

Abstract
The triterpenoid ursolic acid (UA) has been proposed as a potential cancer chemopreventive agent in many preclinical and clinical studies. In the present work, we aimed to characterize the pharmacokinetics (PK) of UA and to quantitatively assess the anti-oxidative and anti-inflammatory effects of UA, which are potentially linked to its chemopreventive efficacy. UA was administered intravenously (i.v., 20 mg/kg) or by oral gavage (100 mg/kg) to male Sprague-Dawley rats, and blood samples were collected at a series of designated time points. The plasma concentration of UA was determined using a validated liquid chromatography-mass spectrometry (LC-MS) approach. A biexponential decline in the UA plasma concentration was observed after i.v. dosing and was fitted to a two-compartmental model. The expression levels of phase II drug metabolism (DM)/anti-oxidant genes and the inflammatory iNos gene in corresponding treatment arms were measured using qPCR as a pharmacodynamic (PD) marker. The expression of phase II DM/anti-oxidant genes increased and peaked approximately 3 h after 20 mg/kg UA treatment. In a lipopolysaccharide (LPS)-induced acute inflammation model, UA inhibited LPS-stimulated iNos expression and that of the epigenetic markers the DNA methyltransferases (DNMTs) and histone deacetylases (HDACs) in leukocytes. A PK–PD model using Jusko's indirect response model (IDR) with transition compartments (TC) was established to describe the time delay and magnitude of the gene expression elicited by UA. The PK-PD model provided reasonable fitting linking the plasma concentration of UA simultaneously with the PD response based on leukocyte mRNA expression. Overall, our results indicate that UA is effective at inducing various phase II DM/anti-oxidant genes and inhibiting pro-inflammatory genes in vivo. This PK-PD modeling approach may provide a conceptual framework for the future clinical evaluation of dietary chemopreventive agents in humans.
Keywords: ursolic acid, anti-oxidant, inflammation, PKPD modeling, indirect response model
1. Introduction
Ursolic acid (UA) is a natural pentacyclic triterpenoid present at abundant levels in fruits and herbs such as cranberry, apple peels, basil, and rosemary 1. Overwhelming evidence has suggested that the consumption of medicinal plants rich in UA provides health benefits against various human diseases 2. In particular, the potential role for UA in prevention and treatment of chronic disease like obesity and cancer has gained increasing attention. Cellular and animal studies have demonstrated that UA can block the development of multiple types of tumors by targeting various signaling pathways 3. Several clinical trials have also been conducted to evaluate the pharmacokinetics of different UA formulations in both healthy volunteers and patients with solid tumors 4-6.
Cancer initiation and development and most other chronic illnesses including diabetes, obesity, cardiovascular disease, and neurologic disease are closely linked to oxidative stress and chronic inflammation 2, 7. Oxidative stress is a consequence of the increased generation of reactive oxygen species (ROS)/reactive nitrogen species (RNS) and/or decreased capacities of anti-oxidant defense systems in the body. Under physiological conditions, endogenous ROS/RNS generated from metabolic processes serve as essential signaling molecules to maintain cellular homeostasis 8. In addition, exogenous challenges such as UV radiation, lipid peroxidation, and inflammation can also trigger ROS/RNS generation 9. In the context of inflammation, ROS/RNS are generated by immune cells to eliminate invading pathogens 10. The secretion of ROS/RNS further attracts more activated immune cells. If the acute immune response fails to remove the exogenous stimuli, excessive cellular ROS/RNS will be produced during prolonged inflammation, thereby inflicting damage on surrounding tissues 11, 12. This exacerbation of oxidative stress and chronic inflammation can result in cellular genetic instability and/or epigenetic alterations, leading to the deregulation of tumor suppressors and oncogenes that drive the initiation of carcinogenesis 13. In addition, significant evidence shows that cancer has the common thread of inflammation and dysregulation of many related pathways with most other chronic diseases 14. Hence, protection against oxidative stress and inflammation is considered an important mechanism contributing to the anti-cancer effect of UA 15, 16.
Cells possess anti-oxidant defense machinery to convert or conjugate hyperactive ROS/RNS into less deleterious molecules. By modulating endogenous substances and xenobiotic elimination/excretion, phase II anti-oxidant and detoxifying enzymes play an essential role in maintaining homeostasis against intracellular or environmental challenges, such as oxidative stress 17. Induction of these phase II drug metabolism (DM)/anti-oxidant enzymes (e.g., NAD(P)H: quinone oxidoreductase, Nqo-1; heme oxygenase-1, Ho-1; UDP-glucuronosyl transferases, Ugt; among others) has been shown to be an effective strategy for chemoprevention using phytochemicals 18. The transcriptional regulation of phase II/anti-oxidant defense genes is typically mediated by nuclear factor (erythroid-derived 2)-like 2 (NFE2L2, Nrf2). When challenged by stress conditions, Nrf2 disassociates from its cytosol-repressive partner Kelch-like ECH-associated protein 1 (Keap1), subsequently accumulating in the nucleus and enhancing the transcriptional activity of cytoprotective phase II/anti-oxidant genes through binding to the anti-oxidant response element (ARE) located in the promoter region of the gene. In addition to activation of the cellular anti-oxidant machinery, Nrf2 can also play an influential role in the suppression of pro-inflammatory signaling pathways 19. For instance, genetic Nrf2 knockout mice are more susceptible to a carcinogen-induced tumorigenesis model, whereas chemopreventive agents show lower effectiveness in Nrf2-deficient mice 20. Considering the above findings, the Nrf2 pathway has been proposed as a primary cellular target for cancer chemoprevention, and Nrf2-mediated phase II gene expression may serve as a valuable surrogate marker in chemoprevention.
Recent studies have indicated that UA exerts a cellular protective effect via the Nrf2 pathway in different experimental models 21, 22. However, to date, no study has been conducted to link the pharmacokinetics (PK) and pharmacodynamics (PD) of UA in vivo. Additionally, gene expression in leukocytes, an easily accessible tissue, may serve as a valuable PD marker for future clinical trials. In this study, we conducted a PK study using rat plasma following the intravenous (i.v.) or oral administration of UA and simultaneously quantitated the corresponding gene expression levels as PD markers in leukocytes.
2. Materials and Methods
2.1 Chemicals and reagents
UA, glycyrrhetinic acid (GA), used as an internal standard (IS), lipopolysaccharide (LPS), ethanol, ethyl acetate, ammonium acetate, formic acid, and red blood cell lysis buffer were purchased from Sigma-Aldrich (St. Louis, MO, USA). HPLC-grade acetonitrile (ACN) and pure water were purchased from Honeywell Burdick & Jackson (Muskegon, MI, USA). Sodium chloride injections (0.9%) and heparin sodium injections (1,000 U/mL) were purchased from Baxter Healthcare Corporation (Deerfield, IL, USA) and Hospira Inc. (Lake Forest, IL, USA), respectively.
2.2 Animal treatment and sample collection
Male Sprague-Dawley rats (weighing 250-300 g) bearing jugular vein cannula were obtained from Hilltop Labs (Scottsdale, PA, USA). The animals were housed in the Animal Care facility at Rutgers University under 12-hour light-dark cycles with free access to food and water. All procedures and protocols were approved by the Institutional Animal Care and Use Committee at Rutgers University. Upon arrival, the animals were fed an anti-oxidant-free AIN-93 diet and were allowed to acclimatize for 3 days. Rats (n=4) were given UA (20 mg/kg) in a vehicle consisting of cremaphor/Tween-80/PEG/ethanol and water (2:1:1:1:5) (i.v.) through an exteriorized cannula in a final injection volume of 0.2 mL, followed by flushing with one volume of heparinized saline solution (10 U/ml). To characterize the inflammation-associated gene expression, 10 μg/kg LPS (i.v.) was given alone or in combination with UA. For oral dosing, rats (n=4) were given UA (100 mg/kg) in a similar vehicle composition by oral gavage in a final volume of 1.0 mL. Blood samples (∼200 μL) were withdrawn at 0, 15, 30, 45 min and 1, 2, 3, 4, 6, 8, 12, and 24 h following the i.v. or oral administration of UA. Plasma samples were separated from whole blood by centrifugation and were frozen at -80°C until analysis. Red blood cell lysis buffer was added into the remaining cell pellets for leukocyte preparation. The resultant cell pellets were dissolved in extraction buffer from the PicoPure® RNA Isolation Kit (Applied Biosystems, Foster City, CA, USA) and were stored at -80°C until analysis.
2.3 Sample preparation and bioanalysis of UA in plasma
Plasma samples (100 μL each) spiked with the internal standard GA (10 μL, 5.0 μg/mL) were precipitated and extracted twice using ethyl acetate: methanol (95/5, v/v). After vortexing, the extraction mixtures were centrifuged at 10,000 g at 4°C for 5 min. The supernatants were transferred to a glass insert and dried under a stream of nitrogen and then were reconstituted with 100 μL of acetonitrile: water (50/50, v/v). Calibration standards were prepared by spiking a series of UA working standard solutions and the IS into 100 μL of blank rat plasma, and then the same extraction procedure was followed as that for the plasma samples above.
The HPLC separation of UA was achieved using a Zorbax Eclipse XDB C18 column (3.5 μm, 4.6×50 mm; Agilent Technologies, Santa Clara, CA, USA). Mobile phase A was composed of 0.1% formic acid in water, mobile phase B was composed of 0.1% formic acid in ACN. The flow rate was 200 μL/min with the following linear gradient elution: 0 to 1.0 min, 85% A; 1.0 to 2.0 min, 85-15% A; 2.0 to 7.0 min, 15% A; 7.0 to 8.0 min, 15%-85% A; and 8.0 to 10.0 min, 85% A. The temperature of the column oven was set as 40°C, and the sample injection volume was 10 μL.
Quantification of UA was performed using the Finnigan LTQ ion-trap mass spectrometer (Thermo Fisher Scientific Inc., San Jose, CA, USA) equipped with an ESI source. The MS system was operated in the negative ion mode. Detection of UA and IS was performed in the selected ion monitoring (SIM) mode following the ions with (m/z)- 455 and 469, respectively. Data acquisition, peak integration and quantitation were performed using the Xcalibur Data System (Thermo Electron, San Jose, CA, USA). The lower and upper limits of quantification were 10 ng/mL and 20 μg/mL, respectively. The plasma calibration curve was linear, with r2>0.99.
2.4 Measurement of mRNA expression in leukocytes
Total RNA was extracted from leukocytes following the protocol of the PicoPure® RNA Isolation Kit. RNA concentrations were quantified using the Infinite M200 NanoQuant spectrophotometer (Tecan, Mannedorf, Switzerland). First-strand cDNA was synthesized from 300 ng of RNA using TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA, USA). Quantitative PCR was conducted using SYBR Green reagent on the QuantStudio5 Real-Time PCR system. The relative mRNA levels were calculated using the ΔΔCt approach, and β-actin was used as a reference for gene expression normalization. PD data were presented as the gene fold change at each time point against their respective expression in the vehicle control arm.
2.5 PK/PD model development
Non-compartment analysis was first conducted on the time-course of UA plasma concentration. Briefly, the area under the curve (AUC) was calculated using the linear trapezoidal rule. The terminal half-life (t1/2) was estimated from the equation t1/2=1n2/λ(z), where λ(z) was defined as the slope of the terminal phase in the concentration-time curve. Clearance (CL) was derived from dividing the dose by the AUC. The mean residence time (MRT) was obtained by dividing the area under the first moment curve (AUMC) by the AUC. The volume of distribution (Vss) was calculated as Vss=CL×AUMC/AUC.
The PK/PD modeling was conducted sequentially: a structural population PK model was firstly established using the nonlinear mixed-effect modeling program (NONMEM version 7.3; ICON Development Solution, MD, USA), with the aim of a good characterization of the absorption and the elimination phase. UA plasma concentration data were natural logarithm-transformed. The model was built using the UA plasma concentration-time profiles following both i.v. and oral dosing with first-order conditional estimation with the interaction option (FOCEI). Based on visual inspection and data-fitting criteria, UA plasma concentration-time data were best characterized by a two-compartment model with delayed first-order absorption (Figure 1).
Figure 1.
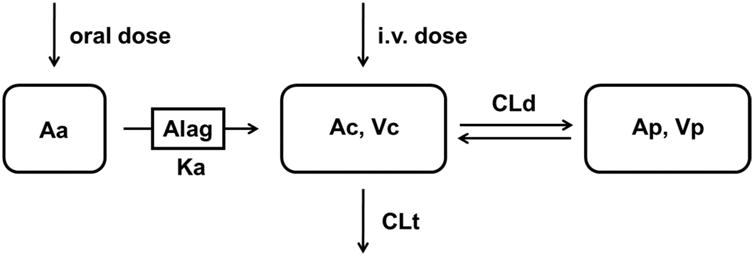
Schematic of final two-compartment model that describes UA PK following both i.v. and oral doses. Vc is the central compartment; Vp is the peripheral compartment; CLd is intercompartmentral clearance between the central compartment and peripheral compartment; CL is the plasma clearance.
| (1) |
| (2) |
| (3) |
Eqs. 1, 2, and 3 show the differential equations for i.v. and oral dosing, where Vc and Vp represent the volume of distribution in the central and peripheral compartment, respectively. CL and CLd represent the total clearance from the central compartment and inter-compartment distribution clearance, respectively. Ac and Ap represent the amount of UA in the central and peripheral compartments; thus, the plasma concentration C equals Ac/Vc.
To quantitatively evaluate the anti-oxidant and anti-inflammatory effects of UA, these two sets of gene mRNAs were measured as the PD response. The relative level of all phase II/anti-oxidant genes in this study was described by an indirect response model incorporated with a time-dependent transduction system (Figure 2) 23.
Figure 2.
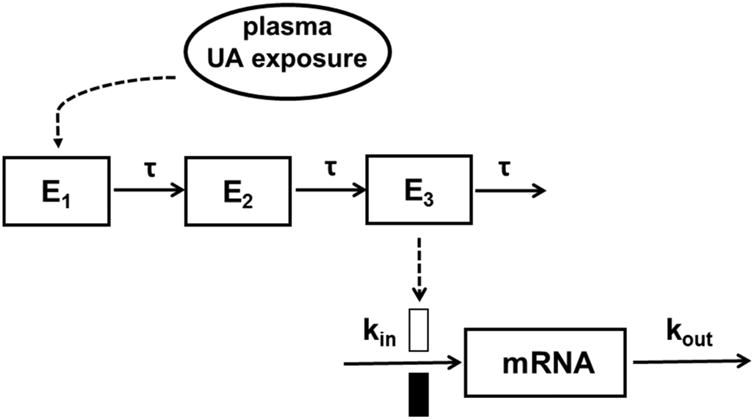
Schematic of the PK-PD model for UA-mediated induction of anti-oxidant phase II genes. The model is defined by Equations 4-8 in the text.
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
The initial biologic signal effector was denoted as E1, where the plasma drug concentration is linked to the rate of change of the signal induction by the Hill function (Eq. 4). Emax is the maximum ability of UA to stimulate signal initiation. EC50 represents the concentration of UA resulting in 50% of the maximum induction. En represents the response in the transit compartments where τ is the mean transit time within each compartment. Finally, an indirect response model with the stimulation of input (kin) by treatment was employed to describe the mRNA formation (PD) (Eq. 8). The relative gene fold change was used as the PD marker with the initial condition defined as mRNA (0) = 1. The typical population parameters obtained from the final PK model were used as input variables to estimate drug concentrations in plasma for PD modeling using the ADAPT5 program (Biomedical Simulations Resource, University of Southern California, Los Angeles, CA) 24.
An LPS-induced acute inflammation model was used to evaluate the anti-inflammatory potential of UA. Similarly, the relative expression of the pro-inflammatory iNos gene was modeled using a time-dependent transduction system (Figure 3). Since the blood concentration of LPS cannot be easily quantified, we set the initial LPS stimulation as S1 and assumed that the loss rate of this effect was linear and described with a rate constant Km. The production of iNos mRNA was induced by signal transduction triggered by LPS and inhibited by UA treatment (Eq. 12), where the maximum inhibition (Imax) was fixed as 1.
Figure 3.
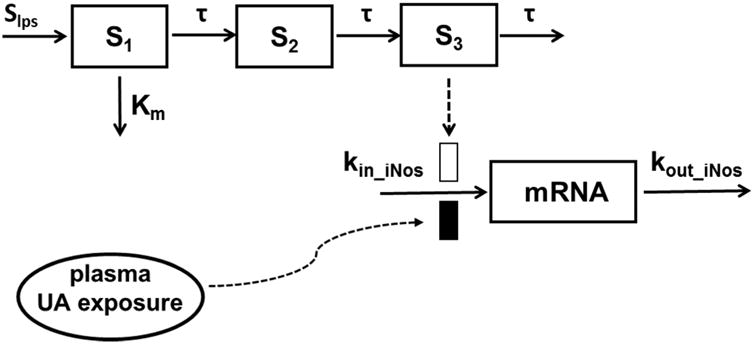
Schematic of the PK-PD model for UA-mediated suppression of LPS-induced iNos expression. The model is defined by Equations 2, 3, and 9-12 in the text.
| (9) |
| (10) |
| (11) |
| (12) |
2.6 Statistical analysis
Statistical significance was tested by one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test between multiple experimental groups and Student's t-test between two experimental groups using GraphPad PRISM software. The values are presented as the means ± SD (standard deviation). p-values of less than 0.05 were considered statistically significant.
3. Results
3.1 Pharmacokinetics of UA
The plasma concentration (Cp)–time (t) profiles of UA following the i.v. administration of 20 mg/kg and oral dosing of 100 mg/kg are shown in Figure 3. The results of non-compartmental analysis are listed in Table 1. The Cp vs. t of the UA profile showed a biexponential decline that could be well-captured by a two-compartment PK model. The two-compartment PK parameter estimates are also summarized in Table 2, and the %CV of these parameters was within a reasonable range. The estimated PK parameters of UA were then used as the input of PK to fit the PK-PD data as described below.
Table 1.
Parameters estimates of UA pharmacokinetics in rat plasma.
| PK parameters | Description | Estimation (CV%) | |||
|---|---|---|---|---|---|
|
| |||||
| i.v. | oral | ||||
|
|
|||||
| Cmax (ng/ml) | Maximum concentration observed | - | 2025(15.3) | ||
| Tmax (h) | Time to reach maximum concentration | - | 0.833(14.1) | ||
| AUC0-12h (ng/ml*h) | Area under the curve 0 to 12h | 8643 (22.8) | 3840 (14.5) | ||
| AUC0-∞(ng/ml*h) | Area under the curve 0 to infinity | 9021 (21.2) | 3891 (14.1) | ||
| AUMC0-12h (ng/ml*h2) | Area under the first moment curve 0 to 12h | 11287 (6.57) | 8239 (14.0) | ||
| AUMC0-∞(ng/ml*h2) | Area under the first moment curve 0 to infinity | 18241 (3.55) | 9012(12.4) | ||
| MRT(h) | Mean residence time | 2.128 (23.9) | 2.328 (7.70) | ||
| MAT(h) | Mean absorption time | - | 0.200 | ||
| CL(L/h/kg) | Total clearance | 2.317 (20.5) | 26.621* (13.5) | ||
| F (%) | Bioavailability | - | 8.72 | ||
These parameters are presented as P/F, where F is the bioavailability of UA.
Table 2.
Final population pharmacokinetics parameters.
| PK parameters | Description | Estimation |
|---|---|---|
| Vc (L/kg) | Volume of distribution in central compartment | 0.553 |
| Vp (L/kg) | Volume of distribution in peripheral compartment | 2.27 |
| CLt (L/h/kg) | Elimination clearance | 2.03 |
| CLd (L/h/kg) | Intercompartment clearance | 0.607 |
| Ka (h-1) | First-order absorption rate constant | 0.925 |
| Alag (h) | absorption delay | 0.221 |
| F (%) | Bioavailability | 8.18 |
3.2 PCR of mRNA in leukocytes and the PK-PD relationship
The mRNA levels of the selected genes were quantified by quantitative reverse transcription polymerase chain reaction (RT-qPCR). The time courses of the relative expression of phase II DM/anti-oxidant genes and the iNos gene in leukocytes are shown in Figures 5 and 6, respectively. The expression levels of Ho1, Nqo1, and Ugt1a1 were increased following the i.v. administration of 20 mg/kg UA. The peak time for gene induction was approximately 3-4 h and for the fold changes in the Ho1, Nqo1, and Ugt1a1 mRNA expression level are about 7, 5, and 4, respectively (P < 0.05); subsequently, the level gradually declined to baseline (Figure 5). The fold changes in the iNos mRNA expression level were determined following treatment with 10 μg/kg LPS alone or LPS combined with 20 mg/kg UA (Figure 6). An indirect response model 25 with transit compartments was used to describe the quantitative aspects of the gene expression profiles. The results of the PK-PD analysis were fitted using ADAPT5. iNos expression was induced by LPS but suppressed by concomitant UA administration. The estimated PD parameters for phase II genes and iNOS are presented in Tables 3 and 4, respectively.
Figure 5.
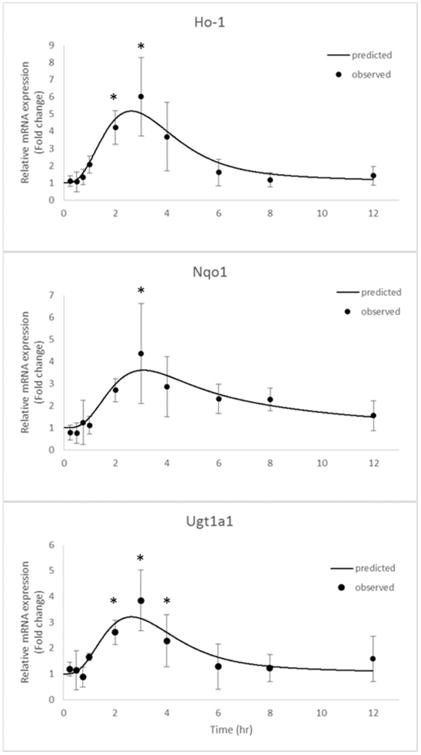
Phase II anti-oxidant gene expression, including Ho1, Nqo1, and Ugt1a1, as described by the designated PK-PD model. The black dots represent the mean of the observed data in rat leukocytes, and the lines represent the model prediction. * The difference was statistically significant when p < 0.05 compared with the first time point.
Figure 6.
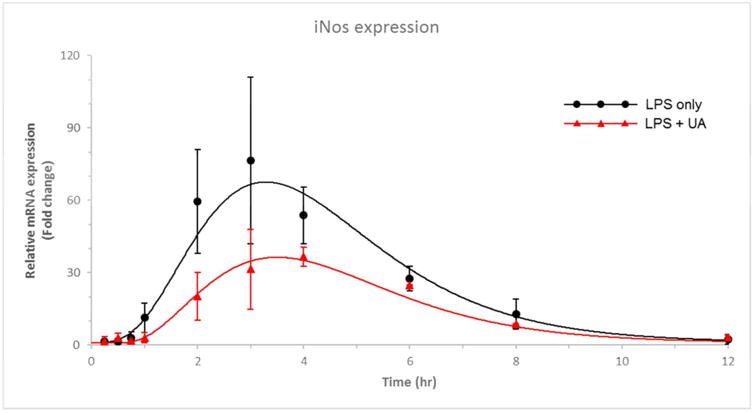
iNos gene expression, as described by the designated PK-PD model. The red and black dots represent the mean of the observed data in rat leukocytes, and the lines represent the model prediction for the LPS alone and LPS plus UA treatment groups.
Table 3.
Pharmacodynamic parameters of phase II antioxidant gene expression estimated from the designated PK-PD model shown in Figure 1.
| mRNA | Emax (CV%) | EC50(μg/mL) (CV%) | τ (h) (CV%) | kin (h-1) (CV%) |
|---|---|---|---|---|
| Ho1 | 13.2 (42.9) | 0.368 (45.7) | 0.767 (fixed) | 1.306 (57.7) |
| Nqo1 | 7.330 (35.6) | 1.738 (42.3) | 0.710 (fixed) | 1.436 (79.3) |
| Ugt1a1 | 8.520 (63.2) | 1.360 (62.2) | 0.751 (fixed) | 1.771 (70.2) |
Table 4.
Pharmacodynamic parameters of iNos expression estimated from the designated PK-PD model shown in Figure 2.
| PD parameter | Estimation | CV(%) |
|---|---|---|
| Slps | 185.8 | 24.03 |
| τ (h) | 0.680 | Fixed |
| Km (h-1) | 0.614 | 20.45 |
| Kin_iNos (h-1) | 1.474 | 38.59 |
| Imax | 1.0 | Fixed |
| IC50 (μg/mL) | 0.423 | 67.33 |
3.3 mRNA gene expression profiles of phase II DM/anti-oxidant genes after oral administration of UA
Following the oral administration of 100 mg/kg UA, the Cp vs. t profiles of UA are shown in Figure 4, and the mRNA gene expression profiles of phase II DM/anti-oxidant genes are shown in Figure 7. Similar to the i.v. condition, the expression levels of Ho1, Nqo1, and Ugt1a1 were increased following the oral administration of 100 mg/kg UA. The peak time for gene induction was approximately 3-4 h and for the fold changes in the Ho1, Nqo1, and Ugt1a1 mRNA expression level are about 5, 3, and 5, respectively (P < 0.05); subsequently, the level gradually declined to baseline (Figure 7).
Figure 4.
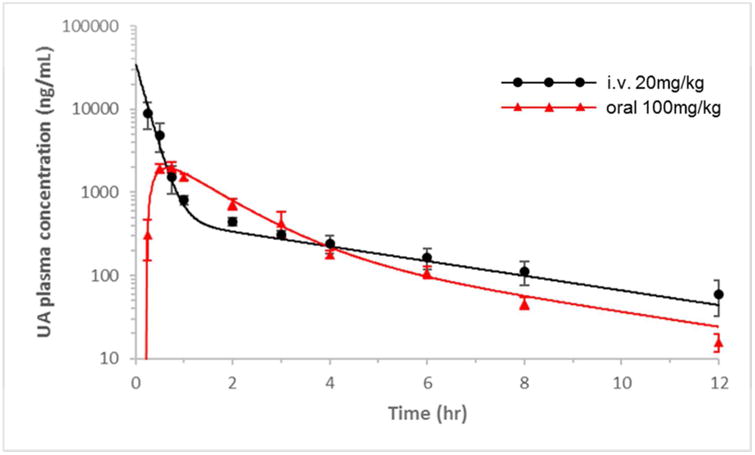
UA plasma pharmacokinetics as described by the two-compartment model. Experimental observations are shown as the mean ± SD. The solid line represents the model prediction for the 20 mg/kg i.v. dose and 100 mg/kg oral dose of UA.
Figure 7.

Changes in the anti-oxidant phase II gene levels, including Ho1, Nqo1, and Ugt1a1, after the oral administration of 100 mg/kg UA. The data are presented as the mean ± SD of the observed data in rat leukocytes. * The difference was statistically significant when p < 0.05 compared with the first time point.
3.4 mRNA gene expression profiles of epigenetic genes after the i.v. administration of UA
Following the i.v. administration of 10 μg/kg LPS and a combination of LPS and 20 mg/kg UA, the mRNA gene expression profiles of epigenetic markers, including DNA methyltransferases (DNMTs) and histone deacetylases (HDACs), are shown in Figure 7. The expression levels of DNMT1, DNMT3a, HDAC1, and HDAC3 were increased significantly at 0.5-1 h following LPS treatment, while UA attenuated the induction of these epigenetic markers mediated by LPS; the fold changes in these 4 genes are about 2 (P < 0.05) (Figure 8).
Figure 8.
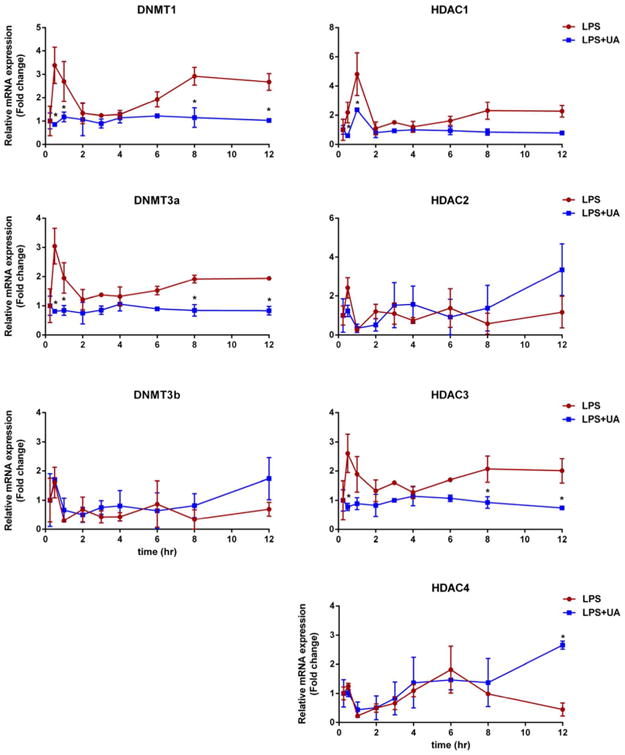
Changes in epigenetic-regulating enzymes, including DNMT1, -3a, and -3b and HDAC1, -2, -3, and -4 after the i.v. administration of LPS alone or LPS plus 20 mg/kg UA. The data are presented as the mean ± SD. * The difference was statistically significant when p < 0.05 compared with the LPS group.
4. Discussion
The anti-oxidant and anti-inflammatory effects of UA confer its cellular protective role against many stress conditions, such as ROS and carcinogens 16, 26. Previously, reports from our laboratory and others have demonstrated that UA could activate Nrf2, a master transcription factor regulating cell protective gene expression 22, 27. In this study, the plasma concentration of UA and the expression of phase II anti-oxidant genes in leukocytes were measured simultaneously. In addition, the iNos gene was evaluated in an LPS-induced acute inflammation model to characterize the anti-inflammatory activity of UA. Moreover, a PK-PD model was established to quantitatively describe the relationship between the PK concentrations of UA and its PD gene expression profiles.
Our preliminary unpublished results showed that UA exhibited linear PK behaviors at the i.v. doses of 5 and 20 mg/kg in rats (data not shown), while a dose higher than 50 mg/kg led to acute toxicity or animal death. Therefore, a tolerable dose of 20 mg/kg was selected in this study to establish the PK-PD relationship in rat leukocytes. According to the US FDA published “Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers (2005), the dose of 20 mg/kg UA in rats corresponds to a human equivalent dose (HED) of 3.3 mg/kg. This number can be converted to ∼130 mg/m2 for a typical human subject with 70 kg body weight and 1.8 m2 body surface area 28.
Following the 20 mg/kg i.v. dose, the plasma disposition of UA exhibited a bi-exponential decline that was well described by a two-compartment model with linear clearance (Figure 1). The apparent volume of distribution was high from the PK parameter estimates, suggesting extensive tissue distribution of UA. This finding was in agreement with a previous report showing that UA had a wide tissue distribution in the liver, lung, and cerebrum 29. The expression of Nrf2 target phase II anti-oxidant genes, including Ho1, Nqo1, and Ugt1a1, was measured in leukocytes, and the expression levels of these genes were increased after the i.v. administration of 20 mg/kg UA. The time course of Nrf2 downstream target gene expression lagged behind the UA plasma concentration. This type of delayed pharmacologic effect is widely seen, and Jusko's indirect response (IDR) model 25 could aptly describe the time delay of this type of PD response. In this context, we first attempted to employ the IDR model to build the PK-PD relationship. However, it appeared that the model prediction somehow underestimated the maximum effects (data not shown). In addition, the change in the mRNA levels at the initial stage was not quite robust, whereas in a typical IDR model, the first derivative of response would be mathematically highest immediately after an i.v. bolus dose. Moreover, the magnitude of the mRNA induction could not be well-captured, likely due to over-simplification of the underlying molecular mechanism of UA. As reviewed previously 30, the activation of gene expression is mediated by the disruption of the Nrf2-Keap1 interaction, an increased cytosol Nrf2 pool, the translocation of Nrf2, and transcription enhancement in the nucleus. This type of cellular signal transduction is not considered in a simple IDR model. Therefore, we introduced a set of time-dependent transduction compartments 23 prior to the IDR of mRNA synthesis (Figure 2). The transit compartments served as the upstream process essential for mRNA activation. Using the same IDR model structure integrated with the transit compartment, the effects of UA on gene induction were well described for all phase II anti-oxidant genes, including Ho1, Nqo1, and Ugt1a1.
In LPS-treated rats, iNos mRNA expression was used as the PD outcome to evaluate the anti-inflammatory effect of UA in this acute inflammation model. Upon LPS dosing alone, iNos expression increased and reached a peak at ∼3 h. When UA was given simultaneously with LPS, the mRNA level of iNos was considerably lowered compared with that elicited by LPS. One challenge in modeling inflammation-associated genes in response to LPS stimulation is that the plasma concentration of LPS cannot be easily quantified. In a previous study regarding the effects of methylprednisolone on the dynamics of lung iNos mRNA expression, an elegant quantitative assessment of iNos induction by LPS and suppression by methylprednisolone (MPL) was conducted using the IDR approach 31. However, in that model, the duration of LPS stimulation was fixed based on experimental observations. In the present study, we incorporated signal transduction and the IDR model to describe the mRNA expression of iNos (Figure 3). We assumed that LPS undergoes first-order elimination and that there is a direct reversible effect of LPS in stimulating the signal initiation 32. Therefore, the loss rate of LPS stimulation is linear and can be denoted as a single parameter Km. The signal triggered by LPS could further stimulate mRNA production (Kin_iNos), whereas UA could antagonize this effect. This model captured and described the time delay and magnitude of iNos gene expression in both the LPS-alone and UA-co-administration arms (Figure 6).
Our current model represents our first attempt to characterize the anti-oxidant and anti-inflammatory effects of UA at the gene transcription level. This model can describe the observed data and provides certain flexibility to capture the dynamics of gene expression in different treatment arms. However, the single dose may have restricted the PK-PD modeling assessment. While this study used the relative gene fold change (normalized to each time point in the control arm) as a PD marker, the circadian effects of mRNA expression may need to be considered in some situations. Although mechanistic consideration was incorporated in the current model, overall, it is still an empirical model. For instance, the total number of transit compartments is determined by a balance between model fitting and the current knowledge of signal activation. The transit compartments are virtual, and the lack of physiological reference means the transit time (τ) was used to reduce the parameter redundancy. A more mechanistic based model could be further developed by measuring the intermediate molecules involved in the signal transduction at a series of time points, as described previously 33.
In this study, we also performed the oral dosing of 100 mg/kg UA to evaluate its bioavailability. This dose selection was based on some commonly available UA nutrition supplements (300 – 600 mg per day). The PK estimate based on AUC data (Table 1) indicates that UA has a very low absolute bioavailability (∼8%), which may due, in part, to its low solubility. The dose we used in establishing the PK-PD model may appear to be high and difficult to achieve by dietary consumption or in the form of nutrition supplements. Thus, efforts have been made to develop UA derivatives with more favorable PK properties 34. In addition, several studies have attempted to improve bioavailability by modifying the formulation of UA 4, 35, 36. Nevertheless, we found that phase II anti-oxidant genes could still be slightly induced in leukocytes following an oral dose of 100 mg/kg UA (Figure 7). Given that the tissue distribution of UA is large, especially in the lung and liver 29, a similar inductive response of phase II enzymes would be possible in humans by UA supplementation. Beyond oxidative stress and chronic inflammation, epigenetic alteration is considered yet another factor that contributes to cancer initiation and development. The epigenetic control of gene expression depends on the dynamic change in chromatin conformation, which is generally determined by DNA methylation and post-translational histone modifications. Multiple levels of interactions between epigenetic regulation and oxidative stress/inflammation have been observed and documented in carcinogenesis 37, 38. Indeed, the role of UA in the epigenetic regulation of gene expression has been recently shown 22. In this study, we measured the mRNA levels of epigenetic modulators, including DNMTs and HDACs, and UA was found to decrease LPS-induced DNMT and HDAC expression. However, the impact of the cancer micro-environment, such as oxidative stress and inflammation, on epigenetic modulators is an interesting but not fully understood area of research.
In summary, we conducted PK and PD assessment after the i.v. or oral administration of UA. The anti-oxidative and anti-inflammatory roles of UA may confer its health benefits, including the chemopreventive effect. In this study, gene expression measured in leukocytes, an easily accessible tissue, was shown to serve as a potential surrogate PD marker. Furthermore, the PK-PD model established in this study could reasonably describe and link the PK and PD of UA. Thus, this study presents a feasible approach to apply PK-PD modeling and simulation in preclinical and clinical studies, particularly in the understanding of a drug's initial acute pharmacological response.
Acknowledgments
We thank all the members of Kong lab for their suggestions and helpful discussion in the preparation of this manuscript. This work was supported in part by institutional funds and R01-AT009152 from the National Center for Complementary and Integrative Health (NCCIH) and R01-CA200129 from the National Cancer Institute (NCI).
References
- 1.Ikeda Y, Murakami A, Ohigashi H. Ursolic acid: an anti- and pro-inflammatory triterpenoid. Molecular nutrition & food research. 2008;52(1):26–42. doi: 10.1002/mnfr.200700389. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochemical pharmacology. 2006;71(10):1397–421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G, Bishayee A. Ursolic acid in cancer prevention and treatment: molecular targets, pharmacokinetics and clinical studies. Biochemical pharmacology. 2013;85(11):1579–87. doi: 10.1016/j.bcp.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Wang XH, Zhou SY, Qian ZZ, Zhang HL, Qiu LH, Song Z, Zhao J, Wang P, Hao XS, Wang HQ. Evaluation of toxicity and single-dose pharmacokinetics of intravenous ursolic acid liposomes in healthy adult volunteers and patients with advanced solid tumors. Expert opinion on drug metabolism & toxicology. 2013;9(2):117–25. doi: 10.1517/17425255.2013.738667. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Z, Qian Z, Yan Z, Zhao C, Wang H, Ying G. A phase I pharmacokinetic study of ursolic acid nanoliposomes in healthy volunteers and patients with advanced solid tumors. International journal of nanomedicine. 2013;8:129–36. doi: 10.2147/IJN.S38271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian Z, Wang X, Song Z, Zhang H, Zhou S, Zhao J, Wang H. A phase I trial to evaluate the multiple-dose safety and antitumor activity of ursolic acid liposomes in subjects with advanced solid tumors. BioMed research international. 2015;2015:809714. doi: 10.1155/2015/809714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free radical biology & medicine. 2010;49(11):1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarti P, Avigliano L, Gorlach A, Brune B. Superoxide and nitric oxide--participation in cell communication. Cell death and differentiation. 2002;9(10):1160–2. doi: 10.1038/sj.cdd.4401099. [DOI] [PubMed] [Google Scholar]
- 9.Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacology & therapeutics. 2010;125(3):376–93. doi: 10.1016/j.pharmthera.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Babior BM. Phagocytes and oxidative stress. The American journal of medicine. 2000;109(1):33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- 11.Kong AN, Mandlekar S, Yu R, Lei W, Fasanmande A. Pharmacodynamics and toxicodynamics of drug action: signaling in cell survival and cell death. Pharm Res. 1999;16(6):790–8. doi: 10.1023/a:1011953431486. [DOI] [PubMed] [Google Scholar]
- 12.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31(1):37–49. doi: 10.1093/carcin/bgp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C, Kong AN. Dietary chemopreventive compounds and ARE/EpRE signaling. Free Radic Biol Med. 2004;36(12):1505–16. doi: 10.1016/j.freeradbiomed.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Mancha-Ramirez AM, Slaga TJ. Ursolic Acid and Chronic Disease: An Overview of UA's Effects On Prevention and Treatment of Obesity and Cancer. Advances in experimental medicine and biology. 2016;928:75–96. doi: 10.1007/978-3-319-41334-1_4. [DOI] [PubMed] [Google Scholar]
- 15.Ramos AA, Pereira-Wilson C, Collins AR. Protective effects of ursolic acid and luteolin against oxidative DNA damage include enhancement of DNA repair in Caco-2 cells. Mutation research. 2010;692(1-2):6–11. doi: 10.1016/j.mrfmmm.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Cho J, Rho O, Junco J, Carbajal S, Siegel D, Slaga TJ, DiGiovanni J. Effect of Combined Treatment with Ursolic Acid and Resveratrol on Skin Tumor Promotion by 12-O-Tetradecanoylphorbol-13-Acetate. Cancer prevention research. 2015;8(9):817–25. doi: 10.1158/1940-6207.CAPR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kensler TW. Chemoprevention by inducers of carcinogen detoxication enzymes. Environ Health Perspect. 1997;105 Suppl 4:965–70. doi: 10.1289/ehp.97105s4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su ZY, Shu L, Khor TO, Lee JH, Fuentes F, Kong AN. A perspective on dietary phytochemicals and cancer chemoprevention: oxidative stress, nrf2, and epigenomics. Topics in current chemistry. 2013;329:133–62. doi: 10.1007/128_2012_340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, Kong AN. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. 2008;76(11):1485–9. doi: 10.1016/j.bcp.2008.07.017. Epub 2008 Jul 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, Conney AH, Kong AN. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer research. 2006;66(16):8293–6. doi: 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 21.Liu W, Tan X, Shu L, Sun H, Song J, Jin P, Yu S, Sun M, Jia X. Ursolic acid inhibits cigarette smoke extract-induced human bronchial epithelial cell injury and prevents development of lung cancer. Molecules. 2012;17(8):9104–15. doi: 10.3390/molecules17089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim H, Ramirez CN, Su ZY, Kong AN. Epigenetic modifications of triterpenoid ursolic acid in activating Nrf2 and blocking cellular transformation of mouse epidermal cells. The Journal of nutritional biochemistry. 2016;33:54–62. doi: 10.1016/j.jnutbio.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mager DE, Jusko WJ. Pharmacodynamic modeling of time-dependent transduction systems. Clin Pharmacol Ther. 2001;70(3):210–6. doi: 10.1067/mcp.2001.118244. [DOI] [PubMed] [Google Scholar]
- 24.D'Argenio D, Schumitzky A, Wang X. ADAPT 5 User's Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. Biomedical Simulations Resource, Los Angeles. 2009 [Google Scholar]
- 25.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. Journal of pharmacokinetics and biopharmaceutics. 1993;21(4):457–78. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma JQ, Ding J, Zhang L, Liu CM. Ursolic acid protects mouse liver against CCl4-induced oxidative stress and inflammation by the MAPK/NF-kappaB pathway. Environmental toxicology and pharmacology. 2014;37(3):975–83. doi: 10.1016/j.etap.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain research. 2013;1497:32–9. doi: 10.1016/j.brainres.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 28.Sacco JJ, Botten J, Macbeth F, Bagust A, Clark P. The average body surface area of adult cancer patients in the UK: a multicentre retrospective study. PloS one. 2010;5(1):e8933. doi: 10.1371/journal.pone.0008933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Q, Luo S, Zhang Y, Chen Z. Development of a liquid chromatography-mass spectrometry method for the determination of ursolic acid in rat plasma and tissue: application to the pharmacokinetic and tissue distribution study. Analytical and bioanalytical chemistry. 2011;399(8):2877–84. doi: 10.1007/s00216-011-4651-x. [DOI] [PubMed] [Google Scholar]
- 30.Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48(2):91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sukumaran S, Lepist EI, DuBois DC, Almon RR, Jusko WJ. Pharmacokinetic/pharmacodynamic modeling of methylprednisolone effects on iNOS mRNA expression and nitric oxide during LPS-induced inflammation in rats. Pharmaceutical research. 2012;29(8):2060–9. doi: 10.1007/s11095-012-0733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibaldi M, Levy G, Weintraub H. Commentary Drug distribution and pharmacologic effects. Clinical pharmacology and therapeutics. 1971;12(5):734–42. doi: 10.1002/cpt1971125734. [DOI] [PubMed] [Google Scholar]
- 33.Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. Modeling endotoxin-induced systemic inflammation using an indirect response approach. Mathematical biosciences. 2009;217(1):27–42. doi: 10.1016/j.mbs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Gao Y, Wang A, Zhou X, Zheng Y, Zhou J. Evolution in medicinal chemistry of ursolic acid derivatives as anticancer agents. European journal of medicinal chemistry. 2015;92:648–55. doi: 10.1016/j.ejmech.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Oliveira Eloy J, Saraiva J, de Albuquerque S, Marchetti JM. Solid dispersion of ursolic acid in Gelucire 50/13: a strategy to enhance drug release and trypanocidal activity. AAPS PharmSciTech. 2012;13(4):1436–45. doi: 10.1208/s12249-012-9868-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang L, Sun Z, Zu Y, Zhao C, Sun X, Zhang Z, Zhang L. Physicochemical properties and oral bioavailability of ursolic acid nanoparticles using supercritical anti-solvent (SAS) process. Food chemistry. 2012;132(1):319–25. doi: 10.1016/j.foodchem.2011.10.083. [DOI] [PubMed] [Google Scholar]
- 37.Rokavec M, Oner MG, Hermeking H. lnflammation-induced epigenetic switches in cancer. Cellular and molecular life sciences: CMLS. 2016;73(1):23–39. doi: 10.1007/s00018-015-2045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Current drug targets. 2015;16(1):13–9. doi: 10.2174/1389450116666150113121054. [DOI] [PubMed] [Google Scholar]