

Abstract
Nanoliposomal irinotecan (nal‐IRI) is a liposomal formulation of irinotecan with a longer half‐life (t1/2), higher plasma total irinotecan (tIRI), and lower SN‐38 maximum concentration (C max) compared with nonliposomal irinotecan. Population pharmacokinetic (PK) analysis of nal‐IRI was performed for tIRI and total SN‐38 (tSN38) using patient samples from six studies. PK‐safety association was evaluated for neutropenia and diarrhea in 353 patients. PK‐efficacy association was evaluated from a phase III study in pancreatic cancer NAPOLI1. Efficacy was associated with longer duration of unencapsulated SN‐38 (uSN38) above a threshold and higher Cavg of tIRI, tSN38, and uSN38. Neutropenia was associated with uSN38 C max and diarrhea with tIRI C max. Baseline predictive factors were race, body surface area, and bilirubin. Analysis identified PK factors associated with efficacy, safety, and predictive baseline factors. The results support the benefit of nal‐IRI dose of 70 mg/m2 (free‐base; equivalent to 80 mg/m2 salt base) Q2W over 100 mg/m2 Q3W.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Liposomal encapsulation extends the half‐lives of irinotecan; however, the association of PK, and the impact of liposomal encapsulation to efficacy or safety endpoints have never been reported from a large number of patients.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study aimed to quantify plasma PK with liposomal irinotecan treatment to discern the differences between derived PK parameters Cavg, C max, and tuSN38>thr and their impact on safety and efficacy, and to identify relevant baseline factors.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ These analyses identified that efficacy was associated with the average concentration of SN‐38 and the duration of SN‐38 above a threshold, while safety was associated with maximum concentrations.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
☑ This study provides an example of PK modification by liposomal encapsulation resulted in ability to differentiate aspects of PK associated to efficacy and safety. The results support the choice of the optimal dose regimen.
Liposomal formulations have been investigated as a drug delivery system to modulate the pharmacological properties of small molecules.1 In cancer therapeutics, liposomal formulations can deposit in tumors through leaky vasculature by the enhanced permeability and retention effect (EPR), creating a local depot for drug release.2 Nanoliposomal irinotecan (nal‐IRI, MM‐398, PEP02, BAX2398) is a liposomal formulation of irinotecan for intravenous injection designed to combine the properties of long plasma circulation and increased delivery of irinotecan to tumor lesions via the EPR effect. The clinical benefit of nal‐IRI was demonstrated in a phase III study in patients with metastatic pancreatic cancer previously treated with a gemcitabine‐based therapy (NAPOLI‐1).3 The results showed that nal‐IRI in combination with 5‐fluorouracil (5‐FU) and leucovorin (LV) significantly increased median overall survival (OS) compared with a 5‐FU/LV control arm (6.1 and 4.2 months, respectively), with an unstratified hazard ratio (HR) of 0.67 (P = 0.012). Additionally, the combination achieved a median progression‐free survival (PFS) that approximately doubled that of the control arm (3.1 and 1.5 months, respectively; HR of 0.56; P = 0.0001). As neutropenia and diarrhea are side effects that are associated with irinotecan, further investigation with nal‐IRI is warranted.4, 5, 6
The clinical pharmacokinetics (PK) of nal‐IRI were previously compared with those of nonliposomal irinotecan (irinotecan HCl) in a phase II study in patients with gastric cancer.7 Reanalysis of the data showed that compared with irinotecan HCl 300 mg/m2 every 3 weeks (Q3W) (n = 27), nal‐IRI 100 mg/m2 Q3W (n = 37; free‐base, equivalent to 120 mg/m2 irinotecan hydrochloride trihydrate salt) had a total irinotecan (tIRI) maximum concentration (C max) that was 13.4‐times higher, a half‐life (t1/2) that was 2.0 times longer, and an area under the concentration‐time curve (AUC0‐∞) that was 46.2‐times greater (reanalysis by calculating geometric means instead of arithmetic means and by reporting the actual values instead of dose‐normalized values). The t1/2 and AUC0‐∞ of SN‐38, the active metabolite of irinotecan, were also increased relative to nonliposomal irinotecan (3.0‐ and 1.4‐times, respectively), while maintaining a 5.3‐times lower SN‐38 C max. In a separate clinical trial, nal‐IRI–mediated tumor delivery was evaluated in tumor biopsies from 13 patients collected 72 h after the administration of 70 mg/m2 nal‐IRI.8 tIRI in the tumor was 0.5‐times those in the plasma; however, the total SN‐38 (tSN38) was 6‐times higher in tumor than in plasma, and the ratio of tSN38:tIRI (a measure of the extent of conversion) was 8‐times higher in tumor than in plasma.
The extended plasma PK of liposomal formulations provides an opportunity to dissect the differences between derived PK parameters, including average concentration (Cavg) and C max, and time above a threshold (tuSN38>thr), and their association with efficacy and safety. With nonliposomal irinotecan, Cavg and C max were highly correlated, and therefore, the dichotomization of the associations with efficacy and safety endpoints have been difficult to elucidate.9 However, these parameters were less correlated with nal‐IRI administration, and therefore, we aim to evaluate the association of these parameters with efficacy and safety in patients treated with nal‐IRI.
RESULTS
Patients
Samples for PK measurements were collected during the first cycle of nal‐IRI treatment in five phase I–II studies and one phase III study. Of the 368 treated patients from the six studies, 353 (96%) had samples analyzed for PK measurement, including 97% (258/266) of patients in the NAPOLI‐1 study. Patient characteristics at baseline are listed in Table 1. Patients with hepatic or renal impairment were excluded from the enrollment; nevertheless, 20 patients were enrolled with bilirubin ≥1 mg/dL (19/20 had bilirubin 1–2 mg/dL; one patient had bilirubin >2 mg/dL). The majority (73%) was obtained from patients with metastatic pancreatic cancer. Most patients received an initial dose of 100 mg/m2 (53%) or 70 mg/m2 (39%). Most patients were either Caucasian (52%) or East Asian (42%).
Table 1.
Patient characteristics at baseline
| Characteristics | Subgroup | N (%)a |
Median (5th and 95th percentile) |
|---|---|---|---|
| Sex | Female | 157 (44) | |
| Male | 196 (56) | ||
| Race | Caucasian | 182 (52) | |
| Others | 21 (6) | ||
| East Asian | 150 (42) | ||
| Liver metastasis (for NAPOLI‐1 only) | No | 87 (34) | |
| Yes | 171 (66) | ||
| Study name | NAPOLI‐1 | 258 (73) | |
| Others | 95 (27) | ||
| UGT1A1*28 (for NAPOLI‐1 only) | Non 7/7 | 244 (95) | |
| 7/7 | 14 (5) | ||
| Treatment (for NAPOLI‐1 only) | nal‐IRI+5FU/LV | 116 (45) | |
| nal‐IRI (mono) | 142 (55) | ||
| Tumor type at diagnosis | Colorectal cancer | 18 (5) | |
| Gastric & GEJ cancer | 37 (10) | ||
| Metastatic pancreatic cancer | 258 (73) | ||
| Solid tumor | 40 (11) | ||
| Initial dose, mg/m2 b | 50 (60) | 4 (1) | |
| 70 (80) | 141 (40) | ||
| 80 (90) | 6 (2) | ||
| 90 (100) | 11 (3) | ||
| 100 (120) | 187 (53) | ||
| 150 (180) | 4 (1) | ||
| Age, y | 353 | 63 (39.8, 79.2) | |
| Albumin, g/L | 349 | 40 (29, 47) | |
| ALT, U/L | 352 | 25 (8.9, 96.3) | |
| AST, U/L | 352 | 29 (14.7, 81.9) | |
| Bilirubin (umol/L) | 352 | 7 (3, 19) | |
| BSA, m2 | 353 | 1.7 (1.3, 2.2) | |
| CrCl, 10‐3 L/s | 352 | 1.36 (0.66, 2.53) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BSA, body surface area; GEJ, gastroesophageal junction.
Percent only included in baseline characteristics with subcategories.
Dose is given based on irinotecan free base with the original protocol dose (based on irinotecan hydrochloride trihydrate is in parentheses.
PK parameter estimates
A total of 1,792 tIRI samples from 355 subjects and 1,765 tSN38 samples from 353 subjects were analyzed. Typical observed and predicted PK with 70 mg/m2 and 100 mg/m2 are shown in Figure 1. The final model sufficiently described the data, as evidenced by the comparison of the data and model fits (Figure S3) and by visual predictive checks for the overall data, and stratified by dose (Figure S4–S5). Final estimated parameters of IRI and SN‐38 in the population PK models are listed in Table S6 and Table S7, respectively. The tSN38 and uSN38 were highly correlated (Kendall τ of 0.81 and 0.70 for Cavg and C max, respectively). The C max and Cavg had lower correlation for tIRI and uSN38 (Kendall τ of 0.31 and 0.44, respectively).
Figure 1.
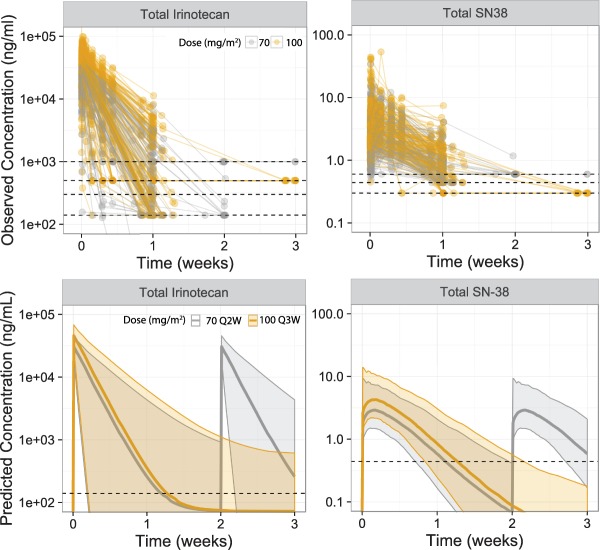
Observed and predicted typical plasma concentration profile of total irinotecan and SN‐38 in patients administered nal‐IRI 70 mg/m2 Q2W or nal‐IRI 100 mg/m2 Q3W.
The estimated PK parameters are provided in Table 2. The estimated initial and terminal t1/2 of tIRI were 38.2 [95% confidence interval (CI) 23.2–56.7] and 12,200 (95% CI 3,990–50,200) h; the t1/2 of SN‐38 was 38.2 (36.5–41.9) h. The estimated terminal t1/2 for tIRI should be treated with cautions because of the limited number of samples measured by assay with a lower limit of quantification. Compared to nal‐IRI 100 mg/m2 Q3W, nal‐IRI 70 mg/m2 Q2W was predicted to have similar tIRI and tSN38 Cavg, 1.5‐fold lower tIRI and tSN38 C max, and longer tuSN38>thr in the first 6 weeks. tIRI was approximately three orders of magnitude higher than tSN38. The estimated volume was 4.58 L, a value comparable to typical blood volume.
Table 2.
Summary of irinotecan and SN‐38 pharmacokinetics parameters by nal‐IRI dose regimen in NAPOLI‐1
| Analyte | Pharmacokinetic parametera |
70 (80) mg/m2
Q2Wb |
100 (120) mg/m2
Q3Wb |
|---|---|---|---|
| Total irinotecan | Cavg, mg/L | 1.19 (0.91–1.55) | 1.66 (1.33–2.05) |
| C max, mg/L | 26.6 (24.1–29.3) | 41.5 (39.8–43.2) | |
| Clearance, L/week | 13.3 (9.17, 22.8) | ||
| Volume, L | 4.58 (4.14, 4.99) | ||
| First‐phase t1/2, h | 38.2 (23.2–56.7) | ||
| Terminal t1/2, h | 12200 (3990–50200) | ||
| Total SN‐38 | Cavg, ng/mL | 0.721 (0.667–0.778) | 0.870 (0.821–0.922) |
| C max, ng/mL | 2.64 (2.47–2.83) | 3.99 (3.77–4.23) | |
| Clearance, L/week | 14.0 (12.7–14.6) | ||
| Terminal t1/2, h | 38.2 (36.5–41.9) | ||
| Unencapsulated SN‐38 | Cavg, ng/mL | 0.589 (0.543–0.639) | 0.702 (0.661–0.745) |
| C max, ng/mL | 2.07 (1.93–2.23) | 3.05 (2.89–3.21) | |
| tuSN38>thr, weeks (first 6 weeks, based on actual doses) | 4.77 (4.59–4.95) | 4.28 (4.12–4.44) | |
| tuSN38>thr, weeks (first 6 weeks, based on simulated doses) | 5.71 (5.64–5.79) | 4.80 (4.69–4.92) | |
Cavg, average concentration; C max, maximum concentration; tuSN38>thr, time uSN38>threshold.
For Cavg, Cmax, and tuSN38>thr, median values and 95% prediction intervals (representing interpatient variabilities) were obtained from NAPOLI1 patients; for Clearance, Volume, and t1/2, median values and 95% confidence intervals (representing precision of parameter estimates) were obtained from bootstrapping.
Dose is given based on irinotecan free base with the original protocol dose (based on irinotecan hydrochloride trihydrate) is in parentheses.
Exposure–efficacy relationships
In the nal‐IRI+5FU/LV arm of NAPOLI‐1, longer OS and PFS were associated with longer tuSN38>thr and higher Cavg of tIRI, tSN38, and uSN38, with the highest association observed for tuSN38>thr. C max of tIRI, tSN38, or uSN38 was not predictive of OS (P = 0.58–0.98). The relationship between OS and quartiles of tuSN38>thr for the nal‐IRI 5‐FU/LV and nal‐IRI monotherapy arms are provided in Figure 2 and Figure S6, respectively. Longer tuSN38>thr was associated with a higher probability of achieving objective response in the nal‐IRI+5‐FU/LV arm (Figure S8). This association was not observed in the nal‐IRI monotherapy arm. The association between OS and uSN38 Cavg is provided in Figure S7, which also shows prolonged OS with higher uSN38 Cavg (uSN38 Cavg and tuSN38>thr is correlated with Kendall τ of 0.48).
Figure 2.
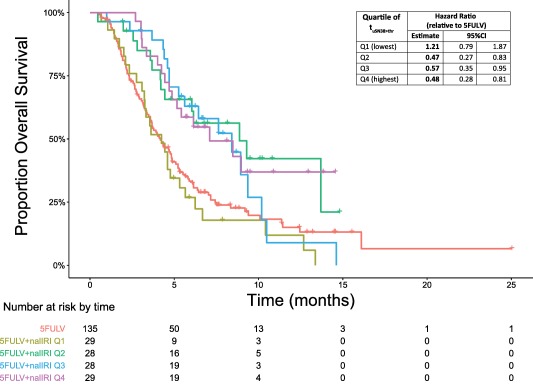
Kaplan–Meier plot of overall survival by quartiles of unencapsulated SN‐38 (uSN38) time above threshold in the nal‐IRI+5‐FU/LV arm of NAPOLI‐1.
Exposure–safety relationships
A total of 353 patients were included in the PK‐safety analysis. Neutropenia was most strongly associated with uSN38 C max (Figure 3). Higher uSN38 was associated with a higher probability of both incidence and severity of neutropenia. The association was observed in both grade ≥1 and grade ≥3 neutropenia. The association with neutropenia was more significant with the uSN38 than tSN38 (for example, for the incidence of neutropenia grade ≥3, the association P‐values were <0.001 and 0.08 for uSN38 and for tSN38, respectively). The association between uSN38 and neutropenia was also greater for C max than for Cavg (e.g., grade ≥3 neutropenia: P = <0.001 vs. P = 0.045 with uSN38 C max and Cavg, respectively). In a multivariate logistic regression analysis of grade ≥3 neutropenia (Table S8), the association between uSN38 C max and neutropenia was still significant (P = 0.00005) even after adding factors known to predict neutropenia (baseline ANC and 5‐FU/LV coadministration). When baseline factors predictive of uSN38 were included (race, bilirubin, and body surface area, BSA), the association with uSN38 C max was only borderline significant (P = 0.068).
Figure 3.
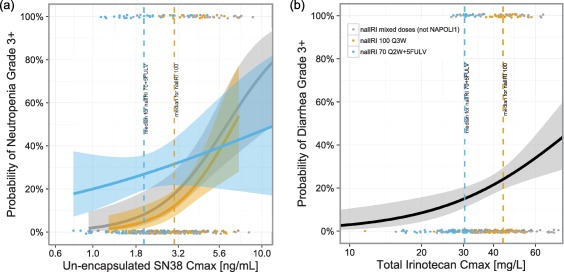
Incidence rates of neutropenia (a) and diarrhea (b) grade ≥3 by plasma PK in patients treated with nal‐IRI.
Diarrhea was most strongly associated with tIRI C max (Figure 3). Higher tIRI C max was associated with a higher incidence and severity of diarrhea. The association was significant for grade ≥3 diarrhea but not for grade ≥1 diarrhea. The association between grade ≥3 diarrhea and tIRI was more significant for C max (P = 0.001) than for Cavg (P = 0.019). The association between tIRI C max and diarrhea was observed in each of the Caucasian and Asian subpopulations. In NAPOLI‐1, this association was observed within the nal‐IRI monotherapy arm, but not within the nal‐IRI+5FU/LV arm. This was likely due to the absence of patients with high tIRI C max in the nal‐IRI+5FU/LV arm and lower nal‐IRI dose. In a multivariate logistic regression analysis of grade ≥3 diarrhea (Table S9), the identified predictive factors were tIRI C max and race (Caucasian vs. East Asian).
Analysis of the NAPOLI‐1 safety data showed that compared with Caucasian patients, East Asian patients who received nal‐IRI+5‐FU/LV had a higher incidence of NCI CTCAE Grade 3 or 4 neutropenia (55% (18/33) vs. 18% (13/73), respectively), yet a lower incidence of Grade 3 or 4 diarrhea (3.0% (1/33) vs. 19.2% (14/73)), respectively.20 Therefore, the differences in the observed rates of neutropenia and diarrhea by race can be explained by the racial differences in the C max of tIRI and uSN38.
Baseline factors predictive of plasma PK
Baseline factors evaluated for associations to plasma PK include: BSA, demographics, hepatic and renal function, pharmacogenomics (UGT1A1*28), and extrinsic factors (Table S3, Figures S9, S10). The significant factors and the corresponding tIRI and uSN38 for nal‐IRI 70 mg/m2 are summarized in Figure 4.
Figure 4.
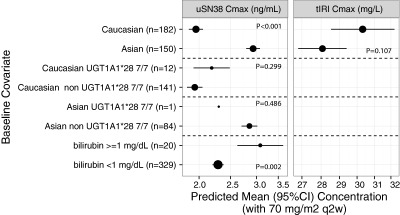
Selected baseline factors and associated plasma total irinotecan and unencapsulated SN‐38 C max with nal‐IRI 70 mg/m2.
Factors with significant association with tIRI PK were race and BSA. Factors with significant association with tSN38 were race, BSA, and bilirubin. Asians had lower tIRI and higher uSN38 compared with Caucasians (7% and 78% lower tIRI C max and Cavg, 50% and 20% higher uSN38 C max and Cavg; all P ≤ 0.001). In the population PK model that accounted for multivariate analysis (including BSA), race remained a significant factor for both tIRI and tSN38 (Tables S6, S7). Comparison of BSA‐based dosing to fixed dosing (70 mg/m2 or an equivalent fixed dose of 116.7 mg) revealed that BSA‐based dosing reduced variability of tIRI and uSN38 C max (3% and 14% less interquartile range, Table S10). This result implies a benefit of BSA‐based dosing in reducing the variability of tIRI and uSN38 C max. While the number of patients with elevated bilirubin was small (n = 20), bilirubin was found to be a significant factor of tSN38: compared with patients with bilirubin <1 mg/dL, patients with bilirubin ≥1 mg/dL had a higher uSN38 Cavg (43% higher) and C max (35% higher).
UGT1A1*28, a pharmacogenomic biomarker, was not a significant predictor of SN‐38 with nal‐IRI administration. In the population PK dataset, the prevalence of UGT1A1*28 7/7 homozygosity in Asians was low (2/129 (1.5%)). Compared with non‐7/7 homozygous Caucasians, 7/7 homozygous Caucasians had similar uSN38 C max if both were dosed at 70 mg/m2 (Figure 4; 2.19 (95% CI 1.92–2.49, n = 12) and 1.94 (95% CI 1.84–2.05, n = 141) ng/mL; P = 0.30; geometric mean ratio: 1.13 (95% CI 0.90–1.42). In NAPOLI‐1, the actual dose homozygous patients received were lower than the dose in nonhomozygous patients). The estimated SN‐38 clearance in UGT1A1 7/7 homozygous was 1.000‐times (0.0% difference) the clearance in non‐7/7 (Table S7). A sensitivity analysis was performed to estimate the SN‐38 clearance by more detailed categories of UGT1A1*28 alleles (separate evaluation for 6/6, 6/7, and 7/7; Table S11). The estimated clearance for UGT1A1*28 6/7 and UGT1A1*28 7/7 were within 0.0% and 2.7% of the clearance for UGT1A1*28 6/6. These results indicate that UGT1A1 is not a significant covariate to SN‐38 clearance.
Other baseline factors evaluated were found not to have significant associations with tIRI or uSN38. Among measures of hepatic functions other than bilirubin, albumin had a weak association with tIRI, but not tSN38 nor uSN38. The direction of the albumin‐tIRI association was the opposite of that expected in hepatic impairment and opposite of the observed diminished clearance reported in patients with hepatic impairment administered with nonliposomal irinotecan.19 Because of the lack of association with the active metabolite SN‐38, the effect of albumin is unlikely to be clinically relevant. Sex and creatinine clearance were not significantly associated with SN‐38 after adjusting for BSA.
DISCUSSION
Similar to the liposomal formulation of doxorubicin, the liposomal formulation of irinotecan modifies the pharmacological properties of irinotecan, resulting in extended half‐lives of plasma total irinotecan and SN‐38. The extended plasma PK observed with nal‐IRI provides a tool to distinguish Cavg and C max, as evidenced by the low correlation between these parameters, and therefore is useful to evaluate pharmacological properties associated with efficacy and safety. The vastly different estimated volumes highlight the different disposition characteristics with liposomal formulation.7
In pancreatic cancer patients treated with nal‐IRI+5‐FU/LV, higher Cavg and longer tuSN38>thr was associated with longer OS and PFS and higher overall response rate. Conversely, C max was not associated with OS. This is consistent with the hypothesis that dividing cells are sensitive to chemotherapy; thus, prolonged duration of chemotherapy drug exposures allow a greater number of tumor cells to be affected.21 The observed association between Cavg and tuSN38>thr with efficacy indicates a strong association between plasma and tumor concentrations. This is consistent with the direct SN‐38 measurements in biopsies during a phase I trial that demonstrated increased tumor SN‐38 PK with nal‐IRI administration.8 Furthermore, the association between these two parameters and efficacy is consistent with the preclinical finding that showed a strong association between the in vivo activity of nal‐IRI and the duration of SN‐38 above a minimum inhibitory concentration.17 This result indicates the potential benefit in extending duration of plasma and tumor exposure via liposomal encapsulation.
Neutropenia and diarrhea are the most prominent adverse events with nal‐IRI treatment. For neutropenia, uSN38 was the analyte that had the highest association, with C max exhibiting a stronger association than Cavg. The association between neutropenia and uSN38 C max appeared to be robust and remained significant in the presence of known factors predictive of neutropenia (e.g., ANC and 5‐FU coadministration). Diarrhea was associated with tIRI C max, and as was seen with neutropenia, the association was stronger with C max than Cavg. The dichotomization of the analytes associated with blood‐ and gut‐related safety events are consistent with reports of differential metabolism occurring in the plasma and in the gut. In particular, it has been reported that SN‐38G can be converted back to SN‐38 in the gut via microflora, but this mechanism is absent in the plasma.22, 23 Because SN‐38G in the plasma is observed at an ∼10‐times higher concentration than SN‐38, the conversion in the gut may result in higher SN‐38 concentrations in the gut compared with the plasma. While the ratio of SN‐38 and SN‐38G would depend on the activity of UGT1A enzymes, the sum of SN‐38 and SN‐38G—both in the gut and plasma—would increase as total drug exposure of irinotecan increased. As total drug exposure of nal‐IRI is linearly proportional to plasma tIRI,20 it can be hypothesized that plasma tIRI is a surrogate measurement of the sum of SN‐38 and SN‐38G in the gut lumen.
Among the baseline factors considered, race (Caucasian vs. East Asian) was the most significant predictive factor for both plasma total irinotecan and SN‐38 PK following the administration of nal‐IRI. Specifically, when compared with Caucasian patients, East Asian patients had lower tIRI and higher SN‐38, and a lower corresponding risk for diarrhea and higher risk for neutropenia. The race‐PK association has not been reported in patients receiving nonliposomal irinotecan. Therefore, the release kinetics of irinotecan from liposome may be linked to the race‐related PK difference. The elimination of liposomal chemotherapy from circulation was hypothesized to follow two pathways: passive leakage from liposomes and active uptake by the mononuclear phagocyte system (MPS).24 The first pathway, passive leakage, is likely to be dependent only on external factors such as manufacturing. Therefore, the second pathway, the uptake by MPS, is the hypothesized pathway that maybe affected by race and provides direction for future research in exploring pharmacogenomic factors.
The plasma SN‐38 depends on both the incoming load of SN‐38 and the activity of UGT1A enzymes. The activity of UGT1A enzymes can be assessed by either baseline bilirubin or by pharmacogenomics (UGT1A1*28). Liposomal encapsulation appears to reduce the incoming load of SN‐38 by controlling the release of irinotecan. Hyperbilirubinemia, a surrogate of reduced UGT activity, has been shown to be predictive to plasma SN‐38 and to neutropenia with administration of nonliposomal irinotecan.19 In patients administered with nal‐IRI described here, baseline bilirubin was also found to be a significant predictor of SN‐38, and SN‐38 concentrations were 44% higher in patients with hyperbilirubinemia. Because of the limited number of patients with bilirubin >1 mg/dL in the dataset, no nal‐IRI dose recommendation is provided, and a lower starting dose may be warranted.
A consistent result is found by pharmacogenomics (UGT1A1*28). In patients treated with nonliposomal irinotecan, the associations between UGT1A1*28 7/7 homozygosity and hematological toxicity were observed only in patients treated with doses >150 mg/m2; however, similar hematological toxicities were observed for both UGT1A1*28 homozygous and nonhomozygous patients with a lower dose of nonliposomal irinotecan of 100–125 mg/m2 every week.25 The association between UGT1A1*28 7/7 homozygosity and SN‐38 concentrations are also dependent on the dose of nonliposomal irinotecan, with much higher SN‐38 concentrations observed for 6/7 and 7/7 (compared to 6/6) when irinotecan was administered at a dose of 300 mg/m2 than when it was administered at a dose of 15–75 mg/m2 daily for 5 days for 2 consecutive weeks.26, 27 With nal‐IRI treatment, SN‐38 PK were similar across UGT1A1*28 polymorphisms. A likely mechanistic explanation is that the liposomal encapsulation protects the majority of irinotecan from being converted into SN‐38 and, therefore, the slow release of irinotecan allows the lower load of SN‐38 to be metabolized by UGT enzymes even in patients with reduced UGT enzyme activities (for example, UGT1A1*28 7/7 homozygous patients). Additional data in phase I–II studies in patients treated with nal‐IRI tested for different UGT1A1 genotypes [UGT1A9*22 (*1b), UGT1A1G‐3156A, UGT1A1*6, UGT1A1*27, UGT1A1T‐3279G, and DPYD*2A] indicate that no difference in SN‐38 concentrations was observed by UGT1A1 genotypes (in preparation). Because of the lack of precision in the comparison between homozygous and nonhomozygous patients (as evidenced by the wide 95% CI range of the ratio), the limited number of patients homozygous for the UGT1A1*28 allele treated with nal‐IRI, and the lower starting nal‐IRI dose used in NAPOLI‐1 for these patients (50 mg/m2), it is recommended that those known to be homozygous for the UGT1A1*28 allele be treated initially with 50 mg/m2, which can be increased to 70 mg/m2 if tolerated. However, UGT1A1*28 testing is not mandated.
In conclusion, the quantification of the plasma PK in patients treated with nal‐IRI showed the benefit of the liposomal formulation in extending the half‐lives of irinotecan and SN‐38. The differential pharmacological parameters associated with efficacy and safety endpoints provide support to the selection of dose regimen for nal‐IRI. Because efficacy is associated with Cavg and tuSN38>thr, and safety is associated with C max, a dose regimen of 70 mg/m2 Q2W would result in improved safety while maintaining efficacy as compared to a dose regimen of 100 mg/m2 Q3W. Therefore, these associations support the benefit in the current dosing of nal‐IRI of 70 mg/m2 Q2W.
METHODS
Patients and treatment
Data were prospectively collected from patients enrolled in six trials on a variety of tumor types, including colorectal, gastric, and pancreatic cancers (Table S1). Detailed eligibilities, methods, and clinical results of these studies have been described previously.3, 7, 8, 10, 11, 12 For example, the eligibility criteria in Study NAPOLI‐1 included adequate bone marrow reserve (absolute neutrophil count (ANC) >1,500 cells/μL, platelet >106 cells/μL, hemoglobin >9 g/dL), adequate renal function (serum creatinine (SCr) ≤1.5 upper limit of normal (ULN)), and adequate liver function (bilirubin ≤ULN, albumin ≥3.0 g/dL; aspartate aminotransferase (AST) and alanine aminotransferase (ALT) of ≤2.5 ULN or ≤5 ULN if liver metastases were present). The nal‐IRI doses in these studies were calculated based on the equivalent doses of irinotecan hydrochloride trihydrate; in this report, the doses described are based on irinotecan as free‐base (i.e., 70 mg/m2 of irinotecan as the free‐base is equivalent to 80 mg/m2 of irinotecan as the hydrochloride trihydrate). The final population PK dataset consisted of 353 subjects. Two patients from NAPOLI‐1 with tIRI but without tSN38 measurements were excluded from the analyses (Table S2).
PK data
PK sample collection during the first cycle consisted of intense sampling in early studies7, 8, 10, 11, 12 and sparse sampling in the phase III study NAPOLI‐1 (Table S1).3 The analytes measured include tIRI (encapsulated plus unencapsulated irinotecan) and tSN38. In the first study, the levels of encapsulated irinotecan were found to be indistinguishable from tIRI10; therefore, only tIRI were measured in the subsequent studies. Samples collected after second dose administrations were excluded because of suspected inaccuracy in the sampling times.
Covariate analysis was conducted using a full covariate approach.13 Baseline information to predict plasma PK included body size (BSA), demographics, hepatic and renal function, pharmacogenomics, and extrinsic factors such as product manufacturing site and coadministration with 5‐FU. Laboratory measurements (ALT, AST, bilirubin, creatinine clearance, and albumin) were log‐transformed (log‐normal distributions were observed) (Table S3). Liver metastasis status was only available from NAPOLI‐1; therefore, the values for the other studies were imputed to “No”; the effect of this imputation was evaluated in a sensitivity analysis. The estimated clearance of IRI was added as a covariate to the SN‐38 input flux. Mechanistically, increased clearance of IRI was hypothesized to generate more release of unencapsulated irinotecan that would be available for conversion to SN‐38 (clearance of nal‐IRI likely results in broken liposome and release of irinotecan).
Population PK modeling analysis methods
Modeling assumptions
Nonlinear mixed effect modeling (NONMEM) was used to analyze the PK data of tIRI and tSN38 in patients administered nal‐IRI. To account for measured values below the detection limit, the M3 method15 was implemented with concentrations in log‐transformed values using the Laplacian estimation method.16
A diagram of the PK models of tIRI and tSN38 is shown in Figure S1. The final model of tIRI was a two‐compartment model with first‐order elimination, and the tSN38 depends on the tIRI model. tSN38 was represented as a sum of unencapsulated SN‐38 (uSN38) and encapsulated SN‐38 (eSN38), with eSN38 as a time‐invariant fraction of tIRI, and uSN38 as a one‐compartment model with first‐order production rate representing the process of release of irinotecan and its conversion to SN‐38. The existence of eSN38 was supported by in vitro measurements and by the observation of delayed metabolism of SN‐38 with nal‐IRI administration. In study PEP0206,7 the delayed appearance of SN‐38G relative to the appearance of SN‐38 was observed after nal‐IRI administration, in contrast to the immediate appearance of SN‐38G and SN‐38 after nonliposomal irinotecan administration (Figure S2). This observation supports the hypothesis that only the uSN38 is bioavailable for glucuronidation. The fraction of eSN38 in tIRI was estimated to be 0.01%, which is comparable to the in vitro measurement of 0.015% and is below the specification limit of irinotecan manufacturing.14 The inclusion of the uSN38 and eSN38 improved the model fitting (Table S4).
Simulation analysis methods
Simulations from post‐hoc estimates were used to derive PK parameters for the first cycle of nal‐IRI, including the Cavg and C max for tIRI, tSN38, and uSN38, as well as tuSN38>thr in the first 6 weeks. In a preclinical study, nal‐IRI activity was strongly associated with tuSN38>thr.17 The threshold of 0.03 ng/mL was chosen based on the median IC50 of SN‐38 in in vitro pancreatic cell lines (different choices of threshold of 0.02–0.3 ng/mL resulted in similar OS concordance indices). For the evaluation of baseline covariates associated with PK, simulations were based on 70 mg/m2 Q2W. For the evaluation of fixed‐ and BSA‐based dosing, simulations were based on 70 mg/m2 Q2W, or 116.7 mg Q2W (equivalent dose for a subject with median BSA).
Exposure–efficacy analysis methods
PK efficacy analysis was performed for each treatment arm of NAPOLI‐1. The associations between PK parameters and survival endpoints were measured using the concordance index.18 The selection of PK parameters was based on the magnitude of the concordance index and the (positive) direction of the association.
Exposure–safety analysis methods
The safety dataset included patients from all six clinical studies (Table S1) and was evaluated for diarrhea and neutropenia. Specialized grouping based on individual MedDRA v. 14.1 terms was used for diarrhea and neutropenia (Table S5) to establish systematic reporting. The reported AEs included any grade and grade ≥3 according to the NCI Common Terminology Criteria for Adverse Events (CTCAE) 4.0.
Software
All data preparation and presentation was performed using SAS v. 9.3 or later (SAS Institute, Cary, NC) and R v. 3.0.2 (Vienna, Austria). PK analysis was performed using NONMEM v. 7.3, with FOCEI and Laplacian estimation method. Package Perl Speaks NONMEM (PSN) v. 3.7.6 was used for interface to NONMEM and for assessing models. R package Xpose4 v. 4.5.0 was used to display diagnostics.
CONFLICT OF INTEREST
Drs. Adiwijaya, J Kim, Fitzgerald, Belanger, and Molnar are employees of Merrimack Pharmaceuticals. Drs. Lang, Csöszi, Cubillo, Chen, JS Kim, Rau, and Gallego Plazas report no conflicts of interest. Dr. Wong reports being a member of a Baxalta advisory board. Dr. Park reports receiving honoraria from Celgene, serving as a consultant for Celgene and Agios Pharmaceuticals, Inc., and receiving research funding from Celgene and AstraZeneca. Dr. Melichar reports receiving honoraria for speeches and advisory roles from Lilly, Sanofi, Roche, Novartis, Pfizer, Janssen, Astellas, BMS, MSD, GSK, Merck, and Amgen. Dr. Ma reports receiving funding for clinical trials and honorarium for advisory boards from Merrimack Pharmaceuticals.
AUTHOR CONTRIBUTIONS
B.A., J.K., J.B.F., I.M., and W.W.M. wrote the article; B.A., J.K., J.B.F., B.B., and W.W.M. designed the research; B.A., J.K., I.L., T.C., A.C., J‐S.C., M.W., J.O.P., J.S.K., K‐M.R., B.M., J.G.P., and W.W.M. performed the research; B.A., J.K., J.B.F., B.B., and W.W.M. analyzed the data.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The first two authors contributed equally to this work. This study was funded by Merrimack Pharmaceuticals. We thank Li‐Tzong Chen, Stephan Klinz, Bart Hendriks, Marc Pipas, Eliel Bayever, and Peter Laivins for contributions to the design and the interpretation of the analysis. Medical writing support (funded by Merrimack Pharmaceuticals) was provided by Beth Kamp.
References
- 1. Gabizon, A. & Martin, F. Polyethylene glycol‐coated (pegylated) liposomal doxorubicin. Rationale for use in solid tumours. Drugs 54(suppl.4), 15–21 (1997). [DOI] [PubMed] [Google Scholar]
- 2. Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 624, 25–37 (2010). [DOI] [PubMed] [Google Scholar]
- 3. Wang‐Gillam, A. et al Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine‐based therapy (NAPOLI‐1): a global, randomised, open‐label, phase 3 trial. Lancet 387, 545–547 (2016). [DOI] [PubMed] [Google Scholar]
- 4. Xie, R. , Mathijssen, R.H. , Sparreboom, A. , Verweij, J. & Karlsson, M.O. Clinical pharmacokinetics of irinotecan and its metabolites in relation with diarrhea. Clin. Pharmacol. Ther. 72, 265–275 (2002). [DOI] [PubMed] [Google Scholar]
- 5. Gupta, E. et al Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res. 54, 3723–3725 (1994). [PubMed] [Google Scholar]
- 6. Fujita, K. & Sparreboom, A. Pharmacogenetics of irinotecan disposition and toxicity: a review. Curr. Clin. Pharmacol. 5, 209–217 (2010). [DOI] [PubMed] [Google Scholar]
- 7. Roy, A.C. et al A randomized phase II study of PEP02 (MM‐398), irinotecan or docetaxel as a second‐line therapy in patients with locally advanced or metastatic gastric or gastro‐oesophageal junction adenocarcinoma. Ann. Oncol. 24, 1567–1573 (2013). [DOI] [PubMed] [Google Scholar]
- 8. Ramanathan, R.K.K. et al Lesion characterization with ferumoxytol MRI in patients with advanced solid tumors and correlation with treatment response to MM398, nanoliposomal irinotecan (nalIRI). 26th EORTC‐NCI‐AACR Symposium on Molecular Targets and Cancer Therapeutics Symposium. Barcelona, Spain; 2014. Abstract 261.
- 9. Chabot, G.G. Clinical pharmacokinetics of irinotecan. Clin. Pharmacokinet. 33, 245–259 (1997). [DOI] [PubMed] [Google Scholar]
- 10. Chang, T.C. et al Phase I study of nanoliposomal irinotecan (PEP02) in advanced solid tumor patients. Cancer Chemother. Pharmacol. 75, 579–586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsai, C.S. , Park, J.W. & Chen, L.T. Nanovector‐based therapies in advanced pancreatic cancer. J. Gastrointest. Oncol. 2, 185–194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chan, L.‐T. et al Phase I study of biweekly liposome irinotecan (PEP02, MM‐398) in metastatic colorectal cancer after first line oxaliplatin‐based chemotherapy. J. Clin. Oncol. 30 (2012). [Google Scholar]
- 13. Gastonguay, M.R. et al Missing data in model‐based pharmacometric applications: points to consider. J. Clin. Pharmacol. 50, 63S–674S (2010). [DOI] [PubMed] [Google Scholar]
- 14. United States Pharmacopeia and National Formulary (USP 39‐NF 34) . Irinotecan HCl. Rockville, MD: United States Parmacopeia Convention; 2010. [Google Scholar]
- 15. Bergstrand, M. & Karlsson, M.O. Handling data below the limit of quantification in mixed effect models. AAPS J. 11, 371–380 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang, Y. Derivation of various NONMEM estimation methods. J. Pharmacokinet. Pharmacodyn. 34, 575–593 (2007). [DOI] [PubMed] [Google Scholar]
- 17. Kalra, A.V. et al Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 74, 7003–7013 (2014). [DOI] [PubMed] [Google Scholar]
- 18. Raykar, V.S. et al On Ranking in Survival Analysis: Bounds on the Concordance Index (Cambridge, MA, MIT Press; 2008). [Google Scholar]
- 19. Camptosar (package insert). New York, NY: Pfizer Injectables; 2014. [Google Scholar]
- 20. Onivyde (package insert). Cambridge, MA: Merrimack Pharmaceuticals; 2015. [Google Scholar]
- 21. Pommier, Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 6, 789–802 (2006). [DOI] [PubMed] [Google Scholar]
- 22. Slatter, J.G. et al Pharmacokinetics, metabolism, and excretion of irinotecan (CPT‐11) following I.V. infusion of [(14)C]CPT‐11 in cancer patients. Drug Metab. Dispos. 28, 423–433 (2000). [PubMed] [Google Scholar]
- 23. Wallace, B.D. et al Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. La‐Beck, N.M. et al Factors affecting the pharmacokinetics of pegylated liposomal doxorubicin in patients. Cancer Chemother. Pharmacol. 69, 43–50 (2012). [DOI] [PubMed] [Google Scholar]
- 25. Hoskins, J.M. , Goldberg, R.M. , Qu, P. , Ibrahim, J.G. & McLeod, H.L. UGT1A1*28 genotype and irinotecan‐induced neutropenia: dose matters. J. Natl. Cancer Inst. 99, 1290–1295 (2007). [DOI] [PubMed] [Google Scholar]
- 26. Iyer, L. et al UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2, 43–47 (2002). [DOI] [PubMed] [Google Scholar]
- 27. Stewart, C.F. et al UGT1A1 promoter genotype correlates with SN‐38 pharmacokinetics, but not severe toxicity in patients receiving low‐dose irinotecan. J. Clin. Oncol. 25, 2594–2600 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information