

Abstract
Nanodiscs offer a very promising tool to incorporate membrane proteins into native‐like lipid bilayers and an alternative to liposomes to maintain protein functions and protein–lipid interactions in a soluble nanoscale object. The activity of the incorporated membrane protein appears to be correlated to its dynamics in the lipid bilayer and by protein–lipid interactions. These two parameters depend on the lipid internal dynamics surrounded by the lipid‐encircling discoidal scaffold protein that might differ from more unrestricted lipid bilayers observed in vesicles or cellular extracts. A solid‐state NMR spectroscopy investigation of lipid internal dynamics and thermotropism in nanodiscs is reported. The gel‐to‐fluid phase transition is almost abolished for nanodiscs, which maintain lipid fluid properties for a large temperature range. The addition of cholesterol allows fine‐tuning of the internal bilayer dynamics by increasing chain ordering. Increased site‐specific order parameters along the acyl chain reflect a higher internal ordering in nanodiscs compared with liposomes at room temperature; this is induced by the scaffold protein, which restricts lipid diffusion in the nanodisc area.
Keywords: lipids, membranes, nanostructures, NMR spectroscopy, solid-state structures
1. Introduction
Despite their ubiquitous presence and participation in the majority of cellular processes, membrane proteins are under‐represented among biophysical investigations and many biological mechanisms that take place at the membrane are still poorly understood. Structural and dynamic characterisation of membrane proteins constitute an extremely challenging task because it requires the use of a suitable membrane‐mimicking environment to maintain the protein three‐dimensional fold and sustain protein functions and activity, which are often based on a specific protein–lipid interplay. High‐resolution biophysical techniques commonly use detergent micelles to mimic a hydrophobic environment because they are easy to handle and very homogeneous in size. However, due to the artificial nature and frequent denaturing capabilities of detergents, as well as a high degree of curvature in micelles, membrane proteins and membrane‐associated peptides might not adapt their natural shape/topology, but might bend unnaturally under steric restraints.7, 30, 47 These structural artefacts can be avoided by using artificial vesicles named liposomes. Apart from their highly tuneable lipid composition to mimic cellular membranes, structural stress due to lipid bilayer bending and lateral pressure are also significantly reduced when compared with micelles.23 However, their micrometre size and very slow molecular tumbling are severe obstacles to performing structural characterisation of embedded membrane proteins at the atomic level by means of conventional techniques, such as electron microscopy, X‐ray crystallography or solution NMR spectroscopy.
Furthermore, it is also difficult to control the stoichiometry of embedded membrane proteins inside liposomes, leading to additional complications during single‐particle analysis. In contrast, lipid nanodiscs, which were introduced by Sligar and co‐workers,2 appear to be a promising solution for the aforementioned drawbacks of micelles and liposomes.9 Nanodisc technology12 has emerged as a new technique to purify and reconstitute proteins in a detergent‐free lipid‐bilayer environment.11, 36 A comparison of membrane protein dynamics in micelles and nanodiscs revealed that micelles compromised the inherent flexibility of the protein, despite conservation of the structure.21, 28 Recent investigations into membrane proteins4, 19, 22, 25 have revealed that nanodiscs are a suitable tool to reconstitute membrane proteins in a native environment to solve their three‐dimensional structure with atomic resolution. They are derived from apolipoprotein A1 (ApoA‐1), which solubilises hydrophobic molecules, such as lipids and cholesterol, to deliver them to the liver through the blood and interstitial fluid as high‐density lipoprotein (HDL) particles.16, 27 A recent study by NMR spectroscopy shows how the structure of ApoA‐1 wraps around the hydrophobic part of the lipids to form a lipid‐bilayer patch in solution.5 The self‐assembly process behind the formation of nanodiscs relies on the amphipathic nature of ApoA‐1, whereas the hydrophobic sides of the ApoA‐1 helices interact with the lipid acyl chains and the hydrophilic sides are exposed to solvent. This process leads, after purification and size‐exclusion chromatography, to a monodisperse solution of highly stable 10 nm nanodiscs containing 120–160 lipid molecules (depending on the nature of the lipid) inside the bilayer.3, 9 ApoA‐1, also called a membrane scaffolding protein (MSP) in the nanodisc field, can be genetically modified to design longer constructions (named MSP2N2 and MSP2N3) that support the formation of larger nanodiscs, with a diameter of 16–17 nm.9, 24, 25 The lipid composition is also tuneable because it is possible to add cholesterol and/or charged lipids.17 Lipid physical properties in nanodiscs have been studied and compared with existing membrane models by different biophysical techniques, such as small‐angle X‐ray scattering (SAXS),9, 10, 35, 40 small‐angle neutron scattering (SANS),35, 41 NMR spectroscopy31, 34 or fluorescence spectroscopy.35, 39 Although a general consensus concerning the driving force of nanodisc assembly and the resulting MSP architecture exists nowadays,5, 11 little is known about the internal dynamics of lipids and membrane proteins embedded in nanodiscs, as well as how the lipid–lipid and lipid–protein dynamic interplay influence the membrane protein activity. A decrease of membrane fluidity along the acyl chain for nanodiscs relative to that of artificial vesicles was indirectly measured by incorporating fluorescent probes into lipid preparation.32, 35 Using solid‐state 13C NMR spectroscopy, Glaubitz and co‐workers reported a rigidifying effect of lipids in nanodiscs, based on only a few resolved carbon positions, C2, C3, C12 and C13, and on the unassigned C4–11 region because the 13C spectral dispersion is limited for most of the acyl chain.34 Recent studies on membrane protein dynamic in nanodiscs suggested that internal lipid dynamics in nanodiscs would influence membrane protein dynamics through lipid–protein interactions to regulate the protein activity.21, 28 It has been shown that lipid dynamics display a broad range of motions in different membrane‐mimicking environments, such as micelles, liposomes and nanodiscs, resulting in different dynamic behaviour of the incorporated protein, as demonstrated for the β2 adrenergic receptor in nanodiscs and detergent micelles,28 the outer membrane protein X21 or the voltage‐dependent anion channel.27 The determination of the internal lipid dynamics in nanodiscs, performed with a direct measurement, that is, without the use of fluorescent probes that might disturb the lipid cooperative behaviour, would be of great interest to understand the role of lipids in nanodiscs and to understand how it might influence the protein activity through specific lipid–protein interaction. Herein, we report the use of solid‐state 2H NMR spectroscopy to investigate internal lipid dynamic and thermotropism in nanodiscs. We determined the lipid‐phase order in nanodiscs and multilamellar vesicles (MLVs; liposomes). The thermotropic behaviour as a function of the temperature was derived from the first spectral moment of deuterium and we measured the order parameters, at site‐specific resolution, through deuterium quadrupolar splittings. Comparative measurements performed for the same lipid composition, as well as in the presence and absence of cholesterol, between liposomes and nanodiscs provide a detailed basis to establish a model of lipid order and thermotropism in nanodisc architectures.
2. Results and Discussion
Nanodiscs and MLVs (liposomes) were prepared as described in the Experimental Section and imaged by means of electron microscopy (EM; Figure 1). In short, nanodisc assemblies were prepared by using established procedures4, 5, 25 based on the production and purification of MSPΔH5 proteins mixed with DMPC. MLVs consisting of DMPC were prepared through a standard freeze–thaw procedure. Solid‐state NMR spectroscopy is a technique that is not restricted by the size of nano‐objects and NMR spectroscopy measurements reported herein were performed on intact nanodiscs and vesicles, without the use of sonication, detergents or organic solvents to solubilise the lipid assemblies.
Figure 1.

Electron micrographs of: A) 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine (DMPC) MSP1ΔH5 nanodiscs (scale bar: 10 nm) and B) multilamellar DMPC liposomes (scale bar: 50 nm). A) The left column shows top views of the nanodiscs, whereas the right column shows side views of nanodiscs that are stacked on top of each other. Stacking arises from EM grid preparation and is a common feature of discoidal nanodiscs.5, 18
Figure 1 A shows electron micrographs of the nanodiscs, showing proper and homogeneous assemblies. Top views (Figure 1 A, left column) display a circular shape with an outer diameter of (10±1) nm. Side views (Figure 1 A, right column) show stacked nanodiscs and a disc rim with a thickness of (5±1) nm, which corresponds well to the thickness of a lipid bilayer. By taking an average area per lipid of 70 Å2[29] and an inner diameter of 8 nm, one may compute that there are about 70 lipids per leaflet. Phospholipid dispersions in the absence of MSP1ΔH5 proteins show a classical onion‐like structure (Figure 1 B), otherwise called MLVs, with external diameters of (500±10) nm. A rough calculation, considering that there are about 10 concentrically nested vesicles, leads to about 15×106 lipids per MLV.
Wide‐line (static) solid‐state 2H NMR (2H‐SSNMR) spectroscopy is a powerful technique to describe the chain ordering of deuterated phospholipids embedded in different structures at atomic and molecular levels. The studied nano‐objects must not tumble too fast and, in the case of nanodiscs, the sample has been centrifuged at 20 000 g to collect sedimented nanodiscs to avoid detection of a sharp isotropic NMR line. A similar procedure was used by the groups of Rienstra31 and Glaubitz34 in previously reported solid‐state NMR spectroscopy measurements of membrane proteins in nanodiscs. We applied 2H‐SSNMR spectroscopy to both nanodiscs and vesicles reconstituted with perdeuterated DMPC chains ([D54]DMPC). The temperature was varied between 283 and 301 K, that is, to test each side of the order–disorder phase transition, which is commonly called the lamellar gel‐to‐fluid phase transition. Deuterium nuclei constitute exquisite probes of lipid dynamics because the detected quadrupolar coupling relies on the orientation of the C−2H chemical bond to the external magnetic field and the dynamic of the vector bond. Each vector bond gives rise to a characteristic Pake doublet, which is assignable to its C−2H pair along the acyl chain.8 Figure 2 shows selected spectra obtained below (Figure 2 A–D, 289 K) and above (Figure 2 E–H, 298 K) the melting temperature (T m) of pure and cholesterol‐doped deuterium‐labelled [D54]DMPC liposomes [T m=(293±1) K].13 At 289 K, that is, below the T m, an axially asymmetric broad spectrum spanning about 120 kHz is observed for DMPC liposomes; this is typical for Lβ′ or gel phases (Figure 2 A). These results depict a situation in which chains are tilted and tightly packed in almost all‐trans conformations due to a considerable reduction of molecular and intermolecular motional rates.44 At 298 K (Figure 2 E), the spectrum is much narrower and individual Pake doublets are well resolved, as in Lα or fluid phases. The widest doublet is only 28.6 kHz and reflects the onset of fast, axially symmetric motions (trans–gauche isomerisation, chain or molecule rotation and wobbling, collective diffusion) that average the quadrupolar interaction over time and space.43 Different behaviour for DMPC lipids in nanodiscs is observed. Even if the spectral quality is not comparable to that of liposomes, due to limited amounts of sample (≈1 mg), below T m at 289 K (Figure 2 B), a reminiscence of the fluid phase is observed, which is characterised by an axially symmetric spectrum with around 43.4 kHz for the outermost doublet. At 298 K (Figure 2 F), the shape of a Lα fluid‐phase spectrum is retained, but individual quadrupolar splittings are less resolved. The outermost doublet is, however, increased to 31.2 kHz compared with liposomes (28.6 kHz). We therefore observed that the spectral shapes were narrower at low temperature and wider at higher temperature for liposomes and nanodiscs, respectively. This is interesting because it indicates that, at high temperatures, lipids are slightly more ordered in nanodiscs, whereas, in contrast, at low temperature, lipids are drastically more mobile in nanodiscs than those in liposomes.
Figure 2.
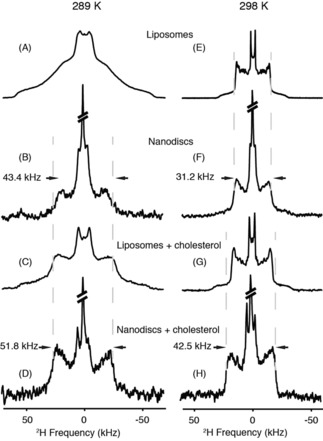
Wide‐line (static) 2H‐SSNMR spectra of deuterium‐labelled [D54]DMPC of various nanodisc and liposome assemblies acquired at 289 and 298 K. Spectra were recorded for samples in the absence (A and E, liposomes; B and F, nanodiscs) and in the presence of 7.5 % molar ratio cholesterol (C and G, liposomes; D and H, nanodiscs). Lorentzian line broadening of 300 (B, D, F and H) and 30 Hz (A, C, E, G) was applied before the Fourier transformation. The isotropic signal (5 % of the total signal) was cut to show the anisotropic contribution. Vertical dashed grey lines help to follow changes in spectral width.
As mentioned above, nanodiscs are derived from HDL particles that play major role in the transport of cholesterol in the blood and interstitial fluid.46 However, the cellular mechanism for cholesterol acceptance during the metabolic pathway, as well as the change of lipid dynamics after cholesterol loading, have not yet been investigated. To understand the role of cholesterol on the lipid fluidity in the context of the nanodiscs, we prepared DMPC liposomes and nanodiscs enriched in cholesterol (7.5 % molar ratio). At 289 K, the spectral shape of DMPC liposomes is globally axially symmetric and results in the addition of contributions from both solid‐ and liquid‐ordered phases in slow exchange15 (Figure 2 C). A reduction in width is observed upon increasing the temperature to 298 K (Figure 2 G), but to a lesser extent than that of liposomes lacking cholesterol. This corresponds to the well‐documented ordering effect of cholesterol on fluid membranes.14 When adding cholesterol to nanodiscs (Figure 2 D and H), the quadrupolar couplings are narrower at T<T m and again wider at T>T m; this is a similar situation to that without cholesterol. We observed that an isotropic NMR signal (at a frequency close to 0 kHz) was always detected in the presence of nanodiscs; it represented about 5 % of the total spectral area. Close inspection shows the presence of two signals: one at the isotropic chemical shift of natural abundance, 1HO2H; the other, of lower intensity, for the isotropic chemical shift of deuterated methylene groups. The latter may arise from nanodiscs that tumble very rapidly in the remaining bulk water. Their concentration is, however, very low because they are not detected by 31P NMR spectroscopy.
Next, we used the first 2H spectral moment, M 1, as a function of the temperature, to investigate the thermotropic behaviour of phospholipids in liposomes and nanodiscs (Figure 3). The first spectral moment is a relevant indicator of the global lipid packing and dynamics because it is proportional to the average order parameter of acyl chain C−2H bonds.44 A higher M 1 value indicates a more rigid lipid system. The M 1 values have been calculated for the entire thermal variation (Figure 3). For liposomes, high M 1 values are observed at low temperature; which reflects the tight packing of Lβ′ phases. Upon increasing the temperature, a steep decrease is detected, which reflects the incoming large membrane dynamics that correspond to the Lα fluid phase. For pure DMPC liposomes, a sharp transition is observed from gel to fluid phases and T m can be unambiguously determined from the inflection point at (20±1) °C, in accordance with the value reported for [D54]DMPC (Figure 3, red line).13 In the presence of cholesterol (Figure 3, red dotted line), the transition is less abrupt, which is also in accordance with the coexistence of several phases (liquid ordered, solid ordered, liquid disordered) over that temperature range. Significantly different values are observed for nanodiscs because spectral moments are almost two times lower (Figure 3, blue line) at low temperatures, but not significantly different from liposomes at high temperatures. The gel‐to‐fluid phase transition of DMPC in nanodiscs appears to be eliminated from 283 to 301 K, which is consistent with a reminiscence of the fluid phase, even below the lipid transition temperature (Figure 2 B). In nanodiscs doped with cholesterol (Figure 3, blue dotted), the transition between rigid and mobile conformations for acyl chains is again abolished, similar to DMPC nanodiscs without cholesterol (Figure 2 D). However, spectral moment values remain higher than those of pure DMPC in nanodiscs and approach similar values to those of cholesterol‐doped liposomes above the T m. In conclusion, spectral moment analysis reveals significantly different lipid packing below the lipid transition temperature between liposomes and nanodisc‐embedded lipids, but rather similar lipid packing above the T m with and without cholesterol.
Figure 3.
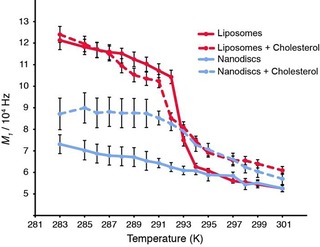
Thermal variation of the first spectral moment (M 1) of [D54]DMPC, as determined from solid‐state 2H NMR spectra for liposomes (red) and nanodiscs (blue) in the presence (dashed lines) and absence (solid line) of cholesterol. Spectra have been acquired from 283 to 301 K and analysed with NMR‐DePaker [1.0rc1 software (Copyright (C) 2009 Sébastien Buchoux)] software to extract the M 1 value for each temperature.
A detailed analysis of carbon–deuterium order parameters |S CD| along the lipid chain can be obtained from back‐calculated 2H NMR spectra. Non‐oriented solid‐state NMR spectra are calculated in the time domain (as free induction decays) and then Fourier transformed. Individual components are built from experimental estimates of quadrupolar splittings, isotropic chemical shifts and individual line widths (line width is considered to be constant throughout the pattern). For perdeuterated lipid chains, the weights are not variable and depend on the number of deuterons per labelled carbon position. The individual time‐dependent signals are then added accordingly, leading to the multicomponent spectrum after Fourier transformation. This procedure was applied to all systems acquired at 298 (Figure 4) and 289 K (Figure S1 in the Supporting Information). The value 2 |S CD| is plotted as a function of labelled carbon position along the DMPC chains (an average is made for both chains) in Figure 4. The factor of two comes from expressing the order along the bilayer normal that is perpendicular to the membrane plane. In such a way, a fully rigid system has a 2 S CD value of one, whereas a fully disordered chain has a value of zero.8 At 298 K, the order parameter profile of pure [D54]DMPC liposomes (Figure 4, red line) as a function of labelled carbon position is characterised by a plateau region corresponding to positions near the membrane interface with a high structural order (carbon atoms 2 to 8–10), followed by a rapid decrease in order parameter towards the terminal methyl group, C14, localised in the middle of the membrane. This profile, which depicts the well‐known gradient of ordering across the bilayer,44 is observed for both liposome and nanodisc samples, with or without cholesterol. However, in nanodiscs (Figure 4, blue line), the acyl chains undergo a slight, but measurable, increase in global ordering for all labelled positions relative to liposomes. Addition of cholesterol to liposomes (Figure 4, red dotted line) induces an ordering effect produced on fluid phases, as indicated by an increased 2 |S CD| value.14 This effect is also observed for nanodiscs containing the same amount of cholesterol (Figure 4, blue dotted line), but to a greater extent. The profile of order parameters of nanodiscs (with and without cholesterol) at 289 K is similar (Figure S1 in the Supporting Information) and we also observe an increase of the local order parameter in the presence of cholesterol.
Figure 4.
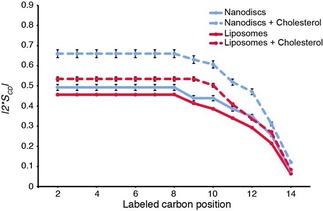
C−2H order parameter as a function of labelled carbon position. Lipid ordering was determined at 298 K in nanodiscs (blue) and liposomes (red) and supplemented with cholesterol (dotted red and blue, respectively). Calculation of oriented‐like spectra from Pake patterns (De‐Pake‐ing) and simulation of 2H‐SSNMR spectra (E. Dufourc, in‐house program) were applied to measure individual quadrupolar splittings for [D54]DMPC and determine order parameters accurately. Error bars due to calculation procedures are estimated to be about 5 %.
Herein, we have compared the lipid dynamics in liposomes and nanodiscs below, at, and above their gel‐to‐fluid‐phase transition temperature, T m. Figure 5 depicts a schematic overview of the lipid‐phase order for both nanodiscs and liposomes. We found that the response of lipid dynamics to a temperature change crossing T m deviates in nanodiscs (Figure 5 C and D) from the classical picture found in liposomes in this study (Figure 5 A and B) and that reported in the literature.6, 33 Specifically, we observed that DMPC molecules in liposomes were in the gel phase below 20 °C and adopted a tilted position with respect to the bilayer normal,26, 42 with very tight packing (Figure 5 A). Lipids in nanodiscs, however, still undergo axially symmetric motions with respect to the membrane normal; this indicates that even significantly below the phase transition temperature a mixture of fluid and gel‐like phases are present (Figure 5 C).
Figure 5.
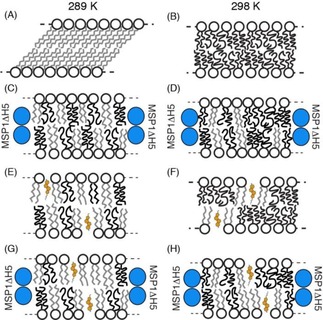
Cartoons representing lipid dynamics at two temperatures (289 and 298 K) for liposome (A, B, E and F) and nanodisc (C, D, G and H) membrane systems. Different results obtained by solid‐state NMR spectroscopy for DMPC are summarised. The MSP1ΔH5 protein helices are represented as blue circles and acyl chain dynamics are indicated in black (for fast motions) and grey (for slow motions). Cholesterol is displayed in orange and the number of sterol molecules is in agreement with the DMPC/cholesterol molar ratio.
Furthermore, the deuterium spectral moment indicates a strongly reduced lipid dynamics jump at T m for DMPC nanodiscs enriched in cholesterol, compared with liposomes, in agreement with previous SAXS and differential scanning calorimetry (DSC) measurements.10, 39 Interestingly, DMPC nanodiscs enriched in cholesterol (Figure 5 G) reach a plateau of order at around 289 K (Figure 3). It is tempting to speculate that this could be due to lipid–MSP1ΔH5 protein hydrophobic attractions. Whereas the area occupied by phospholipids decreases with increasing order (and decreasing temperature), the MSP1ΔH5 protein belt diameter remains the same, which creates a thermodynamically unfavourable hydrophobic vacuum between the lipids and protein belt. This appears to be counteracted by increased lipid dynamics, and therefore, an increased occupied area. It is important to note that an isotropic signal is still detected in 2H‐SSNMR spectra and could come from soluble and fast tumbling nanodiscs in the sample. We hypothesised that part of the line broadening observed in nanodisc samples (in addition to the decrease of motions induced by MSP1ΔH5) could arise from a millisecond timescale chemical exchange between the soluble and gel‐like fractions of nanodiscs. This broadening of NMR signals was previously observed for DMPC in anisotropic bicelles undergoing a millisecond timescale chemical exchange with a soluble fraction of the lipids.1
Interestingly, lipid ordering in nanodiscs is higher at 298 K compared with that of liposomes (Figure 4). This again might be the result of a limited diameter adaptation of the MSP1ΔH5 protein belt, which now restraints free movement of lipids at higher temperatures. Notably, although the acyl chain dynamics are modified by cholesterol, 31P NMR spectroscopy shows that the phosphate found in the lipid head group appears to have the same dynamic properties, regardless of the presence of cholesterol (Figure S2 A in the Supporting Information). This is in agreement with the recently solved 3D structure of HDL particles, in which amphipathic helices surround the lipid disc around the acyl chains and no direct contacts are observed with phospholipid head groups.5, 20, 45
Our data demonstrate that the internal ordering (reflected by individual order parameters) is higher in nanodiscs than that in liposomes. This effect was previously reported by fluorescence spectroscopy on n‐AS (n‐(9‐anthroyloxy)stearic acid) inserted in large unilamellar vesicles (LUVs)35 and solid‐state 13C NMR spectroscopy under magic angle spinning on nanodiscs.34 Whereas lipid signals are sharp for liposomes in the fluid phase, there is an increase of the C−2H bond line widths in nanodiscs at the same temperature. These findings may be explained by the presence of the scaffold protein, which induces a decrease of lipid diffusion and motions inside the nanodisc belt. However, unlike liposomes, these steric restraints are likely to reflect a more realistic environment for lipids because proteins, rafts, glycolipids and sterols influence lipid diffusion in the cell.37 Furthermore, the restricted area in the nanodiscs appears to drastically reduce the cooperative effect of lipid assembly during the phase transition, which increases fluidity above the phase transition temperature.
2.1. Conclusion
Our sets of experiments monitored by wide‐line NMR spectroscopy showed that the lipid bilayer in nanodiscs maintained its fluid properties over a large temperature range. We furthermore showed that the addition of cholesterol provided an easy way to control the dynamics of the membranous disc and its thermotropic properties. Ongoing solid‐state NMR spectroscopy studies in our laboratory will decipher the variability of lipid internal dynamics in different discoidal shapes.
Experimental Section
[D54]DMPC and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL). Deuterium‐depleted water was obtained from Eurisotop (France). Ultrapure water with a nominal resistivity of 18.2 MΩ cm (MilliQ, Millipore, France) was used for lyophilisation.
To prepare MLVs, cholesterol was mixed with the appropriate amount of DMPC powder to adjust the DMPC/cholesterol ratio (92.5:7.5) in organic solvent (chloroform/methanol, 2:1). Solvent was evaporated under a flow of N2 to obtain a thin lipid film. Lipids were rehydrated with ultrapure water before lyophilisation. The lipid powder was hydrated with an appropriate amount of deuterium‐depleted water (80 % hydration ratio) and homogenised by three cycles of shaking in a vortex mixer, freezing (liquid nitrogen, −196 °C, 1 min) and thawing (40 °C in a water bath, 10 min). This protocol led to a milky suspension of micrometre‐sized MLVs. For experiments with DMPC alone, MLV formation was performed by following the same protocol as that outlined above, with only chloroform as the organic solvent. For nanodisc precipitation, 50 % polyethylene glycol (PEG) 4000 was added to a solution of nanodiscs in 20 mm Tris buffer at pH 7.4, with 100 mm NaCl. The sample was homogenised gently with a pipette and incubated overnight at 4 °C. A centrifugation step was performed (30 min, 20 000 g) to recover precipitated nanodiscs in the pellet. This protocol was performed on nanodiscs and nanodisc/cholesterol samples.
For the cryo‐EM micrograph of liposomes, a 0.5 mg mL−1 solution of DMPC MLVs (5 μL) was deposited onto a lacey carbon copper grid submitted to glow discharge. After removing excess solution with filter paper, the grid was rapidly plunged into a liquid ethane bath cooled with liquid nitrogen by using an EM GP instrument (Leica). Specimens were maintained at a temperature of approximately −170 °C, by using a cryo‐holder (Gatan, USA), and observed under low‐dose conditions with a FEI Tecnai F20 electron microscope operating at 200 kV. Images were acquired by using a digital 2k×2k USC1000 camera (GATAN).
The nanodiscs were prepared as described previously.5 For the cholesterol‐containing nanodiscs, a MSP/lipid/cholesterol ratio of 1:40:10 was used. The nanodisc EM micrographs were recorded on the same day and in the same way as described previously.5 In brief, samples were applied to a glow‐discharged thin carbon film covering 200‐mesh copper EM grids and adsorbed for 1 min. Each grid was blotted, washed with three drops of water and negatively stained with two drops of 2 % uranyl acetate. The grids were imaged in a Philips CM10 microscope operated at a voltage of 80 kV. Micrographs were recorded with a charge‐coupled device (CCD) camera (Veleta, Olympus),
2H NMR spectroscopy experiments were performed by using a Bruker Avance II 500 MHz WB (11.75 T) spectrometer. 2H NMR spectroscopy experiments on 2H‐labelled DMPC were performed at 76 MHz with a phase‐cycled quadrupolar echo pulse sequence (90°x‐τ‐90°y‐τ‐acq). 31P NMR spectra were acquired at 202 MHz by using a phase‐cycled Hahn‐echo pulse sequence (90°x‐τ‐180°x/y‐τ‐acq). Acquisition parameters were as follows: spectral window of 250 kHz for 2H NMR spectroscopy and 50 kHz for 31P NMR spectroscopy, π/2 pulse widths ranged from 2.62 to 2.88 μs for 2H and 15 μs for 31P, interpulse delays (τ) were of 40 μs, recycled delays ranged from 1.1 to 2 s for 2H and 5 s for 31P; 2000 to 20 000 scans were used for 2H NMR spectroscopy and 8000 scans were used for 31P NMR spectroscopy, depending on samples. Spectra were processed by using a Lorentzian line broadening of 100 to 300 Hz for 2H NMR spectra before Fourier transform from the top of the echo. Samples were equilibrated for 30 min at a given temperature before data acquisition. All spectra were processed and analysed by using Bruker Topspin 3.2 software. Spectral moments were calculated for each temperature by using the NMR Depaker 1.0rc1 software [Copyright (C) 2009 Sébastien Buchoux]. Orientational order parameters (S CD) were calculated from experimental quadrupolar splittings (Δν Q) according to Equation (1):
| (1) |
in which A Q=(e 2 qQ) h−, the quadrupolar coupling constant for methyl moieties is 167 kHz, and θ is the angle between the magnetic field and the bilayer normal.38 We calculated twice the value of the order parameter as a mean for structural order along the bilayer normal.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was further supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (ERC Starting Grant to A.L., agreement no. 639020), the ANR (ANR‐14‐CE09‐0020‐01 to A.L.), IdEx Bordeaux (Chaire d'Installation to B.H.), the EU COFUND program (FEL‐2012‐1 to S.B.), and the Swiss National Science Foundation (grant 144444 to R.R.). This work has benefited from the facilities and expertise of the Biophysical and Structural Chemistry Platform (BPCS) at IECB, CNRS UMS3033, Inserm US001, Bordeaux University, and financial support from the TGIR‐RMN‐THC Fr3050 CNRS for conducting the research is gratefully acknowledged.
D. Martinez, M. Decossas, J. Kowal, L. Frey, H. Stahlberg, E. J. Dufourc, R. Riek, B. Habenstein, S. Bibow, A. Loquet, ChemPhysChem 2017, 18, 2651.
Contributor Information
Dr. Stefan Bibow, Email: stefan.bibow@unibas.ch
Dr. Antoine Loquet, Email: a.loquet@iecb.u-bordeaux.fr.
References
- 1. Arnold A., Labrot T., Oda R., Dufourc E. J., Biophys. J. 2002, 83, 2667–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bayburt T. H., Carlson J. W., Sligar S. G., J. Struct. Biol. 1998, 123, 37–44. [DOI] [PubMed] [Google Scholar]
- 3. Bayburt T. H., Grinkova Y. V., Sligar S. G., Nano Lett. 2002, 2, 853–856. [Google Scholar]
- 4. Bibow S., Carneiro M. G., Sabo T. M., Schwiegk C., Becker S., Riek R., Lee D., Protein Sci. 2014, 23, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bibow S., Polyhach Y., Eichmann C., Chi C. N., Kowal J., Albiez S., McLeod R. A., Stahlberg H., Jeschke G., Güntert P., Riek R., Nat. Struct. Mol. Biol. 2017, 24, 187–193. [DOI] [PubMed] [Google Scholar]
- 6. Brumm T., Jørgensen K., Mouritsen O. G., Bayerl T. M., Biophys. J. 1996, 70, 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chou J. J., Kaufman J. D., Stahl S. J., Wingfield P. T., Bax A., J. Am. Chem. Soc. 2002, 124, 2450–2451. [DOI] [PubMed] [Google Scholar]
- 8. Davis J. H., Biochim. Biophys. Acta 1983, 737, 117–171. [DOI] [PubMed] [Google Scholar]
- 9. Denisov I. G., Grinkova Y. V., Lazarides A. A., Sligar S. G., J. Am. Chem. Soc. 2004, 126, 3477–3487. [DOI] [PubMed] [Google Scholar]
- 10. Denisov I. G., McLean M. A., Shaw A. W., Grinkova Y. V., Sligar S. G., J Phys Chem. B 2005, 109, 15580–15588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denisov I. G., Sligar S. G., Nat. Struct. Mol. Biol. 2016, 23, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denisov I. G., Sligar S. G., Chem. Rev. 2017, 117, 4669–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dufourc E. J., Mayer C., Stohrer J., Althoff G., Kothe G., Biophys. J. 1992, 61, 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dufourc E. J., Parish E. J., Chitrakorn S., Smith I. C. P., Biochemistry 1984, 23, 6062–6071. [Google Scholar]
- 15. Dufourc E. J. in Chemical Biology: Techniques and Applications (Eds.: B. Larijani, C. A. Rosser, R. Woscholski), Wiley, Chichester, 2006, pp. 113–131. [Google Scholar]
- 16. Duong P. T., Collins H. L., Nickel M., Lund-Katz S., Rothblat G. H., Phillips M. C., J. Lipid Res. 2006, 47, 832–843. [DOI] [PubMed] [Google Scholar]
- 17. Eichmann C., Bibow S., Riek R., Nanotechnol. Rev. 2017, 6, 105–110. [Google Scholar]
- 18. Forte T., Norum K. R., Glomset J. A., Nichols A. V., J. Clin. Invest. 1971, 50, 1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fox D. A., Larsson P., Lo R. H., Kroncke B. M., Kasson P. M., Columbus L., J. Am. Chem. Soc. 2014, 136, 9938–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frauenfeld J., Gumbart J., van der Sluis E. O., Funes S., Gartmann M., Beatrix B., Mielke T., Berninghausen O., Becker T., Schulten K., Beckmann R., Nat. Struct. Mol. Biol. 2011, 18, 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frey L., Lakomek N.-A., Riek R., Bibow S., Angew. Chem. Int. Ed. 2017, 56, 380–383; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 388–391. [Google Scholar]
- 22. Gao Y., Cao E., Julius D., Cheng Y., Nature 2016, 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ge L., Villinger S., Mari S. A., Giller K., Griesinger C., Becker S., Müller D. J., Zweckstetter M., Structure 2016, 24, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grinkova Y. V., Denisov I. G., Sligar S. G., Protein Eng. Des. Sel. 2010, 23, 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hagn F., Etzkorn M., Raschle T., Wagner G., J. Am. Chem. Soc. 2013, 135, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Janiak M. J., Small D. M., Shipley G. G., Biochemistry 1976, 15, 4575–4580. [DOI] [PubMed] [Google Scholar]
- 27. Kingwell B. A., Chapman M. J., Kontush A., Miller N. E., Nat. Rev. Drug Discovery 2014, 13, 445–464. [DOI] [PubMed] [Google Scholar]
- 28. Kofuku Y., Ueda T., Okude J., Shiraishi Y., Kondo K., Mizumura T., Suzuki S., Shimada I., Angew. Chem. Int. Ed. 2014, 53, 13376–13379; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13594–13597. [Google Scholar]
- 29. Kučerka N., Nieh M.-P., Katsaras J., Biochim. Biophys. Acta Biomembr. 2011, 1808, 2761–2771. [DOI] [PubMed] [Google Scholar]
- 30. Laguerre A., Löhr F., Henrich E., Hoffmann B., Abdul-Manan N., Connolly P. J., Perozo E., Moore J. M., Bernhard F., Dötsch V., Structure 2016, 24, 1830–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y., Kijac A. Z., Sligar S. G., Rienstra C. M., Biophys. J. 2006, 91, 3819–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miyazaki M., Tajima Y., Ishihama Y., Handa T., Nakano M., Biochim. Biophys. Acta Biomembr. 2013, 1828, 1340–1346. [DOI] [PubMed] [Google Scholar]
- 33. Morrow M. R., Whitehead J. P., Lu D., Biophys. J. 1992, 63, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mörs K., Roos C., Scholz F., Wachtveitl J., Dötsch V., Bernhard F., Glaubitz C., Biochim. Biophys. Acta Biomembr. 2013, 1828, 1222–1229. [DOI] [PubMed] [Google Scholar]
- 35. Nakano M., Fukuda M., Kudo T., Miyazaki M., Wada Y., Matsuzaki N., Endo H., Handa T., J. Am. Chem. Soc. 2009, 131, 8308–8312. [DOI] [PubMed] [Google Scholar]
- 36. Nath A., Atkins W. M., Sligar S. G., Biochemistry 2007, 46, 2059–2069. [DOI] [PubMed] [Google Scholar]
- 37. O'Leary T. J., Proc. Natl. Acad. Sci. USA 1987, 84, 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seelig J., Q. Rev. Biophys. 1977, 10, 353–418. [DOI] [PubMed] [Google Scholar]
- 39. Shaw A. W., McLean M. A., Sligar S. G., FEBS Lett. 2004, 556, 260–264. [DOI] [PubMed] [Google Scholar]
- 40. Shih A. Y., Denisov I. G., Phillips J. C., Sligar S. G., Schulten K., Biophys. J. 2005, 88, 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Skar-Gislinge N., Simonsen J. B., Mortensen K., Feidenhans'l R., Sligar S. G., Lindberg Møller B., Bjørnholm T., Arleth L., J. Am. Chem. Soc. 2010, 132, 13713–13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tardieu A., Luzzati V., Reman F. C., J. Mol. Biol. 1973, 75, 711–733. [DOI] [PubMed] [Google Scholar]
- 43. Vist M. R., Davis J. H., Biochemistry 1990, 29, 451–464. [DOI] [PubMed] [Google Scholar]
- 44. White S. H., King G. I., Proc. Natl. Acad. Sci. USA 1985, 82, 6532–6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu Z., Wagner M. A., Zheng L., Parks J. S., Shy J. M., Smith J. D., Gogonea V., Hazen S. L., Nat. Struct. Mol. Biol. 2007, 14, 861–868. [DOI] [PubMed] [Google Scholar]
- 46. Zannis V. I., Fotakis P., Koukos G., Kardassis D., Ehnholm C., Jauhiainen M., Chroni A., Handb. Exp. Pharmacol. 2015, 224, 53–111. [DOI] [PubMed] [Google Scholar]
- 47. Zoonens M., Comer J., Masscheleyn S., Pebay-Peyroula E., Chipot C., Miroux B., Dehez F., J. Am. Chem. Soc. 2013, 135, 15174–15182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary