

Abstract
Charcot‐Marie‐Tooth disease (CMT) constitutes a heterogeneous group affecting motor and sensory neurons in the peripheral nervous system. MFN2 mutations are the most common cause of axonal CMT. We describe the clinical and mutational spectra of CMT patients harboring MFN2 mutations in Japan. We analyzed 1,334 unrelated patients with clinically suspected CMT referred by neurological and neuropediatric departments throughout Japan. We conducted mutation screening using a DNA microarray, targeted resequencing, and whole‐exome sequencing. We identified pathogenic or likely pathogenic MFN2 variants from 79 CMT patients, comprising 44 heterozygous and 1 compound heterozygous variants. A total of 15 novel variants were detected. An autosomal dominant family history was determined in 43 cases, and the remaining 36 cases were reported as sporadic with no family history. The mean onset age of CMT in these patients was 12 ± 14 (range 0–59) years. We observed neuropathic symptoms in all patients. Some had optic atrophy, vocal cord paralysis, or spasticity. We detected a compound heterozygous MFN2 mutation in a patient with a severe phenotype and the co‐occurrence of MFN2 and PMP22 mutations in a patient with an uncommon phenotype. MFN2 is the most frequent causative gene of CMT2 in Japan. We present 15 novel variants and broad clinical and mutational spectra of Japanese MFN2‐related CMT patients. Regardless of the onset age and inheritance pattern, MFN2 gene analysis should be performed. Combinations of causative genes should be considered to explain the phenotypic diversity.
Keywords: Charcot‐Marie‐Tooth disease, clinical features, Japanese, MFN2, simultaneous mutation
Introduction
Charcot‐Marie‐Tooth disease (CMT) is one of the most common inherited peripheral neuropathies. The prevalence of CMT was reported to be 1 per 1,215–10,300 persons (Barreto et al., 2016 ). To date, more than 80 causative genes have been reported to be associated with CMT (Timmerman et al., 2014 ). The clinical features of CMT can significantly vary among patients, even among those sharing the same mutation. Generally, CMT cases are classified into a demyelinating type [median MNCV (motor nerve conduction velocity) <38 m/s], an axonal type (median MNCV >38 m/s), and an intermediate type based on the MNCV of the median nerve. Most frequently, the demyelinating type is associated with a mutation in the gene PMP22, while a mutation in MFN2 is linked to the axonal type. MFN2 is a protein present in the mitochondrial outer membrane, which responds to the mitochondrial dynamics through a mitochondrial GTPase. The frequency of MFN2 mutations in CMT2 patients has been reported to be in the range of 17%–23% in Spanish, French, Korean, and Chinese populations (Calvo et al., 2009 ; Casasnovas et al., 2010 ; Choi et al., 2015 ; Xie et al., 2016 ). However, its frequency was reported to be lower, between 8.6% and 11%, in previous Japanese reports (Kijima et al., 2005 ; Abe et al., 2011 ).
MFN2 mutation causes typical CMT2, which is called CMT2A2, and can also present different clinical phenotypes, including hereditary motor sensory neuropathy (HMSN) with pyramidal features (HMSN V), HMSN with optic atrophy (HMSN VIA), AR‐CMT, severe early onset axonal neuropathy, early onset stroke without neuropathy, HMSN with cognitive impairment, and brain mitochondrial dysfunction (Mostacciuolo et al., 2000 ; Zuchner et al., 2006 ; Chung et al., 2008 ; Del Bo et al., 2008 ; Nicholson et al., 2008 ; Polke et al., 2011 ). In this case series, we identified novel pathogenic mutations and investigated variations in the clinical features of CMT patients due to MFN2 variants.
Methods and Materials
Patients
We analyzed 1,334 unrelated patients/families with clinically suspected CMT. The clinical data and DNA samples were collected from neurological and neuropediatric departments throughout Japan between 2007 and 2016. All the demyelinating patients were enrolled in this study after confirming them to be negative for PMP22 duplication/deletion as identified using fluorescence in situ hybridization and multiplex ligation probe amplification. We extracted genomic DNA using QIAGEN's Puregene Core Kit C (Qiagen, Valencia, CA, USA) or Oragene DNA self‐collection kit (DNA Genotech, Ottawa, Ontario, Canada) and carried out mutation screening tests using DNA microarray, targeted resequencing, and whole‐exome sequencing. Candidate variants detected by these methods were validated using Sanger sequencing. If available, segregation analysis was performed for those cases.
The study protocol was reviewed and approved by the institutional review board of Kagoshima University. All patients and family members provided written informed consent to participate in this study, including for the genetic analyses.
Microarray chip sequencing
We designed a customized MyGeneChip® CustomSeq® Resequencing Array (Affymetrix, Inc., Santa Clara, CA, USA) to screen 30 disease‐causing genes for CMT and related diseases. We performed mutation screening using Microarray chip sequencing in patients who were enrolled from 2007 to 2012. The detailed methodology has been described elsewhere (Hashiguchi et al., 2014 ). Table S1, Supporting Information includes the sequences of 30 target genes.
Targeted resequencing
We performed mutation screening of 60 or 72 known/candidate CMT‐related genes using two methods: the Illumina Miseq platform (Illumina Inc., San Diego, CA, USA) and the Ion Proton using a custom Ion AmpliseqM panel and the Ion PI Chip kit v2 BC (ThermoFisher Scientific, Inc., Waltham, MA, USA). We performed mutation screening using Illumina Miseq platform from 2012 to 2014 and Ion Proton platform from 2014 to 2016. After aligning and mapping variant calling, we annotated and filtered variants using the CLC genomics Workbench software program (Qiagen, Hilden, Germany). We filtered out the variants with low read depth (<10) and low quality (<20). The detail methods have been previously described (Maeda et al., 2014 ; Higuchi et al., 2016 ). The symbols of the 60 and 72 target genes are shown in Table S1.
Whole‐exome sequencing
Exome sequences were enriched using a SureSelect V4+UTRs or v5+UTRs Kit (Agilent Technologies, Santa Clara, CA, USA) and were subsequently subjected to sequencing on a hiseq2000 platform (Illumina). Sequence data was aligned to the human genome data (NCBI37/hg19), and variant calling was performed using Burrows Wheeler Aligner and SAM tools (Higuchi et al., 2016; Li and Durbin, 2009 ; Li et al., 2009 ). The called variants were annotated using the CLC genomics Workbench software program and in‐house script, and the variants with low read depth (<10) and low quality (<20) were filtered out.
Data analysis for the determination of pathogenic mutations
We confirmed the previously reported pathogenic mutations by reference to the Human Gene Mutation Database Professional 2016.3 (https://portal.biobase-international.com/hgmd/pro). Moreover, we extracted variants that were not observed in global control databases [dbSNP (https://www.ncbi.nlm.nih.gov/SNP), 1000genome (http://browser.1000genome.org), Exome Sequencing Project (http://evs.gs.washington.edu/EVS), and Exome Aggeregation Consortium (http://exac.broadinstitute.org/)] and Japanese control database [iJGVD; integrative Japanese Genome Variation Database (https://ijgvd.megabank.tohoku.ac.jp), HGVD; Human genetic variation (http://www.hgvd.genome.med.kyoto‐u.ac.jp)] and in‐house not CMT database. Moreover, we performed in silico analysis using SIFT (http://sift.jcvi.org), POLYPHEN2 (http://genetics.bwh.harvard.edu/pph2), PROVEAN (http://provean.jcvi.org/index.php), Mutation Assessor (http://mutationassessor.org), and Condel (http://bg.upf.edu/fannsdb). We evaluated the detected variants using the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (Richards et al., 2015 ).
Results
Epidemiology of MFN2 in Japan
We analyzed 1,334 unrelated patients with clinically suspected CMT. We detected 30 known pathogenic mutations, 1 novel pathogenic mutation, 14 novel likely pathogenic variants of MFN2, from which 44 were heterozygous mutations and 1 was compound heterozygous mutation. Of the CMT patients without PMP22 duplication/deletion, 801 patients (60%) presented the axonal phenotype, and 367 patients (28%) were classified as having the demyelinating type. We could not classify 166 patients due to a non‐recordable median compound motor action potential (CMAP) or no electrophysiological data. These CMT patients with axonal phenotype showed an MFN2 mutation rate of 8%, 63/801 (Fig. 1A).
Figure 1.
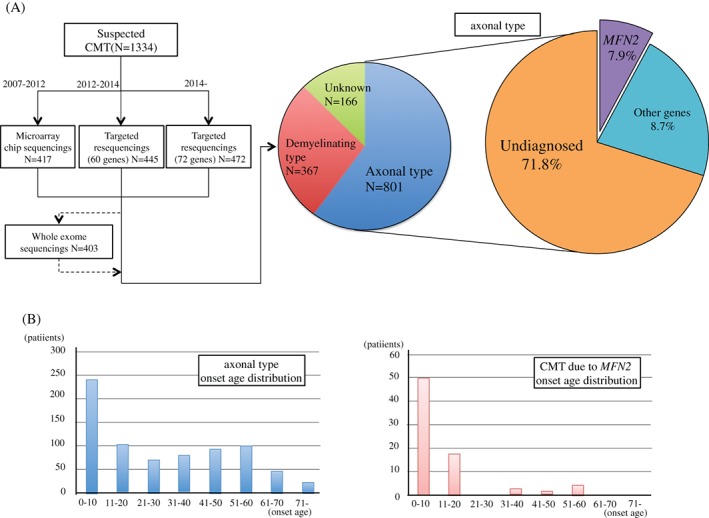
Study flow chart of this and onset age distribution. (A) Study flow chart and the rate of MFN2 mutations in axonal Charcot‐Marie‐Tooth disease (CMT). Onset age distribution for the axonal type and cases with MFN2 mutation.
Clinical features
Data regarding previously reported mutations are included in Table S2. Table 1 shows the clinical data for novel pathogenic variants and likely pathogenic variants. The mutations appeared to be sporadic in 36 patients (46%) and presented an autosomal dominant inherited pattern in 43 patients (54%). The average age of onset was 12 ± 14 (range, 0–59) years and lower than that of the patients presenting the axonal phenotype [30 ± 22 (range, 0–79) years; p < 0.01]. The tendency for juvenile onset is shown in Fig. 1B. The mean onset age of sporadic cases was lower than that of autosomal dominant cases [age of onset for sporadic cases = 7.6 ± 10 years (range 0–59) years; age of onset for autosomal dominant cases = 15 ± 16 (range, 1–57); p value = 0.017].
Table 1.
Clinical information of the cases with novel pathogenic mutations and likely pathogenic variants.

Most patients had developed motor symptoms such as distal leg weakness, foot deformity, gait instability, impossibility to run, or delayed motor milestone by the time of recruitment. Most patients presented with distal weakness, distal atrophy, and hyporeflexia [99% (75/76), 96% (72/75), and 98% (60/61), respectively]. In addition, the frequency of sensory disturbance was lower than that of any other symptoms (61% 33/54). We could evaluate sensory modality in 35 patients of these; 19 showed a decrease in sensory detection only regarding vibration, 8 showed a decrease in both vibration and superficial sensation, and 6 did not show any decrease in either of these parameters.
Only one patient with MFN2 p.Leu710Pro mutation showed HMSN with significant spasticity and increased patellar tendon reflex, which is the HMSN V phenotype, while four patients with MFN2 p.Arg104Trp, p.Arg104Leu, and p.Arg364Trp mutations showed HMSN with optic atrophy. Two patients with MFN2 p.Arg364Trp mutation had vocal cord paralysis. The patient with p.Arg280His and p.Arg250Trp compound heterozygous mutations had an earlier onset than the patients with heterozygous p.Arg280His mutation.
Electrophysiological findings
In the nerve conduction study, the MNCV of the median nerve was 51 ± 7.5 (range, 23–64) m/s and the CMAP was 4.9 ± 4.0 (range, 0–11.7) mV. In addition, the MNCV value for the tibial nerve was 38 ± 8.2 (range, 13–51) m/s and the CMAP was 0.5 ± 1.8 (range, 0–12) mV. Four patients had <38 m/s MNCV and were classified as having demyelinating type; two of them had mild MNCV reduction, 35 and 38 m/s. One patient had significantly decreased MNCV, 23 m/s. The median MNCV of four patients was unknown, and two of them had <38 m/s tibial MNCV. In nine patients, the absence of CMAP did not allow MNCV measurement.
Genetic findings
We detected 1 novel pathogenic variant and 14 novel likely pathogenic variants in 20 patients (Table 2). The novel pathogenic variant was p.Lys109Arg (one patient). The novel likely pathogenic variants were p.Glu52Lys (one patient), p.Thr105Ser (two patients), p.His128Tyr (two patients), p.Thr130Ile (one patient), p.Ala220Thr (one patient), p.Ser245Arg (two patients), p.Gln360Glu (one patient), p.Ala383del (one patient), p.Arg476Pro (one patient), p.Ser546Ala (one patient), p.Leu692Arg (one patient), p.Leu741Ser (one patient), p.Met747Arg (three patients), and p.Thr749Hisfs*14 (one patient). Furthermore, we identified 30 previously reported mutations in 59 unrelated patients. Fig. 2 shows the five pedigrees of segregated novel variants as well as the family with co‐occurrence of a de novo MFN2 mutation and a maternal PMP22 mutation in the proband. All variants of uncertain significance are listed in Table S3.
Table 2.
Genetic information of the novel pathogenic and likely pathogenic variants.
| ACMG | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Amino acid change | SIFT | PP2 | PROVEAN | MA | Condel | Same codon reference | Reference AA change | Strong | Moderate | Support | Classification |
| c.154G>A | p.Glu52Lys | 0.003 | 0.901 | −2.97 | 2.58 | 0.64 | No | — | — | PS4‐modarate, PM2 | PP1, 3, 4 | Likely pathogenic |
| c.313A>T | p.Thr105Ser | 0 | 1 | −3.68 | 3.53 | 0.57 |
Kijima et al. (
2005
)
Sitarz et al. ( 2012 ) Brozkova et al. ( 2013 ) |
Thr105Met Thr105Ala Thr105Arg |
— | PS4‐modarate, PM2, 5 | PP3, 4 | Likely pathogenic |
| c.326A>G | p.Lys109Arg | 0 | 1 | −2.66 | 4.065 | 0.78 | No | — | PS2 | PS4‐modarate, PM2 | PP3 | Pathogenic |
| c.382C>T | p.His128Tyr | 0 | 0.987 | −5.52 | 3.375 | 0.64 | Calvo et al. ( 2009 ) | His128Arg | — | PS4‐modarate, PM2, 5, 6 | PP3 | Likely pathogenic |
| c.389C>T | p.Thr130Ile | 0 | 0.975 | −5.42 | 3.94 | 0.68 | No | — | — | PS4‐modarate, PM1, 2 | PP3, 4 | Likely pathogenic |
| c.658G>A | p.Ala220Thr | 0 | 0.975 | −3.49 | 2.575 | 0.59 | No | — | — | PS4‐modarate, PM2, 6 | PP3 | Likely pathogenic |
| c.733A>C | p.Ser245Arg | 0.003 | 0.57 | −3.18 | 2.23 | 0.55 | No | — | — | PS4‐modarate, PM1, 2 | PP3, 4 | Likely pathogenic |
| c.1078C>G | p.Gln360Glu | 0.003 | 0.81 | −2.44 | 3.07 | 0.62 | No | — | — | PS4‐modarate, PM1, 2 | PP3, 4 | Likely pathogenic |
| c.1147_1149delGCT | p.Ala383del | — | — | −6.62 | — | — | Muglia et al. ( 2007 ) | Ala383Val | — | PS4‐modarate, PM1, 2 | PP4 | Likely pathogenic |
| c.1427G>C | p.Arg476Pro | 0.006 | 0.94 | −2.83 | 2.34 | 0.58 | No | — | — | PS4‐modarate, PM2 | PP1, 3, 4 | Likely pathogenic |
| c.1636T>G | p.Ser546Ala | 0 | 0.999 | −2.64 | 3.08 | 0.58 | No | — | — | PS4‐modarate, PM2, 6 | PP3 | Likely pathogenic |
| c.2075T>G | p.Leu692Arg | 0 | 1 | −4.9 | 2.375 | 0.62 | No | — | — | PS4‐modarate, PM2, 6 | PP3 | Likely pathogenic |
| c.2222T>C | p.Leu741Ser | 0 | 1 | −4.45 | 2.455 | 0.63 | No | — | — | PS4‐modarate, PM1, 2 | PP3, 4 | Likely pathogenic |
| c.2240T>G | p.Met747Arg | 0.53 | 0.001 | −0.72 | −0.05 | 0.48 | Calvo et al. ( 2009 ) | Met747Thr | — | PS4‐modarate, PM2, 5 | PP1, 4 | Likely pathogenic |
| c.2243_4insT | p.Thr749Hisfs*14 | — | — | — | — | — | No | — | — | PS4‐modarate, PM1, 2, 4 | PP4 | Likely pathogenic |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; MA, mutation assessor; PP2, PolyPhen2.
In silico analysis cut off: SIFT <0.05, PP2 >0.9, PROVEAN <−2.5, MA >1.9, and Condel >0.47.
Figure 2.
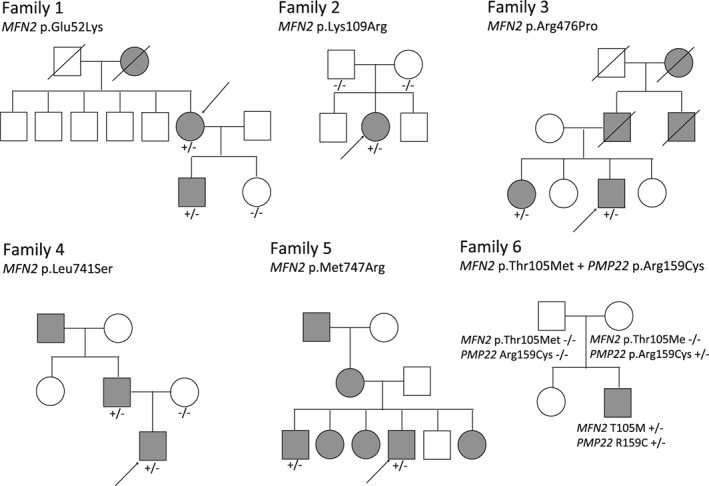
The pedigree of novel variants and simultaneously variants with segregation study. Families 1–5 indicate pedigree chart with novel variants and Family 6 indicate family with simultaneously MFN2 and PMP22 mutation. Arrow indicates probands.
Simultaneous mutations
The presence of simultaneously heterozygous mutations of different CMT disease‐causing genes is noteworthy. We detected the mutations in MFN2 p.Thr105Met and PMP22 p.Arg159Cys. The patient (a female) developed gait disturbances at the age of 1 year and later a mild mental retardation and an IQ of 69. No parental consanguinity or obvious family history was recorded (Fig. 2). Her parents had no neuropathic symptoms and normal cognitive function, but we could not evaluate the electrophysiological findings of her parents. At age 9 years, she had limited dorsiflexion of both feet joints and clubfoot. Due to a deformity in one foot and distal weakness, she could not walk unaided at age 11 years. Her nerve conduction study was normal for the median nerve and presented low CMAP for the tibial nerve. She was diagnosed with CMT2. The genetic analysis showed a p.Thr105Met mutation in MFN2 and a p.Arg159Cys mutation in PMP22. Both mutations had been previously described in other patients; however, none of the parents showed the p.Thr105Met mutation in MFN2, being thus confirmed as a de novo mutation. Her asymptomatic mother presented the p.Arg159Cys mutation in PMP22.
Discussion
In this Japanese case series, we detected MFN2 mutations in 79 of 1,334 CMT patients without a PMP22 deletion/duplication. The MFN2 mutation accounted for 16% of the CMT2 patients in a Spanish cohort study, and 18% of CMT2 without MPZ and GJB1 mutations in French patients (Calvo et al., 2009 ; Casasnovas et al., 2010 ). In Asia, MFN2 was the cause of CMT2 in 23% of Korean CMT2 patients and 18% of Chinese CMT2 patients (Choi et al., 2015 ; Xie et al., 2016 ). The frequency of MFN2 mutations is low, between 9% and 11%, as previously reported in a Japanese population study (Kijima et al., 2005 ; Abe et al., 2011 ). Here, we show a frequency of the MFN2 mutation of 8% (63/801) in Japanese CMT2 patients. A frequency of 28%–34% for sporadic or de novo mutations has been previously reported (Verhoeven et al., 2006 ; Choi et al., 2015 ). However, sporadic cases reached a frequency of 46% in our study. This incidence rate is higher than that reported from other countries. The low frequency of MFN2 mutations and high frequency of sporadic cases might indicate the influence of geographical and social distribution, although there is a possibility of an incomplete family history.
We showed an earlier onset of CMT due to MFN2 mutation than of CMT2. Onset age for CMT2 often show a bimodal distribution with a peak at 0–20 years and another peak at 40–60 years. Onset in MFN2 patients tended to be earlier than the average onset for all CMT patients considered together (Fig. 1B). Some patients showed the characteristic symptoms, for example, spasticity, optic atrophy, and vocal cord paralysis. One of our patients with MFN2 p.Leu710Pro mutation showed spasticity, which has not been previously reported (Verhoeven et al., 2006 ). We found a novel HMSN V phenotype for CMT2 with MFN2 p.Leu710Pro mutation. Patients with MFN2 p.Arg364Trp showed an early onset of the severe phenotype with optic atrophy. In our study, only the patients with this specific mutation presented vocal cord paralysis, similar to that previously reported for this mutation (Zuchner et al., 2006 ). The patient with the novel p.Arg220Thr variant showed the characteristic symptoms of dysphagia and tongue atrophy. Furthermore, we described a case of earlier‐onset CMT phenotype associated with p.Arg280His and p.Arg250Trp compound heterozygous mutation rather than a p.Arg280His heterozygous mutation. This proband was a sporadic case. The p.Arg250Trp heterozygous mutation was reported as causative compound heterozygous mutations with p.Arg400X (Verhoeven et al., 2006 ). This p.Arg250Trp variant might be a genetic burden and make the phenotype more severe.
We presented the electrophysiological findings of CMT patients related to MFN2 mutations. We observed a tendency toward more severe axonal neuropathies in the lower extremities, and the characteristic electrophysiological findings were demyelinating phenotype in some cases. CMT due to MFN2 mutation was known to be mainly axonal phenotype, but some cases classified as demyelinating type or intermediate type were reported (Kijima et al., 2005 ; Braathen et al., 2010 ). We need to consider the possibility that MFN2 mutation will be classified as demyelinating type.
In addition, we describe 1 novel pathogenic variant and 14 likely pathogenic variants. The novel p.Lys109Arg variant was absent in global, Japanese, and in‐house databases (ACMG standards and guidelines; PS4‐Moderate, PM2). This variant was validated as a de novo variant via segregation analysis (PS2), and we classified novel p.Lys109Arg variants as pathogenic. The other novel variants were classified as likely pathogenic variants according to the ACMG standards and guidelines. It is difficult to judge the novel MFN2 variants as pathogenic variants. Without previously reported same amino acid change variant or functional study, novel MFN2 variants with a family history could not be classified as pathogenic variants. Therefore, we reported novel pathogenic and likely pathogenic variants. Eventually, we will need to clarify the level pathogenicity of these variants based on functional or population studies.
We described characteristic cases simultaneously presenting mutations of different CMT disease‐causing genes. Recent advances in genetic techniques, including next‐generation sequencing, have enabled the possibility to analyze a large number of genes from a large number of patients. These data have thus helped in identifying the combined effect of rare variants, their expression, and their contribution to disease burden, included MFN2 and MED25, MFN2 and HSPB1, and MFN2 and WNK1. (Gonzaga‐Jauregui et al., 2015 ). To this end, our study also shows how simultaneous heterozygous mutation may contribute to the level of clinical variability previously described. We presented simultaneous heterozygous mutations in MFN2 p.Thr105Met and PMP22 p.Arg159Cys. Both of these mutations had been reported before, and the mother of the proband appeared to have a mutation in PMP22. The reported onset age for patients with PMP22 p.Arg159Cys mutation is 46 years (Gess et al., 2011 ). Therefore, it is likely that onset had not happened yet for the mother of the proband, and the mutation may present incomplete penetrance. The patient with simultaneous mutations presented with an earlier onset age and mental retardation, which was a novel symptom in a patient with MFN2 p.Thr105Met mutation. Similarly, MFN2 and PMP22 simultaneous mutations may result in a severe phenotype because of a double‐dose effect or genetic burden effect.
Supporting information
Table S1: Target genes analyzed in the study.
Table S2: Clinical data of patients showing mutations previously reported.
Table S3: Variants of uncertain significance in this study.
Acknowledgements
This study was supported, in part, by grants from the research on the Nervous and Mental Disorders and Research Committee for Charcot‐Marie‐Tooth Disease, Neuropathy, Ataxic Disease and Applying Health and Technology of Ministry of Health, Welfare and Labor, Japan. This research is also supported by the research program for conquering intractable disease and Charcot‐Marie‐Tooth Disease (grant number 17929553) from Japan Agency for Medical Research and Development (AMED). And this research was supported by JSPS KAKENHI Grant Numbers . We thank Mrs. Aya Ebina and Tomoko Onishi of our department for her excellent technical assistance, and Enago (www.enago.jp) for the English language review. We authors thank the patients and their families for participating in this study and their physicians for submitting the clinical samples.
References
- Abe A, Numakura C, Kijima K, Hayashi M, Hashimoto T, Hayasaka K (2011). Molecular diagnosis and clinical onset of Charcot‐Marie‐Tooth disease in Japan. J Hum Genet 56:364–368. [DOI] [PubMed] [Google Scholar]
- Barreto LC, Oliveira FS, Nunes PS, de Franca Costa IM, Garcez CA, Goes GM, Neves EL, de Souza Siqueira Quintans J, de Souza Araujo AA (2016). Epidemiologic study of Charcot‐Marie‐tooth disease: a systematic review. Neuroepidemiology 46:157–165. [DOI] [PubMed] [Google Scholar]
- Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB (2010). MFN2 point mutations occur in 3.4% of Charcot‐Marie‐Tooth families. An investigation of 232 Norwegian CMT families. BMC Med Genet 11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozkova DS, Posadka J, Lassuthova P, Mazanec R, Haberlova J, Siskova D, Sakmaryova I, Neupauerova J, Seeman P (2013). Spectrum and frequencies of mutations in the MFN2 gene and its phenotypical expression in Czech hereditary motor and sensory neuropathy type II patients. Mol Med Rep 8:1779–1784. [DOI] [PubMed] [Google Scholar]
- Calvo J, Funalot B, Ouvrier RA, Lazaro L, Toutain A, De Mas P, Bouche P, Gilbert‐Dussardier B, Arne‐Bes MC, Carriere JP, Journel H, Minot‐Myhie MC, Guillou C, Ghorab K, Magy L, Sturtz F, Vallat JM, Magdelaine C (2009). Genotype‐phenotype correlations in Charcot‐Marie‐Tooth disease type 2 caused by mitofusin 2 mutations. Arch Neurol 66:1511–1516. [DOI] [PubMed] [Google Scholar]
- Casasnovas C, Banchs I, Cassereau J, Gueguen N, Chevrollier A, Martinez‐Matos JA, Bonneau D, Volpini V (2010). Phenotypic spectrum of MFN2 mutations in the Spanish population. J Med Genet 47:249–256. [DOI] [PubMed] [Google Scholar]
- Choi BO, Nakhro K, Park HJ, Hyun YS, Lee JH, Kanwal S, Jung SC, Chung KW (2015). A cohort study of MFN2 mutations and phenotypic spectrums in Charcot‐Marie‐Tooth disease 2A patients. Clin Genet 87:594–598. [DOI] [PubMed] [Google Scholar]
- Chung KW, Cho SY, Hwang SJ, Kim KH, Yoo JH, Kwon O, Kim SM, Sunwoo IN, Zuchner S, Choi BO (2008). Early‐onset stroke associated with a mutation in mitofusin 2. Neurology 70:2010–2011. [DOI] [PubMed] [Google Scholar]
- Del Bo R, Moggio M, Rango M, Bonato S, D'Angelo MG, Ghezzi S, Airoldi G, Bassi MT, Guglieri M, Napoli L, Lamperti C, Corti S, Federico A, Bresolin N, Comi GP (2008). Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 71:1959–1966. [DOI] [PubMed] [Google Scholar]
- Gess B, Jeibmann A, Schirmacher A, Kleffner I, Schilling M, Young P (2011). Report of a novel mutation in the PMP22 gene causing an axonal neuropathy. Muscle Nerve 43:605–609. [DOI] [PubMed] [Google Scholar]
- Gonzaga‐Jauregui C, Harel T, Gambin T, Kousi M, Griffin LB, Francescatto L, Ozes B, Karaca E, Jhangiani SN, Bainbridge MN, Lawson KS, Pehlivan D, Okamoto Y, Withers M, Mancias P, Slavotinek A, Reitnauer PJ, Goksungur MT, Shy M, Crawford TO, Koenig M, Willer J, Flores BN, Pediaditrakis I, Us O, Wiszniewski W, Parman Y, Antonellis A, Muzny DM, Katsanis N, Battaloglu E, Boerwinkle E, Gibbs RA, Lupski JR (2015). Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep 12:1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi A, Higuchi Y, Nomura M, Nakamura T, Arata H, Yuan J, Yoshimura A, Okamoto Y, Matsuura E, Takashima H (2014). Neurofilament light mutation causes hereditary motor and sensory neuropathy with pyramidal signs. J Peripher Nerv Syst 19:311–316. [DOI] [PubMed] [Google Scholar]
- Higuchi Y, Hashiguchi A, Yuan J, Yoshimura A, Mitsui J, Ishiura H, Tanaka M, Ishihara S, Tanabe H, Nozuma S, Okamoto Y, Matsuura E, Ohkubo R, Inamizu S, Shiraishi W, Yamasaki R, Ohyagi Y, Kira J, Oya Y, Yabe H, Nishikawa N, Tobisawa S, Matsuda N, Masuda M, Kugimoto C, Fukushima K, Yano S, Yoshimura J, Doi K, Nakagawa M, Morishita S, Tsuji S, Takashima H (2016). Mutations in MME cause an autosomal‐recessive Charcot‐Marie‐Tooth disease type 2. Ann Neurol 79:659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, Ogawa M, Ishizaki Y, Kitamura T, Shozawa Y, Hayasaka K (2005). Mitochondrial GTPase mitofusin 2 mutation in Charcot‐Marie‐Tooth neuropathy type 2A. Hum Genet 116:23–27. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Idehara R, Hashiguchi A, Takashima H (2014). A family with distal hereditary motor neuropathy and a K141Q mutation of small heat shock protein HSPB1. Intern Med 53:1655–1658. [DOI] [PubMed] [Google Scholar]
- Mostacciuolo ML, Rampoldi L, Righetti E, Vazza G, Schiavon F, Angelini C (2000). Hereditary spastic paraplegia associated with peripheral neuropathy: a distinct clinical and genetic entity. Neuromuscul Disord 10:497–502. [DOI] [PubMed] [Google Scholar]
- Muglia M, Vazza G, Patitucci A, Milani M, Pareyson D, Taroni F, Quattrone A, Mostacciuolo ML (2007). A novel founder mutation in the MFN2 gene associated with variable Charcot‐Marie‐Tooth type 2 phenotype in two families from southern Italy. J Neurol Neurosurg Psychiatry 78:1286–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson GA, Magdelaine C, Zhu D, Grew S, Ryan MM, Sturtz F, Vallat JM, Ouvrier RA (2008). Severe early‐onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology 70:1678–1681. [DOI] [PubMed] [Google Scholar]
- Polke JM, Laura M, Pareyson D, Taroni F, Milani M, Bergamin G, Gibbons VS, Houlden H, Chamley SC, Blake J, Devile C, Sandford R, Sweeney MG, Davis MB, Reilly MM (2011). Recessive axonal Charcot‐Marie‐Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology 77:168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitarz KS, Yu‐Wai‐Man P, Pyle A, Stewart JD, Rautenstrauss B, Seeman P, Reilly MM, Horvath R, Chinnery PF (2012). MFN2 mutations cause compensatory mitochondrial DNA proliferation. Brain 135 e219, 1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman V, Strickland AV, Zuchner S (2014). Genetics of Charcot‐Marie‐Tooth (CMT) disease within the frame of the human genome project success. Genes 5:13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven K, Claeys KG, Zuchner S, Schroder JM, Weis J, Ceuterick C, Jordanova A, Nelis E, De Vriendt E, Van Hul M, Seeman P, Mazanec R, Saifi GM, Szigeti K, Mancias P, Butler IJ, Kochanski A, Ryniewicz B, De Bleecker J, Van den Bergh P, Verellen C, Van Coster R, Goemans N, Auer‐Grumbach M, Robberecht W, Milic Rasic V, Nevo Y, Tournev I, Guergueltcheva V, Roelens F, Vieregge P, Vinci P, Moreno MT, Christen HJ, Shy ME, Lupski JR, Vance JM, De Jonghe P, Timmerman V (2006). MFN2 mutation distribution and genotype/phenotype correlation in Charcot‐Marie‐Tooth type 2. Brain 129:2093–2102. [DOI] [PubMed] [Google Scholar]
- Xie Y, Li X, Liu L, Hu Z, Huang S, Zhan Y, Zi X, Xia K, Tang B, Zhang R (2016). MFN2‐related genetic and clinical features in a cohort of Chinese CMT2 patients. J Peripher Nerv Syst 21:38–44. [DOI] [PubMed] [Google Scholar]
- Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J, Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, Bird T, Timmerman V, Shy M, Vance JM (2006). Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 59:276–281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Target genes analyzed in the study.
Table S2: Clinical data of patients showing mutations previously reported.
Table S3: Variants of uncertain significance in this study.