

Abstract
Hybrid poplar genetically engineered to possess chemically labile ester linkages in its lignin backbone (zip‐lignin hybrid poplar) was examined to determine if the strategic lignin modifications would enhance chemical pulping efficiencies. Kraft pulping of zip‐lignin and wild‐type hybrid poplar was performed in lab‐scale reactors under conditions of varying severity by altering time, temperature and chemical charge. The resulting pulps were analyzed for yield, residual lignin content, and cellulose DP (degree of polymerization), as well as changes in carbohydrates and lignin structure. Statistical models of pulping were created, and the pulp bleaching and physical properties evaluated. Under identical cooking conditions, compared to wild‐type, the zip‐lignin hybrid poplar showed extended delignification, confirming the zip‐lignin effect. Additionally, yield and carbohydrate content of the ensuing pulps were slightly elevated, as was the cellulose DP for zip‐lignin poplar pulp, although differences in residual lignin between zip‐lignin and wild‐type poplar were not detected. Statistical prediction models facilitated comparisons between pulping conditions that resulted in identical delignification, with the zip‐lignin poplar needing milder cooking conditions and resulting in higher pulp yield (up to 1.41 % gain). Bleaching and physical properties were subsequently equivalent between the samples with slight chemical savings realized in the zip‐lignin samples due to the enhanced delignification.
Keywords: biomass, delignification, lignin, monolignol ferulate, nuclear magnetic resonance
Introduction
Lignin is one of the principal cell wall polymers in terrestrial plants, which, combined with the other main constituents (cellulose and hemicelluloses), provides structural strength and protection for plants.1 Lignin is generally resistant to enzymatic and chemical degradation, inherently imparting difficulties for biomass utilization as forage,1b, 2 necessitating pretreatments for cellulosic ethanol,3 and requiring harsh chemical conditions for pulp and paper production are needed.4 In pulp production, the cooking and bleaching conditions needed to remove lignin are severe enough to also degrade significant portions of the desired polysaccharides, thus often affecting yield and the overall quality of the cellulosic fibers generated.
It has been shown that lignin is difficult to chemically degrade and remove from plant cell walls, owing to its stable chemical linkages including ether and carbon–carbon bonds,5 as well as its accessibility to chemicals during industrial processing.6 However, researchers hypothesized that if chemically reactive bonds are introduced into the lignin polymer, lignin would be easier to degrade and extract under industrial conditions.7 Recently, the synthesis of monolignol ferulate (ML‐FA) conjugates was engineered into poplar trees during secondary cell wall lignin deposition, which resulted in the introduction of unique ester bonds into the lignin backbone.7a Under industrial acidic or alkaline conditions, the resulting bonds are considerably more reactive than the dominant ether bonds, and should readily “unzip” during chemical processing. Consequently, the new biomass with these unique lignin polymers was coined zip‐lignin biomass,7a which was hypothesized to be less recalcitrant during bioprocessing, including for potential pulping applications. This study is the first to report on the response of zip‐lignin biomass to kraft pulping, and to confirm the speculated impact of the introduction of the ester linkages into the lignin polymer.
Hybrid poplars, selected as the initial wood source species for this genetic modification, are among the fastest growing trees in the temperate forests and are amenable to a variety of climates and geographies. Consequently, they were recognized to have commercial significance as a source of carbohydrates needed to supply a latent biofuels industry in the 1970s during the petroleum embargo.8 Additionally, pulping and papermaking practice with Populus species has a long history.9 Furthermore, biotechnological advances in understanding and manipulating the poplar genome have provided the potential to tailor these trees for specific industrial end uses.10 It should be noted that this study was performed to demonstrate the technology's usefulness in pulping, and could in theory be applied to other hardwood and softwood species.
Kraft pulping was performed on both zip‐lignin hybrid poplar (L7) and wild‐type (WT) poplar as a control, subjecting the materials to varied cooking conditions including chemical usage (active alkali, and sulfidity) and cooking severity (H factor) in a pairwise fashion. The ensuing pulps were washed, air dried, and analyzed to provide information on yield, delignification, and polysaccharide recovery. NMR examinations of the starting lignin and black liquor lignin from both L7 and WT were conducted to investigate potential compositional changes that may arise during the kraft cooks. Statistical models were also created based on these results to assist in identifying the effects of zip‐lignin on pulping efficiency in general, as well as to provide predictions for virtual reactions that achieve identical delignification. Subsequently, more pulp was produced from paired scale‐up cooking of L7 and WT, allowing the bleaching performance and fiber properties to be examined.
Results and Discussion
Raw material characterization
Samples derived from the lines demonstrating that ML‐FA conjugates could be successfully introduced into poplar trees,7a and subjected to gross chemical characterization. The analysis revealed that the main components of hybrid poplar L7 and WT were similar to each other (Table 1) and comparable to those reported in the literature for the natural variation of poplar.11 Although the gross characterization demonstrated similar lignin mass content, the amounts of ML‐FA conjugates in the lignin were also determined using a sensitive derivatization followed by reductive cleavage (DFRC) technique that cleaved lignin ethers but retained esters (Figure 1). The DFRC data demonstrated an increase in releasable ML‐FA conjugates from line 7, validating the desired effect of zip‐lignin modification. Figure 1 indicates the native coniferyl ferulate (G‐FA) to be much higher than the sinapyl ferulate (S‐FA), which is presumably due to the timing of when these moieties are produced during lignification. However, the engineered L7 sample shows a similar incorporation increase over the wild type for both G‐FA (11 to 23 mol g−1) and S‐FA (1 to 17 mol g−1), suggesting that the inserted gene is active for both monomer types and likely during the entire lignification process.
Table 1.
Main chemical components of hybrid poplar L7 and WT.[a]
| Component | L7 [%] | WT [%] |
|---|---|---|
| cellulose | 46.17±0.40 | 46.82±1.18 |
| hemicelluloses[b] | 17.62±0.03 | 16.65±0.35 |
| Klason lignin | 18.93 | 17.43 |
| acid‐soluble lignin | 6.90 | 6.85 |
| ash | 1.47±0.24 | 1.84±0.03 |
| total | 91.09 | 89.59 |
[a] All data are based on OD wood. [b] Hemicellulosic sugars include arabinose, galactose, and xylose, whereas mannose was negligible.
Figure 1.
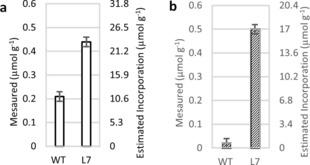
Monolignol ferulate levels releasable by derivatization followed by reductive cleavage (DFRC) from enzymatic lignin samples. a) G‐FA (coniferyl ferulate) and b) S‐FA (sinapyl ferulate). Error bars show the standard error of duplicate measurements.
Kappa number, yield, and cupriethylenediamine viscosity of pulps
Due to limited substrate, each reaction in the experimental design (Table 2) was completed only once, producing one pulp for each cook, except for the center point (Exp. #9), which was conducted twice. Duplicate analysis on Kappa number and cupriethylenediamine (CED) viscosity was completed to reduce errors. The Kappa numbers for all runs (Figure 2) ranged from 19.6 to 93.7, over the range of conventional pulps, but generally fell into three groups corresponding to three levels of active alkali (AA). Exp. #1, #7, #8, and #5 displayed extremely high Kappa levels that would not be useful for conventional pulp production, but did provide insight into delignification under milder conditions. The analysis of variance (ANOVA) results showed that AA had the most significant negative impact on Kappa number, whereas both S (sulfidity) and H factor showed only slight trends. It was expected that the sulfidity effect would be minimal, but the minor effect of H factor10a, 12 was unexpected and believed to be an artifact of the small‐scale cooks.
Table 2.
Experimental design for creating pulping models.
| Experiment | AA [%][a] | S [%][a] | H factor |
|---|---|---|---|
| 1 | 14 | 20 | 800 |
| 2 | 18 | 20 | 800 |
| 3 | 18 | 20 | 1200 |
| 4 | 18 | 30 | 800 |
| 5 | 14 | 30 | 800 |
| 6 | 18 | 30 | 1200 |
| 7 | 14 | 20 | 1200 |
| 8 | 14 | 30 | 1200 |
| 9 | 16 | 25 | 1000 |
[a] % on OD wood.
Figure 2.
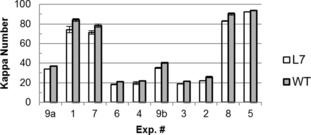
Kappa number of hybrid poplar L7 and WT after kraft cooking (error bars showing the standard error of duplicate measurements).
The most interesting observation, apparent in Figure 2, is the effect of wood type. Visual inspection showed that the Kappa number of L7 was lower than that of WT for each paired run. A paired‐samples t test was subsequently performed on the Kappa number data and showed that the Kappa number of L7 was significantly lower than that of WT (p value=4.6×10−7 from the t test), with an average difference of 4.5. This consistent finding indicates that zip‐lignin improved delignification of hybrid poplar wood during kraft cooking, regardless of the cooking conditions.
Unscreened yields for the pulping experiments were calculated (Figure 3 a) and ranged from 49 to 61 %, with averages of 54.6 and 53.7 % for L7 and WT, respectively. ANOVA showed that AA and H factor had significant negative effects on pulp yield, whereas S had a significant positive effect on pulp yield, as expected.10a, 12 Importantly, inspection of Figure 3 a showed that the yield of L7 was higher than that of WT for each paired run. A paired‐samples t test on yield data confirmed this to be statistically significant with the yield of L7 being significantly higher than that of WT (p value=0.0023), with an average difference of 0.91 %. This was unexpected as the zip‐lignin was hypothesized to provide an increase in delignification, but not in yield for a given pulping condition.
Figure 3.
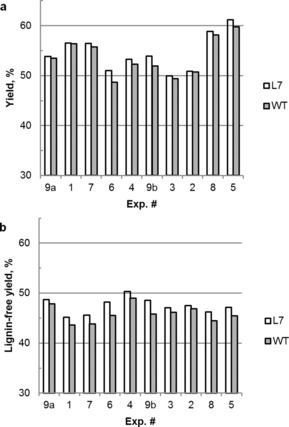
Yields of hybrid poplar L7 and WT after kraft cooking. a) Original unscreened pulp yield; b) estimated lignin‐free yield.
Noting that this yield was affected by residual lignin, we estimated the residual lignin from the Kappa value (lignin wt %≈Kappa#/6.57) and subtracted it from the overall pulp yield to obtain a lignin‐free yield estimate (Figure 3 b). The lignin‐free yield ranged from 43.6 to 50.3 %, with averages of 47.45 and 45.86 % for L7 and WT, respectively. Visual inspection showed that the lignin‐free yield of L7 was still higher than that of WT for each paired run, and the difference was even larger. The paired‐samples t test on lignin‐free yield data also showed that the yield of L7 was significantly higher than that of WT with an even smaller p value (=3.1×10−5≪0.0023) and a larger average difference of 1.59 %. This implies that the improved delignification may also minimize the impact of carbohydrate degradation during the pulping reaction, and therefore improve the overall yield of the pulping reaction, presumably by better liquor penetration as the zip‐lignin is likely being depolymerized more readily during pulping. Additional studies with more mature plants will be needed to further study and confirm this observation, as it has obvious positive ramifications for the pulping industry.
Cellulose degree of polymerization (DP), as determined by CED viscosity values, was only possible for pulps derived from experiments #9, #6, #4, #3, and #2 (Figure 4), as the lignin contents of the pulps from other experiments were too high (Figure 2) for sufficient solubilization and accurate viscosity determination. The measured CED viscosity ranged from 16.3 to 29.2 mPa s, with an overall average of 22.9, which is typical for kraft pulp. Statistical modeling of pulping variables was not done because of the limited data, but visual inspection suggested that the CED viscosity of L7 was higher than that of WT for each paired run, and the paired‐samples t test showed that the CED viscosity of L7 was significantly higher than that of WT (p value=0.0005) with an average difference of 2.1 mPa s. This increase in CED viscosity, denoting an increased cellulose DP, furthers the hypothesis that the zip‐lignin modification has a positive effect on carbohydrates preservation during the delignification process.
Figure 4.
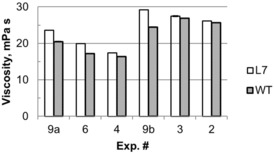
CED viscosity measurements of hybrid poplar L7 and WT pulp from kraft cooking (error bars showing the standard error from duplicate measurements).
Polysaccharides and lignin of pulps
To better understand the reduced Kappa number and improved pulp yield and CED viscosity of L7 compared to WT, chemical component analysis of all pulps was performed (Figure 5). For carbohydrates, only glucan and xylan are shown here because other common wood carbohydrates (arabinan, galactan, and mannan) were either not detected or negligible following pulping, as expected. ANOVA results showed that AA had significant negative effects on residual lignin content, whereas positive effects on xylan, glucan, and total carbohydrates were observed. This is consistent with the effects of AA on Kappa number as previously discussed. Visual inspection of the data showed that the lignin estimates generally agreed with pulp Kappa numbers (Figure 2).
Figure 5.
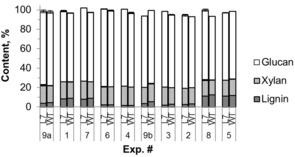
Chemical components of pulps of L7 and WT (all data as percentages of OD pulp; Lignin, Klason lignin; error bars showing the range of duplicate measurements; bottom error bars indicate the xylan content standard errors; top error bars indicate the glucan content standard errors).
Paired‐samples t test showed that the lignin contents of L7 pulps were significantly lower than those of WT pulps (p value=0.00045), with an average difference of 0.73 %, whereas the total carbohydrates contents of L7 pulps were significantly higher (an average difference of 2.40 %) than that of WT pulps (p value=0.002). These results clearly indicated that L7 pulps had lower lignin contents and higher carbohydrate contents compared to WT pulps, supporting our previous results. Paired‐samples t test also showed that the xylan and glucan contents of the L7 pulps were significantly higher than those of WT pulps (p values=0.041 and 0.00072, respectively), with average differences of 0.47 and 1.97 %, respectively, indicating that the yield gain of L7 was largely due to polysaccharide protection.
From the average xylan contents (17.83 % for L7 and 17.36 % for WT), average glucan contents (75.18 % for L7 and 73.21 for WT), and the gains of xylan (0.47 %) and glucan (1.97 %) for L7 pulps compared to WT pulps, it was found that both of these polysaccharide contents of L7 pulps had a same relative gain of 2.7 % (17.83/17.36=1.027, 75.18/73.21=1.027) compared to WT, indicating hemicellulose and cellulose were protected to the same extent owing to the zip‐lignin. These results support the previous observation of improved yield from the zip‐lignin trees, and demonstrate that the yield gain can be ascribed to both hemicellulose and cellulose retention, and the protection of cellulose increased the cellulose DP.
NMR analysis
HSQC NMR spectroscopy was used to determine the extent of compositional change in WT poplar lignin (line P39) and ferulate monolignol transferase (FMT) poplar lignin (line 7) before and after kraft cooking. The enzyme lignin was examined from the original wood, that is, from the poplar before cooking, whereas the kraft lignin was examined for the various samples after cooking, as shown in Figure 6. There were no spectroscopic differences between the two poplar lines (Figure 6, top vs. bottom), either for enzyme lignin or for kraft lignin. Compared to the enzyme lignin (Figure 6 b vs. 6 e), the kraft lignin (Figure 6 c vs. 6 f) had very low levels of β‐ether units, greatly reduced levels of p‐hydroxybenzoate, and the presence of condensed units (G* and S*, substructures with substitution(s) at the 2, 5, and/or 6 positions). Of note is the relative stability of the resinol substructure (R) under kraft pulping. G and S lignin percentages were tabulated for each of the 6 lignins (shown for each in Figure 6). The values indicated the WT and modified L7 wood samples to have similar ratios (G/S of approximately 40:60), which remained throughout the processing. Overall, the WT and L7 lignins were quite similar with the exception of the incorporation of the G‐FA and S‐FA measured by DFRC. It should be noted that are no major NMR signals with the HSQC NMR technique that can be used to uniquely identify ML‐FAs incorporated into the lignin because, as described in a recent paper,7a ferulate coupling into lignins is extraordinarily complex and individual species exist only at low levels. Therefore, the more sensitive DFRC method, which is completely diagnostic for ML‐FA incorporation into lignification,20 was used as previously discussed.
Figure 6.
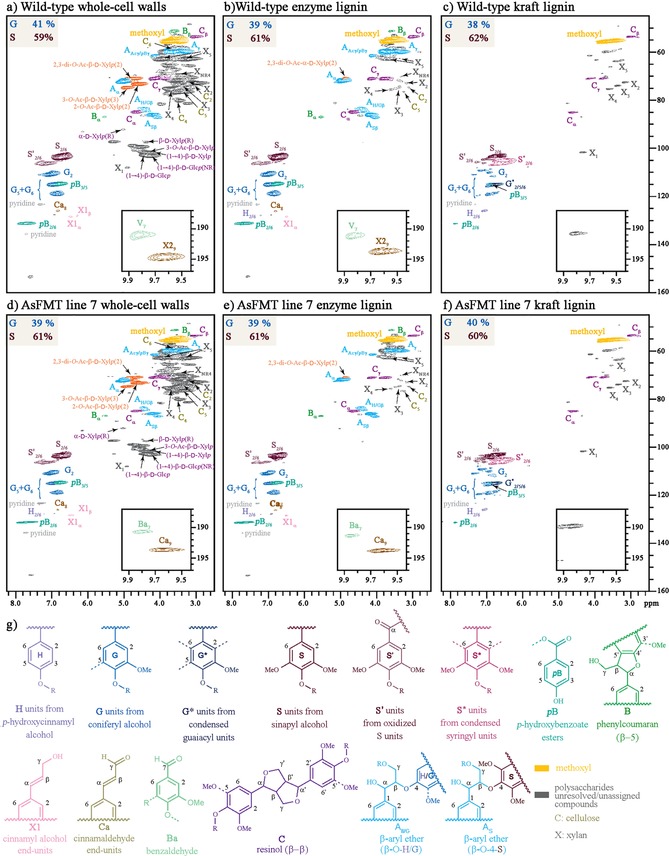
2D 1H‐13C HSQC NMR spectra of lignins from wild‐type (WT) and zip‐lignin (line 7) poplar of (a, d) whole‐cell wall gelled wood, (b, e) enzyme‐isolated before cooking, (c, f) precipitated liquor sample after kraft cooking, and (g) major identified lignin substructures color coded for their NMR contours. Tabulated values are from contour volume integrals only, and are on an S+G=100 % basis. Incorporated ferulate is not readily detected in these experiments (see text).
Pulping models
Linear models based on AA, S, and H factor were generated to predict Kappa number and yield for both L7 and WT separately, using JMP Pro 11 (SAS). The crossed effects of AA, S, and H factor were excluded after they were found to be not significant. The Kappa number (K) model for L7 and WT were:
whereas the yield (Y) models for L7 and WT were:
where the units of AA and S were in wt % based on dry wood, and the unit of H was dimensionless.
These models were plotted to allow visualization of the results. Figure 7 a shows that L7 has a lower Kappa number than WT under the same cooking conditions, which is consistent with the observed experimental results. However, we were interested in another important question: what would the yield difference be if the two wood sources were cooked to the same Kappa number? Using the statistical models provides insight into this question as it is difficult to set conditions to reach the exact Kappa number in practice. Therefore, we used the models to predict the yields of L7 and WT at same Kappa number under varying AA, S, and H factor. These predictive tools indicate, on average, the yield of L7 is approximately 1.41 % higher than that of WT, at a Kappa number range of 9–39 (Figure 7 b). This effect can be ascribed to both the impact of zip‐lignin wood giving a higher yield from some unknown mechanism, as well as the easier delignification allowing lower AA, which in turn provides for higher yield.
Figure 7.
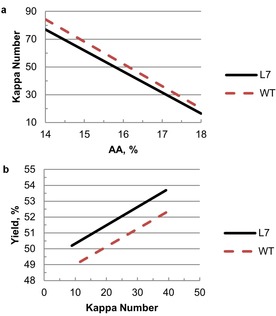
Plots of models to compare the Kappa number (a) and yield (b) between L7 and WT (H=1000, S=25 % for both a and a).
Bleaching properties
The combined and dried pulps from the larger cooks were bleached using an OD(EP)D sequence employing the conditions shown in Table S1 (Supporting Information), which represents a typical sequence used for hardwood pulp. The final D stage varied the chlorine dioxide (ClO2) charge to provide a bleaching response, which was plotted for both pulps (Figure S1). The results demonstrate both WT and L7 pulps have similar bleach response with >88 % ISO brightness achievable with an OD(EP)D sequence. Higher brightness values typical for market pulps (>90 % ISO brightness), were not targeted to allow bleachability differences to be more easily observed, if they existed between the pulps. However, as apparent in Figure S1, no bleaching differences were observed with both pulps achieving similar brightness values as a function of the ClO2 charge. This does not mean there are no advantages to the lower incoming Kappa of L7, which used slightly less ClO2 in the initial stage, as the charge was adjusted to the starting lignin level as determined by Kappa factor, which applies more ClO2 in the first D stage for higher Kappa numbers.
Handsheet properties
The fully bleached pulps were used to produce TAPPI (Technical Association of the Pulp and Paper Industry) standard handsheets to measure key physical properties of both WT and zip‐lignin pulps. Bleached pulp was used to remove any interference from varying lignin contents between the two wood samples common to the unbleached pulps. The produced pulp freeness as well as the paper properties tensile index and density (Table S2) indicate that there were no quality differences between the two pulps, suggesting that the zip‐lignin modification will have no adverse effects on the final papermaking process.
Conclusions
Kraft pulping was conducted in lab‐scale reactors to compare the pulping behaviors of genetically modified zip‐lignin line 7 (L7) and wild‐type (WT) hybrid poplar. Statistical models were generated to predict the Kappa number and yield based on varying reaction conditions (active alkali, sulfidity, and H factor). Lignin changes were examined by NMR and the bleaching performance and fiber properties from handsheets were also investigated. The results showed that the active alkali (AA) was the primary variable to affect the Kappa number and unscreened yield of the kraft pulps, with the zip‐lignin poplar having significantly lower Kappa number, higher yield, and higher cupriethylenediamine (CED) viscosity compared to wild‐type poplar regardless of pulping conditions. Chemical component analysis of ensuing pulps’ polysaccharides and lignin confirmed these results and supported the observation that the zip‐lignin retained polysaccharides derived from both cellulose and hemicelluloses during kraft pulping. Finally, using statistical models, we estimated the effect of the zip‐lignin poplar compared to the control if both were pulped to the same end‐of‐cook Kappa number. The results indicated a gain of 1.41 % in yield compared to the control (at a Kappa number range of 9–39) from both contributions of lower alkali as well as carbohydrates preservation from an unknown mechanism. The lignin differences between L7 and WT were not detected by HSQC NMR, either before or after kraft cooking. Bleaching and handsheet property results indicate that the enhanced delignification did not adversely affect bleaching or papermaking, with a slight chemical savings realized in bleaching due to the overall lower incoming lignin levels.
Overall, the results consistently demonstrate that the zip‐lignin engineered poplars perform better than their wild‐type counterparts under the alkaline kraft cooking conditions, which supports the hypothesis proposed that if chemically reactive bonds are introduced into the lignin polymer, the lignin would be easier to degrade and extract under industrial conditions lowering energy and chemical costs and improving yield. Additionally, as the initial growth data7a does not suggest any detrimental effects, in theory the improvement in delignification should not increase feedstock costs. However, the results are diminished at lower Kappa numbers (higher delignification) as the β‐O‐4 lignin bonds that are replaced are eventually degraded. These initial results are encouraging but suggest additional work will be needed to maximize the economic impact of this technology, including methods to include higher monolignol ferulate content in the engineered wood, modification of different wood species, and techno‐economic analysis of a variety of pulping systems.
Experimental Section
Materials and chemicals
Zip‐lignin hybrid poplar from line 7 (L7) and wild‐type hybrid poplar from line P39 (WT) trees were greenhouse grown from clonally propagated trees. Plant stems were harvested from eight‐month‐old trees, hand‐debarked, air‐dried, and stored at room temperature prior to processing. Sodium hydroxide (NaOH, certified ACS pellets) was purchased from Fisher Scientific (Fair Lawn, NJ, USA). Sodium sulfide (Na2S), sodium thiosulfate solution (Na2S2O3, 1 m), [D6]DMSO and [D5]pyridine were purchased from Sigma–Aldrich (St. Louis, MO, USA). Sulfuric acid (72 % w/w), potassium permanganate (KMnO4, 0.100 N), cupriethylenediamine solution (CED, 1 m), and chlorine dioxide (ClO2) were purchased from Ricca Chemical Company (Arlington, TX, USA). Potassium iodide (KI, 99 %) was purchased from Alfa Aesar (Ward Hill, MA, USA). Starch (soluble potato, powder) was purchased from J. T. Baker (Phillipsburg, NJ, US). Sugars (arabinose, galactose, glucose, mannose, xylose, all 99+%) were purchased from Acros Organics (Geel, Belgium).
Wood chips preparation
Debarked and air‐dried hybrid poplar stems were chipped by an Earthwise 15‐Amp Electric Garden Chipper/Shredder (Model GS70015), then sieved by shaking 15 min in a LABTECH Chip Classifier with round hole screens. The chips that passed the 7 mm round‐hole screen but were retained on the 3 mm round‐hole screen were used to create uniform chips for the small scale cooking (35 g/cooking) for this study, which had total solids of about 95 % for both L7 and WT. The chips retained above the 7 mm round hole screen were used for scale‐up cooking (90 g/cooking) to study the bleaching and handsheet properties.
Chemical component analysis
Chemical component analysis of both raw materials and pulps was conducted. Wood chips or pulps were ground by an IKA MF 10 basic grinder to pass a 1 mm round hole screen. Total solids of the raw material and kraft pulps were determined according to NREL/TP‐510‐42621.13 Ash was determined according to NREL/TP‐510‐42622.14 Carbohydrates and lignin were determined according to our previously modified method,15 and NREL/TP‐510‐42618,16 respectively.
Pulping
Kraft cooking was conducted in 1 L stainless steel bomb reactors in a steam‐heated, rotating digester at the USDA Forest Product Laboratory (Madison, WI). JMP (SAS) software was used to create a box experimental design varying active alkali (AA), sulfidity (S), and H factor, as shown in Table 2. ANOVA of the complete pulping set allowed experimental error to be estimated.
For the small‐scale (35 g) cooking, as designed in Table 2, wood chips (7–3 mm) of transgenic line 7 (L7) and wild‐type (WT) were loaded separately into two of the 1 L stainless steel bombs, then appropriate white liquor (sodium hydroxide, sodium sulfide solution and deionized water) was added at a liquid/solid ratio of 5:1 to reach the active alkali (AA) and sulfidity (S) as designed. The bomb reactors were sealed and loaded into a rotating digester container, rotating 10 min at room temperature to allow mixing. Then, steam was introduced into the digester to heat up the bomb reactors, and maintained at a steam pressure of 80 psig for 10 min, then at 105 psig for 3 min to reach the apparent temperature of 165 °C. The temperature was maintained at 165 °C for defined time (110, 130, 150 min) to reach designed H factor with the ramp time estimated to be about 50 min. At the end of the cook, the steam was released, the digester was opened, and running water was added into the digester to cool the bombs to near room temperature, which took approximately 20 min. Each resultant pulp was carefully transferred to avoid loss, and 100 mL deionized water was used to rinse each bomb reactor. Each ensuing pulp was washed with 2 L tap water for 5 times in a Grainger Felt Filter Bag (polypropylene, pore size 25 μm) to avoid loss, and therefore permit precise yield determination. Washed pulps were not screened due to the small sample size, but were air dried prior to analysis to insure biological stability. Statistical models were created based on these experiments.
The scale‐up (90 g) cooking was conducted by a similar procedure. Wood chips (>7 mm) were cooked at an AA of 18 %, S of 30 %, and liquid/solid ratio of 5. The digester rotated for 30 min before heating up, the steam pressure was kept at 45, 60, 75, 90, 105 psi (gauge) (1.00 psi=6.89 kPa) for 10 min each during the heating up, and the temperature was held at 165 °C for 110 min. Produced pulp was washed until the drain water was colorless. The washed pulps of L7 and WT from four times of same cooking were combined separately, then wet disintegrated, sieved, and air‐dried for bleaching and handsheet testing.
Pulp analysis
Pulp yield, Kappa number, CED viscosity, and carbohydrate and lignin contents of each pulp sample were analyzed. Pulp yield was determined by comparing the total solids of resulted pulps and wood raw materials. The Kappa number was determined according to TAPPI UM 246. CED viscosity was determined according to TAPPI T 230 om‐04. Carbohydrate and lignin analysis were determined according to the previously described NREL procedure.16 To facilitate adequate swelling and hydrolysis during the two‐step acid hydrolysis, the air‐dried pulps were also ground by an IKA MF 10 basic grinder fitted with a 1‐mm round‐hole screen.
Statistical analysis and modeling
The effects of zip‐lignin on kraft cooking results (Kappa number, yield, and CED viscosity) were analyzed by paired‐samples t test as L7 and WT were cooked in pairs, under identical time/temperature conditions during each run, using 2 small reactors in a steam‐filled rotating vessel. The effects of cooking conditions (AA, S, H factor) on pulp properties were analyzed with analysis of variance (ANOVA) by JMP Pro 11 (SAS), and linear models for predicting yield and Kappa number based on AA, S, and H factor were generated separately for L7 and WT, with only primary variables under consideration because their cross effects were not significant. The ensuing models were then used to compare the conditions needed to reach similar Kappa number for L7 and WT, and to predict the yields under these conditions.
Lignin isolation and NMR analysis
HSQC NMR examination was done on poplar lignin for whole‐cell‐wall wood before and after kraft cooking, that is, enzymatic lignin and kraft lignin. The preparation of the extract‐free whole‐cell walls, enzyme lignins, and kraft lignins were performed as described previously.17 Briefly, the ball‐milled extract‐free poplar samples were prepared in 5 mm NMR tubes and swelled/dissolved in [D6]DMSO/[D5]pyridine (4:1 v/v, 500 μL) for NMR analysis. Enzymatic lignin (EL) samples were prepared through digestion of material (200 mg) at room temperature with crude cellulases (20 mg, Cellulysin; Calbiochem), in acetate buffer (40 mL, pH 5.0) on a shaker for 3 days. The samples were then centrifuged (Sorval biofuge primo; 8500 rpm/10 016×g for 10 min) and decanted. The cellulase digestion was then repeated. After decanting the second hydrolysate, the sample was rinsed with reverse osmosis (RO) water (3×40 mL) and lyophilized. From the resulting brown powder (40 mg), 20 mg was transferred to a 5 mm NMR tube and dissolved/swollen in [D6]DMSO/[D5]pyridine “100 %” (4:1, 500 μL). The kraft lignin was prepared from the black liquor of kraft cooking for L7 and WT separately. In brief, the pH of the black liquor (200 mL) was adjusted to 2 with sulfuric acid (72 %), then centrifuged at 10 000×g for 20 min at 4 °C. The solids were washed 5 times with dilute sulfuric acid solution (200 mL, pH 2) and freeze dried, resulting in a dry, brown powder.
The whole‐cell gel was characterized by HSQC spectroscopy under the conditions previously reported.19 NMR spectra were acquired on a Bruker Biospin (Billerica, MA) Avance 700 MHz spectrometer equipped with a 5 mm quadruple‐resonance 1H/31P/13C/15N QCI gradient cryoprobe with inverse geometry (proton coils closest to the sample). The central DMSO solvent peak was used as an internal reference (δ C=39.5, δ H=2.49 ppm). Peak assignments were made by comparison with previously assigned spectra.17d, 18
The two isolated lignin samples were also characterized by HSQC spectroscopy under the conditions previously reported.17d, 19 Briefly, the dry powder (40 mg) was dissolved in [D6]DMSO/[D5]pyridine “100 %” (4:1, 600 μL). The spectroscopic data were acquired on a Bruker Biospin (Rheinstetten, Germany) AVANCE‐III 700 MHz NMR spectrometer equipped with an inverse gradient 5 mm QCI 1H/31P/13C/15N cryoprobe. The central DMSO solvent peak was used as internal reference (δ C=39.5, δ H=2.49 ppm). Peak assignments for syringyl (S), guaiacyl (G), and p‐hydroxyphenyl (H) lignin units, as well as p‐hydroxybenzoate (pB), resinol (R), phenylcoumaran (Co), xylan (X), cinnamyl alcohol (X1), cinnamaldehyde (X2), cellulose (C) and condensed monolignols (G* and S*), were made by comparison with previously assigned spectra.17d, 22
DFRC analysis for ML‐FA content
Incorporation of ML‐FAs into the lignin was determined using the ether‐cleaving ester‐retaining DFRC method previously established for other monolignol conjugates.7a, 20, 21 The DFRC protocol was performed on enzyme lignin (EL) samples (50 mg), which were stirred in 2 dram vials fitted with PTFE pressure release caps with acetyl bromide/acetic acid (1:4 v/v, 4 mL). After heating for 3 h at 50 °C, the solvents were removed and the solids suspended in absolute ethanol (0.5 mL), vacuum (dried on a SpeedVac, 50 °C, 15 min, 6.0 torr, 35 torr min−1; 1 torr=133.322 Pa), and then suspended in dioxane/acetic acid/water (5:4:1 v/v, 5 mL) with nanopowder zinc (250 mg). The vials were then sealed, sonicated to ensure suspension of solids, and stirred in the dark at room temperature for 16–20 h. The reaction mixtures were then quantitatively transferred with dichloromethane (DCM, 6 mL) into separatory funnels charged with a saturated ammonium chloride (10 mL). Organics were extracted with DCM (4×10 mL), combined, dried over anhydrous sodium sulfate, filtered, and the solvents removed by evaporation. Free hydroxyl groups of the DFRC products were then acetylated for 16 h in the dark using a solution of pyridine and acetic anhydride (1:1 v/v, 5 mL), after which the solvents were evaporated to yield acetylated DFRC products. To remove most of the polysaccharide‐derived products, acetylated DFRC products were loaded onto solid‐phase‐extraction (SPE) cartridges (Supelco Supelclean LC‐Si SPE tube, 3 mL, P/N: 505048) with DCM (2×1.0 mL). After elution with hexanes/ethyl acetate (1:1, v/v, 8 mL), the eluted organics were combined and the solvents removed by rotary evaporation. The products were transferred in stages with GC–MS‐grade DCM to GC–MS vials containing a 300 μL insert, with the final sample volumes of 200 μL. Samples were analyzed on a triple‐quadrupole GC–MS (Shimadzu GC/MS‐TQ8030) operating in multiple‐reaction‐monitoring (MRM) mode using synthetic standards for authentication using previously described settings.7a The measured ML‐FAs levels were also used to estimate the incorporated levels in the plants lignin following the work of Karlen et al.20 with 53 μmol of incorporated G‐FA for every μmol measured and 34 μmol of incorporated sinapyl ferulate for every μmol measured.
Bleaching
Bleaching was performed using either a Mark IV Quantum Reactor for oxygen and chlorine dioxide stages or bag bleaching in a water bath for the hydrogen peroxide reinforced alkaline extraction stages. Samples were well‐washed between stages with distilled water with no carryover. Elemental chlorine‐free chlorine dioxide was prepared through sulfuric acid acidification of sodium chlorite and collection of the chlorine dioxide gas into chilled water after passing through a sodium chlorite solution trap. The initial oxygen stage was employed as a low cost method to further delignify, but at conditions mild enough to not further degrade cellulose or lower the yield. Consistency, caustic charge, temperature, and oxygen in this stage were held constant, with conditions and pressure set to have excess oxygen. The final ClO2 stage was varied in the final stage to provide a measure of brightness potential. Brightness pads were made following TAPPI standard T 218 and used to measure ISO brightness following TAPPI standard T 525.
Handsheet preparation and tests
Physical properties were measured on handsheets after they were refined in a Papirindustriens forskningsinstitut (PFI) mill prepared following TAPPI Standards T‐205 and T‐248. Freeness, basis weight, tensile, and caliper were measured using TAPPI Standards T 220, T 410, and T 411, with basis weight and caliper combined to create a specific volume measurement.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to thank the Wisconsin Alumni Research Foundation Accelerator Program and the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE‐FC02‐07ER64494) for funding this work. We also want to thank Gregg Sanford at the Great Lakes Bioenergy Research Center (GLBRC, Madison, WI) for growing poplar raw materials, and Roland Gleisner and Carl Houtman at the USDA Forest Product Laboratory (Madison, WI) for their assistance in kraft cooking and valuable discussions.
S. Zhou, T. Runge, S. D. Karlen, J. Ralph, E. Gonzales-Vigil, S. D. Mansfield, ChemSusChem 2017, 10, 3565.
References
- 1.
- 1a. Bonawitz N. D., Chapple C., Annu. Rev. Genet. 2010, 44, 337–363; [DOI] [PubMed] [Google Scholar]
- 1b. Moore K. J., Jung H.-J. G., J. Range Manage. 2001, 54, 420–430. [Google Scholar]
- 2.
- 2a. Smith L. W., Goering H. K., Gordon C. H., J. Dairy Sci. 1972, 55, 1140–1147; [Google Scholar]
- 2b. Jung H. G., Vogel K. P., J. Anim. Sci. 1986, 62, 1703–1712. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Zhou S., Weimer P. J., Hatfield R. D., Runge T. M., Digman M., Bioresour. Technol. 2014, 170, 286–292; [DOI] [PubMed] [Google Scholar]
- 3b. Zhou S., Yang Q., Runge T. M., Ind. Crops Prod. 2015, 70, 410–416. [Google Scholar]
- 4.
- 4a. Runge T., Zhang C., Tappi J. 2014, 13, 19–24; [Google Scholar]
- 4b. Alonso D. M., Zhou S., Won W., Hosseinaei O., Tao J., Garcia-Negron V., Motagamwala A. H., Mellmer M. A., Huang K. F., Houtman C. J., Labbe N., Harper D. P., Maravealias C. T., Runge T., Dumesic J. A., Sci. Adv. 2017, 3, e1603301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ralph J., Hatfield R. D., Piquemal J., Yahiaoui N., Pean M., Lapierre C., Boudet A. M., Proc. Natl. Acad. Sci. USA 1998, 95, 12803–12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mansfield S. D., Mooney C., Saddler J. N., Biotechnol. Prog. 1999, 15, 804–816. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Wilkerson C. G., Mansfield S. D., Lu F., Withers S., Park J.-Y., Karlen S. D., Gonzales-Vigil E., Padmakshan D., Unda F., Rencoret J., Ralph J., Science 2014, 344, 90–93; [DOI] [PubMed] [Google Scholar]
- 7b. Grabber J. H., Hatfield R. D., Lu F., Ralph J., Biomacromolecules 2008, 9, 2510–2516; [DOI] [PubMed] [Google Scholar]
- 7c. Mottiar Y., Vanholme R., Boerjan W., Ralph J., Mansfield S. D., Curr. Opin. Biotechnol. 2016, 37, 190–200. [DOI] [PubMed] [Google Scholar]
- 8. Morley P. M., Balatinecz J. J., For. Chron. 1993, 69, 46–52. [Google Scholar]
- 9. Durden E. H., Proc. Geol. Polytech. Soc. West Riding Yorkshire 1849, 3, 387–394. [Google Scholar]
- 10.
- 10a. Huntley S. K., Ellis D., Gilbert M., Chapple C., Mansfield S. D., J. Agric. Food Chem. 2003, 51, 6178–6183; [DOI] [PubMed] [Google Scholar]
- 10b. Mansfield S. D., Kang K.-Y., Chapple C., New Phytol. 2012, 194, 91–101. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Luo C., Brink D. L., Blanch H. W., Biomass Bioenergy 2002, 22, 125–138; [Google Scholar]
- 11b. Bura R., Chandra R., Saddler J., Biotechnol. Prog. 2009, 25, 315–322; [DOI] [PubMed] [Google Scholar]
- 11c. Li N., Pan X., Alexander J., Green Chem. 2016, 18, 5367–5376. [Google Scholar]
- 12. Mân Vu T. H., Pakkanen H., Alén R., Ind. Crops Prod. 2004, 19, 49–57. [Google Scholar]
- 13.A. Sluiter, B. Hames, D. Hyman, C. Payne, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton, J. Wolfe in Determination of Total Solids in Biomass and Total Dissolved Solids in Liquid Process Samples, National Renewable Energy Laboratory, Golden, Colorado, 2008, pp. 1–6 www.nrel.gov/docs/gen/fy08/42621.pdf.
- 14.A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton, Determination of Ash in Biomass, National Renewable Energy Laboratory, Golden, Colorado, 2008, pp. 1–5 www.nrel.gov/docs/gen/fy08/42622.pdf.
- 15. Zhou S., Runge T. M., Carbohydr. Polym. 2014, 112, 179–185. [DOI] [PubMed] [Google Scholar]
- 16.A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton, D. Crocker, Determination of structural carbohydrates and lignin in biomass, National Renewable Energy Laboratory, Golden, Colorado, 2011, pp. 1–15, https://www.nrel.gov/docs/gen/fy13/42618.pdf.
- 17.
- 17a. Chang H.-m., Cowling E. B., Brown W., Holzforschung 1975, 29, 153–159; [Google Scholar]
- 17b. Wagner A., Ralph J., Akiyama T., Flint H., Phillips L., torr K., Nanayakkara B., Te Kiri L., Proc. Natl. Acad. Sci. USA 2007, 104, 11856–11861; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Sun R., Tomkinson J., Bolton J., Polym. Degrad. Stab. 1999, 63, 195–200; [Google Scholar]
- 17d. Mansfield S. D., Kim H., Lu F., Ralph J., Nat. Protoc. 2012, 7, 1579–1589. [DOI] [PubMed] [Google Scholar]
- 18. Kim H., Ralph J., Org. Biomol. Chem. 2010, 8, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu F., Ralph J., Plant J. 2003, 35, 535–544 [DOI] [PubMed] [Google Scholar]
- 20. Karlen S. D., Zhang C., Peck M. L., Smith R. A., Padmakshan D., Helmich K. E., Free H. C. A., Lee S., Smith B. G., Lu F., Sedbrook J. C., Sibout R., Grabber J. H., Runge T. M., Mysore K. S., Harris P. J., Bartley L. E., Ralph J., Sci. Adv. 2016, 2, e1600393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Lu F., Ralph J., J. Agric. Food Chem. 1999, 47, 1988–1992; [DOI] [PubMed] [Google Scholar]
- 21b. Lu F., Ralph J. in Lignin: Structural Analysis, Applications in Biomaterials, and Ecological Significance (Ed.: F. Lu), Nova Science Publishers Inc., New York, 2014, pp. 27–65; [Google Scholar]
- 21c. Petrik D. L., Karlen S. D., Cass C. L., Padmakshan D., Lu F., Liu S., Le Bris P., Antelme S., Santoro N., Wilkerson C. G., Sibout R., Lapierre C., Ralph J., Sedbrook J. C., Plant J. 2014, 77, 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.S. A. Ralph, L. L. Landucci, J. Ralph, NMR database of lignin and cell wall model compounds, 2009, https://www.glbrc.org/databases_and_software/nmrdatabase/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary