

Abstract
A polyether antibiotic, ecteinamycin (1), was isolated from a marine Actinomadura sp., cultivated from the ascidian Ecteinascidia turbinata. 13C enrichment, high resolution NMR spectroscopy, and molecular modeling enabled elucidation of the structure of 1, which was validated on the basis of comparisons with its recently reported crystal structure. Importantly, ecteinamycin demonstrated potent activity against the toxigenic strain of Clostridium difficile NAP1/B1/027 (MIC = 59 ng/μL), as well as other toxigenic and nontoxigenic C. difficile isolates both in vitro and in vivo. Additionally, chemical genomics studies using Escherichia coli barcoded deletion mutants led to the identification of sensitive mutants such as trkA and kdpD involved in potassium cation transport and homeostasis supporting a mechanistic proposal that ecteinamycin acts as an ionophore antibiotic. This is the first antibacterial agent whose mechanism of action has been studied using E. coli chemical genomics. On the basis of these data, we propose ecteinamycin as an ionophore antibiotic that causes C. difficile detoxification and cell death via potassium transport dysregulation.
Graphical Abstract

Clostridium difficile infections (CDI) represent a tremendous health threat in both the United States and other industrialized nations, necessitating the clear need for new therapeutic approaches and drug entities. Although precise numbers are not yet available for 2016, recent statistics tell a very grim story, especially in the U.S. In 2015, the Centers for Disease Control and prevention reported that about half a million Americans were afflicted with this deadly bacterial infection and that of these, ~30 000 died within 30 days of initial diagnosis.1 Indeed, the CDC recently listed C. difficile as one of the three most highly ranked dangerous infectious pathogens receiving the designation of “urgent.”1 C. difficile is now widely recognized also as the most common microbial cause of healthcare-associated infections in the U.S., and costs to the national healthcare system exceed $4.8 billion annually. Notably, the U.S. is not alone in facing this health threat. New and hypervirulent strains known as NAP1/B1/027 have emerged and, partly by virtue of their resistance to previously employed therapeutics, have driven epidemics in the U.S., Canada, Britain, and several European nations.2–4
C. difficile is a spore-forming Gram-positive anaerobic bacterium associated with pseudomembraneous colitis and antibiotic-associated diarrhea in patients previously administered antimicrobial drugs.2–4 CDI arises from disruption of normal intestinal microflora, thus creating an opportunity for ingested spores to germinate, colonize the GI tract, and produce toxins able to inflict profound damage. The production of Rho-inactivating toxins A (TcdA) and B (TcdB) in the GI tracts of those afflicted lies at the heart of the bacterium’s toxicity in human hosts; both are proinflammatory and cytotoxic, causing disruption of the actin cytoskeleton and impairment of tight junctions in human intestinal epithelial cells.2–7 The actions of TcdA and TcdB lead to fluid accumulation and extensive damage to the large intestine. More recent studies have shown that, in addition to serving as Rho-glucosylases, TcdA and TcdB trigger release of inflammatory cytokines from mast cells and macrophages as well as from epithelial cells, leading to further fluid secretion and intestinal inflammation.8 Notably, particularly virulent strains, such as NAP1/B1/027, have been noted to generate a third toxin referred to as C. difficile binary toxin (CDT), an actin-specific ADP-ribosyltransferase that disrupts the actin cytoskeleton.9–13 CDT expression and activity is associated with increased C. difficile cell adherence and subsequent pathogenicity.9–13 Taken together, it has become clear that TcdA, TcdB, and CDT are major determinants driving both the lethality and virulence of C. difficile in humans.
Although the advent of fecal transplants14,15 and recent approval of the antidifficile drug fidaxomicin16,17 have helped to stem the tide of CDIs, the emergence of resistance to a wide range of antibiotics, such as clindamycin,18 fluoroquinolones,19 vancomycin, and metronidazole,20–25 and ever increasingly virulent strains such as NAP1/B1/027 underscore the urgent need for new therapeutics. Central to new approaches for combatting C. difficile has been the recognition that toxins A and B enter GI cells via receptor-mediated endocytosis following target cell surface associations with combined repetitive oligopeptides (CROPs) embedded within the toxin C-termini.26–29 Endosomal acidification triggers conformational changes in TcdA and TcdB; this triggers vesicle membrane insertion and translocation of the catalytically active N-terminus into the cytosol. This activation/delivery mechanism is not unlike those of other bacterial toxins, has been hypothesized to represent a unique opportunity for drug targeting, and appears to highlight membrane function as an exploitable point of intervention.30 Importantly, examples of ionophore-driven disruption of assorted ion pumping mechanisms are well established and, in fact, form the basis of a number of commercially crucial anticoccidial feed additives. Monensin (Rumensin), lasalocid (Bovatec), and laidlomycin propionate (Cattlyst) are all polyether antibiotics that disrupt ion concentration gradients (Ca2+, K+, H+, Na+) within microorganisms leading to futile ion cycles.31 Additionally, several reports have shown that monensin can reduce the cytopathogenic effect of C. difficile toxins, and TcdB induced apoptosis relies on ATP-sensitive potassium channels.32–34 Taken together, these data indicate that ionophore antibiotics could play a dual role as CDI antibiotics, by helping clear bacteria and reducing the effect of toxins on epithelial cells in the gut.
In our search for novel antibiotics, we isolated a novel polyether antibiotic, ecteinamycin (1; see Figure 1), from a marine Actinomadura sp. cultivated from the ascidian Ecteinascidia turbinata (Herdman, 1880).35,36 The generation of 13C-enriched samples and acquisition of 13C–13C COSY data along with other spectroscopic methods expedited the determination of the new carbon skeleton. The relative configuration was assigned, in part, using residual dipolar coupling (RDC)-based methods and was validated on the basis of the recently described crystal structure for 1.37 The ability of 1 to serve, as suggested by its structure, as a biologically active ionophore was also validated (Supporting Information).
Figure 1.
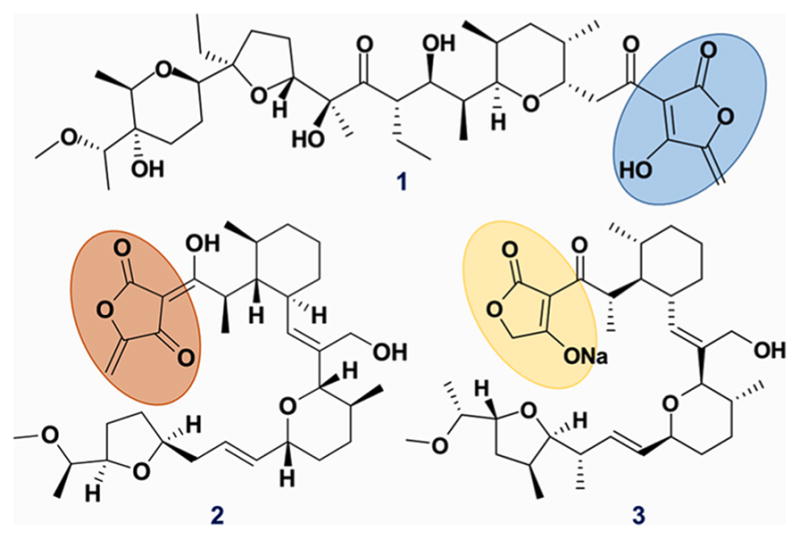
Polyether ionophores sharing the characteristic acyltetramic acid moiety. Ecteinamycin (1), tetronomycin (2), and tetronasin (3).
Bioassays with 1 revealed its potent activity against Gram-positive bacteria. Especially notable, 1 displayed excellent selectivity for C. difficile (MIC 59 ng/mL). The results of chemical genomics studies, combined with its structure and activity in potassium release and membrane depolarization assays, highlight 1 as a new polyether ionophore antibiotic.35 This is, to our knowledge, the first instance of an E. coli chemical genomics study being used to delineate a drug or drug candidate’s mechanism of action (MOA). Notably, alterations in the transport of H+, K+, Na+, Ca+, and/or Mg2+ ions by 1 may lie at the heart of this agent’s toxicity toward C. difficile; indeed, the results of chemical genomics studies support this early hypothesis and identify selectivity for H+/K+. With respect to disrupting endosomal and vesicular trafficking, which we hypothesized was the mechanism by which monensin reduced effects of both C. difficile toxins, we examined the effect of 1 on yeast via chemical genomics. The data clearly indicated that 1 disrupted vesicular trafficking via pH/ion effects providing mechanistic support for how ionophore antibiotics such as monensin abrogate the effects of C. difficile toxins.
Mechanistic studies of pathogenic and virulent processes often unveil new therapeutic opportunities and are therefore crucial to drug discovery. This appears to be the case with TcdA and TcdB as overlaid with our discovery of 1. Several structural features of 1, such as the acyltetramic acid moiety, are found also in other polyether ionophores, such as tetronomycin (2)38 and tetronasin (3).39 However, as a whole, ecteinamycin contains a new carbon skeleton, and its potent selectivity against C. difficile renders 1 a potential antibiotic. Moreover, an in vivo mouse model indicated that 1 has low oral availability. This, combined with its excellent MIC against C. difficile, makes 1 very exciting since antibiotics intended to work in the lower GI would ideally have low oral bioavailability yet potent activity when presented with relevant target organisms.
RESULTS AND DISCUSSION
LCMS-PCA Assisted in Microbial Strain Selection
LCMS profiling of 34 marine-derived bacterial extracts and subsequent multivariate analysis by principal component analysis (PCA)40 led us to pursue the strain WMMB499, which demonstrated unique chemistry compared to other strains by PCA (Supporting Information, Figure S1). WMMB499 was found to produce three classes of novel natural products: (i) halogenated electrophilic polyketides halomadurones A–D,41 (ii) antifungal polyketide forazoline A,42 which have previously been reported, and (iii) the new polyether antibiotic 1.
Structure Elucidation of Ecteinamycin
HRMS data revealed the molecular formula of 1 to be C38H60O12 (Supporting Information, Figure S2), and extensive 1D and 2D NMR data (Supporting Information, Table S1) enabled us to determine the majority of the 2D structure for 1. To improve the overall efficiency of structure elucidation, we used our previously published approach of 13C isotopic enrichment followed by rapid acquisition of 13C–13C COSY data.43,44 In the absence of crystallographically generated structure information at the time, we employed a combination of ROESY correlations (Supporting Information, Figure S11), coupling constants, 13C chemical shifts, molecular modeling, and other spectroscopic methods to partially determine the relative configuration of 1 (see Supporting Information). In addition to these efforts, we also employed residual dipolar couplings (RDC) studies to investigate the stereochemistry of 1. A new method of employing RDCs was developed in parallel and recently published.45 Although the RDC approach was successful, we evaluated extensive DFT calculations and included aspects of those in the Supporting Information. Importantly, our stereochemical assignments have recently been validated by recently reported crystallographic and biological studies of 1 and its chlorinated congener non-thmicin.37
Ecteinamycin Inhibits Growth of Toxigenic C. difficile Strains at Nanogram Levels
Screening of 1 against a panel of Gram-positive and Gram-negative bacteria revealed potent antimicrobial activities for ecteinamycin (Table 1). Ecteinamycin was particularly potent against Gram-positive bacteria, consistent with the established activity of ionophore antibiotics and especially those used as feed additives.33 Given the growing importance of CDIs, we subjected a number of C. difficile strains to 1, including the highly virulent NAP1/B1/027, and MICs were determined. Notably, 1 displayed excellent potency (MIC 59 ng/mL) against NAP1/B1/027, although significant antimicrobial actions were clearly exerted against other C. difficile strains as well. Also noteworthy is that 1, in our hands, was ~20-fold more active against C. difficile than against other bacteria (Table 1). For example, 1 displayed MICs of 16, 0.125, and 8 μg/mL against E. coli, S. aureus (both methicillin sensitive and resistant), and P. aeruginosa, respectively. That 1 is so much more active against C. difficile than other microbes suggests that 1, and possibly other ionophores, may represent extremely important new therapeutics for CDIs. This notion is further strengthened by the fact that 1 was much more effective against C. difficile than the currently employed first-line clinical antibiotics metronidazole (MIC ≤ 2.0 μg/mL)46–48 and vancomycin (MIC = 1.0–2.0 μg/mL).46–48 Notably, nigericin, a well-established ionophore antibiotic, has also been reported to be potent against C. difficile (MIC of 2.5 ng/mL), and a number of other tetramic-acid-containing compounds have shown activity against C. difficile in vitro.49,50 Monensin, despite its established antibiotic activities and ionophore activity, is nowhere near as active (MIC = 0.5 μg/mL) against C. difficile as 1.48
Table 1.
Minimum Inhibitory Concentration (MIC) of 135
| microorganism | MIC (μg/mL) |
|---|---|
| E. coli | 16.0 |
| S. aureus (methicillin-sensitive) | 0.125 |
| S. aureus (methicillin-resistant) | 0.125 |
| Vancomycin-resistant Enterococcus | 0.25 |
| P. aeruginosa | 8.0 |
| C. difficile ATCC 43255a | 0.059 |
| C. difficile BAA-1870a (strain NAP1/B1/027) | 0.059 |
| C. difficile BAA-1382a (strain 630) | 0.059 |
| C. difficile ATCC 700057b | 0.117 |
| C. difficile ATCC 9689a,c | 0.059 |
| C. difficile B6/6 005a,c | 0.117 |
| C. difficile B6/6 006a,c | 0.117 |
| C. difficile ICCD 001a,c | 0.059 |
| C. difficile ICCD 006a,c | 0.059 |
Toxigenic.
Nontoxigenic.
Clinical isolate.
Ecteinamycin Depolarizes Cell Membranes and Interferes with Potassium Cation Homeostasis
The potent and selective activity of 1 against C. difficile inspired us to probe this new natural product’s mechanism of action. A membrane depolarization assay50,51 and E. coli chemical genomic profiling via barcode sequencing were employed to shed light on this issue.52 First, flow cytometry was used to evaluate the impact of 1 on bacterial membrane potentials. Methicillin-sensitive S. aureus (MSSA) cells were subjected to either 6 or 32 μg/mL of 1 (Figure 2). Additionally, all cells, except the DMSO negative control, were treated with DiOC2(3) dye and analyzed by flow cytometry.
Figure 2.

Membrane depolarization assay. Flow cytometry-based analysis of MSSA cells treated with DiOC2(3) dye control (A), ecteinamycin (B), and CCCP (C). (D) The ratio of red to green fluorescence for MSSA cells treated with DiOC2(3) dye and the corresponding compound. Carbonyl cyanide m-chlorophenylhydrazone (CCCP) was used as a positive control. CCCP is a protonophore that disrupts membrane potential.50,51
Compound 1 very rapidly induced membrane depolarization at 6 μg/mL (Figure 2) although the magnitude of depolarization was greater with CCCP, an established depolarizing agent. In parallel, studies with the established anticoccidicidal agent salinomycin revealed that ecteinamicin’s MSSA depolarization activity far surpassed that of salinomycin sodium salt (Supporting Information).53–55 Subsequent potassium-release assays also employing MSSA confirmed salinamycin’s excellent ionophoric activity53,54 and established, for the first time, ecteinamycin’s ionophore activity (Supporting Information). These data highlight a clear and consistent system in which membrane depolarization by 1 aligns with structure-informed expectation and demonstrable potassium release/ionophoric capacity. In total, these data strongly affirm the hypothesis that ionophore function provides at least one basis for the antibacterial activity of 1, albeit at concentrations well above the MIC for 1 against C. difficile. It must also be noted however that the results of these assays do not exclude the potential relevance of other C. difficile specific targets/pathways exploited by 1, particularly at low concentrations.
Alongside depolarization assays, chemical genomic profiling with barcoded E. coli deletion mutants was used to understand the effects of 1 on bacterial cells. Chemical genomics has been used to determine MOAs and molecular targets for many bioactive compounds, including natural products using barcoded S. cerevisiae.56–58 However, E. coli barcoded libraries have not been used. Given the structural resemblance to other ionophore antibiotics as well as the depolaraization data, ecteinamycin was a good candidate for a proof of concept study using E. coli chemical genomics. A library of 6000 E. coli deletion mutants was combined and treated with ecteinamycin. The genomic DNAs were extracted, and mutant-specific DNA barcodes were amplified and sequenced by Illumina sequencing. Ecteinamycin-sensitive and resistant mutants were determined by quantification of DNA barcodes. The resulting chemical genomic profile (Figure 3) provided significant insight into ecteinamycin’s MOA, albeit at concentrations of 1 far higher than necessary to elicit C. difficile activity.52 In general, however, chemical genomic studies require significantly higher concentrations due to the large number of cells required to represent all knockout strains.
Figure 3.

Chemical genomic profile of ecteinamycin. The chemical genomic analysis was performed in triplicate at [1] = 62.5 μg/mL. In green, E. coli mutants deficient in sspA, rseB, cusC, and other genes proved resistant to the actions of 1. In red, E. coli sensitive to 1 are deficient in trkA, sapD, kdpD, and other indicated genes (Table 2).
E. coli deficient in potassium processing, and presumably, membrane function, proved particularly sensitive to the effects of 1. Deletion of trkA, kdpD, and sapD genes drove enrichment of the sensitive mutants following exposure to 1. The loss of TrkA, the NAD binding domain of the TrK potassium transporter; sapD, which is required for ATP-dependence of TrK K+ uptake; and KdpD, a sensor kinase for the Kdp (K+ dependence) operon, characterized particularly susceptible strains, thus indicating the K+ ion as a likely target for 1.59–62 Transport systems and channels, such as the TRK and KDP pathways, are central to regulating potassium accumulation and movement across membranes. Microbial cell membrane integrity is key for homeostasis maintenance, survival, electron transport, and ATP biosynthesis. Taken together, these data firmly support the preceding membrane depolarization assays supporting the idea that, like other ionophores, 1 interferes with bacterial potassium homeostasis and membrane integrity and that this leads to cell killing.59–62 This result is consistent with previous work by Hurdle et al. wherein the membrane-depolarizing reutericyclins were found to kill C. difficile in stationary or nongrowth phases.63 Subsequently, Wu et al. suggested that ion/proton gradients across the C. difficile membrane play key roles in cell viability and concluded that C. difficile is susceptible to antibiotics targeting cell membrane function.48 While we could not rule out a specific target in C. difficile that differs from E. coli, our data were consistent with ecteinamycin acting as an ionophore antibiotic.
Strains resistant to 1 were characterized by alterations in stress response genes (GO: 0033554) and lipid binding (GO: 0008289; Figure 3). Specifically, sspA and rseB deletion mutants displayed resistance to 1. The stringent starvation protein A (SspA), a polymerase-associated protein, is involved in pathogenicity and cellular stress-response under conditions of nutrient deficiency and acidic pH,64–67 whereas RseB antagonizes the activity of the sigma E stress-response (RpoE) transcription factor and binds periplasmic lipopoly-saccharides.68–71 Upon exposure to 1, deletion of the cusC gene was also enriched in E. coli relative to the ~6000 strains. This gene encodes for lipoprotein component factor CusC in the outer membrane, which is part of the Cu+/Ag+ detoxification efflux pump system.72–74 In contrast to chemical genomics data for cells sensitive to 1, the corresponding data related to 1 resistance do not underscore any one particular mechanism or set of mechanisms driving cellular response. Instead, these data suggest that cellular enrichments may have more to do with delayed autolysis mechanisms than with bona fide, long-term resistance to 1.
In parallel, chemical genomic profiling with yeast, Saccharomyces cerevisiae, was used to understand the effects of 1 on eukaryotic cells and to shed further insight into how ionophore antibiotics like monensin attenuate the effects of C. difficile toxins.35,36 Ecteinamycin was screened against over 4000 deletion mutant yeast strains. Genomic DNA was extracted, and mutant-specific DNA barcodes were amplified and sequenced by Illumina as with the E. coli studies.
This resulted in a distinct chemical genomic profile of strains responsive to 1 at 250 μg/mL (Supporting Information Table S3). Relative to our existing data set of known compounds (Piotrowski et al. in review), we found the top two activity correlations of 1 to be with the polyether antibiotics duamycin and nigericin (P < 0.0001).56–58 Both duamycin and nigericin act by generating ion channels in cellular membranes and inhibiting Golgi function in eukaryotic cells. The mutant S. cerevisiae strains most sensitive to 1 (P < 0.0001) had significant gene ontology (GO) enrichment for the biological process “post-Golgi-mediated transport” (P = 1.19 × 10−6), driven by sensitive mutants of genes KES1, APL4, MON2, DRS2, APM2, and VPS9. Further implicating impaired Golgi functioning as an ecteinamycin sensitivity determinant, we identified a hypomorph of SEC14 as the only significantly sensitive (P = 0.03) essential gene mutant in a screen using the DAmP collection.75 SEC14 is a phosphatidylinositol/phosphatidylcholine transfer protein required for correct trans-Golgi network dynamics.76
We then correlated the chemical genomic profile to the genetic interaction network for S. cerevisiae.77 Among the top 20 correlated genetic mutant profiles, we saw significant enrichments for mutants in genes involved in the GO processes “intracellular pH reduction” [P = 7.31 × 10−9, GO: 0051452]. This enrichment was driven by VMA1, VMA8, VMA9, VMA10, and VMA11: all are subunits of a yeast vacuolar ATPase and crucial to maintaining intracellular pH regulation. On the basis of structural and functional similarities to other polyether antibiotics, these data suggest that ecteinamycin works, in eukaryotic systems, by forming ion channels, disrupting Golgi dynamics, and impairing other vesicle-mediated transport systems. It is perhaps noteworthy that significant differences in MOA are apparent for 1 when considering prokaryotic cells versus eukaryotic cells, but that, in all cases, biological activities clearly stem from the ionophoric nature of 1.
Cytotoxicity, Bioavailability, and in Vivo Efficacy
In vitro cytotoxicity of 1 was evaluated in mammalian erythrocytes (sheep red blood cells), in a metabolism-independent manner. The hemolysis assay is based on the survival of erythrocytes upon treatment with 1 for 1 h. The lysis agent, Triton, and amphotericin B were used as positive controls. Hemolysis of the red blood cells was measured by the release of hemoglobin at a wavelength of 570 nm. Notably, 1 proved incapable of inducing hemolysis at any of the concentrations tested (Supporting Information, Figure S18); this contrasts sharply with the positive amphotericin control which induced hemolysis in a dose-dependent manner. Ecteinamycin treatment, even at 20 μM, enabled 99.9% erythrocyte survival. By contrast, exposure to 20 μM amphotericin (positive control) correlated to only 30.1% survival. These data strongly support the notion that 1, although toxic to bacteria, does not display overt toxicity toward mammalian cells.
Bioavailability
A pharmacokinetic study in mice was carried out to determine the oral bioavailability of 1. Mice were dosed orally with 1 at 5 mg/kg, or by retro-orbital intravenous (IV) injection at 2.5 mg/kg. Accounting for this difference in dosage, the oral bioavailability was determined to be 10.9% after 30 min of treatment and 29.3% after 1 h of treatment, indicative of generally poor oral bioavailability (Supporting Information, Figures S19–S23). Such a trait of most agents might be considered disadvantageous. However, given the localization of the C. difficile target organism to the lower GI tract in humans, the intestinal lining in particular, these data are very exciting.78 Notably, neither vancomycin nor fidaxomicin is well absorbed orally, and this trait of both agents has been attributed to their past success in countering CDIs.16 It is perhaps very important to recognize that this report represents the first demonstration of oral bioavailability levels for an ionophoric C. difficile selective antibiotic drug lead. Indeed, the low oral bioavailability of 1 supports earlier hypotheses in the field that ionophores may represent excellent anti-C. difficile therapeutic agents.
In Vivo Efficacy
A preliminary in vivo study in mice was performed using 1 as a therapeutic against C. difficile in a modified murine model for C. difficile infection.79 Three experimental mouse groups were used: a healthy control (n = 5), C. difficile control (n = 5), and a group treated with 30 ng of ecteinamycin (n = 5). The baseline weight loss and the health scores (Figure 4) were collected for all groups. Mice in the C. difficile control group began to decline in health by 30 h postinfection based on the collected health parameters. At 24 h postinfection, each mouse in the ecteinamycin group was dosed with 30 ng of 1. The preliminary data indicate that the average health score values are significantly different at the 0.05 level and the 0.000125 level, as evaluated by one-way ANOVA (overall ANOVA) and the Bonferroni test, respectively. C. difficile control mice lost ≥16% of baseline weight at 33 h postinfection, and ≥20% at 48 h, whereas mice treated with 30 ng of 1 lost ≥9% and 12% at the same time points. The weight loss due to initial diarrhea, which is a characteristic symptom of C. difficile pathogenesis, affected the health scores.
Figure 4.

Effect of 1 on C. difficile challenged mice. Experimental groups: healthy control (uninfected), C. difficile control, and group treated with 30 ng of ecteinamycin. Health scores from 27–48 h were averaged for each experimental group.
In addition, C. difficile clearance was measured in collected feces of experimental mice at seven time points (Figure 5). At 48 h after infection, a 1-log10 reduction in C. difficile burden (CFU/g feces) was observed in the experimental group treated with 30 ng of ecteinamycin compared to that of the C. difficile control group. Nevertheless, there is a need for more in vivo studies in different animal models in order to establish ecteinamycin efficacy.
Figure 5.

C. difficile clearance. Feces samples collected at seven different time points were diluted in PBS and plated onto selective C. difficile-Brucella agar, incubated anaerobically for 48 h, and subsequently enumerated. For healthy controls at all time points, as well as the others at time points 0 and 12 h, the CFU/g was zero, given that the limit of detection of the method is <3.00 × 104 (log10 = 4.4771).
Conclusion
CDIs constitute a global health crisis that continues to grow in both scope and scale. Very few agents are effective at controlling C. difficile, although there exists ample evidence in the literature to suggest that ionophoric feed additives may represent useful therapeutics. Ecteinamycin (1), a polyether antibiotic, was isolated from a marine-derived Actinomadura sp. (Strain WMMB499) and its structure elucidated by 13C–13C COSY NMR of 13C-labeled 1 and other spectroscopic methods including a newly developed and highly useful approach exploiting RDC data. Most importantly, 1 demonstrated potent antibacterial activity, especially against a wide array of C. difficile strains; this was the case both in vitro and in vivo. MOA studies with E. coli and S. cerevisiae barcoded deletion mutants revealed that, indeed, the ability of 1 to serve as an ionophore able to interfere with bacterial ion processing likely explains the compound’s potent activity against C. difficile. Despite its low MICs against C. difficile strains, cell depolarization assays with 1 called for high concentrations due to short exposure times; E. coli chemical genomics also called for high concentrations of 1 due to the large number of mutant strains treated and commensurately high cell density of the assay. Ecteinamycin (1) appears to show no discernible toxicity to noncancerous mammalian cells as reflected by its lack of hemolytic activity. We have also shown that 1 has limited oral bioavailability, a trait highly desirable when attempting to kill/clear C. difficile within the human intestinal lining. Additionally, previous data on monensin as well as the yeast chemical genomics results shown here also establish the hypothesis that ionophore antibiotics might reduce effects of C. difficile toxins by disrupting vesicle-mediated trafficking. Taken together, these findings highlight the newly discovered marine natural product 1 as an extremely promising therapeutic lead agent against C. difficile. These data also underscore the potential use of other ionophores as effective therapeutics for CDIs, an assertion that has been previously suggested by others but perhaps not examined with any one agent in as rigorous a fashion as in the case here for ecteinamycin.
METHODS
See the Supporting Information for details.
Supplementary Material
Table 2.
Ecteinamycin Sensitive E. coli Deletion Mutants
| disrupted gene | fold change | adj. P value | gene function |
|---|---|---|---|
| acrB | 0.086930074 | 1.49e−65 | component of AcrAB-ToIC multidrug efflux system |
| acrA | 0.08687022 | 2.59e−60 | component of AcrAB-ToIC multidrug efflux system |
| trkA | 0.145154921 | 4.19e−27 | NAD-binding component of TrK potassium transporter |
| sapD | 0.196735935 | 4.74e−21 | component of peptide uptake ABC trans-porter, required for ATP-dependence of TrK K+ uptake systems |
| csgE | 0.368679357 | 9.8e−20 | component of curli secretion and assembly complex |
| ldcA | 0.341380178 | 3.45e−18 | L,D-carboxypeptidase A; cytoplasmic protease that cleaves the terminal D-alanine from cytoplasmic muropeptides |
| sapB | 0.241016831 | 1.28e−16 | component of peptide uptake ABC transporter |
| kdpD | 0.373882707 | 2.10e−15 | sensor kinase for the kdp (K+-dependence) operon |
| fis | 0.416051589 | 9.27e−15 | transcriptional activator for rRNA operons, bends DNA |
| rpoS | 0.35004657 | 1.15e−13 | component of RNA polymerase σ 38 |
Acknowledgments
This work was supported by funding from the University of Wisconsin-Madison School of Pharmacy and from the University of Wisconsin Institute for Clinical and Translational Research funded through NIH/NCATS UL1TR000427. This work was also funded by the NIH, NIGMS Grant R01GM104192 (T.S.B.). J.S.P. was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494). C.L.M. was supported by grants from the NIH (1R01HG005084-01A1, 1R01GM104975-01, R01HG005853), a grant from the National Science Foundation (DBI 0953881), and by the CIFAR Genetic Networks Program. For H. Mori: JSPS Kakenhi 25250028, 16H02485 and MEXT Kakenhi 25108716, Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank the Analytical Instrumentation Center at the University of Wisconsin-Madison for the facilities to acquire spectroscopic data. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grant P41GM103399. Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA. We would like to thank G. Ananiev from the UWCCC Small Molecule Screening Facility (SMSF) for performing the hemolysis screening. We also would like to thank D. Demaria for assistance with sample collection.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00388.
1D and 2D NMR spectra, RDC procedures along with considerations of DP4 probability and Pearson’s correlations, experimental data for all membrane depolarization assays, potassium release assays, chemical genomic analyses, and additional experimental data (PDF)
References
- 1.Center for Disease Control and Prevention. [accessed Jan 24, 2014]; http://www.cdc.gov/drugresistance/threat-report-2013/
- 2.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 3.Freeman J, Bauer MP, Baines SD, Corver J, Fawley WN, Goorhuis B, Kuijper EJ, Wilcox MH. The changing epidemiology of Clostridium difficile infections. Clin Microbiol Rev. 2010;23:529–549. doi: 10.1128/CMR.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berg AM, Kelly CP, Farraye FA. Clostridium difficile infection in the inflammatory bowel disease patient. Inflamm Bowel Dis. 2013;19:194–204. doi: 10.1002/ibd.22964. [DOI] [PubMed] [Google Scholar]
- 5.Carter GP, Rood JI, Lyras D. The role of toxin A and toxin B in Clostridium difficile-associated disease: Past and present perspectives. Gut Microbes. 2010;1:58–64. doi: 10.4161/gmic.1.1.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.May M, Wang T, Muller M, Genth H. Difference in F-actin depolymerization induced by toxin B from the Clostridium difficile strain VPI 10463 and toxin B from the variant Clostridium difficile serotype F strain 1470. Toxins. 2013;5:106–119. doi: 10.3390/toxins5010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S, Sun C, Wang H, Wang J. The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins. Toxins. 2015;7:5254–5267. doi: 10.3390/toxins7124874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun X, Savidge T, Feng H. The enterotoxicity of Clostridium difficile toxins. Toxins. 2010;2:1848–1880. doi: 10.3390/toxins2071848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popoff MR, Rubin EJ, Gill DM, Boquet P. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect Immun. 1988;56:2299–2306. doi: 10.1128/iai.56.9.2299-2306.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perelle S, Gibert M, Bourlioux P, Corthier G, Popoff MR. Production of a complete binary toxin (actin-specific ADP-ribosyltransferase) by Clostridium difficile CD196. Infect Immun. 1997;65:1402–1407. doi: 10.1128/iai.65.4.1402-1407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology. 2009;136:1913–1924. doi: 10.1053/j.gastro.2009.02.073. [DOI] [PubMed] [Google Scholar]
- 12.Kok J, Wang Q, Thomas LC, Gilbert GL. Presumptive identification of Clostridium difficile strain 027/NAP1/BI on Cepheid Xpert: interpret with caution. J Clin Microbiol. 2011;49:3719–3721. doi: 10.1128/JCM.00752-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuehne SE, Collery MM, Kelly ML, Cartman ST, Cockayne A, Minton NP. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis. 2014;209:83–86. doi: 10.1093/infdis/jit426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandt LJ. Fecal transplantation for the treatment of Clostridium difficile infection. Gastroenterol Hepatol. 2012;8:191–194. [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly CR, Ihunnah C, Fischer M, Khoruts A, Surawicz C, Afzali A, Aroniadis O, Barto A, Borody T, Giovanelli A, Gordon S, Gluck M, Hohmann EL, Kao D, Kao JY, McQuillen DP, Mellow M, Rank KM, Rao K, Ray A, Schwartz MA, Singh N, Stollman N, Suskind DL, Vindigni SM, Youngster I, Brandt L. Fecal microbiota transplant for treatment of Clostridium difficile infection in immunocompromised patients. Am J Gastroenterol. 2014;109:1065–1071. doi: 10.1038/ajg.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mullane K. Fidaxomicin in Clostrium difficile infection: latest evidence and clinical guidance. Ther Adv Chronic Dis. 2014;5:69–84. doi: 10.1177/2040622313511285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, Gorbach S, Sears P, Shue YK. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364:422–431. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 18.Johnson S, Samore MH, Farrow KA, Killgore GE, Tenover FC, Lyras D, Rood JI, DeGirolami P, Baltch AL, Rafferty ME, Pear SM, Gerding DN. Epidemics of diarrhea caused by a clindamycin-resistant strain of Clostridium difficile in four hospitals. N Engl J Med. 1999;341:1645–1651. doi: 10.1056/NEJM199911253412203. [DOI] [PubMed] [Google Scholar]
- 19.McDonald LC, Killgore GE, Thompson A, Owens RC, Jr, Kazakova SV, Sambol SP, Johnson S, Gerding DN. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 20.Baines SD, Wilcox MH. Antimicrobial Resistance and Reduced Susceptibility in Clostridium difficile: Potential Consequences for Induction, Treatment, and Recurrence of C. difficile infection. Antibiotics. 2015;4:267–298. doi: 10.3390/antibiotics4030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musher DM, Aslam S, Logan N, Nallacheru S, Bhaila I, Borchert F, Hamill RJ. Relatively poor outcome after treatment of Clostridium difficile colitis with metronidazole. Clin Infect Dis. 2005;40:1586–1590. doi: 10.1086/430311. [DOI] [PubMed] [Google Scholar]
- 22.Baines SD, O’Connor R, Freeman J, Fawley WN, Harmanus C, Mastrantonio P, Kuijper EJ, Wilcox MHJ. Emergence of reduced susceptibility to metronidazole in Clostridium difficile. J Antimicrob Chemother. 2008;62:1046–1052. doi: 10.1093/jac/dkn313. [DOI] [PubMed] [Google Scholar]
- 23.Peláez T, Alonso AR, Rodríguez-Créixems M, García-Lechuz JM, Bouza E, Alcala L. Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob Agents Chemother. 2002;46:1647–1650. doi: 10.1128/AAC.46.6.1647-1650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freeman J, Vernon J, Morris K, Nicholson S, Todhunter S, Longshaw C, Wilcox MH. Pan-European longitudinal surveillance of antibiotic resistance among prevalent Clostridium difficile ribotypes. Clin Microbiol Infect. 2015;21:248e9–248e16. doi: 10.1016/j.cmi.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Snydman DR, McDermott LA, Jacobus NV, Thorpe C, Stone S, Jenkins SG, Goldstein EJC, Patel R, Forbes BA, Mirrett S, Johnson S, Gerding DN. U.S.-Based National Sentinel Surveillance Study for the Epidemiology of Clostridium difficile-Associated Diarrheal Isolates and Their Susceptibility to Fidaxomicin. Antimicrob Agents Chemother. 2015;59:6437–6443. doi: 10.1128/AAC.00845-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henriques B, Florin I, Thelestam M. Cellular internalisation of Clostridium difficile toxin A. Microb Pathog. 1987;2:455–463. doi: 10.1016/0882-4010(87)90052-0. [DOI] [PubMed] [Google Scholar]
- 27.Von Eichel-Streiber C, Sauerborn M. Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene. 1990;96:107–113. doi: 10.1016/0378-1119(90)90348-u. [DOI] [PubMed] [Google Scholar]
- 28.Ho JG, Greco A, Rupnik M, Ng KK. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci U S A. 2005;102:18373–18378. doi: 10.1073/pnas.0506391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurdle JG, O’Neill AJ, Chopra I, Lee RE. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat Rev Microbiol. 2011;9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dutton CJ, Banks BJ, Cooper CB. Polyether ionophores. Nat Prod Rep. 1995;12:165–181. doi: 10.1039/np9951200165. [DOI] [PubMed] [Google Scholar]
- 32.Fiorentini C, Malorni W, Paradisi S, Giuliano M, Mastrantonio P, Donelli G. Interaction of Clostridium difficile toxin A with cultured cells: cytoskeletal changes and nuclear polarization. Infect Immun. 1990;58:2329–2336. doi: 10.1128/iai.58.7.2329-2336.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aarestrup FM, Tvede M. Susceptibility of Clostridium difficile toward antimicrobial agents used as feed additives for food animals. Microb Drug Resist. 2011;17:125–127. doi: 10.1089/mdr.2010.0068. [DOI] [PubMed] [Google Scholar]
- 34.Matarrese P, Falzano L, Fabbri A, Gambardella L, Frank C, Geny B, Popoff MR, Malorni W, Fiorentini C. Clostridium difficile toxin B causes apoptosis in epithelial cells by thrilling mitochondria. Involvement of ATP-sensitive mitochondrial potassium channels. J Biol Chem. 2007;282:9029–9041. doi: 10.1074/jbc.M607614200. [DOI] [PubMed] [Google Scholar]
- 35.The novel structure and unique biological activities of 1 provided the basis for the patent: Bugni TS, Wyche TP, Braun DR, Piotrowski JS. WO/2016/069776 A1. Some of the experiments described here and/or their predecessors formed the basis for WO/2016/069776 A1.
- 36.Wyche TP. UW-Madison Dissertation. OCLC; Investigation of marine-derived bacteria for bioactive natural products. ocn904439295. Published: 2014. [Google Scholar]
- 37.For recent structure elucidation, cytotoxicity and related biological studies of 1 and its vinyl chloride congener, see: Igarashi Y, Matsuoka N, In Y, Kataura T, Tashiro E, Saiki I, Sudoh Y, Duangmal K, Thamchaipenet A. Nonthmicin, a Polyether Polyketide Bearing a Halogen-Modified Tetronate with Neuroprotective and Anti-invasive Activity from Actinomadura sp. Org Lett. 2017;19:1406–1409. doi: 10.1021/acs.orglett.7b00318.
- 38.Keller-Juslén C, King HD, Kuhn M, Loosli H, Pache W, Petcher TJ, Weber HP, Wartburg AV. Tetronomycin, a novel polyether of unusual structure. J Antibiot. 1982;35:142–150. doi: 10.7164/antibiotics.35.142. [DOI] [PubMed] [Google Scholar]
- 39.Davies DH, Snape EW, Suter PJ, King TJ, Falshaw CP. Structure of antibiotic M139603; X-ray crystal structure of the 4-bromo-3,5-dinitrobenzoyl derivative. J Chem Soc, Chem Commun. 1981:1073–1074. [Google Scholar]
- 40.Hou Y, Braun DR, Michel CR, Klassen JL, Adnani N, Wyche TP, Bugni TS. Microbial strain prioritization using metabolomics tools for the discovery of natural products. Anal Chem. 2012;84:4277–4283. doi: 10.1021/ac202623g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wyche TP, Standiford M, Hou Y, Braun D, Johnson DA, Johnson JA, Bugni TS. Activation of the nuclear factor E2-related factor 2 pathway by novel natural products halomadurones A-D and a synthetic analogue. Mar Drugs. 2013;11:5089–5099. doi: 10.3390/md11125089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyche TP, Piotrowski JS, Hou Y, Braun D, Deshpande R, McIlwain SJ, Ong IM, Myers CL, Guzei IA, Westler WM, Andes DR, Bugni TS. Forazoline A: marine-derived polyketide with antifungal in vivo efficacy. Angew Chem, Int Ed. 2014;53:11583–11586. doi: 10.1002/anie.201405990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellis GA, Wyche TP, Fry CG, Braun D, Bugni TS. Solwaric acids A and B, antibacterial aromatic acids from a marine Solwaraspora sp. Mar Drugs. 2014;12:1013–1022. doi: 10.3390/md12021013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reibarkh M, Wyche TP, Saurí J, Bugni TS, Martin GE, Williamson RT. Structure elucidation of uniformly (13) C labeled small molecule natural products. Magn Reson Chem. 2015;53:996–1002. doi: 10.1002/mrc.4333. [DOI] [PubMed] [Google Scholar]
- 45.Cornilescu G, Ramos Alvarenga RF, Wyche TP, Bugni TS, Gil RR, Cornilescu CC, Westler WM, Markley JL, Schwieters CD. Progressive Stereo Locking (PSL) - A Residual Dipolar Coupling Force Field Method for Determining the Relative Configuration of Natural Products and other Small Molecules. ACS Chem Biol. 2017 doi: 10.1021/acschembio.7b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pepin J. Vancomycin for the treatment of Clostridium difficile Infection: for whom is this expensive bullet really magic? Clin Infect Dis. 2008;46:1493–1498. doi: 10.1086/587656. [DOI] [PubMed] [Google Scholar]
- 47.Wong SS, Woo PC, Luk WK, Yuen KY. Susceptibility testing of Clostridium difficile against metronidazole and vancomycin by disk diffusion and Etest. Diagn Microbiol Infect Dis. 1999;34:1–6. doi: 10.1016/s0732-8893(98)00139-4. [DOI] [PubMed] [Google Scholar]
- 48.Wu X, Cherian PT, Lee RE, Hurdle JG. The membrane as a target for controlling hypervirulent Clostridium difficile infections. J Antimicrob Chemother. 2013;68:806–815. doi: 10.1093/jac/dks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ueda C, Tateda K, Horikawa M, Kimura S, Ishii Y, Nomura K, Yamada K, Suematsu T, Inoue Y, Ishiguro M, Miyairi S, Yamaguchi K. Anti-Clostridium difficile potential of tetramic acid derivatives from Pseudomonas aeruginosa quorum-sensing autoinducers. Antimicrob Agents Chemother. 2010;54:683–688. doi: 10.1128/AAC.00702-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Novo D, Perlmutter NG, Hunt RH, Shapiro HM. Accurate flow cytometric membrane potential measurement in bacteria using diethyloxacarbocyanine and a ratiometric technique. Cytometry. 1999;35:55–63. doi: 10.1002/(sici)1097-0320(19990101)35:1<55::aid-cyto8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 51.Silverman JA, Perlmutter NG, Shapiro HM. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47:2538–2544. doi: 10.1128/AAC.47.8.2538-2544.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piotrowski JS, Simpkins SW, Li SC, Deshpande R, McIlwain SJ, Ong IM, Myers CL, Boone C, Andersen RJ. Chemical genomic profiling via barcode sequencing to predict compound mode of action. Methods Mol Biol. 2015;1263:299–318. doi: 10.1007/978-1-4939-2269-7_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitani M, Yamanishi T, Miyazaki Y. Salinomycin: a new monovalent cation ionophore. Biochem Biophys Res Commun. 1975;66:1231–1236. doi: 10.1016/0006-291x(75)90490-8. [DOI] [PubMed] [Google Scholar]
- 54.Riddell FG, Tompsett SJ. The transport of Na+ and K+ ions through phospholipid bilayers mediated by the antibiotics salinomycin and narasin studied by 23Na- and 39K-NMR spectroscopy. Biochim Biophys Acta, Biomembr. 1990;1024:193–197. doi: 10.1016/0005-2736(90)90225-d. [DOI] [PubMed] [Google Scholar]
- 55.For an excellent review of salinomycin and its bioactivities and mechanisms see: Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol. 2012;2012:1. doi: 10.1155/2012/950658.
- 56.Ho CH, Piotrowski J, Dixon SJ, Baryshnikova A, Costanzo M, Boone C. Combining functional genomics and chemical biology to identify targets of bioactive compounds. Curr Opin Chem Biol. 2011;15:66–78. doi: 10.1016/j.cbpa.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 57.Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- 58.Fung SY, Sofiyev V, Schneiderman J, Hirschfeld AF, Victor RE, Woods K, Piotrowski JS, Deshpande R, Li SC, de Voogd NJ, Myers CL, Boone C, Andersen RJ, Turvey SE. Unbiased screening of marine sponge extracts for anti-inflammatory agents combined with chemical genomics identifies girolline as an inhibitor of protein synthesis. ACS Chem Biol. 2014;9:247–257. doi: 10.1021/cb400740c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Booth IR, Cairney J, Sutherland L, Higgins CF. Enteric bacteria and osmotic stress: an integrated homeostatic system. J Appl Bacteriol. 1998;65:35S–49S. [PubMed] [Google Scholar]
- 60.Epstein W. The roles and regulation of potassium in bacteria. Prog Nucleic Acid Res Mol Biol. 2003;75:293–319. doi: 10.1016/s0079-6603(03)75008-9. [DOI] [PubMed] [Google Scholar]
- 61.Roosild TP, Castronovo S, Healy J, Miller S, Pliotas C, Rasmussen T, Bartlett W, Conway SJ, Booth IR. Mechanism of ligand-gated potassium efflux in bacterial pathogens. Proc Natl Acad Sci U S A. 2010;107:19784–19789. doi: 10.1073/pnas.1012716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ochrombel I, Ott L, Krämer R, Burkovski A, Marin K. Impact of improved potassium accumulation on pH homeostasis, membrane potential adjustment and survival of Corynebacterium glutamicum. Biochim Biophys Acta, Bioenerg. 2011;1807:444–450. doi: 10.1016/j.bbabio.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 63.Hurdle JG, Heathcott AE, Yang L, Yan B, Lee RE. Reutericyclin and related analogues kill stationary phase Clostridium difficile at achievable colonic concentrations. J Antimicrob Chemother. 2011;66:1773–1776. doi: 10.1093/jac/dkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serizawa H, Fukuda R. Structure of the gene for the stringent starvation protein of Escherichia coli. Nucleic Acids Res. 1987;15(3):1153–1163. doi: 10.1093/nar/15.3.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams MD, Fuchs JA, Flickinger MC. Null mutation in the stringent starvation protein of Escherichia coli disrupts lytic development of bacteriophage P1. Gene. 1991;109:21–30. doi: 10.1016/0378-1119(91)90584-x. [DOI] [PubMed] [Google Scholar]
- 66.Hansen AM, Qiu Y, Yeh N, Blattner FR, Durfee T, Jin DJ. SspA is required for acid resistance in stationary phase by downregulation of H-NS in Escherichia coli. Mol Microbiol. 2005;56:719–734. doi: 10.1111/j.1365-2958.2005.04567.x. [DOI] [PubMed] [Google Scholar]
- 67.Hansen AM, Jin DJ. SspA up-regulates gene expression of the LEE pathogenicity island by decreasing H-NS levels in enterohemorrhagic Escherichia coli. BMC Microbiol. 2012;12:231–240. doi: 10.1186/1471-2180-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Missiakas D, Mayer MP, Lemaire M, Georgopoulos C, Raina S. Modulation of the Escherichia coli sigmaE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol Microbiol. 1997;24:355–371. doi: 10.1046/j.1365-2958.1997.3601713.x. [DOI] [PubMed] [Google Scholar]
- 69.De Las Peñas A, Connolly L, Gross CA. The sigmaE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of sigmaE. Mol Microbiol. 1997;24:373–385. doi: 10.1046/j.1365-2958.1997.3611718.x. [DOI] [PubMed] [Google Scholar]
- 70.Wollmann P, Zeth K. The structure of RseB: a sensor in periplasmic stress response of E. coli. J Mol Biol. 2007;372:927–941. doi: 10.1016/j.jmb.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 71.Lima S, Guo MS, Chaba R, Gross CA, Sauer RT. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science. 2013;340:837–841. doi: 10.1126/science.1235358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kulathila R, Kulathila R, Indic M, van den Berg B. Crystal structure of Escherichia coli CusC, the outer membrane component of a heavy metal efflux pump. PLoS One. 2011;6:e15610. doi: 10.1371/journal.pone.0015610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franke S, Grass G, Nies DH. The product of the ybdE gene of the Escherichia coli chromosome is involved in detoxification of silver ions. Microbiology. 2001;147:965–972. doi: 10.1099/00221287-147-4-965. [DOI] [PubMed] [Google Scholar]
- 74.Franke S, Grass G, Rensing C, Nies DH. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J Bacteriol. 2003;185:3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yan Z, Costanzo M, Heisler LE, Paw J, Kaper F, Andrews BJ, Boone C, Giaever G, Nislow C. Yeast Barcoders: a chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat Methods. 2008;5:719–725. doi: 10.1038/nmeth.1231. [DOI] [PubMed] [Google Scholar]
- 76.Curwin AJ, Fairn GD, McMaster CR. Phospholipid transfer protein Sec14 is required for trafficking from endosomes and regulates distinct trans-Golgi export pathways. J Biol Chem. 2009;284:7364–7375. doi: 10.1074/jbc.M808732200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, Pelechano V, Styles EB, Billmann M, van Leeuwen J, van Dyk N, Lin Z-Y, Kuzmin E, Nelson J, Piotrowski JS, Srikumar T, Bahr S, Chen Y, Deshpande R, Kurat CF, Li SC, Li Z, Usaj MM, Okada H, Pascoe N, San Luis B-J, Sharifpoor S, Shuteriqi E, Simpkins SW, Snider J, Suresh HG, Tan Y, Zhu H, Malod-Dognin N, Janjic V, Przulj N, Troyanskaya OG, Stagljar I, Xia T, Ohya Y, Gingras A-C, Raught B, Boutros M, Steinmetz LM, Moore CL, Rosebrock AP, Caudy AA, Myers CL, Andrews B, Boone C. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353:aaf1420. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van de Waterbeemd H, Smith DA, Beaumont K, Walker DK. Property-based design: optimization of drug absorption and pharmacokinetics. J Med Chem. 2001;44:1313–1333. doi: 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- 79.Theriot CM, Koumpouras CC, Carlson PE, Bergin IL, Aronoff DM, Young VB. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes. 2011;2:326–334. doi: 10.4161/gmic.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.