

Abstract
Psoriasis is a chronic, immune‐mediated disease affecting more than 100 million people worldwide and up to 2.2% of the UK population. The aetiology of psoriasis is thought to originate from an interplay of genetic, environmental, infectious and lifestyle factors. The manner in which genetic and environmental factors interact to contribute to the molecular disease mechanisms has remained elusive. However, the interleukin 23 (IL‐23)/T‐helper 17 (TH17) immune axis has been identified as a major immune pathway in psoriasis disease pathogenesis. Central to this pathway is the cytokine IL‐23, a heterodimer composed of a p40 subunit also found in IL‐12 and a p19 subunit exclusive to IL‐23. IL‐23 is important for maintaining TH17 responses, and levels of IL‐23 are elevated in psoriatic skin compared with non‐lesional skin. A number of agents that specifically inhibit IL‐23p19 are currently in development for the treatment of moderate‐to‐severe plaque psoriasis, with recent clinical trials demonstrating efficacy with a good safety and tolerability profile. These data support the role of this cytokine in the pathogenesis of psoriasis. A better understanding of the IL‐23/TH17 immune axis is vital and will promote the development of additional targets for psoriasis and other inflammatory diseases that share similar genetic aetiology and pathogenetic pathways.
Introduction
Psoriasis is a chronic, immune‐mediated disease1, 2, 3 affecting approximately 100 million people worldwide4 and 2.2% of the UK population.5 Psoriasis affects men and women of all ages6 and can manifest in many different forms, the most common being psoriasis vulgaris (or plaque psoriasis).4 Plaque psoriasis is characterized by patches of erythema covered in a silvery‐white scale,7 the result of rapid hyperproliferation and dysregulated differentiation of epidermal keratinocytes.8
The aetiology of psoriasis is multifactorial and includes a complex interplay of genetic, environmental, infectious and lifestyle factors.9, 10 Genome‐wide association studies have identified numerous psoriasis‐associated gene loci,11, 12, 13 including the HLA‐Cw6 gene,14 specifically the HLA‐class 1 allele, HLA‐C*06:02,13, 15 located within Psoriasis Susceptibility Locus 1 (PSORS1 on 6p21.3).16 Polymorphisms located within this gene locus confer the highest risk of psoriasis (odds ratio [OR] 4.02–16.82).17, 18 Gene loci outside the HLA region mostly represent common genetic variants with low effect sizes, including polymorphisms in the IL‐23/TH17 immune axis such as IL12B (OR 0.78–1.15) and IL23R (OR 0.87–1.10).11, 13, 17, 18, 19, 20, 21 Other variants, independent of HLA‐C*06:02, are related to innate immune pathways, antigen presentation, and T‐cell activation and differentiation.22, 23, 24, 25, 26, 27 When combined with HLA‐C*06:02, single‐nucleotide polymorphisms in IL23A, IL23R, IL12B, NFKB1 and TNIP1 are associated with severe disease.17
Overall, most of the psoriasis‐associated gene loci are related to the innate and/or adaptive immune system. However, as the majority of putative causal variants are located in noncoding regions,28 and coupled with a complex genetic environment, it remains difficult to assign individual gene variants precise roles in the pathogenesis of, and susceptibility to, psoriasis.
Multiple inflammatory cell types are present in plaques, including dendritic cells (DCs), T cells and macrophages, which contribute to disease pathogenesis and drive keratinocyte proliferation.29 T cells are known to be central to the pathogenesis of psoriasis; interfering with T‐cell trafficking and cutaneous T‐cell recruitment improves psoriasis.30, 31, 32 Inhibition of CD8+ T‐cell infiltration and activation into the epidermis prevented the development of psoriasis in a mouse model using human skin transplants.33, 34 More specifically, CD4+ and CD8+ T cells with an interleukin‐17 (IL‐17) secretory phenotype (T‐17 cells) are important contributors owing to their production of the pro‐inflammatory cytokines IL‐17, IL‐22 and tumour necrosis factor (TNF).35, 36 Also, a shift in the T‐cell pool during psoriasis in which regulatory T cells (Tregs) begin expressing IL‐17A has recently been identified.37 Expression of the Treg master transcription factor Foxp3 is progressively lost, whereas expression of the TH17 transcription factor retinoic acid receptor‐related orphan receptor γt (RORγt), is increased by Tregs.37 This process appears to be augmented by IL‐2337 and may be a contributing factor to the chronic inflammation seen in psoriasis. DCs are also important in the pathogenesis of psoriasis owing to their influence on T‐cell activation and cytokine production. Myeloid DCs (CD11c+) are major producers of IL‐23 in the skin,38 Tip‐DCs (a subset of CD11c+ DCs that express inducible nitric oxide synthase) are a source of TNF,39 and plasmacytoid DCs produce high levels of type 1 interferon (IFN).40, 41 CD163+‐activated macrophages are also more abundant in psoriasis compared with normal skin42 and express products typical of classically activated macrophages, including IL‐23p19 and IL‐12/23p40.42 Although their exact role in the pathogenesis of psoriasis remains unclear, IL‐17A–expressing neutrophils are known to aggregate in the epidermis, forming Munro's microabscesses in psoriatic lesions.43 Finally, keratinocytes are a skin‐specific source of IL‐23 and, in health, maintain cutaneous immunity through activation of T‐17 pathways.44
It has been suggested that the localized activation and recruitment of inflammatory cells to plaques are the result of an autoimmune response in the skin.45, 46 The human leucocyte antigen (HLA) class I allele, HLA‐C*06:02, is the main risk allele in psoriasis.17, 18 As HLA‐class I molecules present peptide antigens from intracellular antigens to CD8+ T cells, a HLA‐class I restricted autoimmune response must be directed against a particular target cell.47 An unbiased analysis of epidermal CD8+ T‐cell reactivity unveiled an autoimmune response against melanocytes mediated by HLA‐C*06:02 and identified ADAMTS‐like protein 5 (ADAMTSL5) as a melanocyte autoantigen.45 ADAMTSL5, expressed by epidermal melanocytes, activates CD8+ T cells in the epidermis and has been proposed as an explanation of why psoriasis manifests in the skin.45 ADAMTSL5 stimulation increased production of IL‐17A and IFNγ by peripheral blood mononuclear cells in 62% of patients with psoriasis.45 Cathelicidin (LL‐37) is another likely autoantigen.48 LL‐37 is a cationic peptide involved in antimicrobial defence and is known to stimulate T cells.48 Complexed with nucleic material, LL‐37 has been shown to activate the production of IFN by DCs through ligation of endosomal Toll‐like receptors (TLRs).49, 50 Circulating T cells specific to LL‐37 were present in 46% of tested patients with psoriasis.48 In addition, when complexed to bacterial DNA, IL‐26 (a T‐17 cell‐derived cytokine) also activates DCs to produce IFN via ligation of TLRs.51 More recently, psoriatic T cells have been shown to recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a.52
The IL‐23/TH17 immune axis
When the immune system was initially considered dichotomous, being composed primarily of TH1 and TH2 cells, psoriasis was thought to involve a TH1 response, driven by the cytokines IFNγ and IL‐12.53 However, clinical trials evaluating the efficacy of anti‐IFNγ therapies for the treatment of psoriasis were not successful, indicating that IFNγ does not hold a bottleneck position in the pathophysiology of psoriasis.54 IL‐12 is composed of two subunits, p35 and p40.55 When increased expression of p40 was discovered in psoriatic lesions, this led to the initial conclusion that IL‐12 expression was elevated in psoriasis.56 However, when it was later shown that the p40 subunit of IL‐12 is also found in IL‐23,57 Lee and colleagues were able to attribute the increased expression of p40 in psoriatic skin to IL‐2338 and not IL‐12, as previously suggested.56 As IL‐23 is involved in the TH17 axis,57, 58 while IL‐12 drives TH1 cell development, the IL‐23/TH17 immune axis is now thought to be central to the pathogenesis of psoriasis. The main cytokines involved in psoriasis pathogenesis, IL‐23, TNF and IL‐17, can be subdivided into regulatory and effector cytokines based on their mode of action. IL‐23 exerts regulatory effects on the maintenance of TH17 cells, whereas IL‐17 and TNF mediate effector functions of innate (TNF) and adaptive (TNF, IL‐17) immune cells.
IL‐23: a critical upstream cytokine in the pathogenesis of psoriasis
IL‐23 is a key cytokine involved in protective immune responses to bacterial and fungal infections59; however, dysregulated IL‐23 production also promotes autoimmune inflammation.60, 61 IL‐23 was identified in 2000 as a heterodimer composed of the IL‐12/23p40 subunit and a newly discovered p19 subunit that is exclusive to IL‐23.57 IL‐23 signals through a heterodimeric receptor complex composed of two subunits, IL‐23R and IL‐12Rβ1.62 This complex predominantly activates signal transducer and activator of transcription 3 (STAT3), leading to IL‐23–dependent gene expression.62 IL‐23 is an upstream regulatory cytokine that acts early in the inflammatory cascade in psoriasis (Fig. 1)58, 63, 64 to maintain the TH17 cell phenotype65 and is critical in the production of downstream effector cytokines, such as IL‐17A, IL‐17F and TNF (Table 1).63, 65, 66 It is important to note that IL‐23 cannot directly promote TH17 cell differentiation as the IL‐23 receptor is not expressed on naïve T cells.61, 65, 67, 68 IL‐6 and transforming growth factor β (TGF‐β) released by dermal DCs elicit RORγt‐dependent differentiation of naïve T cells into TH17 cells,67, 68, 69, 70 and the phenotype is then maintained by IL‐23.
Figure 1.
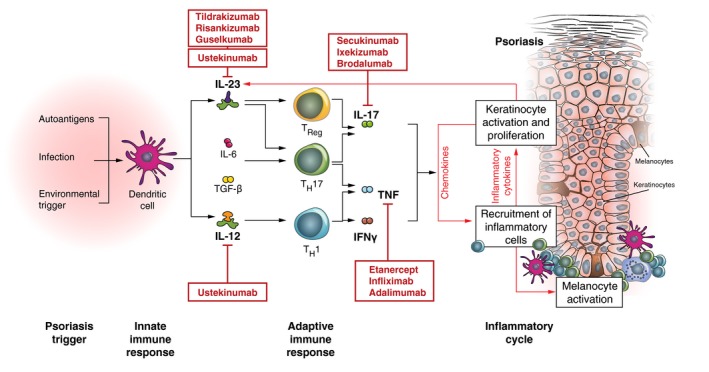
Pathogenesis of psoriasis and diversity of targeted therapies.29, 37, 64, 149, 150, 151 DC, dendritic cell; IFN, interferon; IL, interleukin; TGF, transforming growth factor; TNF, tumour necrosis factor.
Table 1.
Effects of IL‐23 vs IL‐17A in psoriasis
| IL‐23 | IL‐17A | |
|---|---|---|
| Cytokine type | Upstream cytokine | Effector (downstream) cytokine |
| Primary source | Activated monocytes and dendritic cells145, 146 | TH17 cells146, 147 |
| Primary target | TH17 cells58, 146 | Keratinocytes146 |
| Role | Promotes maintenance of TH17 cells37, 103, 147, 148 | Key inflammatory effector cytokine that induces keratinocyte activation and proliferation and reduces differentiation83, 146 |
| Effect | Activation of TH17 cells to produce cytokines including IL‐17A/F, IL‐22, IL‐26, IFNγ, and GM‐CSF, which drives the inflammatory response64, 147 | Stimulates keratinocyte expression of antimicrobial peptides (LL‐37 and β‐defensins), pro‐inflammatory cytokines (TNF, IL‐1β, IL‐6) and chemokines (CXCL8–CXCL11, CCL20), which feed back into the pro‐inflammatory cycle, resulting in a continued immunopathologic progression of psoriasis145, 149 |
| Potential consequences of blockade | Prolonged downregulation of immune activation, potential for a lower risk of AEs compared with IL‐17 blockade | Short‐term interference with effector immune mechanisms, potential for higher risk of AEs/infections compared with IL‐23 blockade |
AEs, adverse events; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; IFN, interferon; IL, interleukin; LL‐37, cathelicidin; TH17, T‐helper 17; TNF, tumour necrosis factor.
Human studies and animal models of psoriasis have confirmed the critical role of IL‐23 signalling in the pathogenesis of psoriasis. For example, IL‐17A and IL‐17F inductions were completely abolished in IL‐23p19 knock‐out mice,71 and intradermal injection of IL‐23–induced skin changes was consistent with human psoriasis in wild‐type mice.63 In humans, expression of IL‐23p19 messenger RNA is increased in psoriatic lesions compared with normal skin.38 In patients with psoriasis, the IL‐23 receptor is overexpressed on dermal DCs and epidermal Langerhans cells72; whereas, in psoriatic lesions, IL‐23 itself is overproduced by dermal DCs and keratinocytes.38, 44, 73 In mice, nociceptors interact with dermal DCs and induce the production of IL‐23, which drives skin inflammation associated with psoriasis.74
IL‐17: a downstream effector cytokine
The IL‐17 cytokine family consists of six isoforms termed IL‐17 A–F.75 Increased expression of IL‐17A, E and F in psoriatic lesions has been described.76, 77, 78 TH17 (CD4+) cells are a major source of IL‐17A, although this cytokine can also be produced by CD8+ T cells and γδ T cells,64 natural killer T cells,79, 80 mast cells and neutrophils.43, 81 IL‐17 is an effector cytokine downstream of IL‐2361 that mediates psoriatic inflammation (Fig. 1).82, 83, 84, 85 In health, TH17 cells act to regulate protective immune responses, promote microbial killing (via IL‐26), and clear bacterial and fungal pathogens by inducing tissue inflammation.51, 86 The IL‐17 receptor is expressed on many cell types, including T cells, epithelial cells and fibroblasts.86 IL‐17 induces IL‐17 receptor–dependent proliferation of keratinocytes and production of pro‐inflammatory cytokines, including IL‐1β, IL‐6 and TNF, and antimicrobial peptides, including β‐defensin and matrix metalloproteinase 9.82, 83, 84, 85, 87 Blockade of either IL‐17A or the IL‐17 receptor has been shown to be an effective therapy in plaque psoriasis.88, 89, 90
Therapeutic targeting in psoriasis
The evolution of biologics for the treatment of psoriasis has mirrored the evolving understanding of the immunopathogenesis of the disease (Fig. 1). Early treatment options centred on broad immunosuppression; however, as our understanding of the pathogenesis has improved, more‐targeted therapies have become available.
TNF inhibitors
Efficacy of TNF blockade in psoriasis was identified in patients with inflammatory bowel disease (IBD) and psoriasis who were prescribed TNF inhibitors for the treatment of IBD.91, 92 This observation initiated the clinical development of TNF inhibitors for psoriasis.93, 94 TNF inhibitors currently approved for the treatment of psoriasis are etanercept,95 adalimumab96 and infliximab.97 These agents have proven efficacy, but their broad mechanism of action is associated with safety issues, including an increase in the risk for severe bacterial and viral infections and potentially cancer.98, 99 Concerns regarding the safety of TNF inhibition have driven a need for new, more‐targeted biologics.
IL‐17A inhibitors
The first IL‐17A inhibitor for the treatment of moderate‐to‐severe plaque psoriasis, secukinumab, was approved for the treatment of moderate‐to‐severe plaque psoriasis in 2015,100 followed by ixekizumab in 2016.101 Both compounds have demonstrated strong efficacy with a fast onset of action. However, they require significant loading doses and frequent dosing to maintain responses.100, 101 In addition, candidiasis occurred more frequently in patients with psoriasis receiving secukinumab compared with those taking etanercept, although the overall incidence was low.102 Increased susceptibility to infections has been identified in mice lacking IL‐17 and the IL‐17 receptor, but this effect has not been reported in humans who have mutations in the IL‐23/TH17 immune axis.103 Targeting IL‐17 and the IL‐17 receptor has also been associated with exacerbated IBD in mice104 and was ineffective in treating patients with moderate‐to‐severe Crohn's disease (CD).105
Brodalumab, which targets the IL‐17 receptor, has recently been approved for the treatment of moderate‐to‐severe plaque psoriasis.106 In three phase 3 studies of brodalumab in moderate‐to‐severe plaque psoriasis (AMAGINE‐1 [NCT01708590], AMAGINE‐2 [NCT01708603], AMAGINE‐3 [NCT01708629]), a ≥ 75% improvement in the Psoriasis Area and Severity Index (PASI 75) was observed at Week 12 compared with baseline.107, 108 An additional biologic, bimekizumab, an antibody targeting IL‐17A, IL‐17F, and the heterodimer IL‐17A/F, is currently in development. A phase 1, first‐in‐human study of bimekizumab in patients with mild‐to‐moderate plaque psoriasis (NCT02529956), showed dose‐dependent improvements in clinical features of plaque psoriasis, including PASI and Physician's Global Assessment scores, compared with placebo.109
IL‐12/23 inhibitors
Ustekinumab is currently the only approved drug that inhibits the IL‐12/23p40 subunit, thus antagonizing both IL‐12 and IL‐23.110 Ustekinumab is generally considered safe and well‐tolerated based on both clinical111, 112 and longitudinal, real‐world studies113 and long‐term follow‐up.114
Development of a second IL‐12/23p40 antibody, briakinumab, was discontinued before all safety data were made available115, 116; some speculate that this decision was associated with cardiovascular safety concerns.117 However, meta‐analyses and long‐term follow‐up of therapies targeting IL‐12/23p40 show an overall reduced risk of cardiovascular events,118, 119, 120 so further data are needed to fully exclude a possible association between major cardiovascular events and the use of anti–IL‐12/23 agents.
Development of biologics that specifically target IL‐23 via IL‐23p19, as opposed to the shared IL‐12/23p40 subunit, may be of clinical benefit as IL‐12 signalling is spared. A protective role for IL‐12 in tumorigenesis has been suggested. In humans, IL‐12 levels are significantly reduced in patients with breast cancer compared with healthy controls,121 and mutations in the IL‐12(p40) gene lead to a higher risk of prostate cancer.122 IL‐12 has also been shown to have a protective role, limiting skin inflammation by restricting the infiltration of IL‐17‐expressing γδ T cells in the imiquimod mouse model of psoriasis.123
IL‐23 inhibitors in development
Three inhibitors specifically targeting IL‐23p19 are currently in active development for the treatment of moderate‐to‐severe psoriasis (Table 2): tildrakizumab, guselkumab and risankizumab. A further antibody, LY3074828 (mirikizumab), is now entering phase 2 development.124
Table 2.
Comparison of efficacy for biologics targeting IL‐23p19a
| Characteristic | Tildrakizumabb 100 mg125 | Tildrakizumabb 200 mg125 | Guselkumabc 100 mg126, 127 | Risankizumab 180 mg130 |
|---|---|---|---|---|
| Phase | Phase 3 | Phase 3 | Phase 3 | Phase 2 |
| Dosing schedule | ||||
| Initial | Weeks 0, 4 | Weeks 0, 4 | Weeks 0, 4 | Week 0 |
| Maintenance | q12w | q12w | q8w | q12w |
| Efficacy, % | Week 12 | Week 12 | Week 16 | Week 12 |
| PASI 75 | 61–64 | 62–66 | 86–91 | 88 |
| PASI 90 | 35–39 | 35–37 | 70–73 | 79 |
| PASI 100 | 12–14 | 12–14 | 34–37 | 48 |
| PGA 0 or 1 | 55–58 | 59 | 84–85d | NR |
| Long‐term efficacy, % | Week 28e | Week 28e | Week 24 | Week 36 |
| PASI 75 | 73–80 | 73–82 | 89–91 | 88 |
| PGA 0 or 1 | 65–66 | 69 | 84d | NR |
Data are not from head‐to‐head comparisons.
Data from reSURFACE1 and reSURFACE2 trials.
Data from VOYAGE 1 and VOYAGE 2 trails.
IGA 0 or 1 reported.
Responder analysis includes only PASI 75 responders at Week 16.
IGA, investigator global assessment; IL, interleukin; NR, not reported; PASI, Psoriasis Area Severity Index; PGA, Physician's Global Assessment; q8w, every 8 weeks; q12w, every 12 weeks.
Two phase 3 studies of tildrakizumab have been completed: reSURFACE 1 (NCT01722331) and reSURFACE 2 (NCT01729754). These studies demonstrated the efficacy of tildrakizumab at Week 12 compared with placebo (reSURFACE 1 and 2) or etanercept (reSURFACE 2 only) (PASI 75: reSURFACE 1: 200 mg 62%, 100 mg 64%, placebo 6%; reSURFACE 2: 200 mg 66%, 100 mg 61%, placebo 6%, etanercept 48%; P ≤ 0.001 in all comparisons).125 Of the patients who responded to tildrakizumab 200 mg at Week 28 (achieved PASI 75 response: reSURFACE 1: 200 mg 82%; reSURFACE 2: 200 mg 73%), 94% and 97% of patients maintained the response at Week 64 (reSURFACE 1) and Week 52 (reSURFACE 2), respectively.125
The results from the guselkumab phase 3 trials, VOYAGE 1 (NCT02207231) and VOYAGE 2 (NCT02207244), showed efficacy in treating moderate‐to‐severe psoriasis compared with placebo and adalimumab at Week 16 (PASI 90 [co‐primary endpoint]: VOYAGE 1: guselkumab 73%, adalimumab 50%, placebo 3%; VOYAGE 2: guselkumab 70%, adalimumab 47%, placebo, 2%), Week 24 (VOYAGE 1: guselkumab 80%, adalimumab 53%; VOYAGE 2: guselkumab 75%, adalimumab 55%), and over a 1‐year period (VOYAGE 1 only: Week 48, guselkumab 76%, adalimumab 48%; P < 0.001 in all comparisons).126, 127, 128
Phase 1 data on risankizumab in moderate‐to‐severe chronic plaque psoriasis (NCT01577550) reveal significant improvements in the proportion of patients achieving PASI 75 at Week 12 (PASI 75: risankizumab 87%, placebo 0%, P < 0.001) and Week 24 (risankizumab 71%, placebo 13%, P = 0.009) compared with baseline.129 In addition, a phase 2 study (NCT02054481) demonstrated superior clinical responses compared with ustekinumab at Week 12 (PASI 75: risankizumab 180 mg 88%, ustekinumab 45/90 mg 73%) and at Week 36 (risankizumab 180 mg 88%, ustekinumab 45/90 mg 55%).130
Overall, preliminary data suggest that anti–IL‐23p19 agents have a favourable safety profile (Table 3). As the p19 subunit is exclusive to IL‐23, whereas the p40 subunit is common to both IL‐12 and IL‐23, therapies that selectively target IL‐23p19 should avoid unnecessary effects associated with IL‐12/23p40 inhibition by sparing the function of IL‐12. This is supported by animal data in which IL‐12–deficient mice are susceptible to chemical carcinogenesis, whereas IL‐23–deficient mice are resistant.131 In patients with solid tumours, 1‐year survival was significantly greater in patients with elevated serum levels of IL‐12.132 However, when targeting IL‐12/23p40, it is not possible to attribute individual adverse events to specific inhibition of either IL‐12 or IL‐23. The relative contributions of each subunit to the safety profile will become apparent as more data from selective IL‐23 inhibitors become available. To date, IL‐23p19 inhibitors currently in development have not encountered signals for opportunistic infections, malignancy or worsening of IBD or CD.129, 133, 134
Table 3.
Most common adverse events and adverse events of special interest for biologics targeting IL‐23p19a
| Characteristic | Tildrakizumabb 100 mg125 | Tildrakizumabb 200 mg125 | Guselkumabc 100 mg126, 127 | Risankizumab 180 mg130 |
|---|---|---|---|---|
| Phase | Phase 3 | Phase 3 | Phase 3 | Phase 2 |
| Dosing schedule | ||||
| Initial | Weeks 0, 4 | Weeks 0, 4 | Weeks 0, 4 | Week 0 |
| Maintenance | q12w | q12w | q8w/q12w | q12w |
| Safety, % | Weeks 0–12 | Weeks 0–12 | Weeks 0–16 | Weeks 0–24 |
| AE | 44–47 | 42–49 | 48–52 | 57 |
| SAE | 1–2 | 2–3 | 2 | 2 |
| Severe infections | <1 | <1 | <1d | NR |
| Malignancies | <1 | <1 | 0 | NR |
| MACE | <1d | 0e | <1f | NR |
| Drug‐related hypersensitivity reactions | <1 | <1 | NR | NR |
| Long‐term safety, % | Weeks 12–28 | Weeks 12–28 | Weeks 16–48g | NR |
| AE | 44–46 | 40–45 | 65 | NR |
| SAE | 2–3 | 2 | 3 | NR |
| Severe infections | ≤1 | ≤1 | 1d | NR |
| Malignancies | <1 | ≤1 | 0 | NR |
| MACE | 0 | 0 | 0 | NR |
Data are not from head‐to‐head comparisons.
Data from reSURFACE1 and reSURFACE2 trials.
Data from VOYAGE 1 and VOYAGE 2 trials.
Serious infections.
Includes non‐fatal myocardial infarction, non‐fatal stroke and CV deaths that are confirmed as ‘cardiovascular’ or ‘sudden’.
Includes sudden cardiac death, myocardial infarction and stroke.
VOYAGE 1 trial only.
AE, adverse event; IL, interleukin; MACE, major adverse cardiac event; NA, not available; NR, not reported; Q8w, every 8 weeks; q12w, every 12 weeks; SAE, serious adverse event.
Dosing regimens
Comparison of treatment regimens across clinical trials suggests that biologics blocking effector cytokines may require more frequent dosing compared with biologics that block upstream cytokines. Approved agents that block IL‐17 require large loading doses with frequent administration within the first 4–12 weeks (secukinumab loading dose of 300 mg administered weekly for 4 weeks; ixekizumab loading dose of 160 mg administered once and 80 mg administered every 2 weeks for 12 weeks), with a reduction to one dose every 4 weeks for response maintenance.100, 101 These dosing intervals correspond to one to two serum half‐lives.100, 101 In comparison, biologics that target IL‐23 require less loading and allow a greater dosing interval for response maintenance (Table 2), corresponding to approximately four serum half‐lives.129, 135, 136 This is probably due to differing pharmacokinetic properties and antibody affinity for IL‐23p19129, 135, 136 but might also suggest that blocking regulatory upstream cytokines may have a longer lasting effect on the pathogenic immune response in psoriasis compared with blocking effector cytokines. We propose that neutralizing IL‐23 prevents the maintenance of the ongoing pathogenic TH17 response without affecting the induction of TH17 differentiation, which depends on IL‐6 and TGF‐β.65, 67, 68, 69, 70
IL‐23 inhibition in other inflammatory diseases
The aetiologies of psoriasis, psoriatic arthritis and CD share several candidate genes and common pathogenetic pathways, which is not surprising considering the clinical overlap of the disorders.26, 137, 138, 139, 140 Mutations in genes upregulated in these conditions, including IL23R, affect the IL‐23/TH17 immune axis,139, 141 suggesting anti–IL‐23 agents may have potential as treatments for CD, psoriatic arthritis and other genetically related disorders. For example, the TNF inhibitors adalimumab and infliximab are approved for the treatment of psoriasis and CD,96, 97 and ustekinumab was recently approved for the treatment of CD.110 In addition, several anti–IL‐23 agents are undergoing clinical development in psoriatic arthritis142, 143 and CD.144
Conclusions
There is robust evidence that the IL‐23/TH17 immune axis is a key driver of psoriatic inflammation, which has led to the development of biologics that specifically target key elements of this pathway. Although there are still gaps in our understanding (Table 4), IL‐23 is now acknowledged to have a critical role in this pathway and is required for the maintenance of inflammatory TH17 cells. Early indications from clinical trials support the use of IL‐23p19–specific inhibitors as a viable treatment option that may bring additional benefits to patients with plaque psoriasis.
Table 4.
Additional research needs
| To fully understand the mechanism of action of IL‐23 and IL‐17 blockade in psoriasis and other chronic inflammatory diseases, there is a need for research that explores: |
|---|
| • Cellular sources of IL‐23 and IL‐17 in psoriasis and conditions for the expression of the IL‐23R and the IL‐17R |
| • The effect of IL‐23 inhibition on TH17 cells in the skin and/or lymph nodes, and the downstream effects on cytokine profile in psoriatic lesions |
| • The role of IL‐23 inhibition in modulating the immune response and the effect on cytokines other than IL‐17 |
| • Contribution of innate immunity (e.g. recruitment of neutrophils to lesions) |
| • Genetic differences between responders and non‐responders to IL‐17 or IL‐23 inhibition |
| • Autoantigens and triggers in psoriasis, including environmental triggers |
IL, interleukin; TH17, T‐helper 17.
Conflicts of interest
GG: Principal investigator in clinical trials sponsored by and/or personal fees (AbbVie, Abiogen, Almirall, Amgen, Bayer, Biogen, Boehringer‐Ingelheim, Celgene, Eli‐Lilly, Galderma, Genzyme, Hospira, Janssen, Leo‐Pharma, MSD, Mundipharma, Novartis, Pfizer, Pierre Fabre, Regeneron, Sandoz, Sanofi, Serono, SUN Pharmaceutical Industries). RS: Speakers bureaus (Lohmann und Rauscher, Meda Pharmaceuticals, Menarini Pharmaceuticals, Pfizer, Schülke and Mayr, Smith & Nephew, Stockhausen); consulting fees (APR Pharmaceuticals, Celgene, Chemomedica, Flen Pharma, Lilly, Lohmann und Rauscher, Novartis, Pantec Biotechnologies, Pfizer, SastoMed, Schülke and Mayr, Stratamed, SUN Pharmaceutical Industries, Urgo, Woulgan); research and educational grants (APR Pharmaceuticals, Chemomedica, Enjo Commercial, Flen Pharma, Lohmann und Rauscher, Montavit, Schülke and Mayr, Smith & Nephew, Stockhausen, Stratamed, Urgo, 3M Wound Care). LP: Consultant, investigator, speaker or advisory board member (AbbVie, Almirall, Amgen, Baxalta, Biogen, Boehringer‐Ingelheim, Celgene, Gebro, Janssen, Leo‐Pharma, Lilly, Merck‐Serono, MSD, Novartis, Pfizer, Regeneron, Roche, Sandoz, SUN Pharmaceutical Industries). HB: Investigator, consultant, speaker or advisory boards (AbbVie, Actelion, Amgen, AnaptysBio, Baxalta, Bayer, Boehringer‐Ingelheim, Celgene, Janssen, Leo‐Pharma, Lilly, MSD, Novartis, Pfizer, SUN Pharmaceuticals, Takeda, UCB); grant support from Pfizer. JB: Speaker at sponsored symposia and advisory boards (AbbVie, Almirall, Boehringer‐Ingelheim, Celgene, Lilly, Janssen, UCB, Sun Pharmaceutical Industries, Novartis). WHB: Speaker and/or advisor (AbbVie, Almirall, Biogen, Celgene, Janssen, MSD, Novartis, Leo, Lilly, Pfizer, SUN Pharmaceutical Industries, UCB). JCP: Consultant, investigator, speaker or advisory boards (Abbott, AbbVie, Biogen‐Idec [formerly Biogen], Centocor, Essex Pharma, Galderma, Janssen‐Cilag/Janssen‐Ortho, Merck‐Serono [formerly Serono], SUN Pharmaceutical Industries, MSD, Novartis, Pfizer, Wyeth).
Funding sources
The authors first discussed this review at an advisory board convened by SUN Pharmaceutical Industries Inc. in July 2016. Following this meeting, SUN Pharmaceutical Industries Inc. agreed to provide financial support to the authors for an independent medical writer and copy editor in the development of this review. These services were provided by Hannah Greenwood, PhD, of Fishawack Communications Ltd., Abingdon, UK.
References
- 1. Bachelez H. Immunopathogenesis of psoriasis: recent insights on the role of adaptive and innate immunity. J Autoimmun 2005; 25(Suppl): 69–73. [DOI] [PubMed] [Google Scholar]
- 2. Boehncke WH, Schon MP. Psoriasis. Lancet 2015; 386: 983–994. [DOI] [PubMed] [Google Scholar]
- 3. Harden JL, Krueger JG, Bowcock AM. The immunogenetics of Psoriasis: a comprehensive review. J Autoimmun 2015; 64: 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization . Global Report in Psoriasis. 2016.
- 5. Seminara NM, Abuabara K, Shin DB, et al Validity of The Health Improvement Network (THIN) for the study of psoriasis. Br J Dermatol 2011; 164: 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hay RJ, Johns NE, Williams HC, et al The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol 2014; 134: 1527–1534. [DOI] [PubMed] [Google Scholar]
- 7. National Psoriasis Foundation . Plaque Psoriasis. 2016. Available at: https://www.psoriasis.org/about-psoriasis/types/plaque (last accessed December 13, 2016).
- 8. Menter A, Korman NJ, Elmets CA, et al Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case‐based presentations and evidence‐based conclusions. J Am Acad Dermatol 2011; 65: 137–174. [DOI] [PubMed] [Google Scholar]
- 9. Huerta C, Rivero E, Rodriguez LA. Incidence and risk factors for psoriasis in the general population. Arch Dermatol 2007; 143: 1559–1565. [DOI] [PubMed] [Google Scholar]
- 10. Di Meglio P, Villanova F, Nestle FO. Psoriasis. Cold Spring Harb Perspect Med 2014; 4: a015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nair RP, Duffin KC, Helms C, et al Genome‐wide scan reveals association of psoriasis with IL‐23 and NF‐kappaB pathways. Nat Genet 2009; 41: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Capon F, Bijlmakers MJ, Wolf N, et al Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum Mol Genet 2008; 17: 1938–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cargill M, Schrodi SJ, Chang M, et al A large‐scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis‐risk genes. Am J Hum Genet 2007; 80: 273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tiilikainen A, Lassus A, Karvonen J, Vartiainen P, Julin M. Psoriasis and HLA‐Cw6. Br J Dermatol 1980; 102: 179–184. [DOI] [PubMed] [Google Scholar]
- 15. Nair RP, Stuart PE, Nistor I, et al Sequence and haplotype analysis supports HLA‐C as the psoriasis susceptibility 1 gene. Am J Hum Genet 2006; 78: 827–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Online Mendelian Inheritance in Man . Susceptibility to Psoriasis 1, PSORS1. 2017. Available at: http://www.omim.org/entry/177900 (last accessed March 01, 2017).
- 17. Nikamo P, Lysell J, Stahle M. Association with Genetic Variants in the IL‐23 and NF‐kappaB Pathways Discriminates between Mild and Severe Psoriasis Skin Disease. J Invest Dermatol 2015; 135: 1969–1976. [DOI] [PubMed] [Google Scholar]
- 18. Zhou F, Cao H, Zuo X, et al Deep sequencing of the MHC region in the Chinese population contributes to studies of complex disease. Nat Genet 2016; 48: 740–746. [DOI] [PubMed] [Google Scholar]
- 19. Di Meglio P, Di Cesare A, Laggner U, et al The IL23R R381Q gene variant protects against immune‐mediated diseases by impairing IL‐23‐induced Th17 effector response in humans. PLoS ONE 2011; 6: e17160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair RP, Ruether A, Stuart PE, et al Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J Invest Dermatol 2008; 128: 1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reich K, Mossner R, Konig IR, Westphal G, Ziegler A, Neumann C. Promoter polymorphisms of the genes encoding tumor necrosis factor‐alpha and interleukin‐1beta are associated with different subtypes of psoriasis characterized by early and late disease onset. J Invest Dermatol 2002; 118: 155–163. [DOI] [PubMed] [Google Scholar]
- 22. Huffmeier U, Uebe S, Ekici AB, et al Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet 2010; 42: 996–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsoi LC, Spain SL, Ellinghaus E, et al Enhanced meta‐analysis and replication studies identify five new psoriasis susceptibility loci. Nat Commun 2015; 6: 7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, Helms C, Liao W, et al A genome‐wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet 2008; 4: e1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stuart PE, Nair RP, Ellinghaus E, et al Genome‐wide association analysis identifies three psoriasis susceptibility loci. Nat Genet 2010; 42: 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ellinghaus D, Ellinghaus E, Nair RP, et al Combined analysis of genome‐wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet 2012; 90: 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun LD, Cheng H, Wang ZX, et al Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat Genet 2010; 42: 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune‐mediated diseases. Nat Rev Genet 2013; 14: 661–673. [DOI] [PubMed] [Google Scholar]
- 29. Lowes MA, Suarez‐Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol 2014; 32: 227–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaclavkova A, Chimenti S, Arenberger P, et al Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet 2014; 384: 2036–2045. [DOI] [PubMed] [Google Scholar]
- 31. Jullien D, Prinz JC, Langley RG, et al T‐cell modulation for the treatment of chronic plaque psoriasis with efalizumab (Raptiva): mechanisms of action. Dermatology 2004; 208: 297–306. [DOI] [PubMed] [Google Scholar]
- 32. D'Ambrosio D, Freedman MS, Prinz J. Ponesimod, a selective S1P1 receptor modulator: a potential treatment for multiple sclerosis and other immune‐mediated diseases. Ther Adv Chronic Dis 2016; 7: 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Conrad C, Boyman O, Tonel G, et al Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med 2007; 13: 836–842. [DOI] [PubMed] [Google Scholar]
- 34. Di Meglio P, Villanova F, Navarini AA, et al Targeting CD8(+) T cells prevents psoriasis development. J Allergy Clin Immunol 2016; 138: 274–276 e276. [DOI] [PubMed] [Google Scholar]
- 35. Infante‐Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL‐17 in Th cells. J Immunol 2000; 165: 6107–6115. [DOI] [PubMed] [Google Scholar]
- 36. Liang SC, Tan XY, Luxenberg DP, et al Interleukin (IL)‐22 and IL‐17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 2006; 203: 2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL‐17A‐producing cells and are found in lesional skin. J Invest Dermatol 2011; 131: 1853–1860. [DOI] [PubMed] [Google Scholar]
- 38. Lee E, Trepicchio WL, Oestreicher JL, et al Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 2004; 199: 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lowes MA, Chamian F, Abello MV, et al Increase in TNF‐alpha and inducible nitric oxide synthase‐expressing dendritic cells in psoriasis and reduction with efalizumab (anti‐CD11a). Proc Natl Acad Sci U S A 2005; 102: 19057–19062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cella M, Jarrossay D, Facchetti F, et al Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 1999; 5: 919–923. [DOI] [PubMed] [Google Scholar]
- 41. Nestle FO, Conrad C, Tun‐Kyi A, et al Plasmacytoid predendritic cells initiate psoriasis through interferon‐alpha production. J Exp Med 2005; 202: 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fuentes‐Duculan J, Suarez‐Farinas M, Zaba LC, et al A subpopulation of CD163‐positive macrophages is classically activated in psoriasis. J Invest Dermatol 2010; 130: 2412–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brembilla NC, Stalder R, Senra L, Bohencke WH. IL‐17A localizes in the exocytic compartment of mast cells in psoriatic skin. Br J Dermatol 2017. DOI: 10.1111/bjd.15358. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 44. Piskin G, Sylva‐Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL‐23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol 2006; 176: 1908–1915. [DOI] [PubMed] [Google Scholar]
- 45. Arakawa A, Siewert K, Stohr J, et al Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 2015; 212: 2203–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krueger JG. An autoimmune “attack” on melanocytes triggers psoriasis and cellular hyperplasia. J Exp Med 2015; 212: 2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol 2008; 8: 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lande R, Botti E, Jandus C, et al The antimicrobial peptide LL37 is a T‐cell autoantigen in psoriasis. Nat Commun 2014; 5: 5621. [DOI] [PubMed] [Google Scholar]
- 49. Lande R, Gregorio J, Facchinetti V, et al Plasmacytoid dendritic cells sense self‐DNA coupled with antimicrobial peptide. Nature 2007; 449: 564–569. [DOI] [PubMed] [Google Scholar]
- 50. Ganguly D, Chamilos G, Lande R, et al Self‐RNA‐antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 2009; 206: 1983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meller S, Di Domizio J, Voo KS, et al T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol 2015; 16: 970–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cheung KL, Jarrett R, Subramaniam S, et al Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med 2016; 213: 2399–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schlaak JF, Buslau M, Jochum W, et al T cells involved in psoriasis vulgaris belong to the Th1 subset. J Invest Dermatol 1994; 102: 145–149. [DOI] [PubMed] [Google Scholar]
- 54. Harden JL, Johnson‐Huang LM, Chamian MF, et al Humanized anti‐IFN‐gamma (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol 2015; 135: 553–556. [DOI] [PubMed] [Google Scholar]
- 55. Kobayashi M, Fitz L, Ryan M, et al Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med 1989; 170: 827–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yawalkar N, Karlen S, Hunger R, Brand CU, Braathen LR. Expression of interleukin‐12 is increased in psoriatic skin. J Invest Dermatol 1998; 111: 1053–1057. [DOI] [PubMed] [Google Scholar]
- 57. Oppmann B, Lesley R, Blom B, et al Novel p19 protein engages IL‐12p40 to form a cytokine, IL‐23, with biological activities similar as well as distinct from IL‐12. Immunity 2000; 13: 715–725. [DOI] [PubMed] [Google Scholar]
- 58. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin‐23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin‐17. J Biol Chem 2003; 278: 1910–1914. [DOI] [PubMed] [Google Scholar]
- 59. Curtis MM, Way SS. Interleukin‐17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 2009; 126: 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cua DJ, Sherlock J, Chen Y, et al Interleukin‐23 rather than interleukin‐12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003; 421: 744–748. [DOI] [PubMed] [Google Scholar]
- 61. Langrish CL, Chen Y, Blumenschein WM, et al IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201: 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Teng MW, Bowman EP, McElwee JJ, et al IL‐12 and IL‐23 cytokines: from discovery to targeted therapies for immune‐mediated inflammatory diseases. Nat Med 2015; 21: 719–729. [DOI] [PubMed] [Google Scholar]
- 63. Chan JR, Blumenschein W, Murphy E, et al IL‐23 stimulates epidermal hyperplasia via TNF and IL‐20R2‐dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 2006; 203: 2577–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Di Cesare A, Di Meglio P, Nestle FO. The IL‐23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol 2009; 129: 1339–1350. [DOI] [PubMed] [Google Scholar]
- 65. Stritesky GL, Yeh N, Kaplan MH. IL‐23 promotes maintenance but not commitment to the Th17 lineage. J Immunol 2008; 181: 5948–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Volpe E, Servant N, Zollinger R, et al A critical function for transforming growth factor‐beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)‐17 responses. Nat Immunol 2008; 9: 650–657. [DOI] [PubMed] [Google Scholar]
- 67. Bettelli E, Carrier Y, Gao W, et al Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441: 235–238. [DOI] [PubMed] [Google Scholar]
- 68. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL‐17‐producing T cells. Immunity 2006; 24: 179–189. [DOI] [PubMed] [Google Scholar]
- 69. Ivanov II, McKenzie BS, Zhou L, et al The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL‐17 + T helper cells. Cell 2006; 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 70. Mangan PR, Harrington LE, O'Quinn DB, et al Transforming growth factor‐beta induces development of the T(H)17 lineage. Nature 2006; 441: 231–234. [DOI] [PubMed] [Google Scholar]
- 71. van der Fits L, Mourits S, Voerman JS, et al Imiquimod‐induced psoriasis‐like skin inflammation in mice is mediated via the IL‐23/IL‐17 axis. J Immunol 2009; 182: 5836–5845. [DOI] [PubMed] [Google Scholar]
- 72. Tonel G, Conrad C, Laggner U, et al Cutting edge: a critical functional role for IL‐23 in psoriasis. J Immunol 2010; 185: 5688–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wilson NJ, Boniface K, Chan JR, et al Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol 2007; 8: 950–957. [DOI] [PubMed] [Google Scholar]
- 74. Riol‐Blanco L, Ordovas‐Montanes J, Perro M, et al Nociceptive sensory neurons drive interleukin‐23‐mediated psoriasiform skin inflammation. Nature 2014; 510: 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kolls JK, Linden A. Interleukin‐17 family members and inflammation. Immunity 2004; 21: 467–476. [DOI] [PubMed] [Google Scholar]
- 76. Zaba LC, Cardinale I, Gilleaudeau P, et al Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med 2007; 204: 3183–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Senra L, Stalder R, Alvarez Martinez D, Chizzolini C, Boehncke WH, Brembilla NC. Keratinocyte‐Derived IL‐17E Contributes to Inflammation in Psoriasis. J Invest Dermatol 2016; 136: 1970–1980. [DOI] [PubMed] [Google Scholar]
- 78. Watanabe H, Kawaguchi M, Fujishima S, et al Functional characterization of IL‐17F as a selective neutrophil attractant in psoriasis. J Invest Dermatol 2009; 129: 650–656. [DOI] [PubMed] [Google Scholar]
- 79. Michel ML, Keller AC, Paget C, et al Identification of an IL‐17‐producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med 2007; 204: 995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Doisne JM, Becourt C, Amniai L, et al Skin and peripheral lymph node invariant NKT cells are mainly retinoic acid receptor‐related orphan receptor (gamma)t+ and respond preferentially under inflammatory conditions. J Immunol 2009; 183: 2142–2149. [DOI] [PubMed] [Google Scholar]
- 81. Lin AM, Rubin CJ, Khandpur R, et al Mast cells and neutrophils release IL‐17 through extracellular trap formation in psoriasis. J Immunol 2011; 187: 490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL‐23‐mediated psoriasis‐like epidermal hyperplasia is dependent on IL‐17A. J Immunol 2011; 186: 1495–1502. [DOI] [PubMed] [Google Scholar]
- 83. Ha HL, Wang H, Pisitkun P, et al IL‐17 drives psoriatic inflammation via distinct, target cell‐specific mechanisms. Proc Natl Acad Sci U S A 2014; 111: E3422–E3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin‐17 and interferon‐gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol 1998; 111: 645–649. [DOI] [PubMed] [Google Scholar]
- 85. Chiricozzi A, Guttman‐Yassky E, Suarez‐Farinas M, et al Integrative responses to IL‐17 and TNF‐alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 2011; 131: 677–687. [DOI] [PubMed] [Google Scholar]
- 86. Patel DD, Kuchroo VK. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 2015; 43: 1040–1051. [DOI] [PubMed] [Google Scholar]
- 87. Adami S, Cavani A, Rossi F, Girolomoni G. The role of interleukin‐17A in psoriatic disease. BioDrugs 2014; 28: 487–497. [DOI] [PubMed] [Google Scholar]
- 88. Papp KA, Leonardi C, Menter A, et al Brodalumab, an anti‐interleukin‐17‐receptor antibody for psoriasis. N Engl J Med 2012; 366: 1181–1189. [DOI] [PubMed] [Google Scholar]
- 89. Langley RG, Elewski BE, Lebwohl M, et al Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med 2014; 371: 326–338. [DOI] [PubMed] [Google Scholar]
- 90. Gordon KB, Blauvelt A, Papp KA, et al Phase 3 Trials of Ixekizumab in Moderate‐to‐Severe Plaque Psoriasis. N Engl J Med 2016; 375: 345–356. [DOI] [PubMed] [Google Scholar]
- 91. Oh CJ, Das KM, Gottlieb AB. Treatment with anti‐tumor necrosis factor alpha (TNF‐alpha) monoclonal antibody dramatically decreases the clinical activity of psoriasis lesions. J Am Acad Dermatol 2000; 42: 829–830. [DOI] [PubMed] [Google Scholar]
- 92. Tan MH, Gordon M, Lebwohl O, George J, Lebwohl MG. Improvement of Pyoderma gangrenosum and psoriasis associated with Crohn disease with anti‐tumor necrosis factor alpha monoclonal antibody. Arch Dermatol 2001; 137: 930–933. [PubMed] [Google Scholar]
- 93. Leonardi CL, Powers JL, Matheson RT, et al Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349: 2014–2022. [DOI] [PubMed] [Google Scholar]
- 94. Reich K, Nestle FO, Papp K, et al Infliximab induction and maintenance therapy for moderate‐to‐severe psoriasis: a phase III, multicentre, double‐blind trial. Lancet 2005; 366: 1367–1374. [DOI] [PubMed] [Google Scholar]
- 95. Amgen . ENBREL® (etanercept) PI. March 2015.
- 96. Abbvie . HUMIRA® (adalimumab) PI. October 2016.
- 97. Janssen Biotech . REMICADE® (infliximab) PI. January 2015.
- 98. Menter A, Gottlieb A, Feldman SR, et al Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008; 58: 826–850. [DOI] [PubMed] [Google Scholar]
- 99. Pathirana D, Ormerod AD, Saiag P, et al European S3‐guidelines on the systemic treatment of psoriasis vulgaris. J Eur Acad Dermatol Venereol 2009; 23(Suppl 2): 1–70. [DOI] [PubMed] [Google Scholar]
- 100. Novartis . COSENTYX® (secukinumab) PI. January 2016.
- 101. Lilly . TALTZ® (ixekizumab) PI. March 2016.
- 102. van de Kerkhof PC, Griffiths CE, Reich K, et al Secukinumab long‐term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol 2016; 75: 83–98 e84. [DOI] [PubMed] [Google Scholar]
- 103. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL‐23‐IL‐17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 2014; 14: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. O'Connor W Jr, Kamanaka M, Booth CJ, et al A protective function for interleukin 17A in T cell‐mediated intestinal inflammation. Nat Immunol 2009; 10: 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hueber W, Sands BE, Lewitzky S, et al Secukinumab, a human anti‐IL‐17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double‐blind placebo‐controlled trial. Gut 2012; 61: 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Valeant Receives FDA Approval Of SILIQ™ (Brodalumab) For Moderate‐To‐Severe Plaque Psoriasis [press release]. 2017.
- 107. Lebwohl M, Strober B, Menter A, et al Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N Engl J Med 2015; 373: 1318–1328. [DOI] [PubMed] [Google Scholar]
- 108. Papp KA, Reich K, Paul C, et al A prospective phase III, randomized, double‐blind, placebo‐controlled study of brodalumab in patients with moderate‐to‐severe plaque psoriasis. Br J Dermatol 2016; 175: 273–286. [DOI] [PubMed] [Google Scholar]
- 109. Glatt S, Helmer E, Haier B, et al First‐in‐human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL‐17A and IL‐17F, in mild psoriasis. Br J Clin Pharmacol 2017; 83: 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Janssen Biotech . STELARA® (ustekinumab) PI. September 2016.
- 111. Papp KA, Langley RG, Lebwohl M, et al Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 52‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 2). Lancet 2008; 371: 1675–1684. [DOI] [PubMed] [Google Scholar]
- 112. Leonardi CL, Kimball AB, Papp KA, et al Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 76‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 1). Lancet 2008; 371: 1665–1674. [DOI] [PubMed] [Google Scholar]
- 113. Kalb RE, Fiorentino DF, Lebwohl MG, et al Risk of serious infection with biologic and systemic treatment of psoriasis: results from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). JAMA Dermatol 2015; 151: 961–969. [DOI] [PubMed] [Google Scholar]
- 114. Papp KA, Griffiths CE, Gordon K, et al Long‐term safety of ustekinumab in patients with moderate‐to‐severe psoriasis: final results from 5 years of follow‐up. Br J Dermatol 2013; 168: 844–854. [DOI] [PubMed] [Google Scholar]
- 115. Gordon KB, Langley RG, Gottlieb AB, et al A phase III, randomized, controlled trial of the fully human IL‐12/23 mAb briakinumab in moderate‐to‐severe psoriasis. J Invest Dermatol 2012; 132: 304–314. [DOI] [PubMed] [Google Scholar]
- 116. Abbott Laboratories . United States Securities and Exchange Commission Form 8‐K. January 14, 2011.
- 117. Schaarschmidt ML, Kromer C, Herr R, et al Patient preferences for biologicals in psoriasis: top priority of safety for cardiovascular patients. PLoS ONE 2015; 10: e0144335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Puig L. Cardiovascular risk and psoriasis: the role of biologic therapy. Actas Dermosifiliogr 2012; 103: 853–862. [DOI] [PubMed] [Google Scholar]
- 119. Ryan C, Leonardi CL, Krueger JG, et al Association between biologic therapies for chronic plaque psoriasis and cardiovascular events: a meta‐analysis of randomized controlled trials. JAMA 2011; 306: 864–871. [DOI] [PubMed] [Google Scholar]
- 120. Tzellos T, Kyrgidis A, Trigoni A, Zouboulis CC. Association of ustekinumab and briakinumab with major adverse cardiovascular events: an appraisal of meta‐analyses and industry sponsored pooled analyses to date. Dermatoendocrinol 2012; 4: 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Youssef SS, Mohammad MM, Ezz‐El‐Arab LR. Clinical significance of serum IL‐12 level in patients with early breast carcinoma and its correlation with other tumor markers. Open Access Maced J Med Sci 2015; 3: 640–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Winchester DA, Till C, Goodman PJ, et al Variation in genes involved in the immune response and prostate cancer risk in the placebo arm of the Prostate Cancer Prevention Trial. Prostate 2015; 75: 1403–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kulig P, Musiol S, Freiberger SN, et al IL‐12 protects from psoriasiform skin inflammation. Nat Commun 2016; 7: 13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Clinicaltrails.gov . A Study of LY3074828 in Participants With Moderate to Severe Plaque Psoriasis. Available at: https://clinicaltrials.gov/ct2/show/NCT02899988 (last accessed January 2017).
- 125. Reich K, Papp K, Blauvelt A, et al Tildrakizumab, a selective IL‐23p19 antibody, in the treatment of chronic plaque psoriasis: results from two randomised, controlled, Phase 3 trials (reSURFACE 1 and reSURFACE 2). Lancet 2017. DOI: 10.1016/S0140‐6736(17)31279‐5. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 126. Blauvelt A, Papp KA, Griffiths CE, et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double‐blinded, placebo‐ and active comparator‐controlled VOYAGE 1 trial. J Am Acad Dermatol 2017; 76: 405–417. [DOI] [PubMed] [Google Scholar]
- 127. Reich K, Armstrong AW, Foley P, et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double‐blind, placebo‐ and active comparator‐controlled VOYAGE 2 trial. J Am Acad Dermatol 2017; 76: 418–431. [DOI] [PubMed] [Google Scholar]
- 128. Gordon KB, Duffin KC, Bissonnette R, et al A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med 2015; 373: 136–144. [DOI] [PubMed] [Google Scholar]
- 129. Krueger JG, Ferris LK, Menter A, et al Anti‐IL‐23A mAb BI 655066 for treatment of moderate‐to‐severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single‐rising‐dose, randomized, double‐blind, placebo‐controlled trial. J Allergy Clin Immunol 2015; 136: 116–124.e117. [DOI] [PubMed] [Google Scholar]
- 130. Papp K, Menter A, Sofen H et al Onset and duration of clinical response following treatment with a selective IL‐23p19 inhibitor (BI 655066) compared with ustekinumab in patients with moderate‐to‐severe plaque psoriasis. Presentation presented at: European Academy of Dermatology and Venerology 2015; Copenhagen, Denmark.
- 131. Langowski JL, Zhang X, Wu L, et al IL‐23 promotes tumour incidence and growth. Nature 2006; 442: 461–465. [DOI] [PubMed] [Google Scholar]
- 132. Lissoni P, Mengo S, Mandala M, et al Physiopathology of IL‐12 in human solid neoplasms: blood levels of IL‐12 in early or advanced cancer patients, and their variations with surgery and immunotherapy. J Biol Regul Homeost Agents 1998; 12: 38–41. [PubMed] [Google Scholar]
- 133. Papp K, Thaci D, Reich K, et al Tildrakizumab (MK‐3222), an anti‐interleukin‐23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo‐controlled trial. Br J Dermatol 2015; 173: 930–939. [DOI] [PubMed] [Google Scholar]
- 134. Sofen H, Smith S, Matheson RT, et al Guselkumab (an IL‐23‐specific mAb) demonstrates clinical and molecular response in patients with moderate‐to‐severe psoriasis. J Allergy Clin Immunol 2014; 133: 1032–1040. [DOI] [PubMed] [Google Scholar]
- 135. Kopp T, Riedl E, Bangert C, et al Clinical improvement in psoriasis with specific targeting of interleukin‐23. Nature 2015; 521: 222–226. [DOI] [PubMed] [Google Scholar]
- 136. Zhuang Y, Calderon C, Marciniak SJ Jr, et al First‐in‐human study to assess guselkumab (anti‐IL‐23 mAb) pharmacokinetics/safety in healthy subjects and patients with moderate‐to‐severe psoriasis. Eur J Clin Pharmacol 2016; 72: 1303–1310. [DOI] [PubMed] [Google Scholar]
- 137. Capon F, Burden AD, Trembath RC, Barker JN. Psoriasis and other complex trait dermatoses: from Loci to functional pathways. J Invest Dermatol 2012; 132: 915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Stuart PE, Nair RP, Tsoi LC, et al Genome‐wide association analysis of psoriatic arthritis and cutaneous psoriasis reveals differences in their genetic architecture. Am J Hum Genet 2015; 97: 816–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut 2011; 60: 1739–1753. [DOI] [PubMed] [Google Scholar]
- 140. Boehncke WH. Psoriasis and psoriatic arthritis: flip sides of the coin? Acta Derm Venereol 2016; 96: 436–441. [DOI] [PubMed] [Google Scholar]
- 141. Filer C, Ho P, Smith RL, et al Investigation of association of the IL12B and IL23R genes with psoriatic arthritis. Arthritis Rheum 2008; 58: 3705–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Clinicaltrails.gov . Efficacy and Safety Study of Guselkumab in the Treatment of Participants With Active Psoriatic Arthritis. 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT02319759?term=NCT02319759&rank=1 (last accessed March 01 2017).
- 143. Clinicaltrials.gov . BI 655066/ABBV066/Risankizumab Compared to Placebo in Patients With Active Psoriatic Arthritis. 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT02719171?term=NCT02719171&rank=1 (last accessed March 01, 2017).
- 144. Clinicaltrails.gov . Efficacy, Safety and Pharmacokinetics of BI 655066/ABBV‐066 (Risankizumab) in Patients With Active, Moderate‐to‐severe Crohn's Disease. 2016. Available at: https://clinicaltrials.gov/ct2/show/NCT02031276?term=NCT02031276&rank=1 (last accessed March 01 2017).
- 145. Gaspari AA, Tyring S. New and emerging biologic therapies for moderate‐to‐severe plaque psoriasis: mechanistic rationales and recent clinical data for IL‐17 and IL‐23 inhibitors. Dermatol Ther 2015; 28: 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71: 141–150. [DOI] [PubMed] [Google Scholar]
- 147. Mease PJ. Inhibition of interleukin‐17, interleukin‐23 and the TH17 cell pathway in the treatment of psoriatic arthritis and psoriasis. Curr Opin Rheumatol 2015; 27: 127–133. [DOI] [PubMed] [Google Scholar]
- 148. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood 2008; 112: 1557–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361: 496–509. [DOI] [PubMed] [Google Scholar]
- 150. Harrington LE, Hatton RD, Mangan PR, et al Interleukin 17‐producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6: 1123–1132. [DOI] [PubMed] [Google Scholar]
- 151. Yoon J, Leyva‐Castillo JM, Wang G, et al IL‐23 induced in keratinocytes by endogenous TLR4 ligands polarizes dendritic cells to drive IL‐22 responses to skin immunization. J Exp Med 2016; 213: 2147‐2166. [DOI] [PMC free article] [PubMed] [Google Scholar]