

Abstract
Ceria (CeO2) supports are unique in their ability to trap ionic platinum (Pt), providing exceptional stability for isolated single atoms of Pt. The reactivity and stability of single‐atom Pt species was explored for the industrially important light alkane dehydrogenation reaction. The single‐atom Pt/CeO2 catalysts are stable during propane dehydrogenation, but are not selective for propylene. DFT calculations show strong adsorption of the olefin produced, leading to further unwanted reactions. In contrast, when tin (Sn) is added to CeO2, the single‐atom Pt catalyst undergoes an activation phase where it transforms into Pt–Sn clusters under reaction conditions. Formation of small Pt–Sn clusters allows the catalyst to achieve high selectivity towards propylene because of facile desorption of the product. The CeO2‐supported Pt–Sn clusters are very stable, even during extended reaction at 680 °C. Coke formation is almost completely suppressed by adding water vapor to the feed. Furthermore, upon oxidation the Pt–Sn clusters readily revert to the atomically dispersed species on CeO2, making Pt–Sn/CeO2 a fully regenerable catalyst.
Keywords: catalyst regeneration, ceria, light alkane dehydrogenation, single atoms, subnanometer Pt–Sn catalysis
Light olefins are important building blocks in the chemical industry. Growing demand for light olefins has led to an increased interest in the dehydrogenation of light alkanes,1 such as ethane and propane,2 which are now much more abundant because of the vast amounts of shale gas deposits.3 The production of light olefins by alkane dehydrogenation is practiced commercially, but because of its endothermicity, it requires high reaction temperatures to achieve economically attractive yields.4 High reaction temperatures cause catalyst deactivation by sintering5 and coke formation.6 While carbon deposits can be effectively oxidized during regeneration in air, it is more difficult to achieve redispersion of Pt.7 In fact, heating Pt/Al2O3 under oxidizing conditions leads to sintering of Pt, forming large Pt particles.8 To achieve redispersion of Pt, it is necessary to add halogens (such as, Cl2, HCl, CCl4, or F2) so that the small particle size of Pt in the initial catalyst can be restored.9 Herein, we explore an alternative support, CeO2, that is unique in its ability to trap Pt atoms in ionic form.10 The CeO2 support facilitates redispersion, restoring the atomic dispersion of Pt without the need for corrosive halogens. Furthermore, the redox properties of CeO2 allow the use of water vapor, which almost completely eliminates the coke deposition traditionally seen during propane conversion at high reaction temperatures.
Synthesis of the catalyst takes advantage of the ability of CeO2 to trap Pt atoms, as explained in our previous work.11 This requires heating Pt/CeO2 to 800 °C for 10 h in flowing air. The scanning transmission electron microscope (STEM) image of Pt/CeO2 (1 wt % Pt) shows atomically dispersed Pt species (Figure 1 b). X‐ray photoelectron spectroscopy (XPS) images of the Pt 4f region for the fresh and spent Pt/CeO2 are shown in Figure 1 a. The only Pt species in the as‐prepared Pt/CeO2 catalyst are Pt2+ and Pt4+, consistent with the STEM image (as confirmed previously by Fourier transform infrared spectroscopy (FTIR) and X‐ray diffractometry (XRD)11). After 3 cycles of propane dehydrogenation at an elevated reaction temperature of 680 °C, the XPS spectrum shows that a portion of the ionic Pt has transformed into metallic Pt. STEM images of the sample after reaction show that atomically dispersed Pt coexists with Pt nanoclusters and the nanoclusters have an average size of about 1.2 nm (Figure 1 c; Supporting Information, Figure S1). The Pt/CeO2 catalyst is very reactive for propane conversion at 680 °C, showing 100 % propane conversion (Table 1). However, the products were CH4 and CO2, and no propylene was detected, indicating that the isolated Pt single atoms are highly efficient catalysts that only promote C−C bond cleavage of the hydrocarbons with no observed selectivity towards dehydrogenation. Pure CeO2 and Sn/CeO2 (3 wt % Sn) were also tested for propane dehydrogenation under the same conditions (680 °C). No dehydrogenation and decomposition reactivity was found on pure CeO2 and Sn/CeO2. This observation is consistent with an earlier report in which Pt catalysts with a low loading (1000 ppm) presented poor activity for propane dehydrogenation.12
Figure 1.

Atomically dispersed Pt on CeO2 (the as‐prepared catalyst) undergoes some reduction to form subnanometer Pt particles after reaction. Isolated Pt single atoms are circled. The monometallic catalyst does not show any selectivity to propylene despite its high activity (100 % conversion at 680 °C). a) XPS spectra of the fresh and spent Pt/CeO2 catalyst after propane dehydrogenation at 680 °C for 6 h. b) STEM images of the as‐prepared Pt/CeO2 catalyst (calcined 800 °C in air). c) STEM image of the spent Pt/CeO2 catalyst after 3 cycles of propane dehydrogenation at 680 °C for 6 h each, with an intermediate oxidation at 580 °C for 2 h after each cycle. Scale bar=5 nm.
Table 1.
Propane dehydrogenation performance of the supported Pt and Pt–Sn catalysts under investigation.
| Catalyst[a] | Propane conversion [%] | Propylene selectivity[b] | Carbon[c] [wt %][d] |
|---|---|---|---|
| Pt/CeO2 | 100 | 0 | 0.9 (3.4) |
| Pt–Sn/CeO2 | 39.5 | 84.5 | 0.5 (2.8) |
| Pt–Sn/Al2O3 | 32.6 | 71.4 | 0.3 (3.0) |
[a] Catalyst (0.1 g), water (0.005 mL min−1), ambient pressure, reaction temperature (T) 680 °C. [b] Calculation based on the hydrocarbons as detected by FID (see formula in the Supporting Information). [c] Carbon content after a 6 h run at 680 °C. [d] The weight loss of the spent catalyst was obtained from TGA carried out between 25–800 °C in flowing air; data for reactions without water vapor are shown in parentheses.
To investigate the causes for the observed lack of selectivity of the single‐atom Pt/CeO2 material towards propylene, we performed DFT calculations for Pt single atoms supported on CeO2 (see Supporting Information). Pt adsorbs strongly at surface Ce4+ vacancies; the adsorption structures of pertinent species are displayed in Figures S2–S4 and the corresponding adsorption energies are listed in Table S1 (Supporting Information). On single‐atom Pt supported by CeO2, the product of the dehydrogenation (propylene) adsorbs strongly at the atomic Pt sites via di‐σ bonds with Pt. The configurations for the initial, transition, and final states along the reaction path are shown in Figure S5 and the barriers and reaction energies are listed in Table S2. Both dehydrogenation steps at the single Pt atoms for the Pt/CeO2 catalyst are facile with relatively low activation energies. The strongly adsorbed propylene is expected to undergo further reactions, leading eventually to C−C bond cleavage. Hence, despite facile dehydrogenation, the observed propylene selectivity is low.
It is known that Pt–Sn alloys (that is, nanoparticles) are highly selective for the dehydrogenation of light alkanes.13 Therefore, to modify the selectivity, we added Sn to the Pt single‐atom catalyst. The Pt–Sn/CeO2 catalyst contains 1 wt % Pt and 3 wt % Sn, with an atomic ratio of about Pt:Sn=1:5 (as confirmed by energy‐dispersive spectroscopy (EDS) and electron probe microanalysis (EPMA)). Co‐impregnation and sequential impregnation yield similar results. The catalysts undergo calcination in flowing air at 800 °C to disperse Pt and Sn. In the as‐prepared Pt–Sn/CeO2 catalyst (Figure 2 b), we see atomically dispersed species as well as subnanometer clusters with an average diameter of 0.6 nm (Figure 2 b; Figure S6). Because Sn and Ce have a similar atomic mass, HAADF (high‐angle annular dark‐field) STEM images cannot provide enough contrast to visualize atomically dispersed Sn on the CeO2 support, but it is easy to detect Pt by HAADF‐STEM because of the high atomic number of Pt. We have used STEM‐EDS mapping to determine the distribution of Pt and Sn on this Pt–Sn/CeO2 sample. The subnanometer clusters in the as‐prepared (calcined at 800 °C in air) catalyst show a Pt:Sn≈1:1 to 5:1, as determined by EDS analysis (Figure S7). The average composition of these subnanometer clusters is Pt:Sn≈3:1. Since the CeO2 can trap single atoms, we also analyzed regions of the support that do not contain any subnanometer clusters. Here we found excess Sn (Sn:Pt from 100:0 to 2.5:1; Figures S7,S8). We infer that the as‐prepared Pt–Sn/CeO2 catalyst contains both Sn and Pt in an atomically dispersed form, with more Sn than Pt, while the subnanometer clusters contain more Pt than Sn, consistent with the overall composition of the catalyst.
Figure 2.
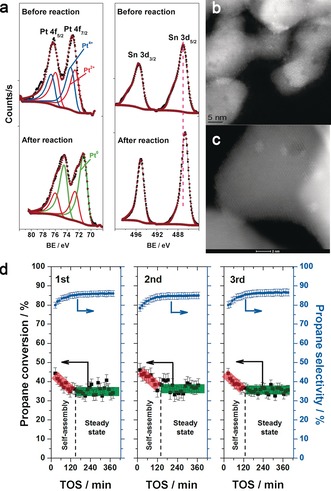
The Pt–Sn/CeO2 catalyst evolves during reaction to convert the atomically dispersed Pt species into Pt–Sn clusters that are selective for propane dehydrogenation. a) XPS spectra of the fresh and spent Pt–Sn/CeO2 catalyst after propane dehydrogenation at 680 °C. b) STEM image of the as‐prepared Pt–Sn/CeO2 catalyst. c) STEM image of the Pt–Sn/CeO2 catalyst after 3 cycles of propane dehydrogenation at 680 °C with intermediate oxidative treatment at 580 °C for 2 h. d) Propane dehydrogenation mediated by Pt–Sn/CeO2 showing 3 cycles of a 6 h run, followed by oxidative treatment in flowing air at 580 °C. The catalyst goes through an activation phase (self‐assembly) where selectivity (based on hydrocarbons) improves and activity drops, but achieves stable performance after this initial phase.
The propane dehydrogenation reactivity of the Pt–Sn/CeO2 catalyst as a function of time on stream at 680 °C is shown in Figure 2 d and the results are summarized in Table 1. The as‐prepared Pt–Sn/CeO2 catalyst starts with a relatively high initial reactivity (ca. 45 % conversion) but a low selectivity to propylene (ca. 78 %). During the first 2 h, the activity drops (to 40 %) and the selectivity towards propylene improves (to 85 %). The selectivity is based on the effluent composition of hydrocarbons detected by a flame ionization detector (FID). After this initial activation phase, the catalyst is remarkably stable during the 6 h run (Figure 2 d) with approximately 39.5 % propane conversion and about 84 % propylene selectivity at the end of the 6 h run (Table 1). The catalyst was subjected to an oxidative regeneration step after 6 h on stream, since similar treatments are used in industrial practice. This involved treating the sample in flowing air at 580 °C for 2 h. The pattern of reactivity seen in the first cycle is reproduced in the second and third cycles. The Pt–Sn/CeO2 catalyst was also tested in a second reactor setup for propane dehydrogenation at reaction temperatures of 550 and 680 °C to verify the stability and carbon balance, and the results show that in addition to the hydrocarbons, we also detect CO and CO2. These data confirm that the initial catalyst has low selectivity for propylene, which improves over the first 2 h and the catalyst is stable over the remainder of the run (Figure S9, Table S3).
STEM images of the Pt–Sn/CeO2 catalyst after propane dehydrogenation at 680 °C are shown in Figure 2 and Figures S10 and S11. We now observe well‐defined nanoclusters with an average diameter of 1.1 nm. The particle size distribution is shown in Figure S11d and EDS analysis shows a Pt:Sn ratio of about 1:3 on the small particles (Figure S11). These results show that the Pt–Sn/CeO2 catalyst is very stable, once the initial activation phase is complete. To understand this initial activation of the catalyst, we can compare the as‐prepared catalyst (after 800 °C calcination, Figure 2 b; Figure S7) with the catalyst after 2 h on stream (Figure S10) and the end‐of‐run catalyst (Figure 2 c; Figures S1c and S1d). The major difference is the presence of atomically dispersed Pt in the as‐prepared catalyst, which is missing in the end‐of‐run catalyst. As already discussed in the context of the monometallic catalyst, the single Pt atoms are very active but only lead to C−C bond cleavage. Another significant change during reaction is the formation of Pt–Sn clusters having a diameter of approximately 1.1 nm and a Pt:Sn atomic ratio of 1:3. As a comparison, on an alumina supported catalyst subjected to the same reaction conditions, the Pt–Sn particles are much larger (ca. 8 nm; Figure S12). We also studied the Pt and Sn species on the Pt–Sn/CeO2 catalyst by XPS (Figure 2 a). The initial catalyst contains only ionic Pt, similar to the fresh Pt/CeO2. After dehydrogenation, the Pt 4f XPS spectra of spent Pt–Sn/CeO2 catalyst show the formation of metallic Pt as well as the existence of Pt2+. This is consistent with the STEM results, where nanoclusters (that is, metallic Pt) are formed on the spent catalyst after dehydrogenation (Figure 2 c; Figure S11). The Sn 3d spectra are broad and correspond to oxidized Sn species in the as‐prepared catalyst, while after reaction the Sn 3d peak shifts to lower binding energy, which is consistent with the transformation of a portion of the oxidized Sn into metallic Pt–Sn nanoparticles.
The coke content on the spent Pt–Sn catalysts was investigated at the end of the first cycle by performing thermogravimetric analysis (TGA) in flowing air and elemental analysis using a CHN analyzer (Table S4). Both techniques gave similar carbon contents. The coke deposited on the sample is reported in Table 1. All of these catalysts show very low coke content of <1 wt % after a 6 h run at 680 °C. This is due to the co‐feeding of water vapor to the catalyst.14 When no water vapor was fed to the Pt–Sn/CeO2 catalyst, the coke content increased from 0.5 to 2.8 wt % and the conversion dropped. The results confirm that the addition of water vapor can suppress the formation of coke during propane dehydrogenation at high temperature.15 The coke formed under the high temperature conditions was investigated by electron microscopy and Raman spectroscopy. As shown in Figure S13, coke layers were found to cover only the support surface for the spent Pt–Sn/CeO2 catalyst. Raman spectra of the spent Pt–Sn/CeO2 and Pt–Sn/Al2O3 catalysts showed the presence of both D and G bands (Figure S14), indicating that the coke layers are composed of graphitic carbon.16 The existence of graphitic carbon on the spent Pt–Sn/CeO2 catalyst was also confirmed by a wide‐area Raman spectroscopy scan (labeled in yellow in Figure S14).
It is commonly accepted that carbon deposits can lead to lowered reactivity during dehydrogenation.17 These catalysts, after the initial activation phase during the first 2 h of reaction, did not show any deactivation during the 6 h run. However, we performed an oxidative treatment to contrast the regenerability of the Pt and Sn species on the CeO2 and alumina supports. On the Al2O3 support, the reactivity of Pt–Sn decreased during reaction and the oxidative treatment was not able to restore reactivity (Figure S12). This suggests that the deactivation of the alumina‐supported catalyst was caused by sintering, which is not reversed during oxidative treatment, rather than the small amount of coke, which is readily removed during oxidation at 580 °C.5b In contrast, the reactivity pattern of the Pt–Sn/CeO2 catalyst was similar after each oxidative regeneration. The initial activity after regeneration was high. Over time, the reactivity dropped and the selectivity improved; during this period a phenomenon we term “self‐assembly” took place. This reactivity pattern was reproducible over the 3 cycles, as shown in Figure 2 d. The STEM images of the regenerated Pt–Sn/CeO2 catalyst, after self‐assembly (that is, after 2 h run), and at the end of a 6 h run, are shown in Figure S10. These results show that the Pt–Sn clusters have formed during self‐assembly and remain stable thereafter.
To gain insight into this self‐assembly process, we carefully examined the Pt–Sn/CeO2 catalyst after oxidative regeneration, which is the state of the catalyst where the reactivity is high but the selectivity is poor (Figure 2 d). The STEM images and STEM‐EDS maps are shown in Figure 3 and Figure S15. The high magnification STEM image in Figure 3 a shows that Pt is present as atomically dispersed, isolated species, similar to the as‐prepared catalyst. The STEM‐EDS maps confirm that Pt is well‐dispersed, but Sn is present in the form of particles that are devoid of Pt (Figure 3 f; Figure S15). One such particle is indicated by an arrow in the inset of Figure 3 a. The lattice images index to SnO2 (Figure 3 a inset; Figure S16). Other evidence of crystalline SnO2 on the CeO2 is provided by Moiré fringes, which result from the interference between the two crystalline phases present on top of each other. The EDS map in Figure 3 f clearly shows high concentrations of Sn, corresponding to the particles seen on the support.
Figure 3.

The Pt–Sn/CeO2 catalyst after regeneration. a) High magnification STEM image of spent Pt–Sn/CeO2 after treatment at 580 °C for 2 h in air showing the Pt species redispersed on the support (Pt species circled) and the SnO2 particles (arrow in inset; scale bar=2 nm). b) Low magnification STEM image of spent Pt–Sn/CeO2 after regeneration in air. c–f) STEM‐EDS maps showing the locations of Ce, O, Pt, and Sn.
Figure 4 a presents a diagram of the Pt–Sn/CeO2 catalyst change from an initial state, after regeneration, to its final steady‐state morphology. The regenerated catalyst contains Pt single atoms dispersed on CeO2 (Figure 4 b) and SnO2 particles. After reaction Pt–Sn clusters are seen (Figure 4 c) instead of atomically dispersed Pt. The composition of these clusters was analyzed by EDS and found to be Pt:Sn=1:3.
Figure 4.
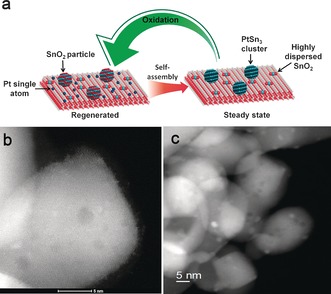
a) Schematic showing the self‐assembly and regeneration processes of Pt–Sn nanoclusters in propane dehydrogenation at 680 °C. b) STEM image of regenerated Pt–Sn/CeO2 catalyst (air, 580 °C). c) STEM image of spent Pt–Sn/CeO2 catalyst after propane dehydrogenation at 680 °C.
As explained in the DFT discussion (Supporting Information), single atoms of Pt on CeO2 are not selective for propane dehydrogenation because the strongly adsorbed propylene is amenable to further reactions. When Sn is added, DFT calculations suggest that it can bind strongly to CeO2. The Pt atoms can bind strongly with Sn embedded in CeO2. Our DFT results also indicate that the Pt–Sn/CeO2 catalyst has a much lower adsorption energy for propylene product than that on the single‐atom Pt/CeO2 catalyst. As a result, formed propylene desorbs easily, which prevents further reaction. This property is similar to that observed with bulk Pt–Sn alloy surfaces, which have been discussed extensively in the literature.6b, 13c, 18 Adding Sn to Pt in the atomically dispersed catalyst improves selectivity, but lowers the rate of propane dehydrogenation because of a higher activation barrier (Table S2, Figure S17). Adding a second Pt atom to Sn causes it to bind strongly, suggesting that Sn helps to serve as a nucleation site for forming Pt–Sn clusters. DFT results help to explain the evolution of the catalyst during reaction, where generated H2 causes transformation of ionic Pt into metallic Pt. The single atoms of Pt are mobile and interact with Sn to form bimetallic Pt–Sn clusters, which grow to a size of about 1.1 nm. The ability of mobile Pt to reduce the dispersed Sn species is similar to that seen previously where Pt was found to reduce PdO to form bimetallic Pt–Pd particles.19
What is unique about the CeO2 support is that these Pt–Sn clusters do not grow in size during reaction at steady state (Figure 2 d). In contrast, the Pt–Sn/Al2O3 catalyst presents particle sizes about 8 nm in diameter (Figure S12). The primary mechanism for sintering of the Pt–Sn catalyst is Ostwald ripening at high temperatures. The CeO2 support slows the process of ripening since it is able to trap the Pt atoms. In a recent study, Moliner et al.20 showed that zeolite cages could trap Pt atoms during oxidative regeneration. However, during reduction, they observed the formation of large metallic Pt particles. The CeO2 support is unique because trapping sites allow the size of the Pt–Sn clusters to remain small even when used at such high temperatures. We also investigated the CO diffuse reflectance infrared Fourier transform (DRIFTS) of the Pt/CeO2 and Pt–Sn/CeO2 catalysts (Figure S18). The initial catalyst shows linearly adsorbed CO at a frequency that is consistent with the isolated single atoms seen in Figures 1 b and Figure 2 b. In the reduced catalyst, we see both linearly and bridge‐bonded CO on both catalysts. The temperature‐programmed desorption (TPD) curves of the two catalysts are consistent with previous literature.21 However, no significant differences were observed in the FTIR spectra of the two catalysts, suggesting that this technique may not be able to distinguish between these two catalysts.22
To further test the stability of the Pt–Sn/CeO2 catalyst we treated it in CH4 at 800 °C. This caused a growth in particle size and a drop in activity, but the sizes of these particles are still smaller than those on the alumina support after reaction at 680 °C (Table S5, Figure S19).
To summarize, Pt–Sn/CeO2 is a thermally stable, active and regenerable catalyst for light alkane dehydrogenation. The atom trapping sites on CeO2 help achieve complete redispersion of Pt atoms under mild oxidative regeneration conditions, without the need for chlorine containing molecules, providing a regenerable, thermally stable catalyst for light alkane activation. The redox properties of CeO2 allow water vapor to be used as the oxidant, suppressing coke formation during the dehydrogenation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support from the US Department of Energy (DE‐FG02‐05ER15712), Center for Biorenewable Chemicals (CBiRC), supported by NSF under grant EEC‐0813570, National Natural Science Foundation of China (21673040), Natural Science Foundation of Fujian Province (2016J01052), National Science Foundation (CHE‐1462019), and European Research Council (ERC Advanced Grant no. 321140). We thank A. Nicholls at the University of Illinois at Chicago for recording some of the aberration corrected STEM images. Part of the research was conducted at the Environmental Molecular Sciences Laboratory located at Pacific Northwest National Laboratory (PNNL), supported by the DOE's Office of Biological and Environmental Research. B.M.W. acknowledges the NWO Gravitation program, Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), and a European Research Council (ERC) Advanced Grant (no. 321140). We thank Fouad Soulimani (Utrecht University) for assistance with the FTIR measurements.
H. Xiong, S. Lin, J. Goetze, P. Pletcher, H. Guo, L. Kovarik, K. Artyushkova, B. M. Weckhuysen, A. K. Datye, Angew. Chem. Int. Ed. 2017, 56, 8986.
Contributor Information
Dr. Sen Lin, Email: slin@fzu.edu.cn
Prof. Dr. Bert M. Weckhuysen, Email: B.M.Weckhuysen@uu.nl
Prof. Dr. Abhaya K. Datye, Email: datye@unm.edu.
References
- 1. Otroshchenko T., Sokolov S., Stoyanova M., Kondratenko V. A., Rodemerck U., Linke D., Kondratenko E. V., Angew. Chem. Int. Ed. 2015, 54, 15880–15883; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16107–16111. [Google Scholar]
- 2.
- 2a. Wu Z., Wegener E. C., Tseng H.-T., Gallagher J. R., Harris J. W., Diaz R. E., Ren Y., Ribeiro F. H., Miller J. T., Catal. Sci. Technol. 2016, 6, 6965–6976; [Google Scholar]
- 2b. Zhu J., Yang M.-L., Yu Y., Zhu Y.-A., Sui Z.-J., Zhou X.-G., Holmen A., Chen D., ACS Catal. 2015, 5, 6310–6319. [Google Scholar]
- 3.National Academies of Sciences, Engineering and Medicine. 2016. The Changing Landscape of Hydrocarbon Feedstocks for Chemical Production: Implications for Catalysis: Proceedings of a Workshop. Washington, DC: The National Academies Press. DOI: 10.17226/23555.
- 4.
- 4a. Sattler J. J. H. B., Ruiz-Martinez J., Santillan-Jimenez E., Weckhuysen B. M., Chem. Rev. 2014, 114, 10613–10653; [DOI] [PubMed] [Google Scholar]
- 4b. Bricker J. C., Top. Catal. 2012, 55, 1309–1314; [Google Scholar]
- 4c. Vora B. V., Top. Catal. 2012, 55, 1297–1308. [Google Scholar]
- 5.
- 5a. Shi L., Deng G.-M., Li W.-C., Miao S., Wang Q.-N., Zhang W.-P., Lu A.-H., Angew. Chem. Int. Ed. 2015, 54, 13994–13998; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14200–14204; [Google Scholar]
- 5b. Im J., Choi M., ACS Catal. 2016, 6, 2819–2826. [Google Scholar]
- 6.
- 6a. Han Z., Li S., Jiang F., Wang T., Ma X., Gong J., Nanoscale 2014, 6, 10000–10008; [DOI] [PubMed] [Google Scholar]
- 6b. Resasco D. E., Encyclopedia of Catalysis 2003, 3, 49. [Google Scholar]
- 7. Pham H. N., Sattler J. J. H. B., Weckhuysen B. M., Datye A. K., ACS Catal. 2016, 6, 2257–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Johns T. R., Goeke R. S., Ashbacher V., Thüne P. C., Niemantsverdriet J. W., Kiefer B., Kim C. H., Balogh M. P., Datye A. K., J. Catal. 2015, 328, 151–164; [Google Scholar]
- 8b. Li W.-Z., Kovarik L., Mei D., Liu J., Wang Y., Peden C. H. F., Nat. Commun. 2013, 4, 2481. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Fung S. C., Tauster S. J., Koo J. Y., US Patent, US005763348A, 1998;
- 9b. Fung S. C. in Studies in Surface Science and Catalysis, Vol. 139 (Eds.: G. W. R. J. J. Spivey, B. H. Davis), Elsevier, Amsterdam, 2001, pp. 399–406. [Google Scholar]
- 10. Bruix A., Lykhach Y., Matolínová I., Neitzel A., Skála T., Tsud N., Vorokhta M., Stetsovych V., Ševčíková K., Mysliveček J., Fiala R., Václavů M., Prince K. C., Bruyère S., Potin V., Illas F., Matolín V., Libuda J., Neyman K. M., Angew. Chem. Int. Ed. 2014, 53, 10525–10530; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10693–10698. [Google Scholar]
- 11. Jones J., Xiong H., DeLaRiva A. T., Peterson E. J., Pham H., Challa S. R., Qi G., Oh S., Wiebenga M. H., Pereira Hernández X. I., Wang Y., Datye A. K., Science 2016, 353, 150–154. [DOI] [PubMed] [Google Scholar]
- 12. Sattler J. J. H. B., Gonzalez-Jimenez I. D., Luo L., Stears B. A., Malek A., Barton D. G., Kilos B. A., Kaminsky M. P., Verhoeven T. W. G. M., Koers E. J., Baldus M., Weckhuysen B. M., Angew. Chem. Int. Ed. 2014, 53, 9251–9256; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9405–9410. [Google Scholar]
- 13.
- 13a. Cortright R. D., Hill J. M., Dumesic J. A., Catal. Today 2000, 55, 213–223; [Google Scholar]
- 13b. Zhou H., Gong J., Xu B., Yu L., Fan Y., Appl. Catal. A 2016, 527, 30–35; [Google Scholar]
- 13c. Hauser A. W., Gomes J., Bajdich M., Head-Gordon M., Bell A. T., Phys. Chem. Chem. Phys. 2013, 15, 20727–20734; [DOI] [PubMed] [Google Scholar]
- 13d. Bariås O. A., Holmen A., Blekkan E. A., J. Catal. 1996, 158, 1–12; [Google Scholar]
- 13e. Cortright R. D., Bergene E., Levin P., Natal-Santiago M., Dumesic J. A. in Studies in Surface Science and Catalysis, Vol. 101 (Eds.: W. N. Delgass, E. Iglesia, J. W. Hightower, A. T. Bell), Elsevier, Amsterdam, 1996, pp. 1185–1194; [Google Scholar]
- 13f. Wu J., Peng Z., Bell A. T., J. Catal. 2014, 311, 161–168; [Google Scholar]
- 13g. Wang G., Zhang H., Wang H., Zhu Q., Li C., Shan H., J. Catal. 2016, 344, 606–608; [Google Scholar]
- 13h. Zhu H., Anjum D. H., Wang Q., Abou-Hamad E., Emsley L., Dong H., Laveille P., Li L., Samal A. K., Basset J.-M., J. Catal. 2014, 320, 52–62; [Google Scholar]
- 13i. Virnovskaia A., Morandi S., Rytter E., Ghiotti G., Olsbye U., J. Phys. Chem. C 2007, 111, 14732–14742. [Google Scholar]
- 14. Shan Y., Sui Z., Zhu Y., Chen D., Zhou X., Chem. Eng. J. 2015, 278, 240–248. [Google Scholar]
- 15. Shan Y.-L., Zhu Y.-A., Sui Z.-J., Chen D., Zhou X.-G., Catal. Sci. Technol. 2015, 5, 3991–4000. [Google Scholar]
- 16. Sattler J. J. H. B., Beale A. M., Weckhuysen B. M., Phys. Chem. Chem. Phys. 2013, 15, 12095–12103. [DOI] [PubMed] [Google Scholar]
- 17. Iglesias-Juez A., Beale A. M., Maaijen K., Weng T. C., Glatzel P., Weckhuysen B. M., J. Catal. 2010, 276, 268–279. [Google Scholar]
- 18.
- 18a. Yang M.-L., Zhu Y.-A., Zhou X.-G., Sui Z.-J., Chen D., ACS Catal. 2012, 2, 1247–1258; [Google Scholar]
- 18b. Hook A., Massa J. D., Celik F. E., J. Phys. Chem. C 2016, 120, 27307–27318. [Google Scholar]
- 19. Xiong H., Peterson E., Qi G., Datye A. K., Catal. Today 2016, 272, 80–86. [Google Scholar]
- 20. Moliner M., Gabay J. E., Kliewer C. E., Carr R. T., Guzman J., Casty G. L., Serna P., Corma A., J. Am. Chem. Soc. 2016, 138, 15743–15750. [DOI] [PubMed] [Google Scholar]
- 21. Qiao B., Wang A., Yang X., Allard L. F., Jiang Z., Cui Y., Liu J., Li J., Zhang T., Nat. Chem. 2011, 3, 634–641. [DOI] [PubMed] [Google Scholar]
- 22. Balakrishnan K., Schwank J., J. Catal. 1992, 138, 491–499. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary