

Abstract
We report pharmacokinetics, pharmacodynamics, and safety of a novel anti‐CD28 domain antibody antagonist (lulizumab pegol) in healthy subjects following single‐ or multiple‐dose administration. A minimal anticipated biological effect level approach was used to select a 0.01 mg starting dose for a single‐ascending‐dose (SAD), double‐blind, first‐in‐human study. Part 1 included 9 intravenous (IV; 0.01‐100 mg) and 3 subcutaneous (SC; 9‐50 mg) doses or placebo. In part 2, a keyhole limpet hemocyanin (KLH) immunization was performed in 16 subjects/panel, who received 1 of 3 IV doses (9‐100 mg) or placebo. In a double‐blind, multiple‐ascending‐dose (MAD) study, subjects received SC lulizumab 6.25 mg every 2 weeks, 12.5 mg weekly, 37.5 mg weekly, or placebo. Among 180 treated subjects, 169 completed the studies. Peak concentrations and areas under the curve from time 0 to infinity increased dose proportionally. Estimated SC bioavailability was 68.2%. Receptor occupancy of approximately ≥80% was maintained for ≥2 weeks at ≥9‐mg doses (SAD) and throughout the dosing interval (MAD). IV doses ≥9 mg inhibited antibody production against KLH for 2 weeks. No significant cytokine or immune cell changes were observed. No immunogenicity responses persisted, and there was no correlation to adverse events. Headache occurred in 21 SAD and 4 MAD subjects receiving lulizumab; in the MAD study 5 lulizumab subjects experienced infections. Lulizumab IV or SC was safe at all doses studied, without evidence of cytokine release.
Keywords: pharmacokinetics and drug metabolism, pharmacodynamics (PDY), clinical trials (CTR), immunopharmacology (imm), biologics, rheumatology
CD28 costimulation is required for T‐cell responses to antigens and for B‐cell responses to T cell–dependent antigens.1, 2 CD28 has been shown to play a role in the pathology of autoimmune diseases, emerging as a promising therapeutic target for treatment of diseases such as systemic lupus erythematosus (SLE).3, 4 A previous publication described the discovery and preclinical characterization of a domain antibody (dAb), lulizumab pegol (hereafter referred to as lulizumab), that binds to the CD28 receptor and blocks this signaling pathway.5 Lulizumab was generated using phage display and affinity maturation through the diversification of a selected subset of amino acid residues. Monomeric anti‐CD28 domain antibodies were formatted with polyethylene glycol (PEG). Lulizumab is a potent inhibitor of T‐cell proliferation and cytokine production. Unlike the first‐generation T‐cell stimulation inhibitor abatacept (a cytotoxic T lymphocyte–associated antigen‐4‐immunoglobulin [CTLA‐4Ig] fusion protein that binds with different affinities to CD80 and CD86 on antigen‐presenting cells),6 lulizumab is equipotent at inhibiting both CD80‐ and CD86‐driven T‐cell proliferation. No agonist activity, as measured by preclinical T‐cell proliferation or cytokine release, was observed with lulizumab.5
Single‐dose administration of lulizumab 0.05, 0.5, and 5 mg/kg was well tolerated in cynomolgus monkeys with no drug‐related effects on plasma cytokine concentrations or profound changes in peripheral blood T‐cell counts.5 Drug‐related effects were restricted to dose‐dependent suppression of primary T cell‐dependent antibody responses (TDAR) to keyhole limpet hemocyanin (KLH) after day‐1 dosing. Lulizumab 0.05 mg/kg showed 87% suppression on day 8; 0.5 mg/kg showed ≥96% suppression on day 8 (decreasing through day 29), and 5 mg/kg suppressed the primary antibody response by ≥90% through day 29. Furthermore, pharmacokinetic (PK)/pharmacodynamic (PD) modeling revealed a strong link between CD28 receptor occupancy (RO) and inhibition of the KLH‐induced immunoglobulin G (IgG) response, with an estimated in vivo CD28 RO half‐maximal effective concentration of 7.6 ± 0.6 nM or 91.2 ± 7.2 ng/mL. Overall, sustained RO >80% for at least 2 weeks is required to produce significant suppression of TDAR to KLH.
An early CD28 agonist, TGN1412, preferentially activated and expanded type 2 helper T cells and, in particular, CD4+CD25+ regulatory T cells in preclinical models, resulting in transient lymphocytosis with no detectable toxic or proinflammatory effects. However, this agent led to life‐threatening cytokine storms in 6 healthy volunteers during first‐in‐human research.7 Subsequent work identified CD4+ effector memory cells—common in human tissues but lacking in CD28 expression among cynomolgus monkeys, which were used in preclinical evaluation—as the source of toxic cytokines.8, 9, 10 Although the safety concerns outlined above were linked to the agonistic properties of TGN1412, given that lulizumab is a first‐in‐class molecule that targets the same receptor, a cautious approach was employed for the clinical characterization of lulizumab.
We report results of a first‐in‐human study of the PK, PD, and safety profile of the novel anti‐CD28 domain antibody antagonist lulizumab from 2 phase 1 studies in healthy subjects following either single‐dose or multiple‐dose administration. In recognition of the risks inherent to targeting the CD28 pathway, we used a minimum anticipated biological effect level (MABEL) approach to determine the safe starting dose for lulizumab.11 The goals of the first‐in‐human study were to (1) design a safe study by using MABEL to determine first‐in‐human dose, intravenous (IV) dosing (allowing immediate termination of the dosing), and sentinel cohorts; (2) evaluate the safety profile of lulizumab in healthy volunteers; (3) establish the range of target engagement and immunosuppressive activities (proof‐of‐mechanism marker); and (4) characterize the PK and the PK/PD relationships of lulizumab in healthy subjects to enable dose selection for patient studies.
Methods
These studies were conducted in accordance with Good Clinical Practice and with the ethical principles of the Declaration of Helsinki. The protocols, amendments, and subject‐informed consents received appropriate approval (Aspire Institutional Review Board, Santee, CA) prior to initiation. All subjects provided informed, written consent before beginning any study procedures.
Trial Design
A double‐blind, randomized, single‐ascending‐dose (SAD) study was conducted in 2 parts. In part 1, 9 healthy subjects were randomized to each dose panel (lulizumab 0.01, 0.05, 0.25, 1, 3, 9, 25, 50, or 100 mg IV, or 9, 25, or 50 mg subcutaneous [SC]). Within each dose panel, subjects were randomized in a 2:1 ratio according to a computer‐generated randomization scheme to receive lulizumab (n = 6) or placebo (n = 3). IV administration was given over approximately 30 minutes using a volumetric pump. The primary point of injection for SC administration was first to each upper arm, then each thigh. In part 2, to test proof of mechanism, the effect of lulizumab on TDAR was evaluated by measuring the percentage inhibition of IgG titers in KLH‐immunized healthy subjects. Sixteen subjects were randomized (3 active:1 placebo) to each panel of 9, 25, or 100 mg IV, with 1 mg KLH administered SC immediately following lulizumab/placebo administration.
In a double‐blind, randomized, multiple‐ascending‐dose (MAD) study, 8 healthy subjects per panel (3 active:1 placebo) received 5 weekly SC doses of lulizumab at 6.25, 12.5, or 37.5 mg (all 6.25‐mg panel subjects received placebo in weeks 2 and 4), or placebo. Treatment groups are thus discussed as 6.25 mg every 2 weeks, 12.5 mg weekly, and 37.5 mg weekly. Subjects were randomized according to a computer‐generated randomization scheme; the first 2 subjects in each panel were randomized such that 1 subject received lulizumab and 1 received placebo. The primary site of injection was each upper arm, then each thigh. Subjects were required to remain in the clinical facility (Parexel, Baltimore, Maryland) for an additional 7 days after dosing on days 1 and 29.
Subjects aged 18 to 45 years with a body mass index of 18 to 30 kg/m2, inclusive, who were healthy, as determined by medical history, physical examination, 12‐lead electrocardiogram (ECG), and clinical laboratory evaluations were eligible. Subjects with positive KLH‐specific antibody titers at screening were excluded from the KLH panels.
Rationale for Dose Selection
MABEL Dose Selection
Available preclinical data demonstrate that lulizumab is an anti‐CD28 antagonist domain antibody and T‐cell costimulation inhibitor without agonist activity. However, as this first‐in‐class molecule targets an immune system cell surface receptor that may elicit a biologic cascade or cytokine release potentially insufficiently controlled by a feedback mechanism, a risk mitigation plan was employed. The plan included sentinel cohorts within each dose panel and MABEL calculation for determining the first‐in‐human dose, selected as 0.01 mg to target RO <10% at maximum predicted plasma concentration (Cmax). Details of MABEL dose selection are published in Yang et al.11 Briefly, the 0.01‐mg dose was derived from the EC10 value (0.32 nM) from the human mixed lymphocyte reaction (MLR) assay and the human plasma volume of 0.04 L/kg. This was approximately 3‐fold greater than the 3.1‐μg MABEL dose calculated for the target 10% CD28 RO using a Kd value of 0.41 nM (from a surface plasmon resonance method, Biacore® T100, GE Healthcare Bio‐Sciences, Pittsburgh, Pennsylvania) and the total CD28 expression of 0.65 nM. The value determined with the MLR assay was used because of the general agreement between in vitro MLR EC50 (2.9 ± 1.2 nM) and CD28 RO EC50 (4.4 ± 0.9 nM), and because the test represents a reasonable approximation of in vivo conditions. The exposure (area under the serum concentration‐time curve [AUC]) of the proposed IV starting dose is projected to be approximately 76,000‐fold below that of no observed adverse effect level (NOAEL; 15 mg/kg per week, IV) in monkeys and approximately 2900‐fold below the human maximum‐recommended starting dose of 29 mg (0.49 mg/kg) based on body surface area scaling method and Good Laboratory Practice toxicity studies from the US Food & Drug Administration and European Medicines Agency Committee for Medicinal Products for Human Use guidance.12, 13
Dose Selection for SC Panels
The potential, predicted, human‐efficacious dose of lulizumab 23 mg SC every 2 weeks would need to achieve a steady‐state trough concentration producing RO of 80%. Three dose levels of lulizumab (9, 25, and 50 mg) were to be administered SC to establish absolute bioavailability, PK linearity, and immunogenicity with SC administration in the efficacious exposure range. The 9‐ and 50‐mg SC doses were selected to test bioavailability, accounting for the variability of the 25‐mg SC dose in patients.
KLH Dose Selection
In part 2 of the SAD study, the effect of lulizumab on the TDAR was evaluated by measuring the percentage inhibition of IgG titers in KLH‐immunized healthy subjects. On day 1, lulizumab 9, 25, and 100 mg, or placebo was administered IV, and immediately afterward all subjects received 1.0 mg KLH (Immucothel®, Biosyn Corp, Carlsbad, California) SC. Both total and KLH‐specific Ig titers were measured predose on day 1 and on days 8, 15, and 29. The dose of 25 mg IV was the predicted efficacious dose; the highest safety dose (expected to be 100 mg) was chosen to increase the probability of success. The highest proposed dose of 100 mg IV was expected to maintain RO >90% through 29 days and provide sufficient safety margin relative to the AUC at the NOAEL in monkeys. This dose enabled further characterization of safety and accounted for potential disparities in case PK and target load differ between healthy volunteers and patients with SLE.
All cohorts in part 2 received an IV formulation for consistency because the SC formulation limits the dose to no higher than 50 mg (4 SC injections). Responses seen in KLH‐immunized monkeys5 suggested a single dose of lulizumab with correlating AUCs ≥500 μg·h/mL could provide sustained (∼90%) RO and significant KLH‐induced IgG inhibition. The predicted areas under the serum concentration‐time curve to end of dosing interval (AUCτ) for the 9‐, 25‐, and 100‐mg IV doses are 139, 774, and 1549 μg·h/mL, respectively. Given similar inhibition with lulizumab in humans and monkeys, a single dose was anticipated to provide similar sustainable inhibition of KLH‐induced IgG. KLH data from an earlier study showed that at therapeutic doses of abatacept, more than half of psoriasis patients had antibody titers >2 standard deviations (SD) below the control group mean, correlating with a ≥50% improvement in Physician's Global Assessment.14 Based on the above data, if ≥50% of the subjects had a response of ≥2 SD below the mean of the control group at 25 mg, development would proceed to the MAD study.
MAD Dose Selection
Three SC dosing regimens were evaluated in the MAD study: 6.25 mg biweekly (every other week), 12.5 mg weekly, and 37.5 mg weekly. These doses were selected to provide increasing projected RO of CD28 by lulizumab at steady state. Based on the findings from the SAD study, along with in vitro and in vivo human and monkey RO, the SC dose range would span the range of anticipated pharmacologic activity and was considered well within the safety margin indicated by AUC and Cmax obtained from the highest dose administered in the SAD study.
Sentinel Cohorts
The risk mitigation plan employed in the SAD study also included a sentinel cohort approach. There were 2 sentinel cohorts for each dose panel: on day 1, 1 subject received lulizumab and another received placebo in a blinded fashion; an additional lulizumab and placebo subject were each randomized on day 2. The remaining subjects of the panel were dosed simultaneously on day 3. Panel 7 (25 mg IV) was dosed simultaneously with panel 8 (9 mg SC); panel 9 (50 mg IV) was dosed simultaneously with panel 10 (25 mg SC), and panel 11 (100 mg IV) was dosed simultaneously with panel 12 (50 mg SC). Safety data from sentinel cohorts were evaluated by the investigator and sponsor before the remaining 5 subjects in each panel were treated, and dose‐escalation decisions were based on safety and laboratory test results from up to day 15 for all subjects within each dose panel. SC dosing did not occur until the respective IV dosing from part 1 completed dosing safely.
In parts 1 and 2 of the SAD study, subjects were admitted to the facility on the morning of day –1 and remained in the clinic for 15 days. Subjects were furloughed following evaluation on day 15 after their laboratory tests from that day were reviewed. Subjects in part 1 returned to the facility on days 22 and 29 for PK, RO, immunogenicity, and/or safety assessments and on days 43 and 57 for additional immunogenicity and/or safety assessments. Subjects in part 2 returned to the clinic on day 29 for PK, immunogenicity, safety, RO, and total/anti‐KLH Ig titers and on days 43 and 57 for immunogenicity and/or safety evaluations. Subjects with ongoing adverse events (AEs) or serious AEs (SAEs) at day 15 remained at the site until the investigator determined that these events resolved or were not clinically significant.
Potential CD28 agonistic activities after lulizumab administration were closely monitored with special attention to any signs of cytokine release syndrome (see Pharmacodynamics, below).
Furthermore, IV administration was selected for the first‐in‐human dose because half an hour is needed for the infusion instead of an instant injection SC, allowing timely discontinuation if needed to mitigate a serious outcome.
Pharmacokinetics
During part 1 of the SAD study, samples were taken on day 1 at 0 (predose), 0.25, 0.5, 2, 6, and 12 hours after dosing; on day 2 at 24 and 36 hours after dosing; and on days 3, 4, 6, 8, 15, 22, and 29; during part 2, blood samples were taken predose and 30 minutes after dosing and on days 8, 15, and 29. The assay used to support the SAD study for quantification of lulizumab was a fully validated plate‐based ligand‐binding assay (LBA) with electrochemiluminescence detection. Individual subject PK‐parameter values were derived by noncompartmental methods using a validated PK analysis program (eToolbox EP v2.7/Kinetica 5.0 [Thermo Fisher Scientific, Waltham, Massachusetts] for SAD and Phoenix 1.3 /WinNonlin 6.3 [Pharsight Corporation, Mountain View, California] for MAD). Parameters included Cmax, time of occurrence of Cmax (Tmax), area under the plasma concentration‐time curve from time 0 to the time with last quantifiable concentration (AUC0‐T), area under the serum concentration‐time curve from time 0 extrapolated to infinite time (AUC∞), half‐life (T1/2), clearance (CL) by IV dosing or apparent CL by SC dosing (CL/F), volume of distribution at terminal phase (Vz) and steady state (Vss) after IV dosing, apparent volume of distribution at terminal phase after SC dosing (Vz/F), and absolute bioavailability (F) after SC dosing.
During the MAD study, samples were taken on day 1 at 0 (predose) and 6 hours after dosing and on days 2, 3, 4, 5, 6, and 7; then at predose on days 8, 15, 22, and 29; then on days 30, 31, 32, 33, 34, 35, and 36; then on days 43 ± 2, 50 ± 2, 57 ± 3, 71 ± 3, and 85 ± 3. The serum samples were analyzed for lulizumab by a fully validated liquid chromatography with tandem mass spectrometry (LC‐MS/MS) method (reasons for the change in method are explained below).15 Parameters included Cmax, Tmax, AUCτ, T1/2, and accumulation index for AUC and Cmax.
The LBA used in the SAD study had a lower limit of quantitation of 80 ng/mL; to better characterize the full PK profile as well as the PK‐RO relationship in the low‐dose cohort of the MAD study, an LC‐MS/MS assay was developed and validated with a lower limit of quantitation of 10 ng/mL. The LC‐MS/MS assay included an acid‐dissociation pretreatment step to overcome interferences from soluble CD28 and antidrug antibodies; this was followed by a protein‐precipitation step. Lulizumab stays in solution after protein precipitation due to its modification with PEG; it is then detected via LC‐MS/MS. A cross‐validation of the LC‐MS/MS and the LBA was conducted through analysis of blinded quality controls and residual samples from the SAD study. A standard equivalence test was used to determine if the 2 methods produced similar predicted concentrations: the 90% confidence intervals (CIs) for the ratios of the geometric means (LC‐MS/MS vs LBA) should fall within the interval of 0.8 to 1.25, with 80% power, and the LC‐MS/MS results were found to be statistically equivalent to the LBA results. However, equivalence between the assays cannot be assumed to apply generally, as the findings of the LBA can be subject to interference if levels of antidrug antibodies and soluble target or other matrix‐interfering factors are sufficiently high.15
Pharmacodynamics
Receptor Occupancy
In parts 1 and 2 of the SAD study as well as in the MAD study following repeated SC dosing, CD28 RO by lulizumab was measured using a flow cytometry assay method in which direct detection of the bound domain antibody was employed. During part 1 of the SAD study, RO samples were taken on day 1 at 0 (predose) and 0.5 hours after dosing and on days 2, 3, 4, 8, 15, 22, and 29. During part 2, samples were taken predose and on days 8, 15, and 29. In the MAD study, RO samples were taken on day 1 at 0 (predose) and 6 hours after dosing and on days 2, 3, 8, 15, 22, and 29; follow‐up samples were taken on days 36, 43 ± 2, 50 ± 2, 57 ± 3, 71 ± 3, and 85 ± 3. Whole blood obtained by venipuncture was collected in tubes (containing a solution of 22.0 g/L trisodium citrate, 8.0 g/L citric acid, and 24.5 g/L dextrose), transported at room temperature, and processed within 24 hours of blood draw. Samples were divided into 2 100‐μL aliquots; to 1 aliquot, a saturating lulizumab concentration was added, and both the treated and test samples were incubated at 37°C for 1 hour. Following sequential washes, blood samples were incubated with mouse IgG to block Fc‐mediated nonspecific binding, followed by incubation with a biotinylated rabbit anti‐PEG monoclonal antibody (Epitomics, Burlingame, California). Samples were washed and incubated with fluorescently labeled anti‐CD3, anti‐CD4, anti‐CD8, and anti‐CD11a antibodies (BD Biosciences, San Jose, California) along with streptavidin PE (Life Technologies, now Thermo Fisher Scientific). A background sample was generated by staining a baseline sample as stated above prior to lulizumab treatment. Following lyse/fix with FACS Lysing Solution (BD Biosciences), samples were analyzed on a BD FACSCanto II. Data were analyzed using FACSDiva software (BD Biosciences), and the median fluorescence intensity was determined by gating on CD8+CD11alow (a subpopulation of CD8+ T cells shown to uniformly express CD28) or CD4+ lymphocytes.
Lulizumab‐positive CD4 and CD8 T cells were used to determine the percentage RO via the following formula:
Safety Biomarkers: Serum Cytokine and Immune Cell Measurements
In the SAD and MAD studies, serum cytokines (interleukin‐1β, ‐2, ‐8, and ‐6; interferon‐γ; and tumor necrosis factor‐α) and frequency of lymphocyte subsets (T, B, or natural killer cells) were also monitored as safety biomarkers to confirm the absence of agonistic activity following lulizumab administration. Cytokines were measured in serum samples using a Meso Scale Discovery Multiplex Assay Kit (Meso Scale Diagnostics, Rockville, Maryland). Lymphocyte subsets were measured using the Multitest TBNK Reagent with BD Trucount™ Tubes (BD Biosciences). In part 2 of the SAD study, lulizumab was evaluated for its effects on the T cell–dependent antibody response following KLH immunization. Blood was drawn at screening and on days 1 (predose), 8, 15, and 29 for the measurement of anti‐KLH IgG and anti‐KLH IgM antibodies using an ELISA method (kits manufactured by Alpha Diagnostics, San Antonio, Texas; assays validated at QLab for Bristol‐Myers Squibb).
Immunogenicity
During the SAD study, immunogenicity samples were taken on days 1 (predose), 15, 29, and 57 for both parts 1 and 2. Samples were analyzed for antibodies to lulizumab by a validated bridging electrochemiluminescence immunoassay on the Meso Scale Discovery platform. When ≥250 ng/mL of positive control antibody was present, the assay could tolerate at least 2 μg/mL of lulizumab and not result in a false‐negative result. Monkey antibodies served as a positive control. This assay provided a semiquantitative assessment of immunogenicity and was performed in 3 tiers (screening, confirmatory, and titer) using statistically determined cutoff points.
During the MAD study, immunogenicity samples were taken on days 1 (predose), 15, 36, 57 ± 3, and 85 ± 3. Serum samples were analyzed for antilulizumab antibodies using the same immunogenicity assay as in the SAD study.
The immunogenicity‐positive population was defined as all subjects who received lulizumab, were predose negative, and had one or more serum samples that were confirmed positive for antilulizumab antibodies, or subjects that were baseline positive and had one or more serum samples that were confirmed positive for antilulizumab antibodies with a titer 4‐fold greater than the baseline titer.
Safety Assessments
Safety assessments included AEs, physical examinations, laboratory evaluations, vital signs, and ECGs.
Statistical Considerations
In part 1 of the SAD study, 108 subjects were randomized with 9 subjects per panel (6 lulizumab:3 placebo). In part 2, 48 subjects were randomized with 16 subjects per panel (12 lulizumab:4 placebo). The number of subjects was not based on statistical power considerations; however, administration of lulizumab to 6 subjects in each panel would provide an 80% probability of observing at least 1 occurrence of any AE that would occur with 24% incidence in the population from which the sample was drawn.
Individual subject PK‐parameter values were derived by noncompartmental methods (sampling schedule and software described under Pharmacokinetics, above). Plots of mean (SD) serum concentration profiles vs time were presented, and summary statistics were tabulated for each PK parameter for each treatment. Dose proportionality was assessed using the power model described by Gough et al.16 A slope (β) equal to 1 would indicate perfect dose proportionality. For each PK parameter (Cmax, AUC), the point estimates and 90% CI of the slopes were provided; in MAD, this was provided for Cmax after the first dose (day 1) and on AUCτ at steady state (day 29), while no dose‐proportionality assessment was conducted on day 1 AUCτ and on day 29 Cmax because the doses were administered using different dosing intervals. For the data used in the power model, a similar approach was applied for nonquantifiable concentrations as done in the PK summary.
In the SAD study to assess the absolute SC bioavailability of lulizumab, a linear fixed‐effect model with dose and route of administration as predictors was fitted to the log‐transformed PK parameters (AUC∞ and AUC0‐T) for use in estimation of effects. Point estimates for the SC‐to‐IV ratio of geometric means on the log scale were exponentiated to obtain estimates for ratios of geometric means on the original scale.
Results
Patient Disposition and Baseline Characteristics
In the SAD study, 156 subjects were randomized and treated; 150 (96.2%) completed the study: 1 subject withdrew consent, and 5 subjects were lost to follow‐up. In the MAD study, 24 subjects were randomized and treated; 19 (79.2%) completed the study: 2 subjects withdrew consent, and 3 discontinued due to AEs.
Demographic and baseline characteristics are shown in Tables S1‐S3.
Pharmacokinetics
During cross‐validation of the assays used in the 2 studies, as the antilog of the lower and upper limits on the 90% CI for the mean difference in log concentrations (LC‐MS/MS to LBA) was 0.96 to 1.05, the LC‐MS/MS results were found to be statistically equivalent to the LBA results. However, Cmax values (after the first dose) generated in analyses of the LC‐MS/MS data from the MAD study were up to 58% higher than the predicted values based on the SAD study. When a subset of 58 samples was reanalyzed by the LBA method in an exploratory manner, differences between the 2 assays ranged up to 44% in absolute percentage terms; the difference was small at low concentrations and increased significantly with increasing concentrations (data not shown). However, as linearity appears to be consistent between the results determined by both assays and the differences in concentration appear to be within 50% of each other, the apparent differences are not considered to be clinically relevant.
Lulizumab serum concentrations increased with increasing dose. The decline in concentrations appears to be biexponential following IV administration but monoexponential following SC administration. Lulizumab exhibited linear PKs after single IV and SC administration. Cmax and AUC∞ of lulizumab administered IV increased approximately in proportion to dose over the range of 3 to 100 mg (Figure 1A‐D); slopes and 90% CIs for Cmax and AUC∞ were 0.972 (0.947; 0.996) and 1.029 (0.992; 1.065). The AUC0‐T for the 0.25‐mg IV dose provided lower values than expected, contributing to a slope above 1 (1.327 [1.221; 1.433]). Dose proportionality assessment could not be performed for the 0.01‐ and 0.05‐mg IV doses because of poorly characterized AUC∞ contributed by lack of measureable concentrations. Cmax and AUC∞ of lulizumab administered SC increased in proportion to dose for the 3 doses tested (9, 25, and 50 mg). Following SC administration, slopes (90% CI) for Cmax and AUC∞ were 0.982 (0.789; 1.175) and 1.052 (0.927; 1.176) respectively; low values for the 9 mg dose were contributed to by a higher slope for AUC(0‐T) (1.121 [1.013; 1.230]). Figure 2 shows that the predicted PK based on monkey‐model allometric scaling based on body weight (assuming monkey weight of 4 kg and human weight of 70 kg) was generally well aligned with the observed clinical data.
Figure 1.
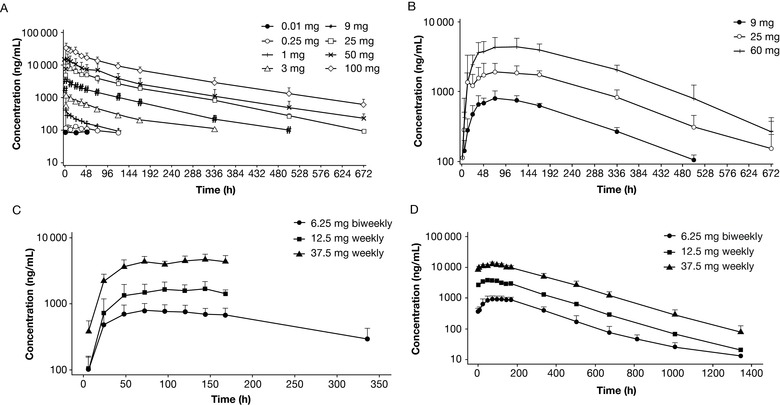
Mean lulizumab serum concentration profile vs time. Mean (SD) lulizumab serum concentration profile vs time is shown on a log‐linear scale. (A) IV treatment in the SAD study; (B) SC treatment in the SAD study; (C) day 1 of the MAD study; (D) day 29 of the MAD study. IV, intravenous; MAD, multiple‐ascending dose; SAD, single‐ascending dose; SC, subcutaneous; SD, standard deviation.
Figure 2.
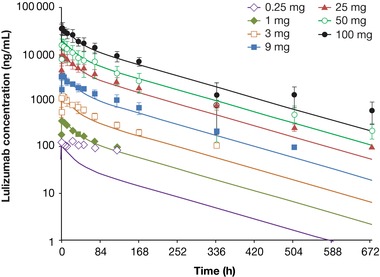
Monkey PK model predicts first‐in‐human PK for lulizumab. Dots with error bars represent observed human PK data from the SAD IV dose panels. Smooth lines represent the simulated profile using the monkey PK model with allometric scaling. IV, intravenous; PK, pharmacokinetic; SAD, single‐ascending dose.
Following single administration of lulizumab, the T1/2 of lulizumab ranged from 98 to 131 hours following IV administration (in the range of 3 to 100 mg) and 118 to 132 hours following SC administration (9, 25, and 50 mg; Table 1). The relatively low T1/2 of 67.6 hours for 1 mg IV was due to lack of measureable concentrations in the terminal elimination phase. As a result, the terminal T1/2 of lulizumab is in the range of 4 to 5.5 days. Following single SC doses, the maximum concentrations of lulizumab occurred at a median Tmax of 60.2 to 120 hours.
Table 1.
Pharmacokinetics in the SAD Study
| SAD (IV) | Cmax (ng/mL), geo mean (%CV) | Tmax (hours), median (min‐max) | AUC(0‐T) (ng·h/mL), geo mean (%CV) | AUC∞ (ng·h/mL), geo mean (%CV) | T1/2 (hours), mean (SD) | CL (mL/min), geo mean (%CV) | Vz (mL), mean (SD) | Vss (mL), mean (SD) |
|---|---|---|---|---|---|---|---|---|
| 0.25 mg | 118 (31) | 0.480 (0.470‐2.02) | 843 (183) | N/A | N/A | N/A | N/A | N/A |
| 1 mg | 390 (9) | 1.99 (0.470‐2.00) | 19 779 (12) | 28 087 (17) a | 67.6 (21.6) a | 0.593 (18) a | 3397 (768) a | 3232 (611) a |
| 3 mg | 1236 (12) | 2.00 (0.470‐6.05) | 91 622 (21) | 114 470 (16) | 102 (32.1) | 0.437 (15) | 3803 (918) | 3412 (706) |
| 9 mg | 3420 (13) | 2.00 (2.00‐6.05) | 318 497 (21) | 345 132 (18) | 97.8 (13.5) | 0.435 (16) | 3698 (659) | 3497 (529) |
| 25 mg | 9613 (30) | 2.06 (0.470‐6.00) | 926 285 (17) | 995 712 (15) | 106 (10.6) | 0.418 (14) | 3906 (779) | 3848 (712) |
| 50 mg | 18 520 (12) b | 6.00 (0.470‐12.1) b | 1 746 031 (14) b | 1 828 687 (13) b | 119 (23.9) b | 0.456 (16) b | 4631 (587) b | 4025 (337) b |
| 100 mg | 39 127 (21) | 2.01 (0.470‐2.02) | 3 562 181 (22) | 3 701 527 (21) | 131 (33.3) | 0.450 (21) | 5121 (1604) | 4509 (1462) |
| SAD (SC) | Cmax (ng/mL), geo mean (%CV) | Tmax (hours), median (min‐max) | AUC0‐T (ng·h/mL), geo mean (%CV) | AUC∞ (ng·h/mL), geo mean (%CV) | T1/2 (hours), mean (SD) | CL/F (mL/min), geo mean (%CV) | Vz/F (mL), mean (SD) | |
|---|---|---|---|---|---|---|---|---|
| 9 mg | 827 (21) | 72.1 (72.0‐120) | 197 840 (14) | 234 288 (8) c | 132 (13.1) c | 0.640 (8) c | 7304 (814) c | |
| 25 mg | 2334 (51) | 60.2 (12.0‐168) | 556 929 (21) | 599 939 (22) b | 119 (13.6) b | 0.695 (24) b | 7327 (2024) b | |
| 50 mg | 4437 (29) | 120 (71.8‐168) | 1 373 820 (17) | 1 421 107 (17) | 118 (16.9) | 0.586 (18) | 6053 (1363) |
%CV, coefficient of variation; AUC, area under the serum concentration‐time curve; AUC0‐T, area under the serum concentration‐time curve from time 0 to the time with last quantifiable concentration; AUC∞, area under the serum concentration‐time curve from time zero extrapolated to infinite time; CL, total body clearance; CL/F, apparent body clearance; Cmax, maximum observed plasma concentration; geo mean, geometric mean; IV, intravenous; N/A, not available; SAD, single‐ascending dose; SC, subcutaneous; SD, standard deviation; T1/2, half‐life; Tmax, time of occurrence of Cmax; Vss, volume at steady state; Vz, volume of distribution at terminal phase; Vz/F, volume of distribution at terminal phase for SC dosing. Dose proportionality assessment could not be performed for the 0.01‐ and 0.05‐mg IV doses because of poorly characterized AUC∞ contributed by lack of measureable concentrations.
Patient number in each treatment group is n = 6, unless stated otherwise:
an = 3; bn = 5; cn = 4.
The mean total CL, Vz, and Vss were in the range of 0.42 to 0.59 mL/min, 3.4 to 5.1 L, and 3.2 to 4.5 L, respectively, and were relatively consistent among all the dose groups following IV administration (Table 1). The mean apparent total CL/F and Vz/F were in the range of 0.59 to 0.70 mL/min and 6.0 to 7.3 L, respectively, and relatively consistent among all the dose groups following SC administration. Bioavailability of lulizumab following SC administration on AUC∞ was 68.2% and is considered reasonable.
Following every‐other‐week and weekly SC administration, the PK of lulizumab was linear over the range of 6.25 mg every other week to 37.5 mg weekly (Table 2). A median Tmax in the range of 72 to 132 hours and a median T1/2 of 6 to 7 days were observed. The slightly longer T1/2 in the MAD study compared to that in the SAD study is likely due to more measurable concentrations leading to better characterized terminal phase PK. The geometric means of accumulation index of AUC were 1.3, 2.4, and 3.0 for 6.25 mg every other week, 12.5 mg weekly, and 37.5 mg weekly, respectively.
Table 2.
Pharmacokinetics in the MAD Study
| MAD (Day 1) (SC) | Cmax (ng/mL), geo mean (%CV) | Tmax (hours), median (min‐max) | AUCτ (ng·h/mL), geo mean (%CV) | |||
|---|---|---|---|---|---|---|
| 6.25 mg biweekly | 798 (22) | 96.0 (47.9‐144) | 177 133 (25) | |||
| 12.5 mg weekly | 1746 (29) | 132 (48.0‐168) | 205 409 (30) | |||
| 37.5 mg weekly | 4812 (15) | 108 (48.1‐144) | 595 335 (16) |
| MAD (Day 29) (SC) | Cmax (ng/mL), geo mean (%CV) | Tmax (hours), median (min‐max) | AUCτ (ng·h/mL), geo mean (%CV) | T1/2 (hours), mean (SD) | AI AUC ([ng·h/mL]/[ng·h/mL]), geo mean (%CV) | AI Cmax ([ng/mL]/[ng/mL]), geo mean (%CV) |
|---|---|---|---|---|---|---|
| 6.25 mg biweekly | 984 (23) | 96.0 (48.0‐168) | 232 163 (26) | 142 (34.0) | 1.311 (20) | 1.234 (25) |
| 12.5 mg weekly | 3857 (19)a | 72.1 (48.0‐96.1)a | 550 924 (17)a | 162 (9.95)a | 2.375 (12)a | 2.000 (13)a |
| 37.5 mg weekly | 12 626 (16)a | 72.2 (48.0‐96.2)a | 1 830 528 (15)a | 156 (19.1)a | 2.992 (18)a | 2.500 (8)a |
%CV, coefficient of variation; AI, accumulation index; AUC, area under the serum concentration‐time curve; AUCτ, area under the serum concentration‐time curve to end of dosing interval; Cmax, maximum observed plasma concentration; geo mean, geometric mean; MAD, multiple‐ascending dose; SC, subcutaneous; SD, standard deviation; T1/2, half‐life; Tmax, time of occurrence of Cmax.
Patient number in each treatment group is n = 6, unless stated otherwise:
n = 5.
Pharmacodynamics
At doses ≥9 mg administered IV or SC, RO of approximately 80% or greater was observed and maintained for ≥2 weeks in the SAD study and throughout the dosing interval in the MAD study with doses ≥6.25 mg SC every 2 weeks. Figure 3 shows CD4 cell RO (results for CD8 cell RO were similar). In the second part of the SAD study, single IV doses of ≥9 mg lulizumab inhibited antibody production against KLH for 2 weeks, demonstrating immunosuppressive activity (Figure 4A). Anti‐KLH IgG antibodies were detectable in approximately half of the individuals receiving placebo by 2 weeks after KLH immunization; levels were maintained or increased for an additional 2 weeks. Complete and sustained suppression of anti‐KLH IgG antibodies was observed in the lulizumab 25‐ and 100‐mg dose groups for 4 weeks in part 2; the 9‐mg IV dose group was the only group to rebound in IgG at day 29. Not surprisingly, at day 15, the 9‐mg IV dose group had a mean RO well below 80% (Figure 4B). As indicated by the IgG response in the placebo group, healthy humans generated antibodies against KLH within roughly 2 weeks; therefore, the rebound of IgG response in the 9‐mg IV dose group at day 29 may be a result of the lower RO at day 15.
Figure 3.
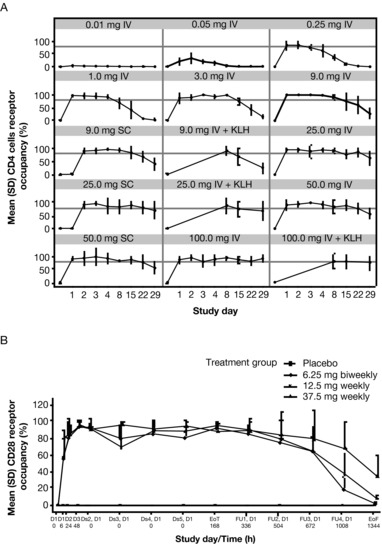
Receptor occupancy. (A) Mean (SEM) CD28 receptor occupancy (%) of CD4 cells by cohort in the SAD study (gray horizontal line indicates 80%) and (B) mean (SD) CD28 receptor occupancy (%) of CD4 cells by cohort in the MAD study. D, day; Ds, dose; EoT, end of treatment; EoF, end of follow‐up; FU, follow‐up; IV, intravenous; MAD, multiple‐ascending dose; SAD, single‐ascending dose; SD, standard deviation; SEM, standard error of the mean.
Figure 4.
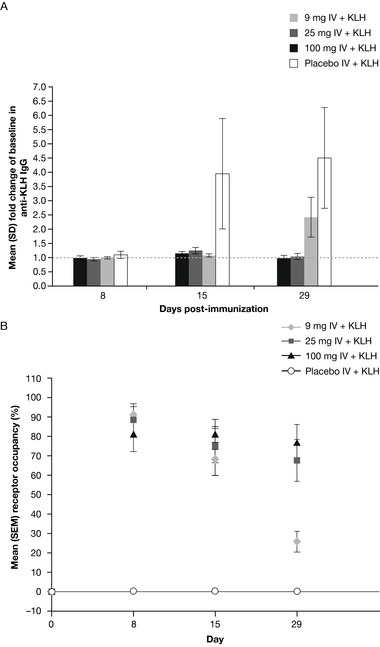
(A) Lulizumab suppression on KLH‐induced IgG response at days 8, 15, and 29 after single IV dose administration in healthy subjects in the SAD study; (B) mean CD4 cell CD28 receptor occupancy in the KLH panels of the SAD study. IgG, immunoglobulin G; IV, intravenous; KLH, keyhole limpet hemocyanin; SAD, single‐ascending dose.
The RO and immunosuppressive activities by KLH immunization translated well from monkey to human. As previously reported,5 preclinical data suggest that 80% RO for 2 weeks is needed for significant immunosuppression as measured by IgG suppression following KLH‐antigen challenge in the 0.5 mg/kg group (Figure S1A,B), and as levels of RO fall well below 80%, the immunosuppressive activity lessens, and anti‐KLH antibody formation rises, in the 0.05 mg/kg group similar to levels seen in the placebo group.
In both the SAD and MAD healthy subject studies, cytokine elevations were few, mild, and transient, with no correlation to dose group. No clinically relevant changes in T‐, B‐, or natural killer cell panels were observed.
Immunogenicity
In the SAD study, 4 subjects were classified as ADA positive (either negative at baseline and had detectable antilulizumab antibodies at time points after drug administration or were baseline positive and had positive samples with a titer 4‐fold greater than the baseline titer). Each subject was in a different dose group (0.05 mg IV; 9 mg IV; 9 mg IV with KLH challenge; and 100 mg IV with KLH challenge). No correlation between positive samples and dose or drug concentration was seen, and cytokine levels in antilulizumab antibody‐positive subjects remained comparable with those with no detectable immunogenic response. In the MAD study, 1 baseline‐negative subject in the 6.25‐mg SC dose group had a positive sample at the end of the follow‐up period (day 84). No subject with a positive sample had a related AE at the time the sample was obtained.
Safety
The AEs that occurred in ≥5% of patients receiving lulizumab are shown in Tables S4‐S6.
The most frequently reported AE in the SAD study was headache (n = 17 [15.7%] and n = 4 [8.3%] for lulizumab and placebo, respectively). Four subjects were discontinued from the study due to AEs of acute infusion reaction after receiving IV administration of lulizumab. These symptoms were reversible on cessation of administration and initiation of standard treatment. Two subjects experienced SAEs. One subject in part 1 experienced acute renal failure due to severe dehydration on day 16 following administration of 25 mg lulizumab SC; this condition fully resolved. One subject in part 2 had a perforated appendicitis on day 8 after having begun receipt of 25 mg lulizumab IV + KLH on day 1. However, in this case, administration of lulizumab was incomplete; infusion was stopped after administration of only 1 mg of lulizumab due to mild to moderate infusion‐related AEs. Both SAE events, ie, acute renal failure and perforated appendicitis, were considered to be unrelated to the study drug by the investigator.
Isolated, asymptomatic alanine transaminase increases (with 41 U/L considered the upper limit of normal) were seen in 20 (18.5%) subjects following administration of lulizumab and 6 (12.5%) subjects following administration of placebo in the SAD study. There were no clinically relevant changes in ECG intervals following administration of lulizumab and no relationship between change in time‐matched ECG intervals and increasing lulizumab serum concentrations. There were no clinically relevant changes in vital signs following administration of lulizumab.
During the MAD study, infections and infestations occurred in 5 (27.8%) subjects following administration of lulizumab (versus 0 in the placebo group). No correlation was observed between exposure and infection rate. One subject in the 12.5‐mg lulizumab weekly group experienced 2 infective episodes: oral herpes on day 40 followed, after 7 days, by an upper respiratory infection; in both episodes the severity was classified as mild. One subject receiving 12.5 mg lulizumab weekly presented with a furuncle of mild severity on day 69. One subject receiving 37.5 mg lulizumab weekly presented on day 89 with a peritonsillar abscess of moderate severity, which required antibiotic treatment with 500 mg amoxicillin 3 times a day for 10 days. One subject receiving 37.5 mg lulizumab weekly had a mild viral infection on day 81. One SAE occurred that was considered possibly related to lulizumab. A subject receiving 6.25 mg lulizumab every 2 weeks required hospitalization on day 49 for cellulitis that developed in his right hand after damage of the skin at the base of his third finger. The hospitalized patient was treated with IV antibiotics, and the lesion was surgically drained. The traumatic skin damage in his right hand is a potential inciting factor for the cellulitis. However, it could not be excluded that lulizumab might have made the subject more susceptible to the subsequent infection. Therefore, the SAE was considered possibly related to the study drug.
Three discontinuations due to AEs were reported. One discontinuation occurred in the placebo group due to a mild increase in transaminases to less than 2 times the upper limit of normal. One subject in the 12.5‐mg and 1 in the 37.5‐mg lulizumab weekly group discontinued due to moderate injection‐site reactions. The most frequently occurring AEs were headache and nausea, and were mild to moderate in severity.
Discussion
These initial, clinical studies with lulizumab in healthy subjects have provided a comprehensive evaluation of the PK, PD, and safety profile of an anti‐CD28 domain antibody. This allows for lulizumab SC administration as infrequently as every other week. In addition, lulizumab has shown high potency, such that it saturates the target receptor at low SC doses. These data were used to develop a PK/PD model that describes the relationship between serum concentrations and RO to assist dose selection for future studies. The safety profile was favorable, without any evidence of cytokine release. The observed trend in increasing infection and transaminases in the MAD study was consistent with immunosuppressant activity and will be monitored closely in our later studies. The safety biomarker results confirmed lack of agonistic activity or any effects on lymphocyte counts in healthy subjects, with no clinically significant changes in proinflammatory serum cytokines or significant changes in T‐, B‐, or natural killer cell counts. These results provide evidence that, in vivo, lulizumab does not induce cytokine storm or cytokine release syndrome in humans. The immunogenicity incidence was low, without any evidence of persistence or impact on the clinical profile. Overall, these results can be used to inform the design of future clinical studies in patients.
Furthermore, it was demonstrated in humans, as well as in monkeys, that near‐maximal target engagement is associated with maximal inhibition of the TDAR. Proof of mechanism was established via KLH data in the SAD study, which provided evidence to assist dose selection for future studies in patients. RO of 80% for immunosuppression was achieved by doses ≥9 mg and resulted in KLH response inhibition. In cynomolgus monkeys, when the CD28 RO was maintained at >80%, the KLH‐induced IgG response was suppressed by >80%, and there was no clinically significant inhibition on the KLH‐induced IgG response when the CD28 RO was well below 30%. The extent and duration of the RO increased with dose, consistent with dose‐dependent increases in drug exposure. Here, in healthy humans, significant suppression of IgG response following KLH immunization was correlated with 80% RO maintained for at least 2 weeks, comparable with predictions from the monkey model. The inhibition of KLH‐induced T cell–dependent antibody generation suggests potent immunosuppressant activity and potential clinical utility. The lowest dose of lulizumab (9 mg IV) maintained complete suppression through day 15; however, presence of anti‐KLH IgG antibodies was detected in some patients at day 29. Notably, the majority of the subjects in whom rebound anti‐KLH IgG was detected had observed RO <80% on day 8 or day 15, indicating that a minimum of 80% RO is required for sufficient suppression of anti‐KLH antibody formation, consistent with preclinical observations. Subsequently, these critical PD targets can effectively be used to guide dose selection for future patient studies.
Notably, the drug levels in the first‐in‐human study were very well predicted using the monkey PK model with allometric scaling. This accurate prediction allowed a safe and informative study design to establish a MABEL first‐in‐human dose and safety, PD, immunosuppressant activity profiles in healthy subjects.
SLE is an extremely complex autoimmune disease. Animal models exist but are of limited predictive value. Tellingly, although many compounds have been found effective in animal models, only 1 compound has been approved for the treatment of human lupus in the last 50 years.17 Corticosteroids and immunosuppressive drugs are generally effective in temporarily controlling flares and disease progression; however, these strategies are insufficiently efficacious, and SAEs significantly limit their prolonged use. Patients with SLE still have a very high unmet medical need. Hyperresponsive T cells with consequential autoantibody production characterize the pathology of SLE. The central role of activated T cells in this disease is supported by the demonstrated efficacy of mycophenolate mofetil and tacrolimus, both potent inhibitors of T‐cell activation.18, 19 Differences in CD28+ T‐cell populations between patients with SLE and controls are not consistently seen. Findings from different studies have included decreased percentages of CD4+CD28+ and CD8+CD28+ T cells among patients with SLE (Alvarado), no significant difference in CD4+CD28+ T‐cell proportion between groups (Bijl), and no difference in overall CD28+ T‐cell proportion but increased proportion of CD8+CD28+ T cells among patients with SLE (Tulunay).20, 21, 22 Additionally, increased CD28 expression may not correlate directly with clinical activity in patients with SLE.23 However, responder T cells from patients with SLE show a loss of CTLA‐4–mediated inhibition of CD3/CD28 costimulation.24 Taken together, these data are indicative of the central role played by the CD28/CTLA‐4–CD80/86 costimulatory pathway in the defective immune response observed in patients with SLE.
In vivo animal efficacy studies further support the usefulness of inhibiting CD28 function and the potential applicability in humans with SLE.5 A powerful and validated CD28 antagonist that directly curtails T‐cell activation might provide a new and much needed therapeutic option for patients with this disease.
Conclusions
The clinical PK of SC lulizumab is favorable, with high bioavailability and the ability to maintain serum concentrations over a 2‐week dosing interval. Following IV and SC administration, lulizumab was well tolerated at all doses studied, without evidence of cytokine release. The most frequently occurring AEs were headache during the SAD study, and headache and nausea during the MAD study; infections occurred but did not show a relationship to dose. Consistent with preclinical and in vitro data, subjects receiving lulizumab showed exposure‐ and time‐dependent RO of CD28. Significant KLH‐response inhibition was achieved over 2 weeks, serving as a proof‐of‐mechanism tool to support further development of lulizumab in patients with SLE.
Disclosures
R. Shi, M. Honczarenko, J. Mora and her spouse, S. K. Lee, R, Wang, X. Liu, and Z. Yang are employees of, and hold stock options and/or bond holdings in, Bristol‐Myers Squibb. S. Zhang and C. Fleener hold stock options and/or bond holdings in Bristol‐Myers Squibb. H. Wang is an employee of Bristol‐Myers Squibb. D. E. Shevell is an employee of, and holds stock options and/or bond holdings in, Bristol‐Myers Squibb and holds stock options and/or bond holdings in Merck & Co. B. Murthy is an employee of, and holds stock options and/or bond holdings in, Bristol‐Myers Squibb and holds stock options and/or bond holdings in Johnson & Johnson.
This study was sponsored by Bristol‐Myers Squibb. Professional medical writing and editorial assistance was provided by Rob Coover, MPH, and Sarah Funderburk, PhD, at Caudex and was funded by Bristol‐Myers Squibb.
The authors would like to thank Guan Xing, formerly of Bristol‐Myers Squibb, for his statistics input for the SAD study during his employment there. G. Xing is currently an employee of Gilead Sciences, Seattle, WA.
The PK and immunogenicity bioanalyses of samples were performed by Janice Gambardella, Sean Crawford, Amy Manney, Shannon Chilewski, Mark Saewert, and Billy Akinsanya. In addition, Hao Jiang and Jonathan Haulenbeek contributed to the method development.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Plasma concentrations (A) and pharmacokinetic/pharmacodynamic modeling of average (B), and KLH‐induced IgG response (C) in cynomolgus monkeys after SC administration of lulizumab. IgG, immunoglobulin G; KLH, keyhole limpet hemocyanin; SC, subcutaneous. Reprinted with permission from Suchard et al. The Journal of Immunology, vol. 191, pp. 4599‐4610, 2013. Copyright 2013. The American Association of Immunologists, Inc.
Supplementary Table S1. Baseline Characteristics (SAD, Part 1)
Supplementary Table S2. Baseline Characteristics (SAD, Part 2)
Supplementary Table S3. Baseline Characteristics (MAD)
Supplementary Table S4. Most Common AEs* Reported During the SAD Study (Incidence During Part 1)
Supplementary Table S5. Most Common AEs* Reported During the SAD Study (Incidence During Part 2)
Supplementary Table S6. AEs Reported in ≥5% of Subjects Receiving Lulizumab (MAD)
References
- 1. Linsley PS, Nadler SG. The clinical utility of inhibiting CD28‐mediated costimulation. Immunol Rev. 2009;229(1):307–321. [DOI] [PubMed] [Google Scholar]
- 2. Ward SG. CD28: a signalling perspective. Biochem J. 1996;318(Pt 2):361–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kow NY, Mak A. Costimulatory pathways: physiology and potential therapeutic manipulation in systemic lupus erythematosus. Clin Dev Immunol. 2013;2013:245928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong CK, Lit LC, Tam LS, Li EK, Lam CW. Aberrant production of soluble costimulatory molecules CTLA‐4, CD28, CD80 and CD86 in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2005;44(8):989–994. [DOI] [PubMed] [Google Scholar]
- 5. Suchard SJ, Davis PM, Kansal S, et al. A monovalent anti‐human CD28 domain antibody antagonist: preclinical efficacy and safety. J Immunol. 2013;191(9):4599–4610. [DOI] [PubMed] [Google Scholar]
- 6. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7‐1 (CD80) and B7‐2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA‐4 receptors. Immunity. 1994;1(9):793–801. [DOI] [PubMed] [Google Scholar]
- 7. Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. [DOI] [PubMed] [Google Scholar]
- 8. Eastwood D, Findlay L, Poole S, et al. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T‐cells. Br J Pharmacol. 2010;161(3):512–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eastwood D, Bird C, Dilger P, et al. Severity of the TGN1412 trial disaster cytokine storm correlated with IL‐2 release. Br J Clin Pharmacol. 2013;76(2):299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stebbings R, Findlay L, Edwards C, et al. "Cytokine storm" in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 2007;179(5):3325–3331. [DOI] [PubMed] [Google Scholar]
- 11. Yang Z, Wang H, Salcedo TW, et al. Integrated pharmacokinetic/pharmacodynamic analysis for determining the minimal anticipated biological effect level of a novel anti‐CD28 receptor antagonist BMS‐931699. J Pharmacol Exp Ther. 2015;355(3):506–515. [DOI] [PubMed] [Google Scholar]
- 12. US Food and Drug Administration (FDA) . Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078932.pdf. Accessed March 3, 2015.
- 13. European Medicines Agency . Guideline on strategies to identify and mitigate risks for first‐in‐human clinical trials with investigational medicinal products. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf. Accessed April 9, 2015.
- 14. Abrams JR, Lebwohl MG, Guzzo CA, et al. CTLA4Ig‐mediated blockade of T‐cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103(9):1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong C, Zeng J, Akinsanya B, et al. Development and validation of an LC‐MS/MS assay for the quantitation of a PEGylated anti‐CD28 domain antibody in human serum: overcoming interference from antidrug antibodies and soluble target. Bioanalysis. 2014;6(18):2371–2383. [DOI] [PubMed] [Google Scholar]
- 16. Gough K, Hutchison M, Keene O, et al. Assessment of dose proportionality: report from the statisticians in the pharmaceutical industry/pharmacokinetics UK joint working party. Drug Information J. 1995;29:1039–1048. [Google Scholar]
- 17. Gatto M, Kiss E, Naparstek Y, Doria A. In‐/off‐label use of biologic therapy in systemic lupus erythematosus. BMC Med. 2014;12:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. 2009;20(5):1103–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mok CC, Tong KH, To CH, Siu YP, Au TC. Tacrolimus for induction therapy of diffuse proliferative lupus nephritis: an open‐labeled pilot study. Kidney Int. 2005;68(2):813–817. [DOI] [PubMed] [Google Scholar]
- 20. Alvarado C, Alcocer‐Varela J, Llorente L, Richaud‐Patin Y, Cerbon M, Alarcon‐Segovia D. Effect of CD28 antibody on T cells from patients with systemic lupus erythematosus. J Autoimmun. 1994;7(6):763–773. [DOI] [PubMed] [Google Scholar]
- 21. Bijl M, Horst G, Limburg PC, Kallenberg CG. Expression of costimulatory molecules on peripheral blood lymphocytes of patients with systemic lupus erythematosus. Ann Rheum Dis. 2001;60(5):523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tulunay A, Yavuz S, Direskeneli H, Eksioglu‐Demiralp E. CD8+CD28–, suppressive T cells in systemic lupus erythematosus. Lupus. 2008;17(7):630–637. [DOI] [PubMed] [Google Scholar]
- 23. Brambila‐Tapia AJ, Gamez‐Nava JI, Salazar‐Paramo M, et al. Increased CD28 serum levels are not associated with specific clinical activity in systemic lupus erythematosus. Rheumatol Int. 2011;31(10):1321–1324. [DOI] [PubMed] [Google Scholar]
- 24. Jury EC, Flores‐Borja F, Kalsi HS, et al. Abnormal CTLA‐4 function in T cells from patients with systemic lupus erythematosus. Eur J Immunol. 2010;40(2):569–578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Plasma concentrations (A) and pharmacokinetic/pharmacodynamic modeling of average (B), and KLH‐induced IgG response (C) in cynomolgus monkeys after SC administration of lulizumab. IgG, immunoglobulin G; KLH, keyhole limpet hemocyanin; SC, subcutaneous. Reprinted with permission from Suchard et al. The Journal of Immunology, vol. 191, pp. 4599‐4610, 2013. Copyright 2013. The American Association of Immunologists, Inc.
Supplementary Table S1. Baseline Characteristics (SAD, Part 1)
Supplementary Table S2. Baseline Characteristics (SAD, Part 2)
Supplementary Table S3. Baseline Characteristics (MAD)
Supplementary Table S4. Most Common AEs* Reported During the SAD Study (Incidence During Part 1)
Supplementary Table S5. Most Common AEs* Reported During the SAD Study (Incidence During Part 2)
Supplementary Table S6. AEs Reported in ≥5% of Subjects Receiving Lulizumab (MAD)