

Abstract
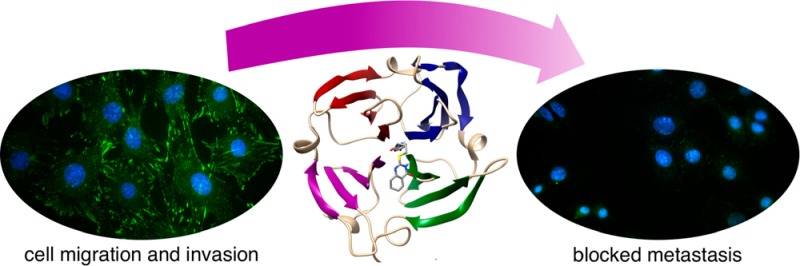
A lack of target specificity has greatly hindered the success of inhibitor development against matrix metalloproteinases (MMPs) for the treatment of various cancers. The MMP catalytic domains are highly conserved, whereas the hemopexin-like domains of MMPs are unique to each family member. The hemopexin-like domain of MMP-9 enhances cancer cell migration through self-interaction and heterointeractions with cell surface proteins including CD44 and α4β1 integrin. These interactions activate EGFR-MAP kinase dependent signaling that leads to cell migration. In this work, we generated a library of compounds, based on hit molecule N-[4-(difluoromethoxy)phenyl]-2-[(4-oxo-6-propyl-1H-pyrimidin-2-yl)sulfanyl]-acetamide, that target the hemopexin-like domain of MMP-9. We identify N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide, 3c, as a potent lead (Kd = 320 nM) that is specific for binding to the proMMP-9 hemopexin-like domain. We demonstrate that 3c disruption of MMP-9 homodimerization prevents association of proMMP-9 with both α4β1 integrin and CD44 and results in the dissociation of EGFR. This disruption results in decreased phosphorylation of Src and its downstream target proteins focal adhesion kinase (FAK) and paxillin (PAX), which are implicated in promoting tumor cell growth, migration, and invasion. Using a chicken chorioallantoic membrane in vivo assay, we demonstrate that 500 nM 3c blocks cancer cell invasion of the basement membrane and reduces angiogenesis. In conclusion, we present a mechanism of action for 3c whereby targeting the hemopexin domain results in decreased cancer cell migration through simultaneous disruption of α4β1 integrin and EGFR signaling pathways, thereby preventing signaling bypass. Targeting through the hemopexin-like domain is a powerful approach to antimetastatic drug development.
Metastasis accounts for 90% of all human cancer related deaths, yet we lack adequate drugs to target this biological process often associated with aggressive cancers.1 Accumulating evidence suggests an important role for matrix metalloproteinases (MMPs) in promoting cancer progression whereby they modify their surrounding environment to promote the growth and spread of tumor cells.2−5 Although MMPs represent the most prominent family of proteinases associated with tumorigenesis, drugs designed to inhibit their proteolytic activities largely failed in clinical trials due to issues with selectivity for individual MMPs.6 The highly conserved catalytic domain within this family of zymogens required a paradigm shift to the development of novel MMP inhibitors (MMPIs) targeting less conserved, noncatalytic functional domain(s) of the proteases to increase target specificity and selectivity.7
The biological functions of MMPs extend beyond just their proteolytic function and include induction of complex signaling cascades.8−10 Homodimerization of secreted proMMP-9 hemopexin domains is sufficient to induce cancer cell migration independent of proteolytic activity.11 Using an shRNA approach, the mechanism behind this phenotype was dissected and found to be dependent on an interaction between MMP-9 and CD44 at the cell surface. This interaction required an intact MMP-9 hemopexin domain (PEX-9). Interaction between CD44 and PEX-9 results in increased epidermal growth factor receptor (EGFR) phosphorylation and subsequent activation of mitogen-activated protein kinase (MAPK)/extracellular signal–regulated kinases (ERK) signaling, thereby enhancing the migratory capacity of proMMP-9 expressing cancer cells.12 Signaling was lost upon deletion or swapping of PEX-9 with the PEX domain of its closest homologue MMP-2.
The X-ray crystal structure of the PEX domain of MMP-9 has been solved. PEX-9 forms a propeller-like structure composed of four blades, which converge and are linked between blades I and IV through disulfide bonds.13 Each blade of the PEX domain is composed of a single α-helix and four antiparallel β-strands. They form a scaffold for substrate recognition and docking14
Published reports have shown that PEX-9 can interact with different integrin subunits to promote enhanced cancer cell migration, invasion, and survival in various cancer cell types.15−20 Of particular interest to our study was a recent finding that peptide sequence FPGVPLDTHDVFQYREKAYFC within the central cavity of PEX9/blade IV is a binding site for α4β1 integrin. Treatment with synthetic peptide was found to prevent B-cell chronic lymphocytic leukemia transendothelial cell migration and survival.21,22 Mutation of either aspartic acid (Asp) residue (Asp-660 and Asp-663) in this peptide sequence abolished inhibition supporting the importance of Asp residues as key recognition sites for integrins.23,24 Furthermore, the LDT motif in this peptide sequence is a highly conserved motif found in a variety of well-known α4β1 integrin ligands.23,25,26
We previously utilized an in silico DOCKing approach for finding small molecules that bind to MMP-9 and identified a substituted pyrimidone, 1a, which docked to the noncatalytic PEX domain of MMP-9.27 Compound 1a inhibited proMMP-9-mediated cancer cell migration and proliferation in vitro. Importantly, this compound did not inhibit MMP-9 enzymatic activity. Pyrimidone 1a specifically binds to the hemopexin domain of MMP-9 and does not bind to closely related MMPs. In addition to inhibiting proMMP9-mediated cell migration, compound 1a effectively prevented cancer metastasis in an in vivo mouse xenograft model.27
In this current study, we generated an in silico library of analogs of compound 1a to find a more potent compound for potential clinical application. After screening for optimal DOCKing scores in silico, the top 14 compounds were synthesized and tested in a 2-D collagen dot migration assay. A high nanomolar inhibitor that maintained MMP specificity was identified (compound 3c). Our data demonstrate that PEX-9 inhibitor 3c selectively inhibits cell migration induced by proMMP-9, leading to reduced cell invasion predominately through disruption of α4β1 integrin focal adhesion complexes. Thus, PEX-9 inhibitors rectify the abnormal migratory phenotype associated with high MMP-9 expression.
Results
Development of a Second Generation Hit Compound
We designed analogs of compound 1a based on possible derivative syntheses that utilized an amide and a thioether coupling. Thus, three moieties were varied: the amine (R1), the linker ([CH2]n), and the sulfhydrylpyrimidone (R2; Scheme 1). To rationally prioritize the synthesis of compound 1a analogs, the University of California San Francisco (UCSF) DOCK (6.7) algorithm was used to rank various structures for predicted binding affinity. The proposed inhibitors were DOCKed to the PEX-9 domain pocket (PDB code 1ITV; Figure 1A and B). This pocket is at the center of the four blades of the hemopexin domain.
Scheme 1. Synthesis of Inhibitors.

Reagents and conditions. (i) 2-Bromoacetic acid, EDC·HCl, DCM, room temp., 24 h: 67% for 6a, 47% for 6c, 28% for 6b. (ii) NaOH, H2O, THF, 70 °C, 2 h: 63% for 1a, 35% for 1b, and 69% for 1c. (iii) Ethyl 2-bromoacetate, K2CO3, MeOH, H2O, rt, 15 min, 67%. (iv) LiOH, MeOH, H2O, rt, 2 days, 28% for 8, 66% for 9. (v, a) EDC·HCl, DMAP, room temp., 24 h: 40% for 1d, 41% for 2a, 39% for 2c, 52% for 3a, 46% for 3b, 70% for 3c, 22% for 3d, 36% for 4b, 46% for 4c, 42% for 4a, 26% for 4d; (b) CDI, room temp., 24 h, 45% for 4e. (vi) Methyl 4-bromobutanoate, K2CO3, MeOH, H2O, reflux: 47% for 9 (overnight), 73% for 12 (overnight), 81% for 14 (1 h). (vii) Thiourea, MeONa, MeOH, reflux, overnight, 93%. (viii) NaOH, EtOH, RT, overnight, 99%. (ix) LiOH, DMSO, H2O, room temp., 2 h, 86%.
Figure 1.

PEX-9 inhibitor DOCKing to PEX-9 and binding site analysis. (A,B) PEX-9 inhibitor derivatives were DOCKed within the central cavity of the hemopexin domain of PEX-9 MMP-9, and hydrogen bonding was visualized with dashed red lines. (C) A cluster of top hit compounds (2c, 3b, 3c, 3a, 4a, 4d) was found to share a similar binding orientation. (D) The binding orientation of compound 1a (green) relative to compound 3c (red).
Among a library of 66 analogs, 14 (compounds 1a–1d, 2a, 2c, 3a–3d, 4a–4e) were chosen for synthesis based on superior DOCK algorithm scores (Scheme 1, Table S1). One motif that demonstrated consistent improvement in predicted binding affinity was modification of the 6-propylpyrimidone group to bicyclic pyrimidone systems.
Chemical Synthesis of PEX-9 Parental Compound 1a and Analogs
First, the early lead compound 1a was resynthesized to confirm the chemical structure based on full characterization, using the synthetic route shown in the Scheme 1. The synthesis commenced with the reaction of aniline 5a with bromoacetic acid in the presence of EDC·HCl to afford bromoacetanilide 6a as a white solid in 67% yield. Subsequently, 6a was reacted with thiouracil 7 in aqueous NaOH and heated to 70 °C for 48 h to give compound 1a in 63% yield as a white solid after recrystallization. In addition, newly designed compounds 1b and 1c were synthesized using the same synthetic process (Scheme 1).
Closely related compound 1d bearing a benzimidazole group was synthesized using a different route. In this synthesis, thiouracil 7 was converted to the corresponding thioacetic acid 8, followed by the amide coupling with 2-aminobenzimidazole to give compound 1d in modest yield. The attempted preparation of 2-bromoacetylaminobenzimidazole, following the synthetic route for compound 1a did not yield a pure compound. Thus, we judged that the synthetic strategy which coupled R1 last would enable a better modular synthesis of analogs.
With this modular synthesis protocol in hand, we undertook synthesis of the analogs based on the original R1 moiety and two bicyclic sulfhydrylpyrimidones, as well as the linkers with different lengths and a variety of aromatic R2 substituents. Synthesis of compounds 2a and 2c followed the same strategy as that for compound 1d by converting 7 to the corresponding thiobutanoic acid 9, followed by amide formation by coupling with 4-substituted anilines 5a and 5c to give 2a and 2c in modest yields.
In a similar manner, compounds 3a–3d were synthesized in moderate yields through the coupling of anilines and 2-aminobenzimidazole with thiobutanoic acid 12, prepared from 2-sulfhydryl-5,6,7,8-tetrahydroquinazolin-4(3H)-one (11) as shown. Compound 11 was readily prepared by reacting keto ester 10 with thiourea and a base. Compounds 4a–4e were synthesized in moderate yields using the same strategy except for starting from 2-sulfhydryltetrahydroquinazolinone (13) and employed anilines and 2-aminoimidazole for the amide formation.
Structure–Activity Relationship Analysis of PEX-9 Derivative Compounds
COS-1 cells were chosen for this set of experiments due to their low migratory capacity and complete lack of MMP-9 expression, thereby minimizing potential artifacts when interpreting the data. Overexpression of proMMP-9 results in enhanced cell migration in a two-dimensional dot migration assay.27 This assay was initially used to test the efficacy of analogs at a single concentration, 50 μM. This concentration was used as a maximal cutoff because concentrations above 50 μM compound 1a did not increase inhibitor effectiveness in COS-1 cells.
Cells were mixed with type I collagen solution with or without the compound, dotted onto a 96-well plate, and allowed to migrate for 18 h. They were then fixed, stained, and scored for the extent of migration (Figure 2A and B). 2-(4-Oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)-N(methyl)-phenylacetamide (1f) was used as a negative control.27 Compound 3c was the most effective and was selected for further study. The migration assay was repeated in HT1080 fibrosarcoma cells, which endogenously express MMP-9, and the same inhibition potency was observed for both compound 1a and 3c relative to control groups (Supporting Information Figure 1A and B).
Figure 2.

Structure–activity relationship analysis of all synthesized PEX-9 inhibitor derivatives. (A) Representative 20× magnification DAPI images of COS-1 cells engineered to stably express either vector control or proMMP-9 cDNA vectors in a two-dimensional migration assay after treatment with 50 μM of 1f, 1a, or 3c. Scale bar = 250 μm. (B) Quantitative analysis of migrated COS-1 vector control and proMMP-9 expressing cells after treatment with 50 μM of inhibitors. (C) Tryptophan fluorescence emission of proMMP-9 or proMMP-9/PEX2 (ex, 280 nm; em, 330 nm) was monitored and inhibitor 3c titrated into the protein solution. A maximal λMax shift of 9 nm was observed after treatment with 25 μM inhibitor. A solid line is a curve fit for Kd for compound 3c that was determined. (D) Kd for each inhibitor was plotted against percent inhibition of proMMP9-mediated cell migration for each inhibitor.
Compound Affinity for PEX-9 Binding
A blue shift in proMMP-9 tryptophan fluorescence was monitored to determine the binding affinity of all derivative compounds as previously described.27 The Kd for compound 3c binding to proMMP-9 is 0.32 μM (Figure 2C), four times tighter than parent compound 1a. Moreover, a clear correlation between binding affinity and inhibition potency in the migration assay was observed (Figure 2D). Thus, the biophysical SAR is maintained in the cellular assay, and increased inhibition of cell migration is MMP9-dependent and due to optimization of the chemical structure for binding the PEX-9 domain.
PEX-9 Binding Specificity
A chimera of proMMP-9 and MMP-2 in which the MMP-9 PEX domain was replaced by that of MMP-2 (proMMP-9/MMP-2PEX)11 was tested for analog binding. The tryptophans in the MMP-9 PEX domain are conserved in the MMP-2 PEX domain. Upon titration of compound 3c with the chimera, no shift in fluorescence was observed suggesting that 3c does not bind to MMP2-PEX (Figure 2C).
As an additional test of binding selectivity, saturated transfer difference (STD) NMR was performed with purified recombinant proMMP-2 and proMMP-9 protein. ProMMP-2 and proMMP-9 are highly similar in structure and function, belonging to the same subfamily of MMPs known as the type IV collagenases and/or gelatinases. Thus, proMMP-2 constitutes the best control for testing the selectivity of the PEX-9 inhibitor.28 Selective saturation of the 1.12 ppm resonance (proMMP-2 and proMMP-9) was used for STD NMR spectra. STD peaks were observed in the presence of proMMP-9, but not in the presence of proMMP-2, indicating that derivative compound 3c is specific for the PEX-9 target (Supplemental Figure 2A–F). Thus, binding specificity is maintained upon increasing binding affinity.
PEX-9 Inhibitor Target Specificity
To test whether compound 3c is specific for inhibiting only proMMP9-mediated migration, a two-dimensional dot migration assay was performed using COS-1 cells engineered to stably express vector control pQCXIP, proMMP-2, proMMP-9, or proMMP-14 cDNA vectors (Figure 3A). An immunoblot was performed to confirm protein expression and proper localization of each MMP (MMP-2/-9 are secreted while MMP-14 is membrane bound; Figure 3B). As expected, expression of these three MMPs resulted in enhanced cellular migration in COS-1 cells.11 Treatment with either compound 1a or 3c only inhibited proMMP9-mediated migration: compound 3c was significantly more effective at inhibiting migration (Figure 3C).
Figure 3.

Determination of PEX-9 inhibitor specificity in the presence of homologous and nonhomologous MMPs. (A) COS-1 cells transiently transfected with either vector control, proMMP-2, proMMP-9, or MMP-14 cDNAs were treated with 50 μM compound 3c and then used in a two-dimensional dot migration assay. Representative 10× DAPI magnification images were taken and are shown. Scale bar = 250 μm. (B) MMP expression and proper localization was confirmed by immunoblot analysis of cell lysates and protein precipitated serum-free medium. pQCXIP is the vector only control. (C) Quantification of inhibition proMMP-9 mediated cell migration by compounds 1a and 3c. Compound 1f was used as a negative control. (D) Assay of MMP-9 catalytic activity using a fluorogenic peptide degradation assay and activated MMP-9. proMMP-9 protein was activated with 1 mM APMA overnight. A total of 100 ng of activated MMP-9 was incubated with 10 μM fluorogenic peptide (excitation, 320 nm; emission, 405 nm) in the presence of buffer; 50 μM compound 1a, 3c, or 1f; or 250 nM broad-spectrum catalytic inhibitor Marimastat. Readings were performed every 10 min over the course of 2 h. (E) Cell viability was assayed with MTT. COS-1 cells engineered to stably express either vector control or proMMP-9 cDNA were treated for 24 h with DMSO control, 50 μM compound 3c, or 100 nM staurosporine (STS) as a positive control for acute cell toxicity.
Effect of PEX-9 Inhibitor on MMP-9 Catalytic Activity
Recombinant proMMP-9 protein was purified and incubated overnight in a 37 °C water bath in TNC buffer + 1 mM APMA to artificially activate the protein.29,30 The next day, the purified protein was incubated with 10 μM fluorogenic peptide (Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2), and catalytic activity was monitored over 2 h in the presence of compounds 1a, 3c, 1f, and marimastat, a broad-spectrum MMP catalytic inhibitor. No inhibition of the catalytic activity was observed with 1a or 3c, corroborating that PEX-9 inhibitors do not modulate the catalytic activity of the protease. In the presence of marimastat (Kd = 80 nM), no substrate cleavage was observed (Figure 3D). Therefore, inhibition of proMMP9-mediated cell migration by compound 1a or 3c is not due to a loss of proteolytic activity.
Cell Viability Analysis
Compound 3c was evaluated for acute cytotoxicity. COS-1 cells stably expressing either vector control or proMMP-9 cDNA were treated with 50 μM 3c for 24 h. As negative and positive controls, cells were treated with DMSO alone or 100 nM staurosporine (STS), an apoptosis inducer, respectively.31 Significant cell death was only observed after treatment with STS. A nonstatistically significant 8% decrease in cell growth occurred after treatment with 50 μM of compound 3c (Figure 3E). These experiments were repeated in endogenous MMP-9 expressing HT1080 fibrosarcoma cells, which were analyzed for chronic toxicity (cells were retreated every 3 days for a total of 9 days). No cell death was observed at 100 μM 3c (data not shown).
PEX-9 Inhibitors Decrease MMP-9 Dimer Formation and Association with EGFR
Human fibroscarcoma HT1080 cells were cotransfected with MMP-9 cDNAs tagged with either a Myc or HA tag as previously reported.12 The next day, cells were switched to serum free media and treated with 50 μM of either compound 1a or 3c overnight. Using the Myc antibody for immunoprecipitation and HA antibody for immunoblotting, the formation of dimeric MMP-9 was monitored. Compound 3c significantly blocked MMP-9 dimer formation as compared to control compound 1f. In addition, inhibition of PEX-9 with 1a or 3c reduced endogenous CD44 and EGFR coimmunoprecipitation (Supporting Information Figure 3A). Moreover, these PEX-9 inhibitors reduced phosphorylation of EGFRTyr1068 and its downstream targets AKTSer473 and Erk1/2Thr202/Tyr204 (Supporting Information Figure 3B).
PEX-9 Inhibitors Disrupt MMP-9 Interaction with α4β1 Integrin
HT1080 cells transiently cotransfected with tagged MMP-9 cDNAs were prepared as described above.12 Inhibition of PEX-9 with 50 μM of either compound 1a or 3c reduced endogenous α4β1 integrin coimmunoprecipitation (Figure 4A). Inhibition of PEX-9 with 50 μM of either compound 1a or 3c reduced SrcTyr418 phosphorylation in addition to phosphorylation of Src downstream targets FAKTyr 576/577 and PAXTyr118 (Figure 4B).
Figure 4.

Treatment with PEX-9 inhibitor resulting in decreased interaction between MMP-9 and α4β1 integrin and attenuated downstream signaling. (A) Co-IP with HT1080 cells transiently overexpressing MMP-9/MYC cDNA was performed in the presence of 50 μM compound 3c. MMP-9/MYC was immunoprecipitated using an anti-Myc antibody and served as an input control while α4 and β1 integrin interactions were probed via immunoblot. A decrease in MMP-9 interacting with both integrin subunits was observed as well as a (B) decrease in SrcTyr418 and downstream target proteins FAKTyr 576/577/PAXTyr118 phosphorylation. (C) 60× confocal images were captured, and representative images of MMP-9 (green) and each integrin subunit (red) were taken after treatment of HT1080 cells with 50 μM of either compound 1a, 3c, or 1f. Scale bar = 100 μm. (D) 60× confocal images were captured, and representative images of p-FAKTyr 576/577 (red) and p-PAXTyr118 (green) were taken after treatment of HT1080 cells with 50 μM of either compound 1a, 3c, or 1f. Scale bar = 100 μm.
Evaluation of MMP-9 Cell Surface Localization upon Treatment with PEX-9 Inhibitors
HT1080 cells were fixed in 4% paraformaldehyde then subsequently incubated with anti-MMP-9 in addition to anti-α4 and/or β1 integrin antibodies to monitor relative levels of localization of these proteins at the cell surface. The cells were counterstained with DAPI nuclear dye. Treatment with either compound 1a or 3c resulted in a loss of MMP-9 from the cell surface (Figure 4C). However, treatment with either solvent control DMSO or negative control compound 1f did not disrupt the localization of MMP-9 on the cell surface.
Evaluation of Focal Adhesion Complexes in the Presence of PEX-9 Inhibitors
HT1080 cells were fixed after overnight treatment with 50 μM PEX-9 inhibitors, probed for either FAKTyr 576/577 or PAXTyr118 and counterstained with DAPI. A decrease in the formation of FAK and PAX adhesion junctions, normally depicted as “large punctae,” was observed in cells after treatment with PEX-9 inhibitor compound 1a or 3c (Figure 4D).
Validation of MMP-9’s Role in Signaling
MMP-9 expression was silenced using an shRNA approach as previously described.12 HT1080 cell lysates were collected and analyzed by immunoblot. Attenuation of MMP-9 expression resulted in a decrease in the phosphorylation of EGFRTyr1068, FAKTyr 576/577, PAXTyr118, SrcTyr418, AKTSer473, and Erk1/2Thr202/Tyr204 (Supporting Information Figure 4A). These findings are corroborated by Kinexus antibody microarray screening of phosphorylation-dependent signaling pathways in MMP-9-transfected COS-1 cells12 in which Src, FAK, and PAX phosphorylation increase in proMMP-9 overexpressing cells (Supporting Information Figure 4B).
Treatment with PEX-9 Inhibitor Prevents Association of EGFR with CD44 and β1 Integrin
HT1080 cells were treated with 50 μM 3c, 1a, 1f, or DMSO alone. Cell lysates were collected and analyzed by a co-IP assay in which endogenous EGFR was captured with an anti-EGFR antibody and served as an input control. CD44 and β1 integrin were individually probed on the immunoblot. Decreased interaction between EGFR-CD44 and EGFR-β1 integrin was observed after treatment with 3c (Supporting Information Figure 5A).
Src Activation is a PEX-9 Dependent Process
Non-MMP-9 expressing MCF-7 breast cancer cells were transiently transfected with either vector control pcDNA3.1, proMMP-9, or chimeric proMMP-9/MMP-2PEX cDNA constructs. Phosphorylation of SrcTyr418 was detected using an immunoblotting assay. Increased SrcTyr418 phosphorylation was observed in proMMP-9 overexpressing cells. PEX domain swapping with MMP-2 has no effect on SrcTyr418 phosphorylation, similar to vector control pcDNA3.1 expressing cells (Supporting Information Figure 5B).
Effect of PEX-9 Inhibitors on Cell Adhesion
A 96-well plate was coated with a thin layer of either collagen or fibronectin (5 μg/mL in PBS) substrate before use. HT1080 cells were then seeded onto wells and incubated at 37 °C for 30 min in the presence of various inhibitors. Wells were washed and cells were fixed in 4% paraformaldehyde and stained with DAPI. Images of each 96-well plate were microscopically captured and counted for the number of cells still adherent to the coated surface using automated computer software. Compound 1a or 3c inhibited cellular adhesion, whereas treatment with Marimastat, a broad-spectrum MMP catalytic inhibitor, had no observable effect on cell attachment. Solvent DMSO and compound 1f also had no effect on cell adhesion (Supporting Information Figure 6A).
Effect of Broad-Spectrum MMP Catalytic Inhibitor Marimastat on Focal Adhesion Complexes
HT1080 cells treated with 80 nM of broad-spectrum MMP inhibitor marimastat were analyzed by immunofluorescence microscopy for both p-FAKTyr 576/577 and p-PAXTyr118. Treatment with marimastat did not prevent the formation of focal adhesion contact sites in HT1080 cells (Supporting Information Figure 6B).
Treatment with PEX-9 Inhibitor Prevents Both Angiogenesis and Invasion in a Chorioallantoic Membrane (CAM) Assay
A CAM assay was carried out to evaluate angiogenic and invasive potential of HT1080 cells after treatment with 3c. HT1080 cells were adsorbed onto a gelatin sponge and implanted onto the surface of the chicken embryo CAM followed by treatment with a 100 μM stock solution of compound 1f, 1a, or 3c (final drug concentration is estimated to be 0.5 μM). After a 4-day incubation, neovascularization was imaged and quantified. Treatment with either compound 1a or 3c significantly reduced the angiogenic potential of HT1080 cells as compared to solvent DMSO and negative compound 1f control treated cells (Figure 5A,B).
Figure 5.

Compound 3c prevents angiogenesis and invasion through the basement membrane in a CAM assay. (A) HT1080 cells were adsorbed onto a gelatin sponge, implanted atop the CAM, and then subsequently treated with DMSO control or 0.5 μM compound 1f, 1a, or 3c. After 4 days of incubation at 37 °C, representative images of the angiogenic bed surrounding the gelatin sponge were taken. (B) Analysis and quantification of the number of blood vessels in the area surrounding the implanted sponge. (C, D) The CAM was inoculated with HT1080 GFP+ cells mixed with a type I collagen solution and treated with DMSO control and 0.5 μM compound 1f, 1a, or 3c. After 7 days of incubation at 37 °C, CAM membranes were isolated, sectioned, H&E stained, and imaged under 20× magnification.
HT1080 cells stably expressing GFP were mixed with a type I collagen solution (3 mg mL–1) and seeded atop the CAM to ensure the cells localized to a specific area. Then, they were treated with a 100 μM stock solution of compound 1f, 1a, or 3c (final drug concentration is estimated to be 0.5 μM). The invasion of cancer cells through the epithelium and basement membrane of the upper CAM into connecting tissue was examined by hematoxylin and eosin staining (Figure 5C) in addition to fluorescence microscopy for GFP+ cells (Figure 5D). Treatment of HT1080 cells with either compound 1a or 3c reduced invasion of CAM tissue compared to treatment with either DMSO or compound 1f control.
Discussion
MMPs are crucial for the degradation of ECM and blood vessel linings. Degradation allows tumor cells to escape their primary location and enter the circulatory system where they can travel to seed secondary tumors elsewhere in the body.32,33 Therefore, targeting MMPs with inhibitors is of interest for cancer drug development.34 Original inhibition attempts focused on synthetic collagen-mimicking hydroxamate inhibitors that chelate the zinc ion in the active site upon binding.35 These drugs largely failed in clinical trials due to lack of selectivity for individual MMPs. Due to the specificity issues inherent in generating inhibitors of MMP proteolytic activity, we developed inhibitors which target MMP-9’s hemopexin domain. MMP hemopexin domains share a common tertiary structure, yet the surface residues of MMP hemopexin domains are distinct, and one hemopexin domain cannot functionally substitute for another.
Identification of a More Potent PEX-9 Inhibitor
To identify compounds that bind more tightly to the hemopexin domain of MMP-9 than our original hit,27 structural diversity was introduced at three positions and computational DOCKing utilized to prioritize synthesis and testing of a small subset. The similarities between their predicted and measured binding affinities indicate that the DOCKed structures may be used to infer important binding modalities. Compounds that displayed the greatest inhibition of proMMP-9 mediated migration had longer linkers with three methylenes between the amide and the sulfide moieties and at least one fluorine in the R1 group. A cluster of derivative compounds (2c, 3a–3c, 4a, 4d) shared a similar binding geometry within the hemopexin domain of MMP-9 (Figure 1C). In all the tightest binding PEX-9 inhibitor derivatives, the aryl fluorine atoms were found deep within the binding pocket in contrast to the original hit 1a, in which the difluoromethoxyphenol moiety remained solvent exposed. The STD-NMR data support the binding pose in which the aniline is deeply buried in the pocket (Supporting Information Figure 2A–F). In addition, fusion of a second ring to the pyrimidone moiety to form a quinazolinone significantly improved inhibitor efficiency and affinity. The length of the aliphatic chain between the two ring moieties is key for optimizing binding interactions. Comparison of DOCKed compounds 1a and 3c highlights differences in binding orientation that may contribute to the higher affinity of 3c (Figure 1D).
Binding affinity for PEX-9 correlates with inhibition of proMMP9-mediated migration (Figure 2D) across our 16-compound series that spans 1.5 logs of binding affinity. These structure–activity results support the mechanism of inhibition of proMMP9-dependent cell migration being through targeting PEX-9 on the cell surface.
proMMP-9 homodimerization is a critical step for interaction with CD44 at the cell surface, which results in decreased phosphorylation of EGFRTyr1068 and its downstream targets AKTSer473 and Erk 1/2Thr202/Tyr204 phosphorylation.12 Compound 3c demonstrated enhanced efficacy of blocking proMMP-9 homodimerization and activation of downstream protein targets (Figure 4, Supporting Information Figure 3). Moreover, our most potent compound, 3c, retained specificity for a single MMP as demonstrated by the lack of inhibition in proMMP-2 or proMMP-14 mediated cell migration (Figure 3C). Our structure–activity correlation (Figure 2D) and the specificity of inhibition for proMMP-9 mediated cell migration corroborate that compound 3c targets PEX-9 in vitro and in vivo and not a downstream signaling target common to other mediators of cell migration.
Recently, Garcia-Pardo and co-workers reported that the central cavity of PEX-9 interacts with α4β1 integrin as a mechanism for cross-talk between MMPs and integrin focal adhesion formation.17,18,21 In turn, Src kinase interacts with the cytoplasmic tail of α4β1 integrin subunits, which is critical for integrin function to promote and regulate focal adhesion turnover.36−42 Interestingly, PEX9-α4β1 integrin downstream signaling increases activation of Fyn, a Src family kinase, and drives chronic lymphocytic leukemia B cell survival.17 Therefore, we examined the consequences of binding 3c to PEX-9 on α4β1 integrin-mediated Src signaling.
Using a coimmunoprecipitation approach, we found that PEX-9 inhibitors interfere with binding between proMMP-9 and α4β1 integrin and can block cross-talk signaling. This disruption is accompanied by decreases in phosphorylation levels of SrcTyr418 and its downstream substrates FAKTyr 576/577 and PAXTyr118 (Figure 4B). Immunofluorescence imaging in HT1080 cells further supported the idea that focal adhesion junctions (“large punctae”) were lost upon treatment with the PEX-9 inhibitor (Figure 4C,D). We also demonstrated that overexpression of proMMP-9 in MCF-7 breast cancer cells enhanced Src activation. Overexpression of proMMP-9/MMP2-PEX in MCF-7 breast cancer cells or silencing of endogenous MMP-9 expression in HT1080 attenuated Src activation. Taken together, the data suggest that the proMMP-9 hemopexin domain is required for enhanced cellular adhesion and migration.
Potential Mechanism by Which Src Is Activated
Our data are in agreement with the literature showing that α4β1 integrin clustering is sufficient for activation of Src kinase.37,43 However, in addition to integrin clustering, EGFR can directly activate Src kinase, and EGFR interacts with β1 integrin.44−46
It is proposed that proMMP-9 forms a large charged surface for complex interactions with multiple surface proteins involved in outside-in signaling.47 The hemopexin domain of proMMP-9 may act as a scaffold to enhance clustering of α4β1 integrin-EGFR-CD44 at the cell surface and result in aberrant Src activation.48 Garcia-Pardo and co-workers demonstrated that a peptide mimicking blade 1 of PEX-9 inhibits interaction with integrin and that a second peptide mimicking blade 4 of PEX-9 inhibits interaction with CD44.21,22 We reasoned that our inhibitor, which binds directly to PEX-9, could simultaneously disrupt both interactions. Indeed, our coimmunoprecipitation experiments demonstrate that the addition of 3c selectively disrupts proMMP-9 interaction with both α4β1 integrin (Figure 4) and CD44 (Supporting Information Figure S3).
Importantly, our inhibitor is specific for MMP-9. Targeting the hemopexin domain to investigate the role of MMP-9 in promoting cellular adhesion has identified a connection to integrin and Src in a complex adhesion network as depicted in Figure 6. There are currently no effective cancer therapeutics designed to target adhesion receptors because cancer cells can change the mechanism by which they migrate depending upon their environmental constraints.49 Our in vivo CAM experiments demonstrate that we can inhibit two key determinants of tumor cell metastatic potential, angiogenesis and invasion (Figure 5), through PEX domain targeting. Our small molecule inhibitor paves the way for designing new therapeutic strategies for the treatment of metastatic disease and opens the door for the future development of new technologies and tools for understanding the role of individual MMPs in cancer progression. Development of PEX inhibitors for other MMPs will provide understanding of their noncatalytic functions and a strategy for reducing undesirable off-target effects previously observed in broad-spectrum MMP catalytic inhibitor clinical trials.
Figure 6.

Overview of the mechanism by which MMP-9 regulates formation of focal adhesion junctions. (A) During exocytosis and upon being secreted into the extracellular space, proMMP-9 forms a large complex with itself that can then act as a scaffold for promoting outside-in signaling. In our model, PEX-9 scaffolding promotes the association of β1 integrin-EGFR-CD44. Interaction with CD44 results in enhanced EGFR activation in addition to increased phosphorylation of its downstream targets AKT and Erk 1 + 2 (through the MAPK/Erk pathway). While complexed to β1 integrin, EGFR then goes on to transactivate Src kinase after it is recruited to the α4 integrin subunit during the generation of a new focal adhesion contact site. After activation, Src can then directly phosphorylate FAK currently associated with the β1 integrin subunit resulting in its maximal catalytic activity. This active Src–FAK complex can then bind and activate PAX, resulting in a mature FAK-PAX complex necessary for the formation of focal adhesion junctions. Formation of a complex between FAK and PAX results in final translocation to ECM–integrin junctions at the cell surface where they regulate cytoskeletal interactions resulting in enhanced cellular adhesion, migration, and invasion of cancer cells. (B) Treatment with PEX-9 inhibitor (depicted as red triangle) prevents MMP-9 scaffolding, thereby preventing downstream signaling driven by EGFR activation.
Experimental Methods
Materials
Collagen type I (acetic-acid-extracted native type I collagen from rat tail tendon) was obtained from BD Bioscience Discovery Labware (Franklin Lakes, NJ). Fibronectin solution was purchased from Sigma-Aldrich (St. Louis, MO). Hoechst nuclear stain was obtained from Invitrogen (Grand Island, NY). Rabbit anti-p-AKTSer473, anti-AKT (total), anti-β1 integrin, anti-p-Erk1/2Thr202/Tyr204, anti-p-FAKTyr 576/577, anti-FAK (total), anti-p-PAXTyr118, anti-PAX (total), and anti-EGFR (total) antibodies were all purchased from Cell Signaling Technology (Danvers, MA). Mouse anti-Erk1/2 (total), anti-β-actin, and anti-α/β-tubulin antibodies were also purchased from Cell Signaling Technology (Danvers, MA). Rabbit anti-p-SrcTyr418 and anti-Src (total) in addition to mouse anti-β1 integrin were obtained from Abcam (Cambridge, MA). Mouse anti-α4 integrin and anti-CD44 in addition to rabbit anti-α4 integrin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-MMP-9 was purchased from Millipore (Darmstadt, Germany). Rabbit p-EGFRTyr1068 was obtained from Enzo Life Sciences (Ann Arbor, MI). Rat anti-CD44 was obtained through BD Pharmingen (San Diego, CA). Mouse anti-HA was purchased from Invitrogen-ThermoFisher Scientific (Waltham, MA). Rabbit anti-MYC and anti-HA in addition to horseradish peroxidase conjugated antirabbit, antimouse, and antirat antibodies were obtained from Rockland Immunochemicals (Gilbertsville, PA). Mouse anti-MYC was purchased from Roche Life Sciences (Branford, CT). Alexa Fluor 488 and 568 antirabbit/-mouse antibodies were purchased from Molecular Probes, Life Technologies (Grand Island, NY). proMMP-9 used in kinetic experiments was purchased from Biolegends (San Diego, CA). Mca-PLGL-Dpa-AR-NH2 Fluorogenic MMP Peptide Substrate was purchased from R&D Systems (Minneapolis, MN). Chemicals were purchased from Sigma-Aldrich, Fisher Scientific, and VWR International and used as received or purified before use by standard methods. Tetrahydrofuran was freshly distilled from sodium and benzophenone. Dichloromethane was also distilled immediately prior to use under nitrogen from calcium hydride. 2-(4-Oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)-N(methyl)-phenylacetamide (1f) was purchased from ChemBridge Corp. (San Diego, CA).
General Methods
1H and 13C NMR spectra were measured on a Bruker 300, 400, 500, or 700 MHz spectrometer. Melting points were measured on a Thomas-Hoover capillary melting point apparatus and are uncorrected. TLC was performed on Sorbent Technologies aluminum-backed Silica G TLC plates (Sorbent Technologies, 200 μm, 20 cm × 20 cm), and column chromatography was carried out on silica gel 60 (Merck, 230–400 mesh ASTM). High-resolution mass spectrometry analysis was carried out on an Agilent LC–UV–TOF mass spectrometer at the Mass Spectrometry facility of the Institute of Chemical Biology and Drug Discovery, Stony Brook University.
Chemical Synthesis of PEX-9 Inhibitor Compounds and Characterization Data
N-(4-Difluoromethoxyphenyl)-2-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)acetamide (1a)
To a solution of bromoacetic acid (170 mg, 1.25 mmol) and EDC·HCl (239 mg, 1.25 mmol) in dichloromethane (6 mL) was added aniline (5a, 200 mg, 1.25 mmol) and was stirred at RT overnight. After the completion of the reaction, the reaction mixture was diluted with dichloromethane and washed twice with 1 N hydrochloric acid, twice with a saturated solution of sodium bicarbonate and twice with brine. The organic layers were collected, dried over magnesium sulfate, filtered, and concentrated to afford an off-white crude product. The crude product was recrystallized from hexanes/dichloromethane to give bromoacetanilide 6a as a white solid (227 mg, 65% yield). 1H NMR (300 MHz, CDCl3): δ 4.27 (s, 2 H), 6.30–6.79 (s, 1 H), 7.21 (d, 2 H, J = 9 Hz), 7.63 (d, 2 H, J = 9 Hz).
To a solution of sodium hydroxide (7.2 mg, 0.18 mmol) in water (2 mL) was added 2-hydrosulfanyl-4-oxo-6-propyl-1,4-dihydropyrimidine (7; 30 mg, 0.18 mmol), and the mixture was stirred until all of 1 was dissolved. To the mixture was added a solution of 6a (50 mg, 0.18 mmol) in tetrahydrofuran (2 mL) and heated at 60 °C for 2 h and then at 70 °C overnight. After the completion of the reaction, a white precipitate was observed, which was collected on a filter and recrystallized from ethanol to afford 1a as white solid (42 mg, 63% yield). Mp: 180 °C (decomp.). 1H NMR (700 MHz, DMSO-d6): δ 0.73 (t, J = 7.0 Hz, 3H), 1.51 (m, 2H), 2.32 (t, J = 7.0 Hz, 2H), 4.05 (s, 2H), 5.97 (s, 1H), 6.97–7.29 (m, 3H), 7.62 (d, J = 8.4 Hz, 2H), 10.44 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 13.4, 20.6, 35.1, 38.2, 116.5.0 (t, J = 256.0 Hz), 119.5, 120.4, 136.4, 146.3, 165.9. 19F NMR (376 MHz, DMSO-d6): δ −81.6. HRMS (ESI+) calcd for C16H18F2N3O3S [M + H]+: 370.1031. Found: 370.1039 (Δ= −2.02 ppm).
In the same manner, 1b and 1c were synthesized.
2-(4-Oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)-N-phenylacetamide (1b)
White solid, 35% yield. Mp: 170 °C (decomposed). 1H NMR (700 MHz, DMSO-d6): δ 0.83 (t, J = 7.0 Hz, 3H), 1.46–1.56 (m, 2H), 2.31 (t, J = 7.0 Hz, 2H), 4.04 (s, 2H), 5.90 (s, 1H), 7.04 (t, J = 7.7 Hz, 1H), 7.30 (t, J = 7.7 Hz, 2H), 7.56 (t, J = 7.7 Hz, 2H), 10.33 (s, 1H), 12.67 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 13.4, 20.5, 35.1, 38.5, 119.0, 123.3, 128.7, 139.0, 165.9. HRMS (ESI+) calcd for C15H18N3O2S [M + H]+: 304.1114. Found: 304.1113 (Δ= 0.29 ppm).
N-(4-Fluorophenyl)-2-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)acetamide (1c)
White solid, 69% yield. Mp: 191–194 °C. 1H NMR (500 MHz, DMSO-d6): δ 0.73 (t, J = 7.0 Hz, 3H), 1.45–1.56 (m, 2H), 2.31 (t, J = 7.0 Hz, 2H), 4.02 (s, 2H), 6.94 (s, 1H), 7.10–7.19 (m, 2H), 7.55–7.62 (m, 2H), 10.30 (s, 1H), 12.54 (s,1H). 13C NMR (500 MHz, DMSO-d6): δ 13.4, 20.5, 35.0, 38.5, 115.3 (d, J = 22.1 Hz), 120.7 (d, J = 7.7 Hz), 135.5 (d, J = 2.5 Hz), 158.0 (d, J = 238.4 Hz), 165.8. 19F NMR (376 MHz, DMSO-d6): δ −119.3. HRMS (ESI+) calcd for C15H17FN3O2S [M + H]+: 322.1020. Found: 322.1020 (Δ= −0.03 ppm).
N-(1H-benzo[d]imidazol-2-yl)-2-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)acetamide (1d)
To a solution of thiouracil 7 (1.70 g, 10.0 mmol) and potassium carbonate (2.07 g, 15.0 mmol) in MeOH (10 mL) and water (20 mL) was added ethyl 2-bromoacetate (1.42 g, 8.5 mmol). The mixture was stirred at RT for 15 min and 50 mL water was added. Then, the reaction mixture was extracted with ethyl acetate (5 × 80 mL). The organic layers were combined and washed with water (100 mL) and brine (100 mL) and dried over MgSO4, and the solvent was removed by rotary evaporator. The crude product was purified by flash column chromatography on silica gel using MeOH/DCM as an eluent to give ethyl 2-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)acetate as a white solid (1.7 g, 67% yield). Mp: 117–118 °C. 1H NMR (300 MHz, CDCl3): δ 0.93 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.59–1.65 (m, 2H), 2.43 (t, J = 7.2 Hz, 2H), 3.93 (s, 2H), 4.20 (q, J = 7.2 Hz, 2H), 6.05 (s, 1H), 13.17 (s, 1H). 13C NMR (175 MHz, CDCl3): δ 13.6, 14.1, 20.8, 32.8, 39.5, 61.9, 108.2, 158.9, 165.5, 168.2, 169.2. HRMS (ESI+) calcd for C11H17N2O3S [M + H]+: 257.0954. Found: 257.0953 (Δ = 0.48 ppm).
To a solution of ethyl 2-((4-oxo-6-propyl-1,4-dihydropyrimidin-2-yl)thio)acetate (1.20 g, 4.68 mmol) in MeOH and water was added lithium hydroxide (0.2 g, 9.36 mmol). The mixture was stirred at RT for 2 days. The reaction mixture was adjusted to pH 1 by 1N hydrochloric acid and extracted with ethyl acetate (5 × 80 mL). The organic layers were combined and dried over MgSO4, and the solvent was removed by rotary evaporator to afford thioacetic acid 8 (0.30 g, 28%) as a white solid. Mp: 154–155 °C. 1H NMR (700 MHz, DMSO-d6): δ 0.86 (t, J = 7.0 Hz, 3H), 1.56 (m, 2H), 2.30 (t, J = 7.0 Hz, 2H), 3.47 (s, 2H), 5.79 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 13.6, 20.7, 36.1, 38.2, 107.4, 163.7, 164.3, 166.3, 171.2. HRMS (ESI+) calcd for C9H13N2O3S [M + H]+: 229.0641. Found: 229.0644 (Δ = −1.11 ppm).
To a mixture of 2-(6-oxo-4-propyl-1,6-dihydropyrimidin-2-ylthio)acetic acid (8; 228 mg, 1.0 mmol), 1H-benzo[d]imidazol-2-amine (5d, 146 mg, 1.1 mmol), and DMAP (134 mg, 1.1 mmol) in DMF (3 mL) was added EDC·HCl (211 mg, 1.1 mmol), and the mixture was stirred at RT for 24 h. A total of 50 mL of water was added, and the resulting precipitate was filtered to give a solid, which was recrystallized from 1,4-dioxane to give compound 1d (136 mg, 40%) as an off-white solid. Mp: 217 °C (decomposed). 1H NMR (700 MHz, DMSO-d6): δ 0.63 (t, J = 7.0 Hz, 3H), 1.50–1.40 (m, 2H), 2.27 (t, J = 7.0 Hz, 2H), 4.14 (s, 2H), 5.93 (s, 1H), 7.05–7.09 (m, 2H), 7.40–7.43 (m, 2H), 12.10 (s, 3H). 13C NMR (175 MHz, DMSO-d6): δ 13.3, 20.5, 34.5, 38.4, 106.5, 111.5, 114.1, 120.3, 121.0, 136.3, 146.7, 161.6, 162.3, 163.9, 167.7. HRMS (ESI+) calcd for C16H18N5O2S [M + H]+ 344.1176. Found: 344.118 (Δ= −1.1 ppm).
N-(4-Difluoromethoxyphenyl)-4-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)butanamide (2a)
To a solution of thiouracil 7 (0.85 g, 5 mmol) and potassium carbonate (1.03g, 7.5 mmol) in methanol (5 mL) and water (10 mL) was added methyl 4-bromobutanoate (1.36g, 7.5 mmol). The solution was heated to reflux overnight. The reaction mixture was cooled to RT and 50 mL of water added, and the mixture was extracted with ethyl acetate (3 × 50 mL). The organic layers were combined and washed with water (50 mL) and brine (50 mL) and dried over MgSO4, the solvent was removed by rotary evaporator. The crude product was purified by flash column chromatography on silica gel using MeOH/DCM as an eluent to give methyl 4-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)butanoate as a white solid (0.64 g, 47% yield). Mp: 129–130 °C. 1H NMR (500 MHz, CDCl3): δ 0.95 (t, J = 7.0 Hz, 3H), 1.64–1.71(m, 2H), 2.06 (m, 2H), 2.46 (m, 4H), 3.24 (t, J = 7.0 Hz, 2H), 3.68 (s, 3H), 6.04 (s, 1H), 12.99 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 13.8, 21.0, 24.7, 29.9, 32.7, 39.7, 51.8, 108.0, 160.2, 165.6, 169.5, 173.4. HRMS (ESI+) calcd for C12H19N2O3S [M + H]+: 271.1111. Found: 271.111 (Δ= 0.51 ppm).
To a solution of methyl 4-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)butanoate (500 mg, 1.84 mmol) in MeOH and water was added lithium hydroxide (88.1 mg, 3.68 mmol), and the mixture was stirred at RT for 2 days. The mixture was adjusted to pH 1 by 1N hydrochloric acid to form a white precipitate. The precipitate was collected on a filter to afford 4-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)butanoic acid 9 as a white solid (308.0 mg, 66% yield). Mp: 139–141 °C. 1H NMR (300 MHz, CDCl3): δ 0.95 (t, J = 7.3 Hz, 3H), 1.68 (m, 2H), 2.01–2.14(m, 2H), 2.49 (m, 4H), 3.28 (t, J = 6.5 Hz, 2H), 6.04 (s, 1H). 13C NMR (175 MHz, CDCl3): δ 13.8, 21.1, 24.5, 29.9, 32.8, 39.7, 107.7, 160.3, 165.9, 170.2, 178.5. HRMS (ESI+) calcd for C11H17N2O3S [M + H]+: 257.0954. Found: 257.0958 (Δ= −1.33 ppm).
To a mixture of 9 (190 mg, 0.74 mmol), 4-(difluoromethoxy)aniline (5a, 130.5 mg, 0.82 mmol), and 4-(dimethylamino)pyridine (DMAP; 99.4 mg, 0.81 mmol) in dimethylformamide (DMF; 3 mL) was added N′-ethylcarbodiimide hydrochloride (EDC·HCl (156 mg, 0.81 mmol)); the mixture was stirred at RT for 1 h. Then, 100 mL of water was added to the reaction mixture to form an off-white precipitate, which was collected on a filter to give an off-white solid. The solid was dissolved in ethyl acetate and dried over MgSO4 and filtered, and the solvent was removed by rotary evaporator. The crude product was purified by recrystallization from ethyl acetate/hexane to afford 2a as a white solid (120.4 mg, 41% yield). Mp: 174–177 °C. 1H NMR (500 MHz, CDCl3): δ 0.92 (t, J = 7.5 Hz, 3H), 1.64 (m, 2H), 2.06–2.22 (m, 2H), 2.42 (t, J = 7.5 Hz, 2H), 2.49 (t, J = 6.9 Hz, 2H), 3.31 (t, J = 5.7 Hz, 2H), 6.01 (s, 1H), 6.45 (t, J = 74.0 Hz, 1H), 7.07 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.6 Hz, 2H), 7.92 (s, 1H), 12.69 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 13.8, 21.1, 24.8, 30.2, 35.5, 39.7, 107.8, 116.1 (t, J = 259 Hz), 120.6, 121.3, 135.6, 147.3, 160.2, 165.5, 169.9, 170.5. 19F NMR (376 MHz, CDCl3): δ −80.7. HRMS (ESI+) calcd for C18H22F2N3O3S [M + H]+: 398.1344. Found: 398.1345 (Δ = −0.03 ppm).
In the same manner, compound 2c was synthesized.
N-(4-Fluorophenyl)-4-(4-oxo-6-propyl-1,4-dihydropyrimidin-2-ylthio)butanamide (2c)
White solid, 39% yield. Mp: 180–182 °C. 1H NMR (300 MHz, CDCl3): δ 0.83–0.96 (t, J = 7.3 Hz, 3H), 1.57–1.69 (m, 2H), 2.05–2.21 (m, 2H), 2.33–2.55 (m, 4H), 3.31 (t, J = 6.3 Hz, 2H), 6.02 (s, 1H), 7.00 (t, J = 8.7 Hz, 2H), 7.49 (m, 2H), 7.64 (s, 1H), 12.45 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 13.4, 20.5, 24.8, 29.2, 34.9, 38.5, 115.2 (d, J = 22.0 Hz), 120.7 (d, J = 7.7 Hz), 135.7 (d, J = 2.3 Hz), 157.1, 158.5, 170.3. 19F NMR (376 MHz, CDCl3): δ −119.7. HRMS (ESI+) calcd for C17H21FN3O2S [M + H]+: 350.1333. Found: 350.134 (Δ = −2.12 ppm).
4-(4-Oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)-N-phenylbutanamide (3b)
Ethyl 2-oxocyclohexane-1-carboxylate 10 (1.70 g, 10.0 mmol) and thiourea (1.52 g, 20.0 mmol) were added to a sodium methoxide solution (0.58 g sodium in 20 mL methanol), and the mixture was refluxed overnight. The solvent was removed, and the residue was dissolved in hot water (50 mL). Glacial acetic acid (6 mL) was added dropwise to make the solution acidic until a white solid was formed. The precipitate was collected by a filter and washed successively with saturated sodium bicarbonate (3 × 10 mL) and water (3 × 10 mL) and recrystallized from ethyl acetate (30 mL) to afford 2-sulfhydryl-5,6,7,8-tetrahydroquinazolin-4(3H)-one (11; 1.70 g, 93%) as a white solid. Mp > 230 °C. 1H NMR (700 MHz, DMSO-d6): δ 1.53–1.76 (m, 4H), 2.16 (t, J = 6.0 Hz, 2H), 2.36 (t, J = 6.0 Hz, 2H), 12.09 (s, 1H), 12.25 (s, 1 H). 13C NMR (175 MHz, DMSO-d6): δ 20.5, 20.7, 20.9, 25.5, 111.6, 149.5, 161.3, 173.9. HRMS (ESI+) calcd for C8H11N2OS [M + H]+: 183.0587. Found: 183.0587 (Δ = −0.44 ppm).
To a solution of 11 (1.59 g, 8.72 mmol) in anhydrous methanol (15 mL) was added methyl 4-bromobutanoate (1.72 g, 9.55 mmol) and potassium carbonate (1.31 g, 9.48 mmol). The reaction mixture was heated to reflux overnight. After being cooled to RT, the reaction mixture was concentrated in vacuo to remove the solvent. To the residue was added water (50 mL), and it was extracted with dichloromethane (4 × 40 mL). The combined organic phases were washed with brine (5 mL), dried over MgSO4, and concentrated in vacuo to give a crude product, which was purified by recrystallization from ethyl acetate to afford methyl 4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanoate (1.80 g, 73%) as a white solid. Mp: 149–150 °C. 1H NMR (700 MHz, CDCl3): δ 1.69–1.79 (m, 4H), 2.01–2.06 (m, 2H), 2.47 (m, 4H), 2.59 (t, J = 7.0 Hz, 2H), 3.22 (t, J = 7.0 Hz, 2H), 3.67 (s, 3H). 13C NMR (175 MHz, CDCl3): δ 21.7, 21.8, 22.2, 24.7, 29.8, 31.6, 32.7, 51.8, 117.6, 156.4, 162.0, 165.0, 173.5. HRMS (ESI+) calcd for C13H19N2O3S [M + H]+ 283.1111. Found: 283.1117 (Δ = −2.09 ppm).
To a mixture of 2N sodium hydroxide (1.51 g in 19 mL water) and ethanol (22 mL) was added methyl 4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanoate (1.34 g, 4.75 mmol) at RT. The mixture was stirred overnight at RT. The reaction mixture was adjusted to pH 5 with 1N hydrochloric acid and extracted with dichloromethane (5 × 40 mL). The combined organic phases were dried over MgSO4 and concentrated in vacuo to give a residue, which was recrystallized from dichloromethane to afford 4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanoic acid 12 (1.26 g, 99% yield) as a white solid. Mp: 176–177 °C. 1H NMR (700 MHz, CD3OD): δ 1.71–1.81 (m, 4H), 1.96–2.03 (m, 2H), 2.39 (t, J = 6.3 Hz, 2H), 2.43 (t, J = 7.0 Hz, 2H), 2.57 (t, J = 6.3 Hz, 2H), 3.23 (t, J = 7.0 Hz, 2H). 13C NMR (175 MHz, CD3OD): δ 22.7, 22.9, 23.1, 26.0, 30.4, 33.4, 176.6. HRMS (ESI+) calcd for C12H17N2O3S [M + H]+: 269.0954. Found: 269.0959 (Δ= −1.54 ppm).
To a mixture of thiobutanoic acid 12 (134 mg, 0.50 mmol), aniline 5b (70 mg, 0.75 mmol), and DMAP (67 mg, 0.55 mmol) in DMF (2 mL) was added EDC·HCl (105 mg, 0.55 mmol), and the mixture was stirred at RT for 48 h. To the reaction mixture was added 50 mL of water with stirring to form a precipitate. The resulting precipitate was collected on a filter and recrystallized from ethyl acetate to give 3b (79 mg, 46% yield) as an off-white solid. Mp: 193–194 °C. 1H NMR (500 MHz, DMSO-d6): δ 1.56–1.67 (m, 4H), 1.91–1.97 (m, 2H), 2.26 (m, 2H), 2.39–2.45 (m, 4H), 3.16 (t, J = 7.0 Hz, 2H), 7.01 (t, J = 8.0 Hz, 1H), 7.27 (t, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 9.90 (s, 1H), 12.42 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 21.4, 21.8, 24.7, 29.0, 31.1, 34.9, 119.0, 122.9, 128.6, 139.3, 170.3. HRMS (ESI+) Calcd for C18H22N3O2S [M + H]+: 344.1427. Found: 344.1430 (Δ = −0.71 ppm).
In the same manner, compounds 3a, 3c, and 3d were synthesized.
N-(4-Difluoromethoxyphenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide (3a)
Off-white solid, 52% yield. Mp: 197–198 °C. 1H NMR (700 MHz, acetone-d6): δ 1.63–1.73 (m, 4H), 2.09 (m, 2H), 2.35 (t, J = 6.3 Hz, 2H), 2.47 (t, J = 6.3 Hz, 2H), 2.53 (t, J = 7.0 Hz, 2H), 3.27 (t, J = 7.0 Hz, 2H), 6.90 (t, J = 74.4 Hz, 1H), 7.11 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 9.27 (s, 1H), 11.46 (s, 1H). 13C NMR (175 MHz, acetone-d6): δ 22.4, 22.5, 23.0, 26.0, 32.2, 35.9, 117.6 (t, J = 255.5 Hz), 117.9, 120.6, 121.3, 137.9, 147.6 (t, J = 2.6 Hz), 157.2, 161.2, 163.4, 171.2. 19F NMR (376 MHz, acetone-d6): δ −82.3. HRMS (ESI+) Calcd for C19H22F2N3O3S[M + H]+: 410.1344. Found: 410.1346 (Δ = −0.33 ppm).
N-(4-Fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide (3c)
White solid, 70% yield. Mp: 211.5–213 °C. 1H NMR (700 MHz, DMSO-d6): δ 1.56–1.66 (m, 4H), 1.94 (m, 2H), 2.26 (t, J = 5.6 Hz, 2H), 2.41 (t, J = 7.0 Hz, 4H), 3.15 (t, J = 7.0 Hz, 2H), 7.10–7.14 (m, 2H), 7.57–7.61 (m, 2H), 9.96 (s, 1H), 12.44 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 21.4, 21.8, 24.7, 29.0, 31.1, 34.8, 115.2 (d, J = 21.9 Hz), 120.7 (d, J = 7.7 Hz), 135.7 (d, J = 2.3 Hz), 157.8 (d, J = 238 Hz), 170.3. 19F NMR (376 MHz, DMSO-d6): δ −119.8. HRMS (ESI+) calcd for C18H21FN3O2S [M + H]+: 362.1333. Found: 362.1337 (Δ = −1.05 ppm).
N-(1H-Benzo[d]imidazol-2-yl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide (3d)
Off-white solid, 22% yield. Mp > 230 °C. 1H NMR (700 MHz, DMSO-d6): δ 1.55–1.66 (m, 4H), 2.09 (m, 2H), 2.21–2.27 (m, 2H), 2.35–2.41 (m, 2H), 3.21 (t, J = 5.6 Hz, 2H), 3.27 (t, J = 5.6 Hz, 2H), 6.93 (t, J = 7.0 Hz, 1H), 7.11–7.22 (m, 2H), 7.38–7.48 (m, 3H), 12.45 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 21.4, 21.8, 23.7, 28.5, 31.0, 36.2, 113.6, 115.7, 116.4, 119.5, 124.1, 130.0, 143.4, 154.6, 156.5, 160.0, 162.4, 173.8. HRMS (ESI+) Calcd for C19H22N5O2S [M + H]+: 384.1489. Found: 384.1494 (Δ = −1.45 ppm).
4-(4-Oxo-3,4-dihydroquinazolin-2-ylthio)-N-phenylbutanamide (4b)
A mixture of 2-sulfhydrylquinazolin-4(3H)-one (13; 1.78 g, 10.0 mmol), methyl 4-bromobutanoate (2.72 g, 15.0 mmol), and K2CO3 (2.07 g, 15.0 mmol) in methanol (10 mL) and water (20 mL) was refluxed for 1 h. After being cooled to RT, 50 mL of water was added, and the reaction mixture was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried over MgSO4, and concentrated in vacuo to give a crude product, which was purified by recrystallization from ethyl acetate/hexanes to give methyl 4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanoate (2.24 g, 81% yield) as a white solid. Mp: 153–154 °C. 1H NMR (700 MHz, DMSO-d6): δ 1.96 (m, 2H), 2.45 (t, J = 7.0 Hz, 2H), 3.23 (t, J = 7.0 Hz, 2H), 3.58 (s, 3H), 7.39 (t, J = 7.0 Hz, 1H), 7.49 (d, J = 7.0 Hz, 1H), 7.73 (t, J = 7.0 Hz, 1H), 8.02 (d, J = 7.0 Hz, 1H), 12.55 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 24.3, 28.9, 32.1, 51.4, 120.0, 125.6, 125.9, 126.0, 134.5, 148.3, 155.5, 161.3, 172.8. HRMS (ESI+) Calcd for C13H15N2O3S [M + H]+: 279.0798. Found: 279.0802 (Δ = −1.42 ppm).
A mixture of methyl 4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanoate (1.39 g, 5.0 mmol) in DMSO (1 mL) and LiOH (aqueous, 1M, 10 mL) was stirred at RT for 2 h. To the reaction mixture was added 100 mL of water, and it was adjusted to pH 1. The resulting precipitate was collected on a filter and washed with water to give 4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanoic acid (14; 1.14 g, 86% yield) as a white solid. Mp: 178–179 °C. 1H NMR (500 MHz, DMSO-d6): δ 1.90–1.96 (m, 2H), 2.37 (t, J = 7.0 Hz, 2H), 3.24 (t, J = 7.0 Hz, 2H), 7.41 (t, J = 7.5 Hz, 1H), 7.52 (d, J = 7.5 Hz, 1H), 7.75 (t, J = 7.5 Hz, 1H), 8.02 (d, J = 7.5 Hz, 1H), 12.37 (s, 2H). 13C NMR (175 MHz, DMSO-d6): δ 24.3, 29.1, 32.5, 120.0, 125.6, 125.9, 126.0, 134.6, 148.3, 155.6, 161.3,173.9. HRMS (ESI+) Calcd for C12H13N2O3S [M + H]+: 265.0641. Found: 265.0646 (Δ = −1.56 ppm).
To a mixture of 4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanoic acid (14; 132 mg, 0.50 mmol), aniline (186 mg, 2.0 mmol), and DMAP (67 mg, 0.55 mmol) in DMF (2 mL) was added EDC·HCl (144 mg, 0.75 mmol), and the mixture was stirred at RT for 18 h. To the reaction mixture was added 20 mL of water with stirring. The resulting precipitate was collected on a filter and recrystallized from 1,4-dioxane to give 4b (61 mg, 36% yield) as a white solid. Mp: > 230 °C. 1H NMR (700 MHz, DMSO-d6): δ 1.56–1.60 (m, 2H), 2.03 (t, J = 7.0 Hz, 2H), 2.84 (t, J = 7.0 Hz, 2H), 6.57 (t, J = 7.0 Hz, 1H), 6.83 (t, J = 7.0 Hz, 2H), 6.95 (t, J = 7.0 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 7.13 (d, J = 7.0 Hz, 2H), 7.25 (t, J = 7.0 Hz, 1H), 7.57 (d, J = 7.0 Hz, 1H), 9.48 (s, 1H), 12.12 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 24.8, 29.2, 35.1, 119.0, 112.0, 123.0, 125.6, 126.0, 128.6, 134.5, 139.3, 148.4, 155.6, 161.2, 170.4. HRMS (ESI+) Calcd for C18H18N3O2S [M + H]+: 340.1114. Found: 340.1121 (Δ= −2.0 ppm).
In the same manner, compounds 4a, 4c, and 4d were synthesized.
N-(4-(Difluoromethoxy)phenyl)-4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanamide (4a)
Off-white solid, 42% yield. Mp: 210–211 °C. 1H NMR (700 MHz, acetone-d6): δ 2.13–2.23 (m, 2H), 2.59 (t, J = 7.0 Hz, 2H), 3.41 (t, J = 7.0 Hz, 2H), 6.90 (t, J = 74.2 Hz, 1H), 7.11 (d, J = 8.4 Hz, 2H), 7.40 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.69 (m, 3H), 8.09 (d, J = 7.7 Hz, 1H), 9.25 (s, 1H), 11.22 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 24.7, 29.2, 34.9, 116.5 (t, J = 256 Hz), 119.5, 120.0, 120.4, 125.6, 126.0, 134.5, 136.7, 146.1, 148.40, 155.6, 161.3, 170.4. 19F NMR (376 MHz, DMSO-d6): δ −81.4. HRMS (ESI+) Calcd for C19H18F2N3O3S [M + H]+: 406.1031. Found: 406.1033 (Δ = −0.44 ppm).
N-(4-Fluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)butanamide (4c)
White solid, 46% yield. Mp: 216–217 °C. 1H NMR (500 MHz, DMSO-d6): δ 1.20–2.05 (m, 2H), 2.47 (t, J = 7.0 Hz, 2H), 3.29 (t, J = 7.0 Hz, 2H), 7.09–7.14 (m, 2H), 7.38–7.41 (m, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.57–7.60 (m, 2H), 7.68–7.72 (m, 1H), 8.02 (dd, J = 8.0, 1.5 Hz, 1H), 9.97 (s, 1H), 12.56 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 24.7, 29.2, 34.9, 115.2 (d, J = 21.9 Hz), 112.0, 120.7 (d, J = 7.7 Hz), 125.6, 126.0, 126.0, 134.5, 135.7 (d, J = 2.4 Hz), 148.4, 155.5, 157.8 (d, J = 237.9 Hz), 161.2, 170.3. 19F NMR (376 MHz, DMSO-d6): δ −119.8. HRMS (ESI+) Calcd for C18H17FN3O2S [M + H]+ 358.1020. Found: 358.1027 (Δ = −1.99 ppm).
N-(1H-Benzo[d]imidazol-2-yl)-4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanamide (4d)
White solid, 25% yield. Mp > 230 °C. 1H NMR (700 MHz, DMSO-d6): δ 2.05–2.09 (m, 2H), 2.61 (t, J = 7.0 Hz, 2H), 3.31 (t, J = 7.0 Hz, 2H), 7.07 (m, 2H), 7.33–7.50 (m, 4H), 7.58 (t, J = 7.0 Hz, 1H), 8.00 (d, J = 7.0 Hz, 1H), 11.52 (s, 1H), 12.06 (s, 1H), 12.57 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 24.5, 29.0, 31.1, 111.5, 116.9, 120.0, 121.8, 125.5, 126.0, 132.5, 134.4, 140.5, 146.6, 148.4, 155.6, 161.3, 171.8. HRMS (ESI+) Calcd for C19H17N5O2S [M + H]+: 380.1176. Found: 380.1178 (Δ = −0.54 ppm).
N-Hexyl-4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanamide (4e)
To a mixture of 4-(4-oxo-3,4-dihydroquinazolin-2-ylthio)butanoic acid (14; 79 mg, 0.30 mmol) in 1,4-dioxane (2 mL) was added carbonyldiimidazole (CDI; 73 mg, 0.45 mmol), and the mixture was stirred at RT for 1 h. 1-Hexylamine (5e, 61 mg, 0.60 mmol) and DMF (5 mL) were added to the mixture and heated at 60 °C for 48 h. After being cooled to RT, 40 mL of water was added, and the reaction mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with water (20 mL) and brine (20 mL), dried over MgSO4, and concentrated in vacuo to give a yellow oil. Then, 20 mL of hexane was added to this oil with stirring to form a precipitate. The resulting precipitate was collected on a filter to give 4e (46.7 mg, 45% yield) as a white solid. Mp: 168–169 °C. 1H NMR (700 MHz, DMSO-d6): δ 0.81 (t, J = 5.6 Hz, 3H), 1.20 (m, 6H), 1.35 (m, 2H), 1.92 (m, 2H), 2.22 (t, J = 7.0 Hz, 2H), 3.01 (m, 2H), 3.20 (t, J = 7.0 Hz, 2H), 7.40 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.73 (t, J = 7.7 Hz, 1H), 7.82 (s, 1H), 8.02 (d, J = 7.7 Hz, 1H), 12.55 (s, 1H). 13C NMR (175 MHz, DMSO-d6): δ 13.9, 22.1, 25.0, 26.1, 29.1, 29.3, 31.0, 34.2, 38.5, 120.0, 125.5, 125.9, 126.0, 134.5, 148.4, 155.7, 161.3, 171.0. HRMS (ESI+) Calcd for C18H26N3O2S [M + H]+: 348.1740. Found: 348.1742 (Δ = −0.54 ppm).
Cell Culture and Transfection
COS-1 monkey epithelial, human fibrosarcoma HT-1080, and breast MCF-7 cancer cell lines were purchased from American Type Culture Collection (ATCC) and were maintained in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) containing 10% fetal calf serum under a 5% CO2 atmosphere. Transfection of plasmid DNA (human) into cells was achieved using polyethylenimine (Polysciences), and the transfected cells were incubated for 48 h at 37 °C followed by biochemical and biologic assays unless otherwise stated.
Two-Dimensional Dot Migration Assay
Transfected cells were mixed with an equal volume of neutralized type I collagen (3 mg mL–1) on ice. The cell–collagen mixture (1 μL of 1 × 107 cell/mL) was then dotted onto a 96-well plate. After solidification of cell–collagen dots, the cell–collagen hemispheres were covered with 100 μL of complete media and incubated for 8–18 h (incubation time varies by cell type), followed by staining with DAPI nuclear dye and counting of the migrated cells using a Nikon Elements Basic Research Software analysis tools.50
Co-immunoprecipitation
Twenty-four hours following transfection, the conditioned medium in the absence of serum was collected from the transfected HT1080 cells. The cells were then lysed in RIPA buffer containing 10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, and a protease inhibitors mixture (Sigma). Both the conditioned medium and the cell lysates were incubated with specific antibodies overnight (18 h) at 4 °C. Antigen antibody complexes were precipitated with protein A-agarose beads followed by a brief centrifugation, washing, and then electrophoresis in a 10% SDS-polyacrylamide gel. Western blotting was then followed using a corresponding antibody as previously described.12
Immunofluorescence
Cultured cells were fixed with 4% paraformaldehyde/phosphate-buffered saline (PBS) followed by blocking with 5% bovine serum albumin (BSA)/PBS. Both FAK and PAX were detected with an anti-p-FAKTyr 576/577 and anti-p-PAXTyr118 antibody followed by secondary antibodies conjugated with antirabbit Alexa 568 or Alexa 488, respectively (Invitrogen). For colocalization experiments, MMP-9 was detected with an anti-MMP-9 antibody followed by a secondary antibody conjugated with antirabbit Alexa 488, while α4 and β1 integrins were detected with an anti-α4 integrin and anti-β1 integrin, respectively, followed by a secondary antibody conjugated with antimouse Alexa 568. Nuclei were counterstained with DAPI (Invitrogen). Images were captured using a Zeiss LSM 510 META NLO Two-Photon Laser Scanning Confocal Microscope System (Central Microscopy Imaging Center, Stony Brook University).
Fluorogenic Assay of Enzyme Activity
Mca-PLGL-Dpa-AR-NH2 fluorogenic peptide substrate (final concentration of 10 μM in DMSO) was incubated with the compounds and p-aminophenylmercuric acetate (APMA)-activated proMMP-9 for 30 min at 25 °C before detection. Fluorescence emission at 405 nm with excitation at 320 nm was measured in a fluorescent plate reader (Gemini EM; Molecular Devices).51
Cell Viability
HT1080 cells were cultured in complete media with or without compound 3c. Media and drugs were changed on a bidaily basis, and cell viability was monitored by MTT assay [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Promega, Madison, WI). Each day, cells were exposed to MTT and incubated at 37 °C for 4 h. The reaction was stopped, and formazan crystals were solubilized; the resultant solution was subject to colorimetric spectrophotometry and read at a wavelength of 570 nm.52
Chorioallantoic Membrane Angiogenesis and Invasion Assay
The chorioallantoic membrane (CAM) assay was performed as previously described.53 Fertilized white chicken eggs (SPF Premium, Charles River Laboratory, North Franklin, CT) were incubated at 37 °C in 70% humidity for 3 days. The embryos were then incubated ex ovo in a sterile Petri dish for 7 days. Gelatin sponges adsorbed with HT1080 cells treated with or without compound 3c were implanted on the CAM surface, and neovasculature was counted on day 4 postimplantation.54,55 For histochemical analysis of the chorioallantoic membrane, embryos were treated as for the angiogenesis assay, except at day 10 the embryos were inoculated with pretreated HT1080 cells mixed with a type IV collagen solution (3 mg mL–1). After a 7-day incubation, CAM segments containing the cell dot collagen mixture were isolated, formalin fixed, and sectioned by microtome into 6-μm sections after embedding in OCT. Sections were then stained with hematoxylin and counterstained with eosin.56
proMMP-9 Protein Purification
For purification of proMMP-2 and proMMP-9 protein, COS-1 cells stably expressing each respective MMP were incubated overnight in 10 mL of serum-free medium until 500 mL was collected for purification via a gelatin-sepharose column. Both proMMP-2 and proMMP-9 have three fibronectin repeats present in their gelatin binding domain, which allowed us to capture and collect the secreted protein present in the serum-free medium fraction. Protein was then eluted off the gelatin sepharose column using 5% DMSO and then dialyzed to remove any organic contaminants. A coomassie blue gel was then run to check the purity of the samples to ensure no protein contamination was present in the final solution. All methods were followed as previously described.57
Fluorescence Spectroscopy
Binding of compound 3c to proMMP-9 was assayed by observing the change of tryptophan emission upon binding. Purified recombinant proMMP-9 (0.5 μM) or MMP-9/MMP-2PEX (0.5 μM) was diluted in TNC buffer (50 mM Tris-HCl at pH 7.5, 150 nM NaCl, 10 mM CaCl2; pH 7.5) in the presence or absence of compound 3c. As a control for protein stability and loss, an analogous buffer solution was added to the protein. The protein sample was excited at 280 nm, and emission scans were collected from 300 to 425 nm, using slit widths of 0.3 nm on a QM-4/200SE spectrofluorimeter with double excitation and emission monochromators. Between each titration, the protein/inhibitor mixture was gently stirred for 1 min at RT before collecting each reading. Three emission scans were collected and averaged at each concentration. The Kd was determined using the Prism software package (GraphPad V5) to fit the data to the equation ΔF/ΔFMax, in which ΔF is the nanometer shift for a given titration and ΔFMax is the maximal nanometer shift observed overall.
Small Molecule DOCKing
To DOCK the MMP-9 PEX domain inhibitors and predict the binding mode, the inhibitors were first created via the Advanced Chemistry Development Chemsketch software (ACD Laboratories, Toronto, Canada) in the MOL format. The molecules were then loaded into the program Avogadro to undergo energy minimization and were then saved as PDB format files. Once in PDB format, the molecules were individually loaded into the AutoDock Tools (The Scripps Research Institute, La Jolla, California) program and assigned charges, rotatable bonds, and a root. The molecules were saved as a pdbqt files and then used for DOCKing. The crystal structure of the PEX domain of MMP-9 was downloaded from rcsb.org (PDB code 1ITV). Chain B and all sulfate ions were deleted, followed by a brief energy minimization of the receptor via the AMBER force field. The parameters are as follows.
Steepest descent steps: 100
Steepest descent size (Angstroms): 0.01
Conjugate gradient steps: 30
Conjugate gradient step size (Ångstroms): 0.02
Force field: AMBER ff14SB (AM1-BCC)
The compounds docked underwent energy minimization via the Merck Molecular Force Field 94s (MMFF94s). DOCKing was performed via AutoDock Vina with the following parameters (untransformed coordinates of protein):
Box center (x axis) = −42.494
Box center (y axis) = −27.748
Box center (z axis) = 5.00
Box size (x axis) = 22
Box size (y axis) = 22
Box size (z axis) = 20
Exhaustiveness = 64
The results were loaded in UCSF Chimera (UCSF, San Francisco, California) and saved as JPEG images.
Statistical Analysis
Data are expressed as the mean ± standard error of triplicates. Each experiment was repeated at least three times. The Student t test and analysis of variants (ANOVA) were used to assess differences with P < 0.05, **P < 0.01, and ***P < 0.001.
Acknowledgments
This work was supported by National Institutes of Health 1R01CA166936 (to J.C. and N.S.S.), T32GM092714 (to V.M.A.) and 1R35GM119437 (to M.A.S.) and a seed grant from the Institute of Chemical Biology and Drug Discovery (to I.O.). We are grateful to Weiyi Li for all her hard work and guidance in helping use the Zeiss LSM 510 META NLO Two-Photon Laser Scanning Confocal Microscope System at SBU’s CMIC core facility. We also thank all members of the Cao and Sampson laboratory for insightful discussion and support given throughout the duration of this project.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00758.
Supplemental experimental procedures, figures, data, and spectra for compounds are described (PDF)
Author Contributions
J.C. and N.S.S. conceived the project. V.M.A. performed and contributed to designing all protocols and experiments. I.O, A.K., X.R., K.K., and Q.G. contributed through designing and synthesizing all PEX-9 parental and derivative compounds. M.A. contributed through performing the DOCKing analysis of all derivative compounds. M.T. and M.A.S. contributed through helping design and perform STD NMR experiments in addition to kinetic experiments to measure Kd values. V.M.A. prepared all figures and tables. All authors contributed to writing, reviewing, commenting on, and approving the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Weigelt B.; Peterse J. L.; van ’t Veer L. J. (2005) Breast cancer metastasis: markers and models. Nat. Rev. Cancer 5, 591–602. 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- Curran S.; Murray G. I. (1999) Matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 189, 300–308. . [DOI] [PubMed] [Google Scholar]
- Curran S.; Murray G. I. (2000) Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur. J. Cancer 36, 1621–1630. 10.1016/S0959-8049(00)00156-8. [DOI] [PubMed] [Google Scholar]
- Deryugina E. I.; Quigley J. P. (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 25, 9–34. 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K.; Plaks V.; Werb Z. (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67. 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens L. M.; Fingleton B.; Matrisian L. M. (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392. 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Cathcart J.; Pulkoski-Gross A.; Cao J. (2015) Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2, 26–34. 10.1016/j.gendis.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J., Chan M. F., and Werb Z. (2016) Metalloproteinases: a Functional Pathway for Myeloid Cells, Microbiol. Spectrum 4, DOI: 10.1128/microbiolspec.MCHD-0002-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez D.; Morrison C. J.; Overall C. M. (2010) Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta, Mol. Cell Res. 1803, 39–54. 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Fanjul-Fernandez M.; Folgueras A. R.; Cabrera S.; Lopez-Otin C. (2010) Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta, Mol. Cell Res. 1803, 3–19. 10.1016/j.bbamcr.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Dufour A.; Sampson N. S.; Zucker S.; Cao J. (2008) Role of the hemopexin domain of matrix metalloproteinases in cell migration. J. Cell. Physiol. 217, 643–651. 10.1002/jcp.21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour A.; Zucker S.; Sampson N. S.; Kuscu C.; Cao J. (2010) Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J. Biol. Chem. 285, 35944–35956. 10.1074/jbc.M109.091769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha H.; Kopetzki E.; Huber R.; Lanzendorfer M.; Brandstetter H. (2002) Structural basis of the adaptive molecular recognition by MMP9. J. Mol. Biol. 320, 1065–1079. 10.1016/S0022-2836(02)00558-2. [DOI] [PubMed] [Google Scholar]
- Sternlicht M. D.; Werb Z. (2001) How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17, 463–516. 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadag A.; Fedarko N. S.; Fisher L. W. (2005) Dentin matrix protein 1 enhances invasion potential of colon cancer cells by bridging matrix metalloproteinase-9 to integrins and CD44. Cancer Res. 65, 11545–11552. 10.1158/0008-5472.CAN-05-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai B.; Rogers M. J.; Chellaiah M. A. (2007) Mechanisms of osteopontin and CD44 as metastatic principles in prostate cancer cells. Mol. Cancer 6, 18–42. 10.1186/1476-4598-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo-Munoz J.; Ugarte-Berzal E.; Terol M. J.; Van den Steen P. E.; Hernandez del Cerro M.; Roderfeld M.; Roeb E.; Opdenakker G.; Garcia-Marco J. A.; Garcia-Pardo A. (2010) Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia b cell survival through its hemopexin domain. Cancer Cell 17, 160–172. 10.1016/j.ccr.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Redondo-Munoz J.; Ugarte-Berzal E.; Garcia-Marco J. A.; del Cerro M. H.; Van den Steen P. E.; Opdenakker G.; Terol M. J.; Garcia-Pardo A. (2008) Alpha4beta1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 112, 169–178. 10.1182/blood-2007-08-109249. [DOI] [PubMed] [Google Scholar]
- Stefanidakis M.; Bjorklund M.; Ihanus E.; Gahmberg C. G.; Koivunen E. (2003) Identification of a negatively charged peptide motif within the catalytic domain of progelatinases that mediates binding to leukocyte beta 2 integrins. J. Biol. Chem. 278, 34674–34684. 10.1074/jbc.M302288200. [DOI] [PubMed] [Google Scholar]
- Bjorklund M.; Heikkila P.; Koivunen E. (2004) Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J. Biol. Chem. 279, 29589–29597. 10.1074/jbc.M401601200. [DOI] [PubMed] [Google Scholar]
- Ugarte-Berzal E.; Bailon E.; Amigo-Jimenez I.; Vituri C. L.; del Cerro M. H.; Terol M. J.; Albar J. P.; Rivas G.; Garcia-Marco J. A.; Garcia-Pardo A. (2012) A 17-residue sequence from the matrix metalloproteinase-9 (MMP-9) hemopexin domain binds alpha4beta1 integrin and inhibits MMP-9-induced functions in chronic lymphocytic leukemia B cells. J. Biol. Chem. 287, 27601–27613. 10.1074/jbc.M112.354670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte-Berzal E.; Bailon E.; Amigo-Jimenez I.; Albar J. P.; Garcia-Marco J. A.; Garcia-Pardo A. (2014) A novel CD44-binding peptide from the pro-matrix metalloproteinase-9 hemopexin domain impairs adhesion and migration of chronic lymphocytic leukemia (CLL) cells. J. Biol. Chem. 289, 15340–15349. 10.1074/jbc.M114.559187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries M. J.; Komoriya A.; Akiyama S. K.; Olden K.; Yamada K. M. (1987) Identification of two distinct regions of the type III connecting segment of human plasma fibronectin that promote cell type-specific adhesion. J. Biol. Chem. 262, 6886–6892. [PubMed] [Google Scholar]
- Garcia-Pardo A.; Wayner E. A.; Carter W. G.; Ferreira O. C. Jr. (1990) Human B lymphocytes define an alternative mechanism of adhesion to fibronectin. The interaction of the alpha 4 beta 1 integrin with the LHGPEILDVPST sequence of the type III connecting segment is sufficient to promote cell attachment. J. Immunol. 144, 3361–3366. [PubMed] [Google Scholar]
- Wayner E. A.; Kovach N. L. (1992) Activation-dependent recognition by hematopoietic cells of the LDV sequence in the V region of fibronectin. J. Cell Biol. 116, 489–497. 10.1083/jcb.116.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyano J. V.; Carnemolla B.; Dominguez-Jimenez C.; Garcia-Gila M.; Albar J. P.; Sanchez-Aparicio P.; Leprini A.; Querze G.; Zardi L.; Garcia-Pardo A. (1997) Fibronectin type III5 repeat contains a novel cell adhesion sequence, KLDAPT, which binds activated alpha4beta1 and alpha4beta7 integrins. J. Biol. Chem. 272, 24832–24836. 10.1074/jbc.272.40.24832. [DOI] [PubMed] [Google Scholar]
- Dufour A.; Sampson N. S.; Li J.; Kuscu C.; Rizzo R. C.; Deleon J. L.; Zhi J.; Jaber N.; Liu E.; Zucker S.; Cao J. (2011) Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 71, 4977–4988. 10.1158/0008-5472.CAN-10-4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkedal-Hansen H.; Moore W. G.; Bodden M. K.; Windsor L. J.; Birkedal-Hansen B.; DeCarlo A.; Engler J. A. (1993) Matrix metalloproteinases: a review. Crit. Rev. Oral Biol. Med. 4, 197–250. 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- Nagase H.; Suzuki K.; Morodomi T.; Enghild J. J.; Salvesen G. (1992) Activation mechanisms of the precursors of matrix metalloproteinases 1, 2 and 3. Matrix Suppl. 1, 237–244. [PubMed] [Google Scholar]
- Galazka G.; Windsor L. J.; Birkedal-Hansen H.; Engler J. A. (1996) APMA (4-aminophenylmercuric acetate) activation of stromelysin-1 involves protein interactions in addition to those with cysteine-75 in the propeptide. Biochemistry 35, 11221–11227. 10.1021/bi960618e. [DOI] [PubMed] [Google Scholar]
- Belmokhtar C. A.; Hillion J.; Segal-Bendirdjian E. (2001) Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20, 3354–3362. 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- Lu P.; Takai K.; Weaver V. M.; Werb Z. (2011) Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harbor Perspect. Biol. 3, a005058. 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C.; Chou J.; Werb Z. (2014) Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801. 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky E. S.; Raeeszadeh-Sarmazdeh M.; Radisky D. C. (2017) Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell. Biochem. 118, 3531–3548. 10.1002/jcb.26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtowicz-Praga S. M.; Dickson R. B.; Hawkins M. J. (1997) Matrix metalloproteinase inhibitors. Invest. New Drugs 15, 61–75. 10.1023/A:1005722729132. [DOI] [PubMed] [Google Scholar]
- Palacios E. H.; Weiss A. (2004) Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000. 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- Hsia D. A.; Lim S. T.; Bernard-Trifilo J. A.; Mitra S. K.; Tanaka S.; den Hertog J.; Streblow D. N.; Ilic D.; Ginsberg M. H.; Schlaepfer D. D. (2005) Integrin alpha4beta1 promotes focal adhesion kinase-independent cell motility via alpha4 cytoplasmic domain-specific activation of c-Src. Mol. Cell. Biol. 25, 9700–9712. 10.1128/MCB.25.21.9700-9712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Okura M.; Imamoto A. (2002) Focal adhesions require catalytic activity of Src family kinases to mediate integrin-matrix adhesion. Mol. Cell. Biol. 22, 1203–1217. 10.1128/MCB.22.4.1203-1217.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. K.; Cheng Y.; Liang Cheng M.; Yu L.; Mu M.; Li H.; Liu Y.; Zhang B.; Yao Y.; Guo H.; Wang R.; Zhang Q. (2016) Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Sci. Rep. 6, 19276–19288. 10.1038/srep19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff M. A.; Serrels B.; Fincham V. J.; Frame M. C.; Carragher N. O. (2004) SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol. Cell. Biol. 24, 8113–8133. 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. C.; Cary L. A.; Jamieson J. S.; Cooper J. A.; Turner C. E. (2005) Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol. Biol. Cell 16, 4316–4328. 10.1091/mbc.E05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S. K.; Schlaepfer D. D. (2006) Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523. 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Wu L.; Bernard-Trifilo J. A.; Lim Y.; Lim S. T.; Mitra S. K.; Uryu S.; Chen M.; Pallen C. J.; Cheung N. K.; Mikolon D.; Mielgo A.; Stupack D. G.; Schlaepfer D. D. (2008) Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene 27, 1439–1448. 10.1038/sj.onc.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen B.; Johnson F. M. (2011) Regulation of SRC family kinases in human cancers. J. Signal Transduction 2011, 865819–865833. 10.1155/2011/865819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osherov N.; Levitzki A. (1994) Epidermal-growth-factor-dependent activation of the src-family kinases. Eur. J. Biochem. 225, 1047–1053. 10.1111/j.1432-1033.1994.1047b.x. [DOI] [PubMed] [Google Scholar]
- Mao W.; Irby R.; Coppola D.; Fu L.; Wloch M.; Turner J.; Yu H.; Garcia R.; Jove R.; Yeatman T. J. (1997) Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 15, 3083–3090. 10.1038/sj.onc.1201496. [DOI] [PubMed] [Google Scholar]
- Vandooren J.; Born B.; Solomonov I.; Zajac E.; Saldova R.; Senske M.; Ugarte-Berzal E.; Martens E.; Van den Steen P. E.; Van Damme J.; Garcia-Pardo A.; Froeyen M.; Deryugina E. I.; Quigley J. P.; Moestrup S. K.; Rudd P. M.; Sagi I.; Opdenakker G. (2015) Circular trimers of gelatinase B/matrix metalloproteinase-9 constitute a distinct population of functional enzyme molecules differentially regulated by tissue inhibitor of metalloproteinases-1. Biochem. J. 465, 259–270. 10.1042/BJ20140418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Miyamoto S.; Mekada E. (2000) Integrin alpha 2 beta 1-dependent EGF receptor activation at cell-cell contact sites. J. Cell Sci. 113 (12), 2139–2147. [DOI] [PubMed] [Google Scholar]
- Friedl P.; Wolf K. (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374. 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- Alford V. M.; Roth E.; Zhang Q.; Cao J. (2016) A Novel Collagen Dot Assay for Monitoring Cancer Cell Migration. Methods Mol. Biol. 1406, 181–187. 10.1007/978-1-4939-3444-7_15. [DOI] [PubMed] [Google Scholar]
- Knight C. G.; Willenbrock F.; Murphy G. (1992) A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 296, 263–266. 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Deryugina E. I.; Quigley J. P. (2008) Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem. Cell Biol. 130, 1119–1130. 10.1007/s00418-008-0536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribatti D.; Nico B.; Vacca A.; Roncali L.; Burri P. H.; Djonov V. (2001) Chorioallantoic membrane capillary bed: a useful target for studying angiogenesis and anti-angiogenesis in vivo. Anat. Rec. 264, 317–324. 10.1002/ar.10021. [DOI] [PubMed] [Google Scholar]
- Ribatti D.; Nico B.; Vacca A.; Presta M. (2006) The gelatin sponge-chorioallantoic membrane assay. Nat. Protoc. 1, 85–91. 10.1038/nprot.2006.13. [DOI] [PubMed] [Google Scholar]
- LeBleu V. S.; Macdonald B.; Kalluri R. (2007) Structure and function of basement membranes. Exp. Biol. Med. (London, U. K.) 232, 1121–1129. 10.3181/0703-MR-72. [DOI] [PubMed] [Google Scholar]
- Imai K.; Okada Y. (2008) Purification of matrix metalloproteinases by column chromatography. Nat. Protoc. 3, 1111–1124. 10.1038/nprot.2008.74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.