each data point represents the average of assays performed in triplicate, with error bars representing the S.E.
Fig. 4.

Kinetics of testosterone hydroxylation by the recombinant CYP3A7 enzyme. Testosterone 6β-hydroxylation (A), 2β-hydroxylation (B), and 2α-hydroxylation (C) by CYP3A7 Supersomes fitted using the Hill equation. Each data point represents the average of assays performed in triplicate, with error bars representing the S.E.
TABLE 1.
Kinetic parameters of 6β-OH-T, 2β-OH-T, and 2α-OH-T metabolites for recombinant CYP3A4, CYP3A5, and CYP3A7 enzymes
| Testosterone Metabolites | CYP3A4 |
CYP3A5 |
CYP3A7 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Fit (n) | Vmax (nmol/min/nmol P450) | S50 (µM) | Fit (n) | Vmax (nmol/min/nmol P450) | S50 (µM) | Fit (n) | Vmax (nmol/min/nmol P450) | S50 (µM) | |
| 6β-OH-T | Hill (1.0) | 95.5 ± 8.0 | 26.1 ± 6.4 | Hill (1.4) | 17.4 ± 0.6 | 41.6 ± 4.1 | Hill (1.4) | 0.72 ± 0.03 | 11.8 ± 1.2 |
| 2β-OH-T | Hill (0.9) | 5.4 ± 0.4 | 25.8 ± 6.5 | Hill (1.6) | 4.5 ± 0.1 | 40.3 ± 3.5 | Hill (1.2) | 0.15 ± 0.01 | 12.2 ± 1.7 |
| 2α-OH-T | NA | ND | NA | NA | ND | NA | Hill (1.5) | 0.51 ± 0.03 | 6.6 ± 0.9 |
Hill, Hill equation; n, Hill coefficient; NA, not applicable; ND, not detected; P450, cytochrome P450.
Fig. 5.

Metabolite ratios for recombinant CYP3A4, CYP3A5, and CYP3A7 as a function of testosterone concentration. 2β-OH-T/6β-OH-T metabolite ratios were calculated as a function of testosterone concentration for the three CYP3A enzymes (solid line), and the 2α-OH-T/6β-OH-T metabolite ratio was calculated for CYP3A7 (dashed line).
Testosterone Hydroxylation in Human Adult and Fetal Liver Microsomes.
The testosterone metabolite formation rate was determined for two lots of human male adult HLMs and fHLMs at three discrete concentrations of testosterone (50, 100, and 250 µM) (Table 2). The fHLMs used in this work were previously characterized by mass spectrometry for their CYP3A content (Shuster et al., 2014). In fHLMs lots 5 and 18, no CYP3A4/5 protein was detected, and CYP3A7 protein concentrations were 79.7 ± 7.3 and 17.9 ± 1.3 pmol/mg protein, respectively. Representative MRM chromatograms of the hydroxylated testosterone metabolites formed in the incubations of the adult HLMs and fHLMs are shown in Fig. 6. The 6β-, 2β-, and 2α-testosterone hydroxylation products were detected in all of the incubations, with the exception of the 2α-OH-T product in HLM lot RGS at the 50 and 250 µM substrate concentrations. Additionally, due to variations in enzymatic activity, some metabolites were below the limit of quantification (BLQ), and their formation rate was not determined. Although there was substantial interindividual variation in the absolute metabolite formation rate between the two adult HLM lots examined, the 2α-OH-T/6β-OH-T metabolite ratio was near zero (lot DDV) or BLQ (lot RGS). Although the 6β- and 2β-hydroxylation activity of testosterone metabolism decreased considerably in the fHLMs compare with the adult HLMs, a similar formation rate was obtained for the 6β- and 2α-hydroxylation products in both lots of fHLMs tested. However, in contrast to the adult HLMs, the 2α-OH-T/6β-OH-T ratio was decreased at the higher concentration of testosterone for both lots of fHLMs, paralleling the trend that we observed with the recombinant enzymes (Fig. 5).
TABLE 2.
Testosterone 6β-, 2β-, and 2α-hydroxylation rate in human adult and fetal liver microsomes
The testosterone incubations were performed in duplicate and the data correspond to the formation rate mean.
| HLMs | [Substrate] (µM) | Testosterone Metabolite Formation (pmol/min/mg protein) |
2α-OH-T/6β-OH-T Ratio | ||
|---|---|---|---|---|---|
| 6β-OH-T | 2β-OH-T | 2α-OH-T | |||
| Adult | |||||
| Lot DDV | 50 | 1167.5 | 67.5 | 2.5 | 0.002 |
| 100 | 3377.6 | 284.9 | 8.6 | 0.003 | |
| 250 | 1805.7 | 134.3 | 3.7 | 0.002 | |
| Lot RGS | 50 | 55.4 | 3.1 | ND | NA |
| 100 | 182.7 | 14.3 | BLQ | NA | |
| 250 | 101.7 | 6.5 | ND | NA | |
| Fetal | |||||
| Lot 5 | 50 | 6.7 | BLQ | 7.0 | 1.054 |
| 100 | 14.9 | BLQ | 19.9 | 1.336 | |
| 250 | 12.9 | BLQ | 4.5 | 0.351 | |
| Lot 18 | 50 | 5.4 | BLQ | 5.4 | 0.993 |
| 100 | 11.6 | BLQ | 10.5 | 0.905 | |
| 250 | 8.5 | BLQ | 2.1 | 0.246 | |
NA, not applicable; ND, not detected.
Fig. 6.

Testosterone hydroxylation by adult HLMs and fHLMs. Representative MRM chromatograms for hydroxylated testosterone metabolites (MRM transitions: 305 > 269 atomic mass unit) formed in incubations of testosterone (100 µM) with HLMs (lot DDV; A) and fHLMs (lot 5; B) for 20 minutes in the presence (continuous black trace) or absence (dotted gray trace) of the NADPH-generating system.
Testosterone Docking into the CYP3A4 Crystal Structure and CYP3A7 Homology Model.
In an effort to obtain a better understanding of the structural elements of each individual enzyme involved in ligand binding, we performed an in silico docking study with testosterone and the respective enzyme receptors using the UCSF DOCK 6.7 software suite. The DOCK 6.7 algorithm allows for flexible ligand docking while searching for the lowest-energy binding mode between multiple potential poses in a rigid receptor binding pocket. Although this docking method likely does not capture all the conformational nuances that may be involved in testosterone interaction with the individual CYP3A enzymes, given the possibilities for induced fit and interaction with potential allosteric sites in the substrate-binding pocket, it does serve as a coarse grain means by which we can directly compare a ligand-receptor interaction between two structures given a similar starting conformation. Initially, to assess the robustness of our docking algorithm, we removed the MDZ ligand from the CYP3A4 active site, prepared the empty receptor, and redocked the ligand. The MDZ ligand was observed to dock in an almost identical position (<0.01 Å root mean square deviation) and orientation as that found in the CYP3A4-MDZ crystal structure (data not shown). Once the validity of the docking procedure was established, we proceeded to dock the testosterone ligand in the empty CYP3A4 receptor active site (Fig. 7, A–C). The most energetically favorable binding pose found for docking testosterone into the CYP3A4 structure positioned the testosterone C6 atom approximately 5.97 Å away from the heme iron, with the Fe-6βH-C6 angle equal to 115° (optimal angle, 180° ± 45) (Szklarz and Halpert, 1997) (Fig. 7A). A hydrogen bond was found to be present between the carbonyl of C3 and the side chain hydrogen of S119. Additionally, E374 was in close proximity to the substrate hydroxyl group of C17, which may provide an opportunity for hydrogen bonding to occur between these species within the dynamic active site environment. Since the testosterone substrate was oriented “methyl-side-down” (with the β-side of the molecule facing toward the plane of the heme), this allowed for favorable hydrophobic interactions to occur between the previously identified CYP3A4 “phenylalanine cluster” (F215, F220, F304) (Harlow and Halpert, 1997, 1998; Domanski et al., 1998, 2000; Stevens et al., 1999; Khan et al., 2002; Fishelovitch et al., 2009) and the substrate. A somewhat less energetically favorable binding pose for testosterone binding to CYP3A4 was also observed, orienting testosterone for a 2β attack by the activated oxygen species (Fig. 7B). Although the distance between the C2 atom and the heme iron is slightly closer, 4.81 Å versus 5.97 Å, the hydrogen bond between the carbonyl oxygen and S119 is no longer present due to the increased distance between these two moieties. In this binding orientation, the Fe-2βH-C2 angle was determined to be 145°. Both the 6β and the 2β docking poses were obtained with 50 maximum binding orientations. However, reducing the constraints to allow for 150 maximum binding orientations produced three energetically equivalent docking poses, two of which oriented the hydroxyl group of the C17 carbon toward the heme iron (data not shown). The third orientation positioned the C2 atom for α attack by the activated oxygen species with a distance of 4.69 Å from the heme iron and a Fe-2αH-C2 angle of 148° (Fig. 7C). This pose also indicated the presence of a hydrogen bond from the side chain of E374 to the hydroxyl oxygen of C17.
Fig. 7.

In silico docking results for testosterone ligand docking to CYP3A4 (A–C) and CYP3A7 (D–F). (A and D) 6β-OH-T binding orientation. (B and E) 2β-OH-T binding orientation. (C and F) 2α-OH-T binding orientation. Testosterone ligand is colored in purple, and heme in orange.
When docking testosterone to the CYP3A7 homology model, the most energetically favorable docking pose oriented the substrate in position for α attack on the C2 carbon atom, with a distance of 4.84 Å to the heme iron and 88° for the Fe-2αH-C2 bond angle (Fig. 7F). Remarkably, the 2α docking pose obtained for CYP3A7 positioned testosterone in the active site in an inverted position compared with that seen previously (i.e., “methyl-side-up”) (the α-side of the molecule facing the plane of the heme), in direct contrast to what was observed with CYP3A4 (Fig. 7, A–C). Although the distance between the hydroxyl side chain of S119 and the C3 carbonyl of the testosterone ligand was too great to form a stable hydrogen bond, a hydrogen bond was present between E374 and the hydroxyl group of testosterone C17 (Fig. 7F), with a distance of 2.30 Å, as observed with the 2α attack pose obtained for CYP3A4. Two other, lower energy conformations were obtained by increasing the number of maximum binding orientations by a factor of 2-fold. The slightly more energetically favorable of the two placed the substrate in position for β attack on the C6 carbon atom of testosterone (Fig. 7D), which was also observed with CYP3A4. However, in contrast with CYP3A4, the C6 atom is positioned slightly farther away from the heme iron, at a distance of 6.55 Å compared with 5.97 Å for CYP3A4. The Fe-6βH-C6 bond angle is also decreased to 109° with CYP3A7 versus 115° for CYP3A4. As found in the case of CYP3A4, a hydrogen bond is again present between the S119 side chain and the carbonyl oxygen of testosterone C3; however, at a slightly longer distance (2.77 Å) compared with the 6β binding pose for CYP3A4 (2.59 Å). The third, and least energetically favorable, binding pose obtained for CYP3A7 oriented the testosterone substrate in position for β-hydroxylation at the C2 atom (Fig. 7E). In this scenario, the distance from the carbon to the heme iron was determined to be 4.88 Å (slightly farther away than in the CYP3A4 pose), and the Fe-2βH-C2 bond angle was 124° (smaller than the 145° obtained for CYP3A4).
Finally, in an effort to understand how the most energetically favorable CYP3A7 docking pose might affect the binding of a second ligand in the case of multiple ligand binding, a docking study was conducted with the singly occupied species of CYP3A7 where a single testosterone ligand was bound to the enzyme in the most energetically favorable orientation, the 2α pose in the case of CYP3A7 (Supplemental Fig. 1). The ligand was observed to occupy a position above the 2α pose, in a largely perpendicular orientation in relationship to the 2α ligand. It was found to be in close proximity (<5 Å) to E374 and the aforementioned phenylalanine cluster, located in the F/G loop region.
Discussion
The 6β-carbon hydroxylation of testosterone is a well-known and characteristic reaction performed by the CYP3A family (Waxman et al., 1988). In addition to 6β-OH-T, it was previously reported that CYP3A4 can produce several other testosterone metabolites, albeit in lower abundance than the 6β metabolite (Waxman et al., 1988; Krauser et al., 2004). Although testosterone hydroxylation by CYP3A4 (Lu et al., 2001; Williams et al., 2002; He et al., 2003; Krauser et al., 2004) and CYP3A5 (Williams et al., 2002; Huang et al., 2004) has been extensively studied, less is known about testosterone hydroxylation by CYP3A7, the CYP3A enzyme predominantly expressed in fetuses and neonates (Stevens et al., 2003; Stevens, 2006).
To address this knowledge gap, and to further assess the possibility of using the 2α-OH-T/6β-OH-T ratio as an endogenous biomarker of CYP3A7, we undertook a complete kinetic characterization of testosterone metabolism by CYP3A7. Additionally, we sought to confirm our results in fHLMs whose CYP3A7 concentration had been accurately determined from LC-MS/MS proteomic studies (Shuster et al., 2014).
Our results largely confirmed those obtained previously by others (Williams et al., 2002; Leeder et al., 2005), with 6β-OH-T and 2α-OH-T being the two major metabolites formed by both recombinant CYP3A7 and fHLMs, whereas CYP3A4/5 and adult HLMs predominately formed the 6β-OH-T and 2β-OH-T metabolites (Figs. 1–4; Tables 1–2). Although there is no known physiologic role for 2α-OH-T in fetal development, it is striking that this is a predominant metabolite and may hint at some yet undiscovered function for this compound. Additionally, although the 2α-OH-T/6β-OH-T metabolite ratio observed for recombinant CYP3A7 (Fig. 5) at 10 µM testosterone was similar to that reported by Leeder et al. (2005) (∼1), this ratio only held true in CYP3A7 incubations for concentrations of testosterone of 12.5 µM or less. Indeed, we observed a dramatic decrease in the ratio of the 2α-OH-T/6β-OH-T metabolites at higher substrate concentrations, eventually reaching a ratio of 0.5 with the recombinant CYP3A7 enzyme at a concentration of 250 µM testosterone (Fig. 5), and between 0.25 (lot 18) and 0.35 (lot 5) for the fHLMs tested. Given that the 2α-OH-T/6β-OH-T metabolite ratio decreases with both the recombinant CYP3A7 enzyme and the fHLMs, it indicates that the substrate-dependent modulation of metabolite ratios is a characteristic that is inherent to the enzyme itself and not a product of its particular environment. Although there are differences observed in the absolute magnitude of the metabolite ratio, which may be due to the local environment of the enzyme, the trend remains the same with both the recombinant enzyme and the fHLMs. It should be noted that a change in the regiospecificity of oxidation with substrate concentration has been previously observed for CYP3A4/5 with the drug MDZ (Williams et al., 2002) as well as with CYP3A7 for estrone metabolism (Lee et al., 2003). However, to our knowledge, this is the first report of it occurring with testosterone and CYP3A7.
Two major conclusions can be drawn from this: 1) it is unlikely that the 2α-OH-T/6β-OH-T metabolite ratio will be useful as an endogenous biomarker of CYP3A7 activity, as it varies dramatically with substrate concentration; and 2) the shift in the regiospecificity of oxidation implies that a reorientation of the substrate in the CYP3A7 active site occurs with increasing substrate concentration. The Hill coefficient of 1.5 obtained from the fit of the kinetic parameters for the 2α-OH-T oxidation by CYP3A7 (Table 1) suggests a significant amount of cooperativity and may imply that more than one testosterone molecule is bound to the enzyme simultaneously.
It has been known for some time that the CYP3A enzyme family can exhibit atypical, sometimes referred to as “allosteric,” kinetics resulting in an activation of metabolism for certain substrates (Ueng et al., 1997; Atkins et al., 2001; Tracy, 2003; Davydov and Halpert, 2008; Lampe et al., 2008). Testosterone has previously been reported to be both a heterotropic (Kenworthy et al., 2001) and a homotropic (Harlow and Halpert, 1998) allosteric effector for CYP3A4, CYP3A5 (Patki et al., 2003), and CYP3A7 (Williams et al., 2002; Nakamura et al., 2003). One possible explanation for the shift in the regiospecificity of testosterone oxidation in CYP3A7 is that higher substrate concentrations increase the likelihood for multiple ligand binding to occur, and the multiple ligand–bound enzyme alters the orientation of testosterone within the active site, which leads to a shift in regiospecificity.
To explore this possibility further, we performed in silico docking studies with testosterone to CYP3A4 and a CYP3A7 homology model. The most energetically favorable docking pose for CYP3A4 positioned testosterone for 6β-hydroxylation (Fig. 7A). This docking pose fits well with, and is predicted by, the kinetic data we obtained. The second most energetically favorable conformation oriented testosterone for 2β oxidation (Fig. 7B), which is also consistent with our kinetic data (Table 1). In both instances, the testosterone molecule was positioned with methyl groups pointing toward the heme (i.e., β-face “down”), decreasing the potential for steric clashes to occur between the testosterone methyl groups and phenylalanine residues located in the F/G loop. Similarly, the hydrogen bond formed between the ligand and the S119 residue in the 6β binding pose likely stabilizes the substrate in the correct orientation for oxidation at C6. S119 has proven critical for oxidative activity against a number of CYP3A4 substrates, including diazepam (He et al., 2003), carbamazepine (Müller et al., 2015), and testosterone (Roussel et al., 2000). Interestingly, the S119-C3 carbonyl hydrogen bond does not occur in the 2β binding pose due to the increased distance between the testosterone carbonyl and the hydroxyl side chain of S119, despite 2β-OH-T being a major CYP3A4 metabolite. This is consistent with previous site-directed mutagenesis studies of this residue, which indicate a shift in the 2β-OH-T/6β-OH-T metabolite ratio when the serine is mutated to an alanine, valine, or leucine (Supplemental Table 1) (Fowler et al., 2000; Roussel et al., 2000).
The least energetically favorable conformation observed with CYP3A4 positioned testosterone in the active site such that the 2α-carbon could potentially be oxidized (Fig. 7C). Although no hydrogen bond to S119 is present, the substrate is in a favorable position for a hydrogen bond to form between residue E374 of the protein and the C17 hydroxyl of testosterone. This hydrogen bond may serve to pull the substrate out of optimal alignment with the heme iron, thus resulting in a decreased opportunity for 2α-hydroxylation. These structural constraints may provide an explanation as to why the 2α-OH-T metabolic product is not generated at appreciable amounts by CYP3A4/5.
In contrast, the most energetically favorable docking pose obtained with the CYP3A7 structure placed testosterone in an ideal position for attack on C-2α (Fig. 7F). This required that the substrate was orientated in a β-side “up” position, with the methyl groups pointed away from the plane of the heme. In this position, the 2α-carbon is in both close proximity and a proper angle for oxidation by the activated oxygen of the heme iron. This may be a more facile orientation for CYP3A7 to achieve given that two of the phenylalanines (F215 and F220) from the F/G loop phenylalanine cluster are absent in CYP3A7. Formation of the hydrogen bond between the substrate and E374 may be particularly important for stabilizing the substrate in the active site with the α-side of the molecule facing the heme iron.
As observed with CYP3A4 and CYP3A5 above, CYP3A7 also forms significant amounts of the 6 β-OH-T metabolite, and this was reflected in the second most energetically favorable pose retrieved from our docking study (Fig. 7D). The S119-C3 carbonyl hydrogen bond is again present in this docking pose, whereas the E374 bond is absent, which may imply that the S119 hydrogen bond is necessary for the 6β oxidation event to occur. Indeed, there may exist a dynamic “push-pull” between the substrate and residues S119 and E374, the end result of which may determine the likelihood of oxidation of the substrate at either the C6 or the C2 position, respectively.
The least energetically favorable pose obtained from our CYP3A7 docking study positioned the substrate in the active site such that 2β-carbon oxidation might occur (Fig. 7E). It should be noted that this pose was only observed after substantially relaxing the docking constraints (1000 maximum possible orientations). Hence, it is significantly less likely to occur than the 2α or 6β orientations.
When a second testosterone molecule was docked into the CYP3A7 homology model, it was easily accommodated by the large open region of the active site reaching toward the surface (the substrate access channel) (Supplemental Fig. 1). This arrangement forces the second testosterone into a position approximately perpendicular to the first 2α molecule. The second testosterone is sandwiched between the F/G loop and the B/C loop, both of which are regions known to be involved in substrate recognition and cooperativity. Moreover, the docking results demonstrate that a second testosterone can be accommodated in the CYP3A7 active site, which may give a structural explanation for the cooperative effects observed with substrate oxidation.
In summary, here we have demonstrated that the metabolite profile of testosterone oxidation by CYP3A7 is substantially distinct from that of CYP3A4/5 and that the type and concentration of metabolites produced varies with substrate concentration, which likely is due to homotropic cooperativity. Therefore, it is unlikely that the 2α-OH-T/6β-OH-T ratio would serve as an effective endogenous biomarker of CYP3A7. Furthermore, the unique features of the CYP3A7 active site reveal structural elements that may give rise to particular steroid metabolites and hint at the unique role of the enzyme in fetal and neonatal growth and development. This current endeavor, and subsequent studies, will continue to give us more insight into why the CYP3A family members metabolize steroids with vastly different rates, affinities, stereospecificities, and regiospecificities.
Acknowledgments
S.E.K. and J.N.L. thank Dr. Robert Winefield and the COBRE-supported Analytical Core Facility in the Department of Pharmacology, Toxicology and Therapeutics at the University of Kansas Medical Center for their support with this project.
Abbreviations
- 2α-OH-T
2α-hydroxytestosterone
- 2β-OH-T
2β-hydroxytestosterone
- 6β-OH-T
6β-hydroxytestosterone
- BLQ
below the limit of quantitation
- CE
collision energy
- DHEA
dehydroepiandrosterone
- DHEA-S
dehydroepiandrosterone sulfate
- fHLM
fetal human liver microsome
- HLM
human liver microsomes
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- MDZ
midazolam
- MRM
multiple reaction monitoring
- S50
substrate concentration occupying half of the binding sites
- UCSF
University of California, San Francisco
Authorship Contributions
Participated in research design: Kandel and Lampe.
Conducted experiments: Kandel.
Contributed new reagents or analytical tools: Han and Mao.
Performed data analysis: Kandel and Lampe.
Wrote or contributed to the writing of the manuscript: Kandel, Han, Mao, and Lampe.
Footnotes
This work was supported by the National Institutes of Health (NIH), Institute of General Medical Sciences [Grant P20-GM103549-07], National Center for Research Resources [Grant P20-RR021940] and Kansas IDeA Network of Biomedical Research Excellence [Grant QH846868-K-INBRE] to J.N.L. This study was additionally supported in part by an NIH Clinical and Translational Science Award Grant [UL1 TR000001, formerly UL1RR033179] awarded to the University of Kansas Medical Center, and an internal Lied Basic Science Grant Program award of the University of Kansas Medical Center Research Institute to J.N.L. The mass spectrometric data presented in this work was collected on an instrument belonging to the Analytical Core Laboratory (University of Kansas Medical Center), which was supported in part by grants from the NIH, Institute of General Medical Sciences [Grants P20-GM103549 and P30-GM118247].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC. (2015) DOCK 6: impact of new features and current docking performance. J Comput Chem 36:1132–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. (2006) The swiss-model workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. [DOI] [PubMed] [Google Scholar]
- Atkins WM, Wang RW, Lu AY. (2001) Allosteric behavior in cytochrome p450-dependent in vitro drug-drug interactions: a prospective based on conformational dynamics. Chem Res Toxicol 14:338–347. [DOI] [PubMed] [Google Scholar]
- Benkert P, Biasini M, Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, et al. (2014) Swiss-model: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodin K, Andersson U, Rystedt E, Ellis E, Norlin M, Pikuleva I, Eggertsen G, Björkhem I, Diczfalusy U. (2002) Metabolism of 4 beta -hydroxycholesterol in humans. J Biol Chem 277:31534–31540. [DOI] [PubMed] [Google Scholar]
- Dai Y, Iwanaga K, Lin YS, Hebert MF, Davis CL, Huang W, Kharasch ED, Thummel KE. (2004) In vitro metabolism of cyclosporine A by human kidney CYP3A5. Biochem Pharmacol 68:1889–1902. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Halpert JR. (2008) Allosteric P450 mechanisms: multiple binding sites, multiple conformers or both? Expert Opin Drug Metab Toxicol 4:1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanski TL, He YA, Harlow GR, Halpert JR. (2000) Dual role of human cytochrome P450 3A4 residue Phe-304 in substrate specificity and cooperativity. J Pharmacol Exp Ther 293:585–591. [PubMed] [Google Scholar]
- Domanski TL, Liu J, Harlow GR, Halpert JR. (1998) Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: role in steroid hydroxylase activity and alpha-naphthoflavone stimulation. Arch Biochem Biophys 350:223–232. [DOI] [PubMed] [Google Scholar]
- Ekroos M, Sjögren T. (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci USA 103:13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fæste CK, Ivanova L, Uhlig S. (2011) In vitro metabolism of the mycotoxin enniatin B in different species and cytochrome p450 enzyme phenotyping by chemical inhibitors. Drug Metab Dispos 39:1768–1776. [DOI] [PubMed] [Google Scholar]
- Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A. (2009) Molecular docking screens using comparative models of proteins. J Chem Inf Model 49:2512–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishelovitch D, Shaik S, Wolfson HJ, Nussinov R. (2009) Theoretical characterization of substrate access/exit channels in the human cytochrome P450 3A4 enzyme: involvement of phenylalanine residues in the gating mechanism. J Phys Chem B 113:13018–13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler SM, Riley RJ, Pritchard MP, Sutcliffe MJ, Friedberg T, Wolf CR. (2000) Amino acid 305 determines catalytic center accessibility in CYP3A4. Biochemistry 39:4406–4414. [DOI] [PubMed] [Google Scholar]
- Gorski JC, Hall SD, Jones DR, VandenBranden M, Wrighton SA. (1994) Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem Pharmacol 47:1643–1653. [DOI] [PubMed] [Google Scholar]
- Harlow GR, Halpert JR. (1997) Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P450 3A4. Role of residues 210 and 211 in flavonoid activation and substrate specificity. J Biol Chem 272:5396–5402. [DOI] [PubMed] [Google Scholar]
- Harlow GR, Halpert JR. (1998) Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc Natl Acad Sci USA 95:6636–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YA, Roussel F, Halpert JR. (2003) Analysis of homotropic and heterotropic cooperativity of diazepam oxidation by CYP3A4 using site-directed mutagenesis and kinetic modeling. Arch Biochem Biophys 409:92–101. [DOI] [PubMed] [Google Scholar]
- Honda A, Miyazaki T, Ikegami T, Iwamoto J, Maeda T, Hirayama T, Saito Y, Teramoto T, Matsuzaki Y. (2011) Cholesterol 25-hydroxylation activity of CYP3A. J Lipid Res 52:1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Shoichet BK, Irwin JJ. (2006) Benchmarking sets for molecular docking. J Med Chem 49:6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Lin YS, McConn DJ, II, Calamia JC, Totah RA, Isoherranen N, Glodowski M, Thummel KE. (2004) Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab Dispos 32:1434–1445. [DOI] [PubMed] [Google Scholar]
- Isin EM, Guengerich FP. (2006) Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4. J Biol Chem 281:9127–9136. [DOI] [PubMed] [Google Scholar]
- Kenworthy KE, Clarke SE, Andrews J, Houston JB. (2001) Multisite kinetic models for CYP3A4: simultaneous activation and inhibition of diazepam and testosterone metabolism. Drug Metab Dispos 29:1644–1651. [PubMed] [Google Scholar]
- Khan KK, He YQ, Domanski TL, Halpert JR. (2002) Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: an evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Mol Pharmacol 61:495–506. [DOI] [PubMed] [Google Scholar]
- Kitada M, Kamataki T, Itahashi K, Rikihisa T, Kanakubo Y. (1987a) P-450 HFLa, a form of cytochrome P-450 purified from human fetal livers, is the 16 alpha-hydroxylase of dehydroepiandrosterone 3-sulfate. J Biol Chem 262:13534–13537. [PubMed] [Google Scholar]
- Kitada M, Kamataki T, Itahashi K, Rikihisa T, Kanakubo Y. (1987b) Significance of cytochrome P-450 (P-450 HFLa) of human fetal livers in the steroid and drug oxidations. Biochem Pharmacol 36:453–456. [DOI] [PubMed] [Google Scholar]
- Kittler K, Fessard V, Maul R, Hurtaud-Pessel D. (2014) CYP3A4 activity reduces the cytotoxic effects of okadaic acid in HepaRG cells. Arch Toxicol 88:1519–1526. [DOI] [PubMed] [Google Scholar]
- Krauser JA, Guengerich FP. (2005) Cytochrome P450 3A4-catalyzed testosterone 6beta-hydroxylation stereochemistry, kinetic deuterium isotope effects, and rate-limiting steps. J Biol Chem 280:19496–19506. [DOI] [PubMed] [Google Scholar]
- Krauser JA, Voehler M, Tseng LH, Schefer AB, Godejohann M, Guengerich FP. (2004) Testosterone 1 beta-hydroxylation by human cytochrome P450 3A4. Eur J Biochem 271:3962–3969. [DOI] [PubMed] [Google Scholar]
- Kumar S, Liu H, Halpert JR. (2006) Engineering of cytochrome P450 3A4 for enhanced peroxide-mediated substrate oxidation using directed evolution and site-directed mutagenesis. Drug Metab Dispos 34:1958–1965. [DOI] [PubMed] [Google Scholar]
- Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. (1982) A geometric approach to macromolecule-ligand interactions. J Mol Biol 161:269–288. [DOI] [PubMed] [Google Scholar]
- Lampe JN, Fernandez C, Nath A, Atkins WM. (2008) Nile red is a fluorescent allosteric substrate of cytochrome P450 3A4. Biochemistry 47:509–516. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Usmani KA, Chanas B, Ghanayem B, Xi T, Hodgson E, Mohrenweiser HW, Goldstein JA. (2003) Genetic findings and functional studies of human CYP3A5 single nucleotide polymorphisms in different ethnic groups. Pharmacogenetics 13:461–472. [DOI] [PubMed] [Google Scholar]
- Leeder JS, Gaedigk R, Marcucci KA, Gaedigk A, Vyhlidal CA, Schindel BP, Pearce RE. (2005) Variability of CYP3A7 expression in human fetal liver. J Pharmacol Exp Ther 314:626–635. [DOI] [PubMed] [Google Scholar]
- Lu P, Lin Y, Rodrigues AD, Rushmore TH, Baillie TA, Shou M. (2001) Testosterone, 7-benzyloxyquinoline, and 7-benzyloxy-4-trifluoromethyl-coumarin bind to different domains within the active site of cytochrome P450 3A4. Drug Metab Dispos 29:1473–1479. [PubMed] [Google Scholar]
- Mäenpää J, Pelkonen O, Cresteil T, Rane A. (1993) The role of cytochrome P450 3A (CYP3A) isoform(s) in oxidative metabolism of testosterone and benzphetamine in human adult and fetal liver. J Steroid Biochem Mol Biol 44:61–67. [DOI] [PubMed] [Google Scholar]
- Michaels S, Wang MZ. (2014) The revised human liver cytochrome P450 “Pie”: absolute protein quantification of CYP4F and CYP3A enzymes using targeted quantitative proteomics. Drug Metab Dispos 42:1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KK, Cai J, Ripp SL, Pierce WM, Jr, Rushmore TH, Prough RA. (2004) Stereo- and regioselectivity account for the diversity of dehydroepiandrosterone (DHEA) metabolites produced by liver microsomal cytochromes P450. Drug Metab Dispos 32:305–313. [DOI] [PubMed] [Google Scholar]
- Müller CS, Knehans T, Davydov DR, Bounds PL, von Mandach U, Halpert JR, Caflisch A, Koppenol WH. (2015) Concurrent cooperativity and substrate inhibition in the epoxidation of carbamazepine by cytochrome P450 3A4 active site mutants inspired by molecular dynamics simulations. Biochemistry 54:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Torimoto N, Ishii I, Ariyoshi N, Nakasa H, Ohmori S, Kitada M. (2003) CYP3A4 and CYP3A7-mediated carbamazepine 10,11-epoxidation are activated by differential endogenous steroids. Drug Metab Dispos 31:432–438. [DOI] [PubMed] [Google Scholar]
- Ohmori S, Nakasa H, Asanome K, Kurose Y, Ishii I, Hosokawa M, Kitada M. (1998) Differential catalytic properties in metabolism of endogenous and exogenous substrates among CYP3A enzymes expressed in COS-7 cells. Biochim Biophys Acta 1380:297–304. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR. (2015) Cytochrome P450: Structure, Mechanism, and Biochemistry. Springer International Publishing, New York. [Google Scholar]
- Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. (2006) The human intestinal cytochrome P450 “pie”. Drug Metab Dispos 34:880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patki KC, Von Moltke LL, Greenblatt DJ. (2003) In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: role of cyp3a4 and cyp3a5. Drug Metab Dispos 31:938–944. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. (2004) UCSF chimera--a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- Rendic S, Guengerich FP. (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem Res Toxicol 28:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel F, Khan KK, Halpert JR. (2000) The importance of SRS-1 residues in catalytic specificity of human cytochrome P450 3A4. Arch Biochem Biophys 374:269–278. [DOI] [PubMed] [Google Scholar]
- Schuetz JD, Kauma S, Guzelian PS. (1993) Identification of the fetal liver cytochrome CYP3A7 in human endometrium and placenta. J Clin Invest 92:1018–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel IH. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, John Wiley & Sons, Inc., New York. [Google Scholar]
- Sevrioukova IF, Poulos TL. (2017) Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc Natl Acad Sci USA 114:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Fitzloff JF, Cook CS. (2004) Differential enantioselectivity and product-dependent activation and inhibition in metabolism of verapamil by human CYP3As. Drug Metab Dispos 32:186–196. [DOI] [PubMed] [Google Scholar]
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423. [PubMed] [Google Scholar]
- Shuster DL, Risler LJ, Prasad B, Calamia JC, Voellinger JL, Kelly EJ, Unadkat JD, Hebert MF, Shen DD, Thummel KE, et al. (2014) Identification of CYP3A7 for glyburide metabolism in human fetal livers. Biochem Pharmacol 92:690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim SC, Edwards RJ, Boobis AR, Ingelman-Sundberg M. (2005) CYP3A7 protein expression is high in a fraction of adult human livers and partially associated with the CYP3A7*1C allele. Pharmacogenet Genomics 15:625–631. [DOI] [PubMed] [Google Scholar]
- Sohl CD, Cheng Q, Guengerich FP. (2009) Chromatographic assays of drug oxidation by human cytochrome P450 3A4. Nat Protoc 4:1252–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JC. (2006) New perspectives on the impact of cytochrome P450 3A expression for pediatric pharmacology. Drug Discov Today 11:440–445. [DOI] [PubMed] [Google Scholar]
- Stevens JC, Domanski TL, Harlow GR, White RB, Orton E, Halpert JR. (1999) Use of the steroid derivative RPR 106541 in combination with site-directed mutagenesis for enhanced cytochrome P-450 3A4 structure/function analysis. J Pharmacol Exp Ther 290:594–602. [PubMed] [Google Scholar]
- Stevens JC, Hines RN, Gu C, Koukouritaki SB, Manro JR, Tandler PJ, Zaya MJ. (2003) Developmental expression of the major human hepatic CYP3A enzymes. J Pharmacol Exp Ther 307:573–582. [DOI] [PubMed] [Google Scholar]
- Strougo A, Yassen A, Monnereau C, Danhof M, Freijer J. (2014) Predicting the “first dose in children” of CYP3A-metabolized drugs: evaluation of scaling approaches and insights into the CYP3A7-CYP3A4 switch at young ages. J Clin Pharmacol 54:1006–1015. [DOI] [PubMed] [Google Scholar]
- Szklarz GD, Halpert JR. (1997) Molecular modeling of cytochrome P450 3A4. J Comput Aided Mol Des 11:265–272. [DOI] [PubMed] [Google Scholar]
- Torimoto N, Ishii I, Toyama K, Hata M, Tanaka K, Shimomura H, Nakamura H, Ariyoshi N, Ohmori S, Kitada M. (2007) Helices F-G are important for the substrate specificities of CYP3A7. Drug Metab Dispos 35:484–492. [DOI] [PubMed] [Google Scholar]
- Tracy TS. (2003) Atypical enzyme kinetics: their effect on in vitro-in vivo pharmacokinetic predictions and drug interactions. Curr Drug Metab 4:341–346. [DOI] [PubMed] [Google Scholar]
- Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. (1997) Cooperativity in oxidations catalyzed by cytochrome P450 3A4. Biochemistry 36:370–381. [DOI] [PubMed] [Google Scholar]
- Wang H, Dick R, Yin H, Licad-Coles E, Kroetz DL, Szklarz G, Harlow G, Halpert JR, Correia MA. (1998) Structure-function relationships of human liver cytochromes P450 3A: aflatoxin B1 metabolism as a probe. Biochemistry 37:12536–12545. [DOI] [PubMed] [Google Scholar]
- Waxman DJ, Attisano C, Guengerich FP, Lapenson DP. (1988) Human liver microsomal steroid metabolism: identification of the major microsomal steroid hormone 6 beta-hydroxylase cytochrome P-450 enzyme. Arch Biochem Biophys 263:424–436. [DOI] [PubMed] [Google Scholar]
- Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K, Hamman MA, Hall SD, Wrighton SA. (2002) Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos 30:883–891. [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305:683–686. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Shaw PM, Guengerich FP, Shimada T. (1998) Roles of cytochromes P450 1A2 and 3A4 in the oxidation of estradiol and estrone in human liver microsomes. Chem Res Toxicol 11:659–665. [DOI] [PubMed] [Google Scholar]
- Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. (2004) The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem 279:38091–38094. [DOI] [PubMed] [Google Scholar]
- Zanger UM, Schwab M. (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138:103–141. [DOI] [PubMed] [Google Scholar]
- Zhao C, Gao Q, Roberts AG, Shaffer SA, Doneanu CE, Xue S, Goodlett DR, Nelson SD, Atkins WM. (2012) Cross-linking mass spectrometry and mutagenesis confirm the functional importance of surface interactions between CYP3A4 and holo/apo cytochrome b(5). Biochemistry 51:9488–9500. [DOI] [PMC free article] [PubMed] [Google Scholar]