

Abstract
A highly diastereoselective carbon–carbon bond-forming reaction involving the tandem coupling of benzyltriboronates, enoates, and alkyl halides is described. This method was enabled by the discovery of α-diimine nickel catalysts that promote the chemoselective triborylation of benzylic C(sp3)–H bonds using B2Pin2 (Pin = pinacolate). The C–H functionalization method is effective with methylarenes and for the diborylation of secondary benzylic C–H bonds, providing direct access to polyboron building blocks from readily available hydrocarbons. Combination of the benzylic perborylation with a new deborylative conjugate addition–alkylation method enables a one-pot procedure in which multiple simple precursors are combined to generate diastereopure products containing quaternary stereocenters.
Organoboron compounds are versatile reagents for carbon–carbon and carbon–heteroatom bond formation.1 Arylboronates are the most widely used nucleophilic partner in Suzuki–Miyaura cross couplings2,3 and the most commonly deployed in the pharmaceutical industry due to their ease of handling, synthetic accessibility, and reliability.4 Alkylboronates, typically synthesized by alkene hydroboration5 or Miyaura-type borylation of alkyl halides,6 are likewise valuable reagents for C–C bond formation whose utility will continue to grow as methods for C(sp3)–C(sp3) cross coupling mature.7 Polyboron compounds such as gem-diboronates are beginning to emerge as useful reagents for C–C bond formation as well and have been applied to alkylation reactions,8 boron–Wittig olefinations,9 1,2-additions to carbonyls10 and imines,11 addition to pyridine-N-oxides,12 allylic substitution,13 and enantioselective cross couplings.14 The use of 1,1,1-alkyltriboronates for C–C bond formation offers similar, if not greater, potential but is less explored and limited to deborylative boron–Wittig olefination15 or deborylative alkylation (Scheme 1A).16
Scheme 1.
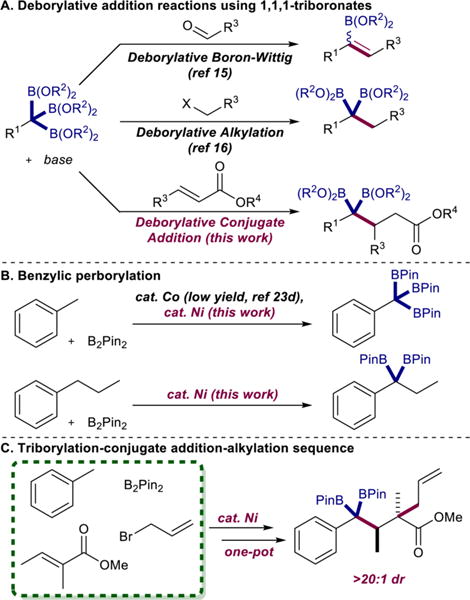
Utility of Alkyltriboronates in C–C Bond Formation, Synthesis of Benzylpolyboronates by C–H Borylation, and Combination of These Methods To Provide a Diastereoselective Tandem Coupling Sequence
The full synthetic potential of geminal alkylpolyboronates has yet to be realized principally due to the paucity of straightforward methods for their preparation. The direct, metal-catalyzed polyborylation of C(sp3)–H bonds offers a potentially transformative route as readily available, minimally functionalized hydrocarbons may be used as starting materials. Although C–H borylation is one of the most widely used C–H functionalization methods, most precious17 and base metal catalysts18 favor C(sp2)–H sites, a result of established kinetic preference for oxidative addition.19 Metal-catalyzed methods for alkane borylation have been reported17,20 including recent notable examples of iridium- and rhodium-catalyzed borylation of methane at high pressures,21 but chemoselective C(sp3)–H borylation in the presence of C(sp2)–H bonds is far less common and more challenging. Strategies to overcome this selectivity bias rely primarily on the use of directing groups,22 while a handful of nondirected methods selectively functionalize benzylic C–H bonds.23 In most of these examples, only one C–H bond is converted to a C–B bond.24 New catalytic methods that convert multiple C–H bonds on the same carbon to boron functional groups would be valuable and enable exploration of the full synthetic potential of alkyl polyboronates.
Our laboratory previously reported α-diimine cobalt dialkyl and bis(carboxylate) compounds that are active for the catalytic borylation of C(sp3)-H bonds of methylarenes with B2Pin2 as the boron source.23d The 1,1-diboronates were the major products but, with toluene as the substrate, a small amount (18% yield) of the 1,1,1-triboronate product was isolated when excess boron reagent and high cobalt loadings of 50 mol% were used. The forcing conditions, low yield, and difficulty in product isolation precluded reactivity studies of this interesting benzylpolyboronate and inspired development of a synthetically useful catalytic method. Here we describe α-diimine nickel dialkyl and bis(carboxylate) complexes that enable the selective preparation of benzyltriboronates via perborylation of benzylic C–H bonds (Scheme 1B). Combination of the Ni-catalyzed perborylation method with a new deborylative conjugate addition protocol (Scheme 1A) provides a convenient one-pot, tandem procedure for the diastereoselective synthesis of value-added products from simple, abundant precursors (Scheme 1C).
Inspired by the observation of benzylboronate products by Tobisu, Chatani, and co-workers in Ni catalysis18f and our own success with N-alkyl-substituted α-diimine (RADI) cobalt dialkyl complexes,23d the analogous Ni(II) dialkyl complexes were targeted for evaluation in catalytic C–H borylation. (CyADI)Ni-(CH2SiMe3)2 (1) was isolated as a teal, diamagnetic solid in 79% yield following pyridine displacement by addition of the free α-diimine to (py)2Ni(CH2SiMe3)2 (py = pyridine).25 The solid-state structure of 1 established a distorted square planar geometry (see Supporting Information (SI), Figure S2). Because both cobalt and nickel bis(carboxylate) compounds have proven to be readily synthesized, bench-stable precursors for a host of catalytic reactions,18e,23d,26 (CyADI)Ni(OPiv)2 (2) (Piv = pivaloyl) was also prepared by addition of the free α-diimine to a well-defined Ni(II) pivalate precursor, [(NEt3)Ni(OPiv)2]2, a compound first reported by Eremenko and co-workers.27
With excess (4.0 equiv.) B2Pin2 and 25 mol% of 1, the catalytic borylation of toluene in cyclopentylmethyl ether (CPME) at 80 °C resulted in isolation of the benzyltriboronate 4 in 84% yield (Scheme 2A). Repeating this procedure with 25 mol% of 2 in combination with 100 mol% of methylaluminoxane (MAO) activator provided a quantitative yield of 4. Notably, the yields of 4 using 1 and 2 were significantly higher with lower precatalyst loadings and temperatures than with cobalt, highlighting the advantage of the nickel-catalyzed method.
Scheme 2. Benzylic Perborylation of Substituted Methylarenes and Linear Alkylarenes Using α-Diimine Nickel Dialkyl and Bis(pivalate) Precatalystsa.
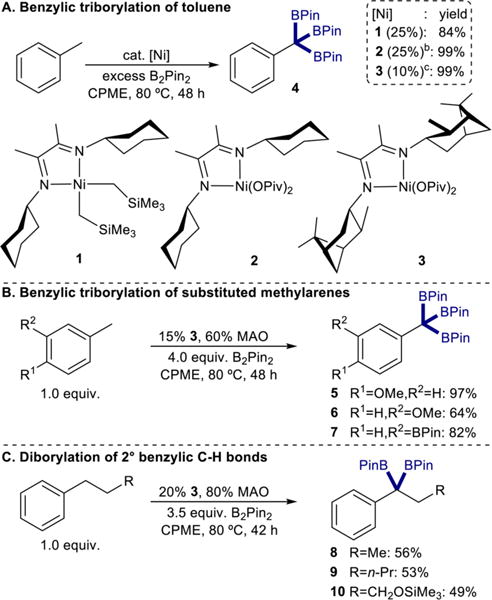
aIsolated yields. Reactions conducted with 0.55 mmol methylarene or alkylarene in 0.55 mL of CPME at 80 °C. See SI for experimental details. b100 mol% MAO as catalyst activator. 4 was isolated with 13% impurity of remaining B2Pin2. c40 mol% MAO activator used.
Nickel sources, α-diimine ligands, and catalytic conditions were evaluated to lower the amount of Ni required (see SI, Table S1). Because formation of bis(chelate) nickel complexes was believed to be a deactivation pathway (see SI, Scheme S1), the nickel bis(pivalate) complex bearing a bulkier diimine ligand was synthesized. Pale green (ipcADI)Ni(OPiv)2 (3, Scheme 2A) was isolated in 89% yield and was active for the triborylation of toluene. With only 10 mol% of 3 in combination with 40 mol% MAO, quantitative formation of 4 was obtained. This reaction was also successfully scaled; starting with 2.17 mmol of toluene, 1.00 g (99% yield) of 4 was isolated.
With the improved performance of 3, the generality of benzylic triborylation was examined with a series of methylarenes (Scheme 2B). High yields of the benzyltriboronate products derived from 3-methylanisole, 4-methylanisole, and m-tolylboronic acid pinacol ester were obtained. Although these products were isolated following acidic aqueous workup with no additional purification, the benzyltriboronates underwent protodeborylation to mono- or diboronates upon prolonged exposure to aqueous conditions or by attempted purification by column chromatography. Extended exposure to air also resulted in decomposition, likely by autoxidation.
The polyborylation of the benzylic positions of linear alkyl arenes was also explored with 3. C(sp3)–H borylation of this substrate class has only been achieved previously using silyl directing groups22d or vast excess of substrate and long reaction times.23b,e Using 20 mol% of 3 and excess (3.5 equiv.) B2Pin2, the benzylic diborylation of n-propylbenzene, n-pentylbenzene, and trimethylsilyl-protected 3-phenylpropanol was accomplished to afford 8, 9, and 10, respectively (Scheme 2C). Precatalyst 3 was also effective for the synthesis of benzyldiboronates from methylarenes (see SI, Scheme S2) at superior yields and lower catalyst loadings than previously reported with cobalt.23d
The development of a direct method for the preparation of 1,1,1-alkyltriboronates from C(sp3)–H functionalization offers a new opportunity to explore the utility of these products in C–C bond-forming reactions. Known deborylative boron–Wittig olefination15 and deborylative alkylation16 protocols were demonstrated (see SI, Scheme S3). Further efforts focused on development of new C–C bond-forming transformations. Conjugate addition of α-boryl nucleophiles to unsaturated carbonyls has previously been reported for α-boryl radicals28 and α-boryl-zirconocenes.29 Benzylic α-diboryl anions were postulated to be sufficiently soft nucleophiles to exhibit similar reactivity. To test this, a THF solution of 4 and (E)-methylcrotonate was treated with NaOEt at room temperature, providing 11 (Scheme 3A) in 77% yield following aqueous workup. Repeating this procedure with methyl methacrylate and methyl tiglate provided 12 and 13, respectively, in good yield and, in the case of 13, excellent diastereoselectivity (19:1). Notably, products resulting from the deborylative conjugate addition to phenyl-substituted (E)-methyl cinnamate (14) and the α,β,γ,δ-unsaturated ethyl sorbate (15) were also obtained, albeit at reduced yields, predominantly due to competing 1,2-addition to the more electron deficient enoates.
Scheme 3. Base-Mediated Deborylative Conjugate Addition Using Isolated Benzyltriboronate 4a.
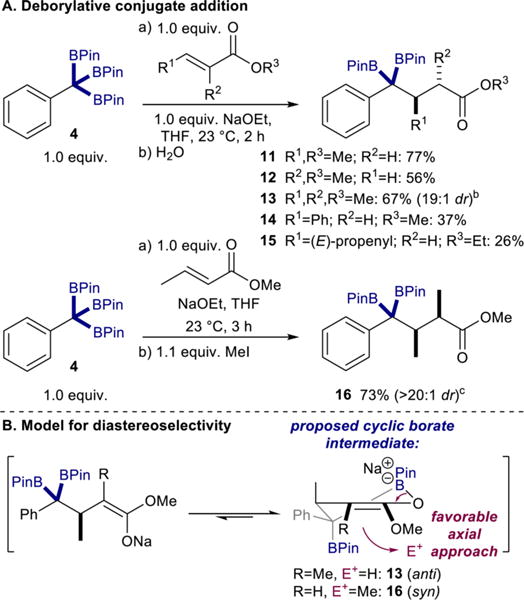
aIsolated yields. Reactions conducted with 0.28 mmol of benzyltriboronate in 5.6 mL of THF. See SI for experimental details. Diastereomeric ratios (dr) determined by gas chromatography of crude reaction mixtures and confirmed by 1H NMR spectroscopy of isolated products. bMajor diastereomer 13 assigned as opposite of that assigned for 16 (minor diastereomer). cDiastereomer of 16 determined by X-ray crystallography (see SI, Figure S4).
Given that the intermediate in the deborylative conjugate addition of 4 to methyl tiglate proceeds through a sodium enolate, the stereocontrol at the α-position of the ester in 13 likely occurs during protonation when the reaction is quenched. Intramolecular coordination of the enolate to a boronate substituent and subsequent axial approach of an electrophile was hypothesized to explain this phenomenon (Scheme 3B).30 To test this, the deborylative conjugate addition of 4 and (E)-methylcrotonate was carried out, and the reaction was quenched with iodomethane (Scheme 3A). The resulting product, 16, was isolated in >20:1 dr as the syn diastereomer opposite that of 13, consistent with the stereochemical model. These results highlight the unique ability of the boronate groups to act not only as masking groups for soft nucleophilic anions but also elements for substrate-controlled diastereoselectivity.
The possibility of generating these benzyltriboronates in situ and directly applying them toward further chemistry was also investigated. This would not only obviate the need for difficult isolation of these sensitive reagents, but it would provide a convenient one-pot method for rapidly building molecular complexity directly from toluene or other simple substituted methylarenes. The synthesis of 16 was conducted directly from toluene, B2Pin2, (E)-methylcrotonate, and iodomethane without isolating 4 (Scheme 4A). Following the Ni-catalyzed triborylation of toluene, the crude triboronate product was exposed to the conjugate addition–alkylation conditions to provide 16 in 70% yield and >20:1 dr. The method was then successfully extended to other substituted methylarenes. Generation of 5, 6, and 7 in situ, followed by conjugate addition and alkylation, yielded products 17, 18, and 19, respectively, in good to modest yields with high diastereoselectivity (>20:1). The one-pot procedure was also applied using p-cymene, 4-tolyltrimethylsilane, and N,N-dimethyl-m-toluidine, resulting in isolation of 20, 21, and 22, respectively, following column chromatography without the need for purification of the 1,1,1-triboronate intermediates.
Scheme 4. Diastereoselective, One-Pot Tandem Triborylation–Conjugate Addition–Alkylationa.
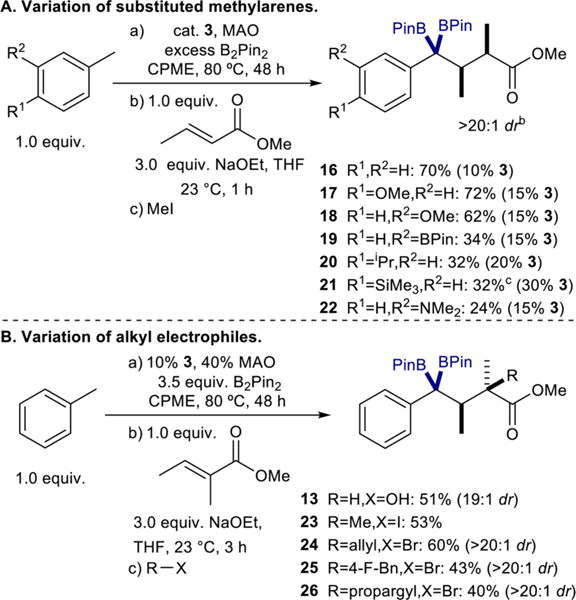
aIsolated yields. See SI for experimental details. Diastereomeric ratios (dr) determined by gas chromatography of crude reaction mixtures and confirmed by 1H NMR spectroscopy of isolated products. bDiastereomers assigned by analogy to that of 16. cNMR yield; 22% isolated yield.
The scope of the alkyl electrophile was also explored. Using toluene, B2Pin2, methyltiglate, and quenching with water, 13 was isolated (19:1 dr) in 51% yield (Scheme 4B). Quenching the reaction with iodomethane provided the methylated product 23 in 53% yield. Further variation of the alkyl electrophile might provide access to diastereomerically enriched products with quaternary stereocenters. This was successfully demonstrated using allyl bromide, 4-fluorobenzyl bromide, and propargyl bromide to give products 24, 25, and 26, respectively, with high degrees (>20:1) of diastereoselectivity. The solid state structure of 25 was confirmed by X-ray crystallography (see SI, Figure S5), verifying the identity of the major diastereomer as that predicted by the stereochemical model in Scheme 3B and allowing for assignment of the diastereomers of 24 and 26 by analogy.
In summary, an α-diimine nickel catalyst has been developed that is effective for the perborylation of benzylic C(sp3)–H bonds of methyl- and alkylarenes. The unique products are useful building blocks for a diastereoselective conjugate addition–alkylation sequence, highlighting the ability of the multiple boronate groups to act simultaneously as soft carbanion masking groups and intramolecular stereocontrol elements. Combination of the C(sp3)–H polyborylation with deborylative C–C bond formation into a one-pot procedure allowed an effective method for rapid elaboration of simple and abundant precursors to complex products poised for further elaboration with remaining C–B bond functionality.31
Supplementary Material
Acknowledgments
W.N.P. acknowledges the National Science Foundation (CHE-1265988) and the 2016 Eli Lilly Graduate Research Fellowship Award for financial support. C.Z. acknowledges MINECO for a FPU fellowship. We thank M. J. Bezdek and I. Pappas for crystallographic studies, and we also thank Allychem for a generous gift of B2Pin2. Financial support was provided by NIH (R01 GM121441).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b12896.
X-ray crystallographic data (CIF)
Complete experimental details, precatalyst optimization studies, and characterization data of nickel complexes and boronate products (PDF)
ORCID
Paul J. Chirik: 0000-0001-8473-2898
Notes
The authors declare no competing financial interest.
References
- 1.Hall DG. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 2.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 3.Lennox AJJ, Lloyd-Jones G. Chem Soc Rev. 2014;43:412. doi: 10.1039/c3cs60197h. [DOI] [PubMed] [Google Scholar]
- 4.Roughley SD, Jordan AM. J Med Chem. 2011;54:3451. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 5.(a) Burgess K, Ohlmeyer MJ. Chem Rev. 1991;91:1179. [Google Scholar]; (b) Beletskaya I, Pelter A. Tetrahedron. 1997;53:4957. [Google Scholar]; (c) Crudden CM, Edwards D. Eur J Org Chem. 2003;24:4695. [Google Scholar]
- 6.(a) Yang CT, Zhang ZQ, Tajuddin H, Wu CC, Liang J, Liu JH, Fu Y, Czyzewska M, Steel PG, Marder TB, Liu L. Angew Chem, Int Ed. 2012;51:528. doi: 10.1002/anie.201106299. [DOI] [PubMed] [Google Scholar]; (b) Ito H, Kubota K. Org Lett. 2012;14:890. doi: 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]
- 7.(a) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leonori D, Aggarwal VK. Angew Chem, Int Ed. 2015;54:1082. doi: 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]
- 8.Hong K, Liu X, Morken JP. J Am Chem Soc. 2014;136:10581. doi: 10.1021/ja505455z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Endo K, Hirokami M, Shibata T. J Org Chem. 2010;75:3469. doi: 10.1021/jo1003407. [DOI] [PubMed] [Google Scholar]; (b) Coombs JR, Zhang L, Morken JP. Org Lett. 2015;17:1708. doi: 10.1021/acs.orglett.5b00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Joannou MV, Moyer BS, Meek SJ. J Am Chem Soc. 2015;137:6176. doi: 10.1021/jacs.5b03477. [DOI] [PubMed] [Google Scholar]; (b) Joannou MV, Moyer BS, Goldfogel MJ, Meek SJ. Angew Chem, Int Ed. 2015;54:14141. doi: 10.1002/anie.201507171. [DOI] [PubMed] [Google Scholar]; (c) Murray SA, Green JC, Tailor SB, Meek SJ. Angew Chem, Int Ed. 2016;55:9065. doi: 10.1002/anie.201603465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park J, Lee Y, Kim J, Cho SH. Org Lett. 2016;18:1210. doi: 10.1021/acs.orglett.6b00376. [DOI] [PubMed] [Google Scholar]
- 12.Jo W, Kim J, Choi S, Cho SH. Angew Chem, Int Ed. 2016;55:9690. doi: 10.1002/anie.201603329. [DOI] [PubMed] [Google Scholar]
- 13.(a) Zhan M, Li RZ, Mou ZD, Cao CG, Liu J, Chen YW, Niu D. ACS Catal. 2016;6:3381. [Google Scholar]; (b) Kim J, Park S, Park J, Cho SH. Angew Chem, Int Ed. 2016;55:1498. doi: 10.1002/anie.201509840. [DOI] [PubMed] [Google Scholar]; (c) Shi Y, Hoveyda AH. Angew Chem, Int Ed. 2016;55:3455. doi: 10.1002/anie.201600309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Sun C, Potter B, Morken JP. J Am Chem Soc. 2014;136:6534. doi: 10.1021/ja500029w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Potter B, Szymaniak AA, Edelstein EK, Morken JP. J Am Chem Soc. 2014;136:17918. doi: 10.1021/ja510266x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Matteson DS, Moody RJ, Jesthi PK. J Am Chem Soc. 1975;97:5608. [Google Scholar]; (b) Matteson DS, Moody RJ. J Org Chem. 1980;45:1091. [Google Scholar]
- 16.(a) Matteson DS, Thomas JR. J Organomet Chem. 1970;24:263. [Google Scholar]; (b) Zhang L, Huang Z. J Am Chem Soc. 2015;137:15600. doi: 10.1021/jacs.5b11366. [DOI] [PubMed] [Google Scholar]
- 17.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 18.(a) Dombray T, Werncke CG, Jiang S, Grellier M, Vendier L, Bontemps S, Sortais JB, Sabo-Etienne S, Darcel C. J Am Chem Soc. 2015;137:4062. doi: 10.1021/jacs.5b00895. [DOI] [PubMed] [Google Scholar]; (b) Obligacion JV, Semproni S, Chirik PJ. J Am Chem Soc. 2014;136:4133. doi: 10.1021/ja500712z. [DOI] [PubMed] [Google Scholar]; (c) Schaefer BA, Margulieux GW, Small BL, Chirik PJ. Organometallics. 2015;34:1307. [Google Scholar]; (d) Obligacion JC, Semproni SP, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:10645. doi: 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]; (e) Léonard NG, Bezdek MJ, Chirik PJ. Organometallics. 2017;36:142. [Google Scholar]; (f) Furukawa T, Tobisu M, Chatani N. Chem Commun. 2015;51:6508. doi: 10.1039/c5cc01378j. [DOI] [PubMed] [Google Scholar]
- 19.Jones WD, Feher FJ. Acc Chem Res. 1989;22:91. [Google Scholar]
- 20.Chen H, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]
- 21.(a) Cook AK, Schimler SD, Matzger AJ, Sanford MS. Science. 2016;351:1421. doi: 10.1126/science.aad9289. [DOI] [PubMed] [Google Scholar]; (b) Smith KT, Berritt S, González-Moreiras M, Ahn S, Smith MR, Baik MH, Mindiola DJ. Science. 2016;351:1424. doi: 10.1126/science.aad9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Kawamorita S, Miyazaki T, Iwai T, Ohmiya H, Sawamura M. J Am Chem Soc. 2012;134:12924. doi: 10.1021/ja305694r. [DOI] [PubMed] [Google Scholar]; (b) Kawamorita S, Murakami R, Iwai T, Sawamura M. J Am Chem Soc. 2013;135:2947. doi: 10.1021/ja3126239. [DOI] [PubMed] [Google Scholar]; (c) Mita T, Ikeda Y, Michigami K, Sato Y. Chem Commun. 2013;49:5601. doi: 10.1039/c3cc42675k. [DOI] [PubMed] [Google Scholar]; (d) Cho SH, Hartwig JF. J Am Chem Soc. 2013;135:8157. doi: 10.1021/ja403462b. [DOI] [PubMed] [Google Scholar]; (e) Cho SH, Hartwig JF. Chem Sci. 2014;5:694. [Google Scholar]
- 23.(a) Shimada S, Batsanov AS, Howard JAK, Marder TB. Angew Chem, Int Ed. 2001;40:2168. doi: 10.1002/1521-3773(20010601)40:11<2168::AID-ANIE2168>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T, Ishida K, Takagi J, Miyaura N. Chem Lett. 2001;30:1082. [Google Scholar]; (c) Larsen MA, Wilson CV, Hartwig JF. J Am Chem Soc. 2015;137:8633. doi: 10.1021/jacs.5b04899. [DOI] [PubMed] [Google Scholar]; (d) Palmer WN, Obligacion JV, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:766. doi: 10.1021/jacs.5b12249. [DOI] [PubMed] [Google Scholar]; (e) Manna K, Ji P, Lin Z, Greene FX, Urban A, Thacker NC, Lin W. Nat Commun. 2016;7:12610. doi: 10.1038/ncomms12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For examples of general methods for C(sp3)–H polyborylation, see refs 22c and e (directed) or 23d (non-directed).
- 25.Carmona E, González F, Poveda ML, Atwood JL, Rogers RD. J Chem Soc, Dalton Trans. 1981:777. [Google Scholar]
- 26.(a) Scheuermann ML, Johnson EJ, Chirik PJ. Org Lett. 2015;17:2716. doi: 10.1021/acs.orglett.5b01135. [DOI] [PubMed] [Google Scholar]; (b) Noda D, Tahara A, Sunada Y, Nagashima H. J Am Chem Soc. 2016;138:2480. doi: 10.1021/jacs.5b11311. [DOI] [PubMed] [Google Scholar]; (c) Schuster CH, Diao T, Pappas I, Chirik PJ. ACS Catal. 2016;6:2632. [Google Scholar]; (d) Pappas I, Treacy S, Chirik PJ. ACS Catal. 2016;6:4105. [Google Scholar]
- 27.Eremenko IL, Nefedov SE, Sidorov AA, Golubnichaya MA, Danilov PV, Ikorskii VN, Shvedenkov YG, Novotortsev VM, Moiseev II. Inorg Chem. 1999;38:3764. [Google Scholar]
- 28.Batey RA, Pedram B, Yong K, Baquer G. Tetrahedron Lett. 1996;37:6847. [Google Scholar]
- 29.Pereira S, Srebnik M. Tetrahedron Lett. 1995;36:1805. [Google Scholar]
- 30.(a) Molander GA, Bobbitt KL, Murray CK. J Am Chem Soc. 1992;114:2759. [Google Scholar]; (b) Curtis ADM, Mears RJ, Whiting A. Tetrahedron. 1993;49:187. [Google Scholar]
- 31.The geminal diboronate functionality in the conjugate addition products in Schemes 3 and 4 can likely be further elaborated using the methods described in refs 8–14.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.