

Abstract
The single nucleotide polymorphism, rs1990760, in the cytosolic viral sensor, IFIH1, results in an amino-acid change (p.A946T) and is associated with multiple autoimmune diseases. The impact of this polymorphism on both viral-sensing and autoimmune pathogenesis remains poorly understood. Here, we find that human PBMCs and cell lines with the risk variant, IFIH1T946, exhibit heightened, basal and ligand-triggered type I interferon (IFN-I) production. Consistent with these findings, IFIH1T946 knock-in mice display enhanced basal IFN-I expression, survive a lethal viral challenge, and exhibit increased penetrance in autoimmune models including a combinatorial impact with other risk variants. Further, IFIH1T946 mice manifest an embryonic survival defect consistent with enhanced responsiveness to RNA self-ligands. Together, our data support a model wherein autoimmune risk variant-driven, ligand-triggered IFN-I production functions to protect against viral challenge, likely accounting for its selection within human populations, but provides this advantage at the cost of modestly promoting the risk for autoimmunity.
Autoimmune diseases arise from a complex interplay of genetic and environmental factors1. The anti-viral immune system includes several pathways where the nexus between genetics, the environment, and autoimmunity has become evident. Type I interferon (IFN-I) and viral infection have each been associated with autoimmune diseases, including systemic lupus erythematosus (SLE) and type-1 diabetes (T1D)2-5 and genome-wide association studies (GWAS) have linked genes important for anti-viral immunity to these disorders. This association network highlights the delicate balance in selective pressure for mediating a more effective anti-viral response while limiting the risk for autoimmune sequelae. A striking example is interferon-induced helicase C domain-containing protein 1 (IFIH1; also known as MDA5) - a critical, upstream component in the innate response in nearly all cell types to RNA viruses including those in the Picornaviridae and Flaviviridae families6-8. IFIH1 contains two N-terminal caspase activation recruitment domains (CARDs) and a DExD/H-box helicase domain followed by a C-terminal domain (CTD) 9. While the full repertoire of ligands recognized by IFIH1 remain to be defined, previous work has shown that these viral families generate both long double-stranded RNAs (dsRNA) and branched high-molecular weight RNAs that contain both dsRNA and single-stranded RNAs (ssRNA) that stimulate signaling10,11. Upon coordinate recognition of dsRNA by the CTD and helicase domains, IFIH1 undergoes conformational changes leading to its oligomerization, assembly into filaments8 and association of its CARD domain with the signaling effector mitochondrial anti-viral signaling protein (MAVS). This initiates signaling events that ultimately promote transcription of IFN-I and hundreds of interferon stimulated genes (ISGs).
Gain-of-function missense mutations in the helicase domain of IFIH1 have been identified in subjects with the rare, early onset, auto-inflammatory disorder, Aicardi-Goutieres syndrome (AGS), a type I interferonapathy 12. The various mono-allelic mutants identified consistently target the dsRNA or ATP binding sites within the helicase domain. Activating IFIH1 helicase domain mutations have also been identified in Singleton-Merten Syndrome, a rare disorder characterized by variable penetrance of dental and skeletal abnormalities and aortic and cardiac calcifications. In addition, an activating mutation in IFIH1 (R779H), previously described in AGS, was identified in a single subject with early onset SLE and IgA deficiency13. Similarly, ENU mutagenesis in mice led to identification of an IFN-dependent inflammatory disease resulting from a gain-of-function Ifih1 helicase domain mutation14 although this specific mutation has not been associated with human disease.
In contrast to these penetrant, rare autosomal dominant disorders, GWAS studies have identified both protective and risk variants in IFIH1 in multiple common human autoimmune diseases15-18. One study reported a statistically significant allelic association with risk for T1D at rs1990760, a non-synonymous coding variant in IFIH115. Subsequent studies have reported similarly significant associations at this SNP in other autoimmune diseases including SLE as well as suggestive findings or regional associations in many additional disorders including psoriasis, rheumatoid arthritis, multiple sclerosis (MS), and vitiligo, implicating IFIH1 as playing a basic role in the genesis of autoimmunity17-21. Subsequent re-sequencing of IFIH1 in T1D cases and controls revealed that rare loss of function variants (including rs35667974; leading to an I923V amino-acid substitution) were associated with protection from disease16. In distinction to either rare loss-of-function variants or dominant mutations that promote auto-inflammatory disease, the rs1990760 risk allele is common, with an allele frequency of ~57% in European populations22 and predicted to result in an amino acid change from alanine to threonine at codon 946 (A946T) within the CTD of IFIH1 (IFIH1T946) of unknown functional significance.
While multiple studies have replicated and expanded the range of disorders associated with the IFIH1A946T autoimmune risk variant, little information exists with respect to its impact on protein function and/or signaling activity. Most importantly, its mechanistic impact(s) on viral control and immune tolerance in vivo remains unknown. In this study, we define the role of IFIH1T946 as a partial gain-of-function variant that establishes the capacity for enhanced protection against specific viral challenges while concurrently promoting the risk for autoimmune disease via heightened basal and ligand-dependent signaling. We demonstrate that IFIH1T946 promotes increased IFN-I production in vitro and in vivo. Consistent with these findings, IFIH1T946 enhances antiviral responses in vitro and viral control in vivo. In parallel, using two models of autoimmune disease, we show that the variant leads to an inflammatory state that promotes autoimmunity, and can function in concert with an additional common human autoimmune risk allele.
Results
PBMCs expressing IFIH1T946 display increased IFN signaling
To evaluate both the role of genetic variation at IFIH1 on T1D risk and the relationship of the different polymorphic sites reported to be associated with autoimmunity at this locus, we reanalyzed genotyping data from the Type 1 Diabetes Genetics Consortium23,24. Four rare variants (rs35744605, rs72650663, rs35667974 and rs3573203416) as well as these two common non-synonymous coding variants (rs1990760 and rs3747517 encoding A946T and H843R, respectively) were tested separately and as haplotypes for association with T1D. Of note, the common coding variant rs3747517 has also previously been reported to be associated with T1D, MS, and SLE17,25,26. Family-based single-marker analysis indicated that the G allele at rs3747517 and the A allele at rs1990760 were each associated with increased risk for T1D (Table 1). Haplotype analysis identified three haplotypes (H1, H2 and H4) that were significantly associated with T1D (Table 2). The H1 haplotype carrying the rs3747517 G allele and the rs1990760 A allele conferred risk for T1D, whereas the H2 and H4 haplotypes carrying the non-risk alleles of rs3747517 (A) and rs1990760 (G) protected against T1D (Table 2). In summary, the H1 risk haplotype encodes for arginine at position 843 and threonine at position 946 of the IFIH1 protein. Additionally this risk haplotype was found to be in a moderate linkage disequilibrium and associated with psoriasis16,27,28.
Table 1.
Single-Marker Family-Based Association Analysis of IFIH1 Markers
| dbSNP ID (137) | Exon/Intron | cDNA Change | Amino Acid Change | MAF | Familya (n) | Zb | P Valueb |
|---|---|---|---|---|---|---|---|
| rs35744605 | Exon 10 | c.1879G>T | p.Glu627Ter | 0.0034 | 36 | -0.792 | 0.43 |
| rs72650663 | Exon 11 | c.2105C>T | p.Thr702Ile | 0.0029 | 26 | -0.854 | 0.39 |
| rs3747517 | Exon 13 | c.2528A>Gc | p.His843Arg | 0.27(A) | 1592 | -3.925(A) | 0.000087 |
| rs35667974 | Exon 14 | c.2767A>G | p.Ile923Val | 0.0087 | 84 | -2.902 | 0.0037 |
| rs35732034 | Intron 14 | c.2807+1G>A | -- | 0.0061 | 66 | -1.300 | 0.19 |
| rs1990760 | Exon 15 | c.2836G>Ad | p.Ala946Thr | 0.41(G) | 1755 | -4.369(G) | 0.000012 |
Abbreviations are as follows: MAF, minor allele frequency; n, number.
Variant locations are based on reference gene accessions NM_022168.2 and NP_071451.2.
T1D sibpair/trio families collected by the Type 1 Diabetes Genetics Consortium.
Z scores and p values were calculated for the minor alleles.
A is the minor allele, which is opposite to dbSNP
G is the minor allele, which is opposite to dbSNP
Table 2.
Family-Based Haplotype Association Analysis of IFIH1
| Haplotype | rs35744605 | rs72650663 | rs3747517 | rs35667974 | rs35732034 | rs1990760 | Freq. | Familya (n) | Z | P Value |
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | 2 G | 2 C | 2 G Arg | 2 A | 2 G | 2 A Thr | 0.571 | 1678 | 4.797 | 1.61×10-6 |
| H2 | 2 G | 2 C | 1 A His | 2 A | 2 G | 1 G Ala | 0.269 | 1500 | -3.309 | 9.35×10-4 |
| H3 | 2 G | 2 C | 2 G Arg | 2 A | 2 G | 1 G Ala | 0.141 | 1009 | -1.119 | 0.26 |
| H4 | 2 G | 2 C | 1 A His | 1 G | 2 G | 1 G Ala | 0.009 | 86 | -3.022 | 0.0025 |
| H5 | 1 T | 2 C | 2 G Arg | 2 A | 2 G | 2 A Thr | 0.003 | 36 | -0.815 | 0.42 |
| H6 | 2 G | 2 C | 2 G Arg | 2 A | 1 A | 2 A Thr | 0.003 | 40 | -0.980 | 0.33 |
| H7 | 2 G | 1 T | 2 G Arg | 2 A | 1 A | 2 A Thr | 0.003 | 26 | -0.854 | 0.39 |
| H8 | 2 G | 2 C | 1 A His | 2 A | 2 G | 2 A Thr | <0.001 | 2 | -0.999 | 0.32 |
Abbreviations are as follows: 1, minor allele; 2, major allele; freq., frequency, n, number.
T1D sibpair/trio families collected by the Type 1 Diabetes Genetics Consortium.
To directly assess the impact of rs1990760 risk variant expression in primary human hematopoietic cells, we obtained peripheral blood mononuclear cells (PBMCs) from a number of healthy donors with (R/R) or without (NR/NR) the risk A allele at rs1990760. PBMCs were isolated and cultured with media alone or stimulated with the artificial dsRNA ligand, poly(I:C). PBMCs from all subjects had similar basal levels of IFIH1 mRNA (Supplementary Fig. 1a) that increased with poly(I:C) treatment. We found no differences in the ability to up-regulate IFIH1 transcripts levels in response to poly(I:C) between the IFIH1 genotypes (Fig. 1a; Supplementary Fig. 1a-b). Following poly(I:C) stimulation, homozygous risk donors, expressing the IFIH1T946 protein, exhibited increased Interferon-β (IFNB1) mRNA (Fig. 1b). Subjects expressing the IFIH1R843/T946 variant protein, representing the product of the identified risk haplotype H1, also displayed increased IFNB1 mRNA relative to R843 alone. This increase is thus likely attributable to the co-occurrence of the R843 and T946 substitutions (Fig. 1c). Based on these observations, for simplicity we refer to the H1 haplotype encoding for IFIH1R843/T946, as IFIH1R. Please refer to reference table for further explanation of detailed genotypes in this and all subsequent figures (Supplementary Table 1).
Figure 1. IFIH1R mediates a modest increase in poly(I:C) triggered IFN-β production and leads to a basal IFN-I signature.
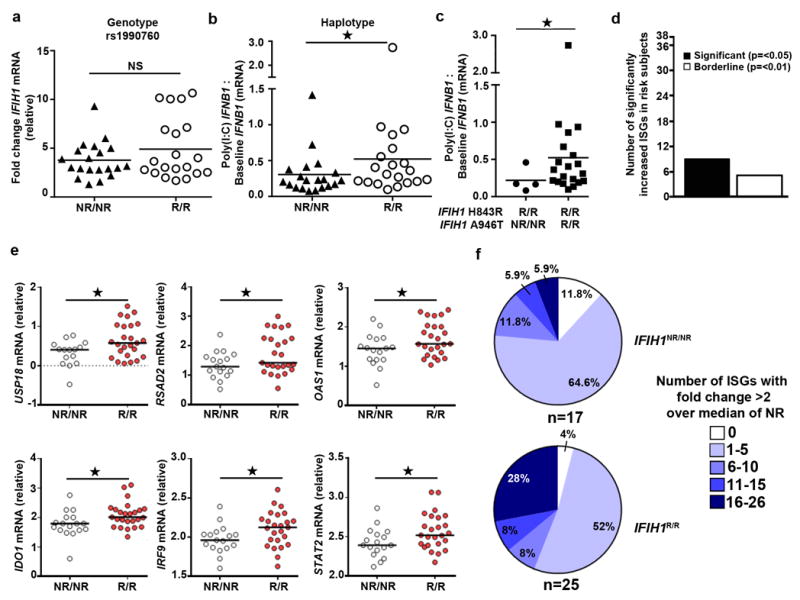
(a-c) Frozen PBMCs from healthy donors were thawed and stimulated with transfection of 1μg/ml of poly(I:C) for 24 hours. Quantitative RT-PCR was performed for IFNB1 and IFIH1 mRNA expression. Statistical analysis was performed using the Mann-Whitney U test (one-tailed). Each data point represents an individual. Bars represent median. (a) IFIH1 mRNA levels were normalized to POLR2A mRNA. Data is displayed as fold change post-stimulation relative to baseline. (b) Post-poly(I:C) IFNB1 mRNA expression was normalized to IFIH1 baseline mRNA levels. Assays (a-b) were performed twice with a total of 20 IFIH1NR/NR and 20 IFIH1R/R individuals based upon rs1990760 genotype. (c) Post-poly(I:C) IFNB1 mRNA expression normalized to IFIH1 baseline mRNA levels for subject according to non-risk or risk haplotype (n=4 for non-risk and n=20 risk haplotype, respectively). (d-f) PBMCs were thawed and rested for 24 hours. mRNA was analyzed using a custom high-throughput qPCR assay for ISG expression. Assay was performed twice with a combined total of n=17 IFIH1NR/NR and n=25 IFIH1R/R individuals. All subjects were homozygous for IFIH1R843. (d) Number of ISGs with a significant increase in expression level (vs. borderline change (p<0.1)) among 38 candidate ISGs. (e) Representative ISGs with significant differential expression. Data points show log-transformed relative expression. Welch’s T test was used for statistical analysis. Each data point represents an individual. (f) Percent of individuals with indicated number of ISGs with a fold change of >2 over the median expression in NR subjects. NR=non-risk (encoding IFIH1H843 and IFIH1A946 or); R=risk (encoding IFIH1R843 and IFIH1T946). NS=not significant. * p<0.05.
We next sought to determine if expression of IFIH1R/R was associated with evidence for enhanced IFN-I signaling in vivo. Using a custom high-throughput qPCR assay to measure mRNA expression of candidate ISGs, we found that PBMCs from healthy donors homozygous for the risk haplotype showed significantly elevated basal expression of 9 of 38 candidate ISGs (Fig. 1d-e; Supplementary Fig. 1c-d); none of the ISGs were significantly down-regulated relative to the IFIH1NR/NR PBMCs.
Furthermore, we examined the number of genes that were elevated per individual to assess the presence of an IFN-I signature marked by ISGs. Individuals carrying the risk haplotype exhibited an increased number of IFN signature genes compared to individuals carrying the non-risk haplotype. Hence IFIH1R/R leads to an IFN signature in healthy human subjects (Fig. 1f). Taken together, these results show that individuals carrying the IFIH1 risk haplotype exhibit both basal and post-ligand enhanced IFIH1 activity and basal IFN-I signaling.
IFIH1R promotes basal and ligand-triggered activity
In anticipation of establishing a knock-in murine model (described below), we generated a series of murine IFIH1 cDNA expression constructs to assess the function of candidate variant alleles in vitro following overexpression in HEK293T cells (Fig. 2a-c). The Ifih1 gene in C57Bl/6 mice encodes for the amino acid, R843, thus the murine IFIH1R cDNA precisely models the human H1 risk haplotype encoding for IFIH1R843/T946 (Fig. 2a). Control constructs included mIFIH1NR (with the naturally occurring R843 variant) and mIFIH1P, a cDNA with the I923V amino-acid substitution16,29 encoded by the rare rs35667974 variant that protects against T1D (Table 1). While all constructs yielded similar amounts of mIFIH1 protein (Fig. 2b-c; Supplementary Fig. 2a-b), transfection with the mIFIH1R construct led to a 2-3 fold increase in basal IFNB1 mRNA levels in comparison with the control, non-risk construct (mIFIH1NR) (Fig. 2d). This result was consistent with a previous report showing the human IFIH1T946 variant is a gain-of-function mutation14. Next, we explored the ability of mIFIH1R to respond to RNA ligands. Cells expressing each alternative murine IFIH1 construct were transfected with poly(I:C), and IFN-I levels were analyzed. Cells expressing the mIFIH1R construct exhibited a modest but consistent increase in inducible IFNB1 mRNA compared to mIFIH1NR transfected cells (Fig. 2e-f). In contrast, the rare variant rs35667974, whose minor allele protects against T1D (Table 1) and leads to an I923V amino-acid substitution, referred to here as mIFIH1P, ablated both basal (Fig 2d) and poly(I:C) triggered IFNB1 mRNA (Supplementary Fig. 2c-d)16,29.
Figure 2. Murine IFIH1T946 displays increased basal and heightened ligand-dependent signaling in vitro.
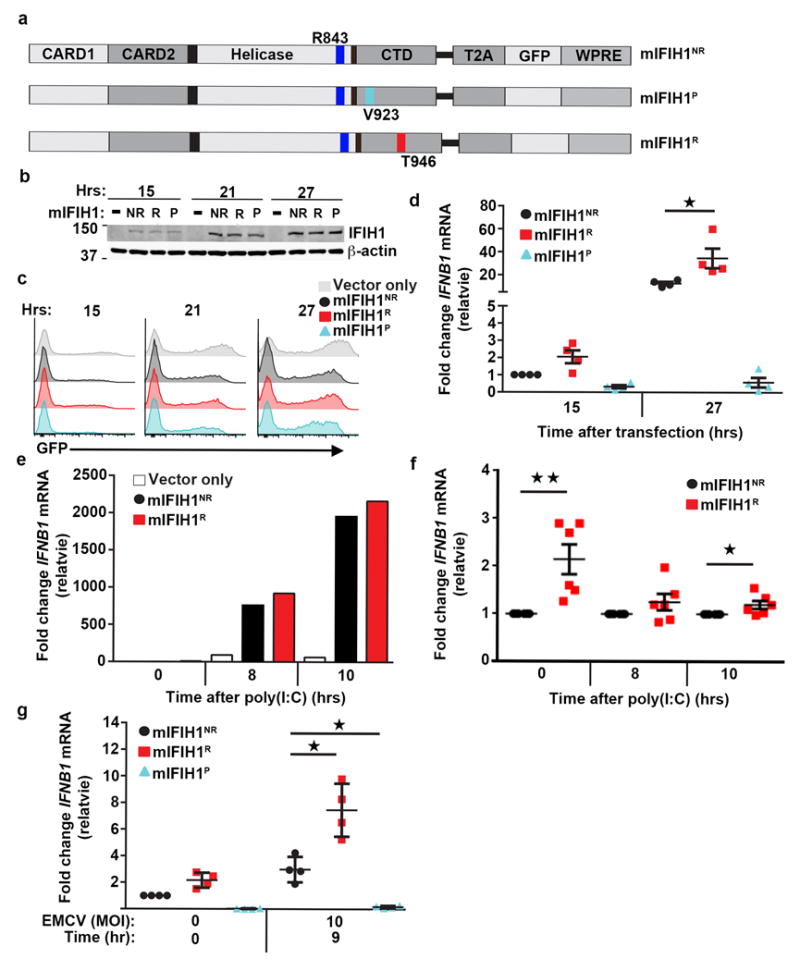
(a) Schematic of murine (m)IFIH1 constructs indicating domains, location of relevant amino-acid substitutions, and T2A-peptide cis-linked green fluorescent protein (GFP) marker. (b-g) HEK293T cells were transfected with 1 μg of plasmid co-expressing either empty vector (vector only) or indicated mIFIH1 constructs for the times indicated. (b) Whole cell lysates were resolved by SDS-PAGE and immune-blotted with indicated antibodies. Blots were cropped for images. (c) Histograms of time course of GFP expression for samples shown in (b). Data are representative of four biological replicates. (d) Quantitative RT-PCR was performed for IFNB1 mRNA expression and results were normalized to the reference gene, HPRT, and subsequently normalized to mIFIH1NR at 15 hrs. Data are displayed as a mean of four biological replicates. (e-f) Transfected cells were stimulated with 1.25 μg of poly(I:C) at 15 hours post-transfection and quantitative RT-PCR was performed for IFNB1 mRNA expression and normalized as in (d). (e) Representative data from one of 6 experiments. (f) Data displayed as a mean ratio of R vs. NR IFNB1 mRNA from six biological replicates. (g) Cells were infected with EMCV at 15 hours post-transfection. Quantitative RT-PCR was performed for IFNB1 mRNA expression and normalized as in (d). Data are the mean of four biological replicates. (d-f) Statistical analysis was performed using a one-way ANOVA (d,g) or one-tail student T test (f). Each data point represents a biological replicate. Error bars represent ± SEM. P=protective (IFIH1V923). *p<0.05, **p<0.01.
We next examined IFIH1 activity in response to viral challenge. Following transfection with alternative mIFIH1 constructs, HEK293T cells were infected with encephalomyocarditis virus (EMCV), a picornavirus specifically recognized by IFIH1.30 Cells expressing the mIFIH1R construct exhibited a 2-3 fold increase in viral-triggered IFNB1 mRNA relative to cells expressing the mIFIH1NR construct at 9 hours post-infection (Fig. 2g); in contrast, mIFIH1P exhibited no response to viral challenge. In a second test, cells expressing mIFIH1R exhibited increased IFN-I concomitant with reduced viral load following infection with West Nile virus (WNV; Supplementary Fig. 2e-g) which also triggers IFN-I induction in part through IFIH16. These combined results demonstrate that mIFIH1R promotes heightened basal and ligand-triggered signaling and enhanced responsiveness against RNA viruses that express IFIH1 ligands during their replication cycle.
IFIH1R mice exhibit increased IFN-I and viral protection
To gain a better understanding of the functional role of IFIH1R in both autoimmune disease and viral control, we generated a novel murine strain that has an Ifih1 allele that precisely mimics the human IFIH1R (IFIH1R843/T946) haplotype. We utilized homologous recombination to generate founder animals in the non-autoimmune prone C57Bl/6 genetic background (Supplementary Fig. 3a-b). Gene targeting achieved the variant-specific coding change (A946T) in exon 15 of Ifih1 without impacting Ifih1 mRNA (Supplementary Fig. 3c). When we intercrossed heterozygous Ifih1NR/R mice, we observed partial embryonic lethality based upon a substantial reduction in the expected number of homozygous Ifih1R/R offspring (Fig. 3a). While newborn heterozygous and homozygous IFIH1 risk animals appeared healthy, exhibited normal weight gain and a normal frequency of lymphoid and myeloid subsets (data not shown), Ifih1NR/R and Ifih1R/R mice exhibited a trend for increased splenocyte numbers in comparison with littermate controls at 2-12 months of age (Supplementary Fig. 3d). Similar to our findings in human PBMCs ex vivo, splenocytes from mice with the H1 haplotype allele (Ifih1R/R) had basally higher mRNA levels of Ifnb1 and multiple ISGs including Mx1, Ifit1 and Isg15 (Fig. 3b-c).
Figure 3. Ifih1R variant knock-in mice exhibit increased embryonic loss and enhanced basal IFIH1 activity.
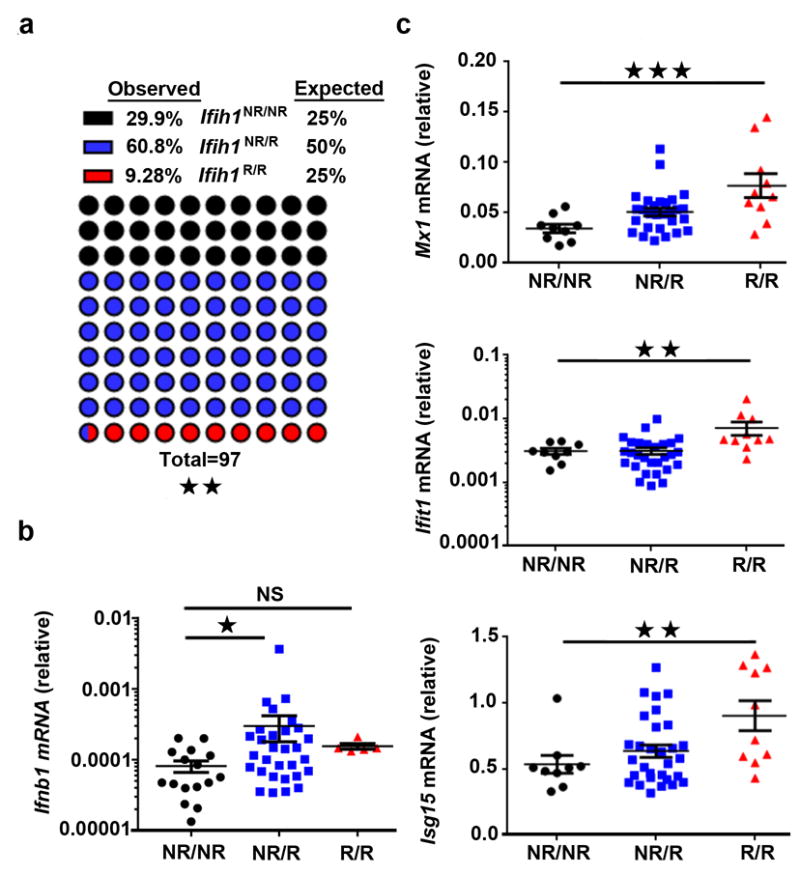
(a) Genotyping results (97 pups) obtained following breeding of heterozygous Ifih1NR/R mice. Significance was determined by chi-square goodness-of-fit. (b-c) Splenocyte mRNA was isolated for quantitative RT-PCR measurement of (b) Ifnb1 mRNA or (c) candidate ISG expression (Mx1, Ifit1, Isg15). Ct values were normalized to Hprt. Each dot represents results from an individual animal with Ifih1NR/NR=16, Ifih1NR/R=30, Ifih1R/R=5 (in b) and Ifih1NR/NR=9, Ifih1NR/R=30, Ifih1R/R=10 (in c). Error bars represent ± SEM and significance was assessed using Kruskal-Wallis test (b) or one-way ANOVA (c). NR/NR=Ifih1NR/NR, NR/R=Ifih1NR/R, R/R=Ifih1R/R. *p<0.05, **p<0.01, ***p<0.001.
In order to test if the H1 haplotype confers an in vivo advantage against viral pathogens, we assessed the ability of IFIH1R mice to control an EMCV challenge. After i.p. injection with EMCV, ~75% of the Ifih1NR/NR mice succumbed to EMCV infection within 8 days. However both heterozygous and homozygous IFIH1R mice were significantly protected from EMCV compared to the non-risk littermates (Fig. 4). Taken together, our findings show that IFIH1R knock-in mice – whether heterozygous or homozygous manifest elevated basal IFIH1 activity, leading to an ISG signature and a modest auto-inflammatory state. In parallel, they have an increased resistance to challenge with an IFIH1-specific, RNA viral pathogen.
Figure 4. Ifih1R variant mice exhibit protection from EMCV challenge.
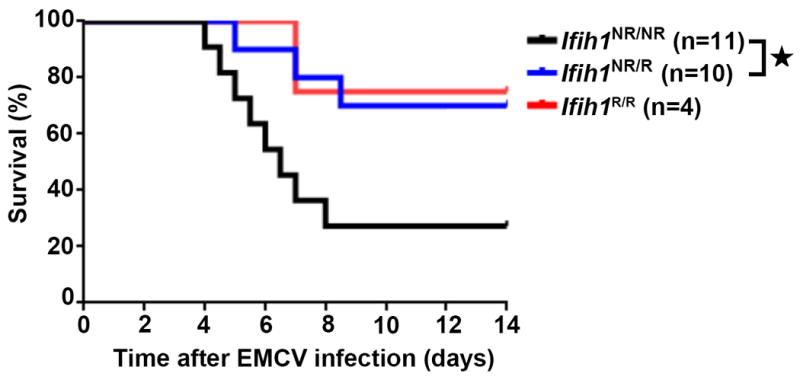
Male mice of indicated genotypes were injected intraperitoneally with 100 pfu of EMCV-K strain on day 0 and evaluated twice daily for clinical symptoms for 14 days. Mice were euthanized at a clinical score of 4. Graph displays combined data from three independent experiments with significance assessed using a Mantel-Cox test. Total numbers of animals: Ifih1NR/NR =11, Ifih1NR/R =10, Ifih1R/R =4. *p<0.05.
IFIH1R promotes disease in murine autoimmune models
We were interested in determining whether IFIH1R has a causal role in the promotion of autoimmune disease. Streptozocin (STZ), a DNA alkylating agent that is selectively toxic to pancreatic β cells, can induce T1D in C57Bl/6 mice. We utilized an STZ dosing regimen that did not affect the blood glucose levels of littermate control Ifih1NR/NR animals. In contrast, homozygous Ifih1R/R mice treated with this regimen had increased incidence of STZ-induced diabetes, while the heterozygous Ifih1NR/R mice showed an intermediate effect (Fig. 5a-b). As human variants do not occur in isolation, we next intercrossed the IFIH1R mice with another murine risk model described by our laboratory consisting of a single nucleotide change in the phosphatase Ptpn22 (Ref.31). This strain, referred to hereafter as PTPN22R, mimics the extensively studied human autoimmune risk variant rs2476601 that leads to an amino-acid substitution in a key interacting domain of the phosphatase. PTPN22R animals on a mixed 129/BL6 background exhibit increased STZ-induced diabetes31. For these experiments, we utilized PTPN22R animals that had been fully backcrossed into the C57/Bl6 background and challenged animals of single or combined genotype with STZ in order to assess potential synergy in T1D development. Ifih1NR/NRPtpn22NR/R animals exhibited a low rate of diabetes development, while Ifih1NR/RPtpn22NR/NR mice and control Ifih1NR/NRPtpn22NR/NR animals failed to develop disease. In contrast, double heterozygous, Ifih1NR/RPtpn22NR/R animals displayed increased incidence and rate of diabetes (Fig. 5c-d). Compound heterozygous Ifih1NR/RPtpn22NR/R mice displayed both early and late stage, focal or focally-extensive, pancreatitis and pancreatic atrophy, not seen in diabetic heterozygous Ifih1NR/NRPtpn22NR/R mice (compare Supplementary Fig. 4b vs 4a respectively). Compound heterozygous animals also exhibited atrophic and sclerotic pancreatic tissue that contained remnants of pancreatic ducts with damaged islets embedded within fibrous tissues (Supplementary Fig. 4b upper panels) or scattered among clusters of fibroblasts and inflammatory cells (Supplementary Fig. 4b lower panels). Together, these data provide the first direct demonstration of an additive effect of two human risk alleles in promoting autoimmune disease in vivo.
Figure 5. Ifih1R variant mice exhibit enhanced triggering of autoimmune disease.
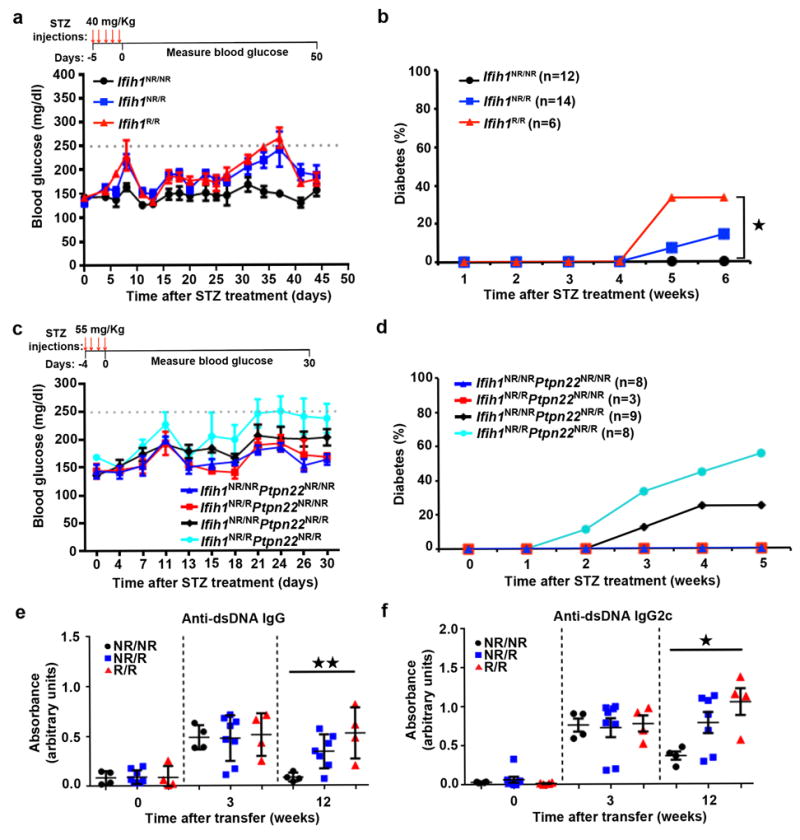
(a-b) Mice were injected with daily with streptozocin (STZ; 40 mg/kg) for 5 days. (a) Representative data from one out of two experiments. Average blood glucose level displayed with error bars representing ± SEM. Day 0 represents start of glucose monitoring. Red arrows indicate STZ injection. (b) Combined data from two experiments with indicated total numbers of animals/genotype. Animals with glucose levels above 250 mg/dl for two consecutive measurements or in 3 of 5 measurements were considered diabetic. (c-d) Ifih1R/R mice were crossed with Ptpn22R mice to generate single or compound heterozygous Ifih1NR/RPtpn22NR/R mice. Mice were injected with STZ (55 mg/kg) for 4 days. (c) Representative data from one experiment. (d) Combined data from two experiments with indicated total numbers of animals/genotype. Disease was determined as in (a). (e-f) 4.7×106 BM12 CD4+ T cells were adoptively transferred into mice of indicated genotypes by retro-orbital injection. ELISAs were performed on serum collected at the indicated times for (e) IgG anti-dsDNA and (f) IgG2c anti-dsDNA autoantibodies showing representative data from one of two experiments. Graphs display optical density (OD) at 450 nm that is normalized to a blank well. Each data point represents an individual mouse. Error bars represent ± SEM. Statistical significance was assessed by Mantel-Cox test (b) or one-way ANOVA (e-f). *p<0.05, **p<0.01.
As IFIH1R is also associated with risk for SLE in humans, we next tested whether the murine allele directly promotes lupus using the BM12 lupus model. The C57Bl/6 derived, BM12 strain contains a three amino acid change in the H2-Ab1b class II allele32. Adoptive transfer of BM12 CD4+ T cells into C57/BL6 recipients initiates autoimmune germinal center responses that generate anti-dsDNA and anti-smRNP autoantibodies (autoAb) within ~3 weeks of cell transfer 33,34. We transferred CD4+ T cells from BM12 mice into heterozygous or homozygous IFIH1R or littermate control mice (Fig. 5e-f; Supplementary Fig. 4c). All strains displayed similar levels of autoAb at 3 weeks. In contrast, both IgG and IgG2c autoAb directed against dsDNA and smRNP remained elevated in homozygous Ifih1R/R animals at 12 weeks whereas levels returned to baseline in control animals. Similar trends were observed in heterozygous Ifih1NR/R mice. Additionally, we observed a trend toward increased spleen size in Ifih1R/R mice (Supplementary Fig. 4d). This sustained, BM12 triggered, autoAb response observed in Ifih1R/R animals may mimic autoAb responses in SLE subjects with an IFN-I signature and is consistent with clinical features of SLE subjects where anti-dsDNA titers were shown to correlate with the IFIH1 rs1990760 risk allele35. Taken together, our findings demonstrate that IFIH1R can facilitate autoimmune disease in response to a triggering event and can function coordinately with additional risk alleles in altering tolerance.
IFIH1R exhibits increased activity toward self-RNA ligands
In addition to viral dsRNA and ssRNA ligands, previous work has demonstrated that IFIH1 may be triggered by dsRNA structures within endogenous RNAs36-39 . We hypothesized that the observed embryonic survival defect (Fig. 3a) and increased basal IFN-I production associated with the IFIH1R allele in our SPF-housed mice (Fig. 3c-d) could be due to IFIH1R having enhanced responsiveness toward self-RNA ligands. We tested this hypothesis in a cell line engineered to limit the processing of self-RNA, dsRNA-specific adenosine deaminase 1 (ADAR1)-null HEK293T cells. ADAR1 performs adenosine-to-inosine RNA editing that is proposed to destabilize duplexes formed from inverted repetitive elements within self-RNAs and thereby prevents IFIH1 from sensing these cytoplasmic RNAs36,37. Consistent with this role, biallelic missense mutations in human ADAR1, like dominant IFIH1 mutations, cause a severe congenital interferonopathy40, and disruption of ADAR1 in HEK293T cells leads to a marked increase in signaling following overexpression of IFIH137. We therefore assessed the impact of the candidate mIFIH1 constructs shown in Fig. 2a in control vs. ADAR1-null 293T cells (Fig. 6a-b). Recapitulating our result above, control HEK293T cells expressing mIFIH1R exhibited increase basal IFNB1 mRNA compared to the cells expressing mIFIH1NR (Fig. 6c, left). However, IFNB1 mRNA was significantly enhanced (~50-fold) in cells ADAR1-null cells expressing mIFIH1NR and these events were further amplified (~100-fold, relative to control 293T cells) in cells expressing mIFIH1R (Fig. 6d,e), consistent with recognition of accumulated self-dsRNA ligands. Taken together, these results indicate that IFIH1R is hypersensitive to RNA self-ligands present in the ADAR1-null cells thereby generating a stronger IFN-I signal.
Figure 6. mIFIH1R mediates increased sensitivity to self-RNA ligands.
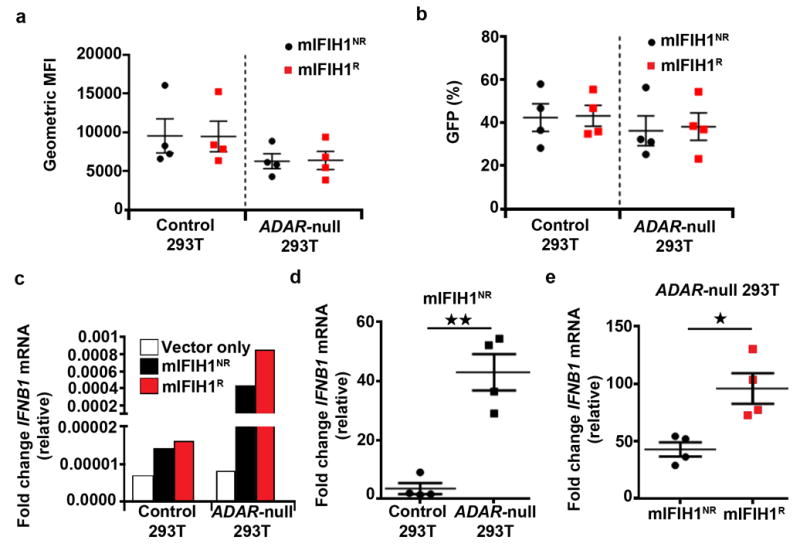
(a-e) Control or ADAR-null HEK293T cells were generated by lenti-CRISPR technology. Cell lines were transfected with 1 μg of mIFIH1 risk or non-risk constructs shown in Fig 2. Cells were analyzed by flow cytometry at 27 hours post-transfection for (a) geometric mean fluorescent intensity (MFI) and (b) percent GFP expression and combined data from four biological replicates are shown. (c-e) Quantitative RT-PCR for IFNB1 mRNA expression in control vs. ADAR-null cells transfected with mIFIH1NR construct. (c) Representative data from one experiment. (d) Combined data from four biological replicates. (e) Relative levels of IFNB1 mRNA expression in mIFIH1NR vs. mIFIH1R transfected ADAR-null cells showing combined data from four biological replicates. Results were normalized by the Livak method as in Fig 2d. Statistical analysis performed using a two-tail student T test. Each data point represents one biological replicate. Error bars represent ± SEM. *p<0.05, **p<0.01.
Discussion
While IFIH1R has been linked to multiple autoimmune disorders, the functional impact of the protein isoform encoded by this haplotype on immune systems has been largely unexplored. Notably, despite strong evidence for autoimmune risk, the frequency of this risk haplotype is relatively high in some human populations suggesting it may also provide a selective advantage in the setting of infectious challenge. In this study, we comprehensively assessed the functional role(s) of IFIH1R both in vitro and in vivo in murine and human PBMCs. Our findings demonstrate that IFIH1R facilitates increased, ligand-dependent, viral sensing while also increasing the risk for autoimmunity. First, we show that PBMCs from healthy donors with IFIH1R exhibit increased ligand-triggered IFN-I production. Strikingly, homozygous risk individuals also showed a significant increase in baseline ISG expression consistent with a basal IFN-I signature. Previous work identified an IFN-I signature in both asymptomatic and affected subjects from a family with a rare gain-of-function mutation in the IFIH1 helicase domain 12,41. Here we expand on these observations showing an IFN-I signature in the majority of healthy subjects defined on the basis of IFIH1R. Notably, previous work implicated rs3747517 leading to an amino-acid change within the helicase domain (R843) in altered protein function28. Our data clearly show that IFIH1T946 plays a dominant role in the enhanced activity driven by the IFIH1R843/T946 risk haplotype. While an effect of the R843 allele on some aspects of IFIH1 function cannot be ruled out, we note that the R843 allele is only associated with risk for T1D when it occurs on the same haplotype as T946, supporting our conclusion that IFIH1T946 is driving the phenotype. Second, using carefully matched expression of risk and non-risk proteins, we show that IFIH1R exhibits heightened basal and ligand-triggered activity, which is independent of potential variant-mediated alterations in IFIH1 protein expression as suggested in earlier studies accounting for the impact of this amino-acid change42,43. Moreover, consistent with a protective effect to an in vivo viral challenge, we show that IFIH1R expressing cells display increased activity in response to two relevant RNA viruses. Third, using a novel murine model, we show that IFIH1R leads to a gene-dose dependent increase in splenic IFN-I mRNA and ISGs. Most notably, the risk variant also mediates in vivo protection against a lethal viral challenge - providing a mechanism whereby selection could act on the variant, increasing its frequency in the population. Fourth, consistent with its association with human autoimmunity, we show that IFIH1R promotes disease in both diabetes and lupus murine models. Importantly, our study also provides the first example wherein two human autoimmune GWAS risk variants are modeled in unison, displaying an additive effect in promoting disease. Finally, we show that homozygous IFIH1T946 expression leads to increased fetal loss and that this phenotype is likely due to an enhanced ability to recognize RNA self-ligands.
In distinction from earlier work, our combined data demonstrate that the IFIH1T946 variant exhibits enhanced ligand-dependent signaling in vivo and in vitro. Consistent with our findings, a recent report found overexpression of human IFIH1T946 promotes increased basal levels of IFN-I compared with the IFIH1A946 14. However, in contrast, they reported this variant was unable to recognize RNA ligands and trigger signaling due to a lack of ATPase activity. Our data show IFIH1T946 recognizes both synthetic dsRNA and viral RNA ligands leading to an increase in IFN-I mRNA as assessed in three different systems in vitro and/or in vivo. The differences between our work and this earlier study may reflect alternative time course and/or reagents. Of note, the requirement for ligand-triggering of IFIH1 activity may reflect the location of missense mutation within IFIH1 with constitutive activation from helicase domain mutations44 vs. CTD mutation (IFIH1T946) that functions to modulate RNA ligand responsiveness. Additional structural modeling will be required to address these possibilities.
Importantly, in addition to viral RNAs, our data also implicate an enhanced response to RNA self-ligands with respect to the biological impact of IFIH1R. Using ADAR-null cells, that generate increased endogenous self-RNAs, we provide evidence that IFIH1R recognizes and displays heightened activity toward self-RNA37. Correspondingly, we observed basal increases in ISGs in both the knock-in mice and in healthy subjects with IFIH1R. While the relative increase in IFN-I production associated with the risk allele is modest, low-level elevation in interferon correlated to disease phenotype. Similarly, IFN-I is frequently too low to be consistently measured leading to use of an IFN signature as biomarker for AGS45. Whether IFIH1R is recognizing different or a greater breadth of ligands remains to be determined. Overall, our data support the hypothesis that IFIH1R leads to an increased IFN state through recognition of self-RNAs and that this may increase risk for autoimmune disease with or without concomitant viral challenge.
Our viral challenge data support the possibility that the IFIH1T946/R843 haplotype may have been subject to positive selection in humans based on its capacity to promote improved response against viral infection. Prior population genetics studies have noted a high degree of nucleotide diversity and strong population differentiation at IFIH1. The action of positive selection on IFIH1 has been proposed, with the SNP rs10930046 (R460H)46. More recently, a study of RIG-I-like receptors reported a second signature of positive selection at rs3747517 (R843H) in African and Asian populations47. Further, haplotypes at IFIH1 encoding His at position 843 and Thr at position 946 were reported to correlate with the resolution of hepatitis C virus (HCV)28. Notably, we found IFIH1H843/T946 was very rare in our data, consistent other recent reports48,49. Thus, at the population level, the impact of the common risk haplotype (IFIH1T946/R843) upon response to viral challenge and autoimmune disease risk is likely to be important; whereas the impact of the rare IFIH1H843/T946 haplotype would be negligible. Overall, our functional and population data suggests that non-synonymous substitutions at IFIH1 can play the role of a double-edged sword, protecting and promoting illnesses at the same time and that this may affect their frequency in the population.
Interestingly, we identified a partial embryonic survival defect in the homozygous Ifih1R/R mice, raising the possibility of a similar impact on human fertility. However, IFIH1R allele frequency suggests that this is unlikely. Furthermore, increased fetal loss has not been reported for more severe gain-of-function variants leading to early-onset interferonopathies. In contrast, embryonic lethality has been described in several murine strains with alterations in the IFIH1 signaling cascade37,50. Thus, we anticipate the threshold for IFN-I to mediate embryonic lethality differs between mice and humans.
In summary, we have identified the functional role of IFIH1T946 risk variant in regulation of a key antiviral sensing pathway where it both defends against viral infection and bolsters response toward self-RNA. Our data provide a compelling demonstration of the power of a combinatorial approach that makes use of studies of healthy human subjects in association with murine modeling to assess the impact of candidate GWAS variants and functionally model combinatorial impacts of additional variants, an important consideration as disease-associated genetic variants are unlikely to function in isolation. Our combined work provides new insight into functional mechanism(s) in the setting of endogenous gene expression and candidate disease initiators. Application of this approach to other risk or protective variants is likely to promote a more rapid advance in understanding of human disease mechanism.
Methods
Association analyses
Genotypes for IFIH1 SNPs were obtained from the Type 1 Diabetes Genetics Consortium23. The Family-Based Association Test (FBAT) program (version 2.0.4) was used for single-marker association tests and haplotype analyses, as well as for checking Mendelian inconsistency in genotyping51 Minor allele frequencies were estimated using PLINK (v.1.07)52.
Plasmids and cultured cells
Murine IFIH1 cDNA corresponding to nucleotides 326-3403 of the National Center for Biotechnology Information Genbank accession number NM_027835.3. This was cloned into pRRL.MND.T2A.green fluorescent protein (GFP), a plasmid backbone for lentivirus that also effectively expresses proteins when transiently transfected. Point mutations for mIFIH1P and mIFIH1R were made to correspond to rs35667974 and rs1990760 using Quikchange II XL Site-Directed Mutagenesis Kit (Agilent Technologies).
ADAR-null- and untargeted- HEK293T cells37 were obtained from D. Stetson.
Cryopreserved PBMC were obtained from 85 participants in the Benaroya Research Institute (BRI) Healthy Control Registry and Biorepository. Subjects were selected based on IFIH1 genotype and the absence of autoimmune disease or any family history of autoimmunity. 38 individuals were homozygous for the IFIH1A946 non-risk (NR) variant, and 47 were homozygous for the IFIH1T946 risk (R) variant. Subjects were age matched (mean age: NR group, 35.3 ± 18.8 years; R group, 35.2 ± 19.2 years) and gender matched as far as possible (NR group, 22 males and 16 females; R group, 25 males and 22 females). Genotyping for the IFIH1 SNPs rs1990760 C/T (Ala946Thr) and rs3747517 G/A (His843Arg) was performed using major groove binding Eclipse genotyping probes (ELITech Group, Inc., Logan UT). All genotyping assays were validated using HapMap DNAs of known genotype. Samples were blinded for analysis but were provided in a manner that guaranteed that samples from both groups would be tested on each assay day. DNA from control subjects was genotyped for the IFIH1 SNPs rs1990760 C/T (Ala946Thr) and rs3747517 G/A (His843Arg) using major groove binding Eclipse genotyping probes (ELITech Group, Inc., Logan UT). All genotyping assays were validated using HapMap DNAs of known genotype. Informed consent was obtained from all participants and the research protocols were approved by the Institutional Review Board at BRI.
All cell lines and primary cell were cultured at 37°C in a humidified environment with 5% CO2. HEK293T cells were cultured in DMEM, 10% fetal calf serum (FCS), and 1% Glutamax (Gibco). PBMCs were cultured in RPMI with 10% FCS, 1%Glutamax. HEK293T cells were a gift from St. Jude’s Research institute and are annually tested for mycroplasm.
Cell transfection and stimulation with IFIH1 ligands
1.5 × 106 HEK 293T cells in 1 ml media transient transfection with 1 μg plasmid DNA using Fugene 6 (Promega) (1 μg DNA to 3 μl of Fugene). After 15 hours, cells were either transfected with (1.25 μg) high molecular weight poly(I:C) (InvivoGen) using TransIT-mRNA Transfection kit (Mirus Bio) in serum-free DMEM following manufacture instructions or infected virus as previously described6. Briefly, EMCV-K strain at 10 MOI or WNV-TX02 at 5 MOI was added to serum-free RPMI for 1 hour, then virus- containing media was removed and cells were cultured for indicated time points using standard conditions. Cells were collected and RNA or protein extracted (see below).
For primary human PBMCs, stimulations were preformed in 96-well round bottom tissue culture plates (2 × 106 cells in media/well). HEK293T cells were transfected as described above. For poly(I:C) stimulations, cells were either untreated (media alone) or were transfected with 1 μg poly(I:C) using the TransIT-mRNA transfection kit (Mirus Bio) according to the manufacturer’s instruction, followed by standard culturing. 24 hours post-stimulation cells were collected and RNA extracted (see below).
Quantitative RT-PCR
RNA was extracted from cell pellets or murine splenocytes (see below) using the RNeasy Mini Kit (Qiagen) and the manufacturer’s instructions. For murine splenocytes and HEK293T cells, cDNA was made from RNA using Maxima First Strand cDNA Synthesis Kit (ThermoFIsher Scientific). Real-Time PCR was preformed on the cDNA using iTaq Universal Syber Green Supermix (Bio-Rad) using the BioRad C1000 Thermal Cycler (Supplementary Table 2). For Primary PBMCs, cDNA was generated using Superscript III Reverse transcriptase and Oligo(dT)20 Primer (Invitrogen). TaqMan multiplex real-time PCR was performed for expression of human IFN-β and IFIH1 transcripts using FAM-labeled probes (Thermo-Fisher) Hs01077958_s1 (IFNB1) and Hs0170332_m1 (IFIH1). Transcript levels of RNA polymerase II were determined as endogenous control using VIC-labeled probe Hs001722187_m1. PCR reactions were run on a 7500 Fast Real-Time PCR system (Applied Biosystems).
Fluidigm assay
Total RNA from human PBMCs were converted to cDNA (High Capacity RT kit, Life Technologies) and preamplified by limited PCR (PreAmp Master Mix, Life Technologies) with a pool of 96 primers (DeltaGene, Fluidigm). We looked at 38 ISGs in this pool of 96 primers53-56. Preamplified cDNAs were treated with ExonucleaseI (New England Biolabs) and diluted to remove unused primers and dNTPs and loaded onto a 96.96 Dynamic Array IFC for real-time PCR analysis on a BioMarkHD (Fluidigm). Analysis was performed, using Fluidigm’s Real Time PCR Analysis Software to determine Ct values, using linear (derivative) baseline correction and auto-detected, assay-specific threshold determination. Ct values were standardized to 3 housekeeping genes (DOCK2, EEF1A1, and FAM105B) that showed high correlation across samples to correct for sample input differences57. Four samples from same donor and same draw were run on both Fluidigm assays. The mean of the Ct values were used for analysis. Human sample size was based on availability of IFIH1NR/NR and IFIH1R/R subjects.
Immunoblotting, and Antibodies
HEK293T cells were lysed using RIPA buffer (50 mM Tris HCL, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Igepal, 1 mM EDTA, and 0.1% sodium dodecyl sulfate (SDS)) with 1 μM okadaic acid and 1 μM phosphatase inhibitor mixture II (both EMD-Millipore) and 10 μM protease inhibitor cocktail (P-8340, Sigma-Aldrich). Whole-cell lysates were separated on a 10% SDS-polyacrylamide gel electrophoresis gel, transferred to Immobilon x (EMD-Millipore) and immunoblotted by the following primary antibodies: rabbit anti-MDA5 (Cell Signaling-5321), mouse anti-β-actin (Cell signaling-3700), and goat anti-WNV NS3 (R&D Systems-AF2907). Anti-goat, mouse, or rabbit IRDye (LI-COR Biosciences) were used as secondary antibodies. Membranes were imaged by the Odyssey Infrared Imager (LI-COR Biosciences) and quantified by ImageJ software. Blots were cropped for figures (Supplementary Fig. 5a-b)
Mice
A construct designed to generate a T946A mutation in exon 15 of Ifih1 by homologous recombination in C57Bl/6j mice was generated and injected by Biocytogen as follows. The construct contained a neomycin selection cassette between two FRT sequences that would be inserted in intron 16 after successful recombination. LoxP sites were also added between intron 15 and 16 to allow for lineage specific deletion that were not used. The construct was introduced into C57Bl/6j embryonic stem (ES) cells and clones were obtained by limited dilution and G418 selection. Clones with successful integration of the knock-in template into the locus were confirmed by southern blot and PCR of gDNA. Successfully targeted clones were injected into BALB/c blastocysts and subsequently transferred into pseudopregnant females. One clone gave rise to a line with germline transmission of the allele. The mutation was confirmed by sequencing exon 15 (Supplemental Figure 2) and PCR was used to genotype all litters (using the following primers: 5’ AGAATCTCATTTCTTGTGTCGGC-3’ and 5’-GGTCTTCTTTGATGTCTGCTGATG-3’). BM12 mice (B6(C)-H2-Ab1bm12/KhEgJ) were purchased from Jackson Laboratory. All mice used in the BM12 transfer experiments were females at 11-20 weeks old. Ptpn22R mice have been previously described31. No statistical methods were used to determine sample size. No randomization or blinding was done during experimental setup. All strains were maintained in a specific pathogen-free facility, and studies were performed in accordance with procedures approved by the Institutional Animal Care and Use Committees of Seattle Children’s Research Institute or the University of Washington.
Analyses of mouse tissues
Murine serum was tested by ELISA for reactivity to calf thymus dsDNA (Sigma-Aldrich), and sm-RNP (ATR1-10; Arotech Diagnostics Limited) which has previously been described58.
To obtain RNA samples for quantitative PCR, murine spleens were digested with collagenase Type 4 (Worthington Biochemical), followed by calcium chelation, RBC lysis and made into single cell suspensions.
Insulin was detected by immunohistochemistry in paraffin embedded pancreas tissue sections using a Leica Bond MAX Automated Immunostainer (Leica Microsystems) and guinea pig anti-insulin (Dako-A0564) primary antibody with rabbit anti-guinea pig (Abcam) and goat anti-rabbit poly-HRP (Leica Microsystems) secondary antibodies. Visualization was with DAB-Bond Polymer Refine (Leica Microsystems).
In vivo infection
Mice were infected with 100 pfu of EMCV K strain diluted in phosphate buffered saline (PBS) in 200 μL total volume i.p. Recipients were monitored twice daily for 14 days and scored for clinical symptoms as follows: 1 = lethargic; 2 = hunched and/or scruffy coat; 3 = weakness in 1 limb; 4-weakness in >1 limb; 5-moribund; mice were euthanized at a clinical score of 4 or more. Recipients were all males and 6-22 weeks old in three independent experiments.
STZ-induced diabetes
Mice received daily i.p. injections of STZ (Sigma Aldrich) at a dose of 40 mg/Kg for 5 consecutive days, or 55 mg/Kg for 4 consecutive days. Following the final injection, peripheral blood glucose levels were measured 2-3 times per week throughout the experiment using a Contour Glucose Meter (Bayer). Mice with blood glucose levels above 250 mg/dL for two consecutive reads, or three out of five consecutive reads were considered diabetic. Any mouse that exhibited blood glucose levels >500 mg/dL were euthanized. All mice were 7-12 weeks old.
Statistical Analysis
All statistical analysis were performed using graphPad Prism version 6.01 unless specified above. All specific statistical tests and P-values are indicated in the relevant figures. To assess statistical significance between two groups with not normally distributed data sets, the Mann Whitney U test was used. For normally distributed data sets, the Student’s t-test and the Welch’s t-test (for non-equal standard deviations were used). When three groups were analyzed, we used either a one-way ANOVA or Kruskal-Wallis test.
Data Availability
The Data that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
Acknowledgments
The authors would like to thank: K. Sommer for laboratory management and assistance with manuscript editing; the University of Washington Histology and Imaging Core and Benaroya Clinical Core; M. Kinsman for technical assistance with RNA preparation and E. Whalen and V. Gersuk for assistance with Fluidigm assays. This work was supported by grants from the NIH: DP3-DK097672 (to J.H.B.), DP3-DK111802 (to D.J.R), R01-AI084914 (to D.B.S.), R01-AI104002, R01-AI060389, R01-AI127463, (to M.G.), U19-AI083019 (to M.G.), U19-AI100625 (to M.G.), R01-DK106718 (to P.C.), T32-AR007108 (to J.A.G.), T32-GM007270 (to K.P.) and from the Juvenile Diabetes Research Foundation 3-APF-2016-177-A-N (to Y.G.). Additional support was provided by the Children’s Guild Association Endowed Chair in Pediatric Immunology and the Benaroya Family Gift Fund (to D.J.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions
J.A.G., C.H., A.S., and Y.G.: designed and performed experiments, analyzed data and wrote and/or edited the manuscript. J.E: designed and performed experiments and analyzed data. X.D., E.A, T.A., C.C., S.K., K.P., K.C., and M.O.: developed required models, strains or reagents and/or performed experiments. D.L., D.B.S., R.G.J, P.C., M.G.: analyzed data and edited the manuscript. J.H.B.: designed and interpreted human subject studies. D.J.R.: conceived and supervised the study, interpreted data and edited the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Wandstrat A, Wakeland E. The genetics of complex autoimmune diseases: non-MHC susceptibility genes. Nat Immunol. 2001;2:802–809. doi: 10.1038/ni0901-802. [DOI] [PubMed] [Google Scholar]
- 2.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira RC, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes. 2014;63:2538–2550. doi: 10.2337/db13-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steed AL, Stappenbeck TS. Role of viruses and bacteria-virus interactions in autoimmunity. Curr Opin Immunol. 2014;31:102–107. doi: 10.1016/j.coi.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Münz C, Lünemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol. 2013;87:11416–11425. doi: 10.1128/JVI.01488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loo Y-M, Gale M. Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.del Toro Duany Y, Wu B, Hur S. MDA5-filament, dynamics and disease. Curr Opin Virol. 2015;12:20–25. doi: 10.1016/j.coviro.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung DW, Amarasinghe GK. Structural insights into RNA recognition and activation of RIG-I-like receptors. Curr Opin Struct Biol. 2012;22:297–303. doi: 10.1016/j.sbi.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato H, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pichlmair A, et al. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rice GI, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Eyck L, et al. Brief Report: IFIH1 Mutation Causes Systemic Lupus Erythematosus With Selective IgA Deficiency. Arthritis Rheumatol. 2015;67:1592–1597. doi: 10.1002/art.39110. [DOI] [PubMed] [Google Scholar]
- 14.Funabiki M, et al. Autoimmune Disorders Associated with Gain of Function of the Intracellular Sensor MDA5. Immunity. 2014 doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 15.Smyth DJ, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 16.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enevold C, et al. Multiple sclerosis and polymorphisms of innate pattern recognition receptors TLR1-10, NOD1-2, DDX58, and IFIH1. J Neuroimmunol. 2009;212:125–131. doi: 10.1016/j.jneuroim.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Cunninghame Graham DS, et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 2011;7:e1002341. doi: 10.1371/journal.pgen.1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, et al. Carriers of rare missense variants in IFIH1 are protected from psoriasis. J Invest Dermatol. 2010;130:2768–2772. doi: 10.1038/jid.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martínez A, et al. Association of the IFIH1-GCA-KCNH7 chromosomal region with rheumatoid arthritis. Ann Rheum Dis. 2008;67:137–138. doi: 10.1136/ard.2007.073213. [DOI] [PubMed] [Google Scholar]
- 21.Jin Y, Andersen GHL, Santorico SA, Spritz RA. Multiple Functional Variants of IFIH1, a Gene Involved in Triggering Innate Immune Responses, Protect against Vitiligo. J Invest Dermatol. 2016 doi: 10.1016/j.jid.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 22.Lek M, Karczewski K, Minikel E, Samocha K, Banks E. Analysis of protein-coding genetic variation in 60,706 humans. BioRxiv. 2016 doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Onengut-Gumuscu S, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet. 2015;47:381–386. doi: 10.1038/ng.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Concannon P, et al. Genome-wide scan for linkage to type 1 diabetes in 2,496 multiplex families from the Type 1 Diabetes Genetics Consortium. Diabetes. 2009;58:1018–1022. doi: 10.2337/db08-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, et al. Contribution of IKBKE and IFIH1 gene variants to SLE susceptibility. Genes Immun. 2013;14:217–222. doi: 10.1038/gene.2013.9. [DOI] [PubMed] [Google Scholar]
- 26.Liu S, et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet. 2009;18:358–365. doi: 10.1093/hmg/ddn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen G, et al. Genetic variants in IFIH1 play opposite roles in the pathogenesis of psoriasis and chronic periodontitis. Int J Immunogenet. 2012;39:137–143. doi: 10.1111/j.1744-313X.2011.01068.x. [DOI] [PubMed] [Google Scholar]
- 28.Hoffmann FS, et al. Polymorphisms in melanoma differentiation-associated gene 5 link protein function to clearance of hepatitis C virus. Hepatology. 2015;61:460–470. doi: 10.1002/hep.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chistiakov DA, Voronova NV, Savost’Anov KV, Turakulov RI. Loss-of-function mutations E6 27X and I923V of IFIH1 are associated with lower poly(I:C)-induced interferon-β production in peripheral blood mononuclear cells of type 1 diabetes patients. Hum Immunol. 2010;71:1128–1134. doi: 10.1016/j.humimm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 31.Dai X, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest. 2013;123:2024–2036. doi: 10.1172/JCI66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry D, Sang A, Yin Y, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol. 2011;2011:271694. doi: 10.1155/2011/271694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klarquist J, Janssen EM. The bm12 Inducible Model of Systemic Lupus Erythematosus (SLE) in C57BL/6 Mice. J Vis Exp. 2015:e53319. doi: 10.3791/53319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Z, Chen F, Madaio MP, Cohen PL, Eisenberg RA. Modulation of autoimmunity by TLR9 in the chronic graft-vs-host model of systemic lupus erythematosus. J Immunol. 2006;177:7444–7450. doi: 10.4049/jimmunol.177.10.7444. [DOI] [PubMed] [Google Scholar]
- 35.Robinson T, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J Immunol. 2011;187:1298–1303. doi: 10.4049/jimmunol.1100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liddicoat BJ, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pestal K, et al. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity. 2015;43:933–944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heraud-Farlow JE, Walkley CR. The role of RNA editing by ADAR1 in prevention of innate immune sensing of self-RNA. J Mol Med. 2016;94:1095–1102. doi: 10.1007/s00109-016-1416-1. [DOI] [PubMed] [Google Scholar]
- 39.Schlee M, Hartmann G. Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol. 2016;16:566–580. doi: 10.1038/nri.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rice GI, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niewold TB, Hua J, Lehman TJA, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Downes K, et al. Reduced expression of IFIH1 is protective for type 1 diabetes. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0012646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lincez PJ, Shanina I, Horwitz MS. Reduced expression of the MDA5 Gene IFIH1 prevents autoimmune diabetes. Diabetes. 2015;64:2184–2193. doi: 10.2337/db14-1223. [DOI] [PubMed] [Google Scholar]
- 44.Buers I, Nitschke Y, Rutsch F. Novel interferonopathies associated with mutations in RIG-I like receptors. Cytokine Growth Factor Rev. 2016;29:101–107. doi: 10.1016/j.cytogfr.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 45.Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med. 2016;213:2527–2538. doi: 10.1084/jem.20161596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fumagalli M, et al. Population genetics of IFIH1: ancient population structure, local selection, and implications for susceptibility to type 1 diabetes. Mol Biol Evol. 2010;27:2555–2566. doi: 10.1093/molbev/msq141. [DOI] [PubMed] [Google Scholar]
- 47.Vasseur E, et al. The selective footprints of viral pressures at the human RIG-I-like receptor family. Hum Mol Genet. 2011;20:4462–4474. doi: 10.1093/hmg/ddr377. [DOI] [PubMed] [Google Scholar]
- 48.Vergara C, Thio CL, Thomas D, Duggal P. Polymorphisms in melanoma differentiation-associated gene 5 are not associated with clearance of hepatitis C virus in a European American population. Hepatology. 2016;63:1061–1062. doi: 10.1002/hep.27954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedergnana V, et al. Refined association of melanoma differentiation-associated gene 5 variants with spontaneous hepatitis C virus clearance in Egypt. Hepatology. 2016;63:1059–1061. doi: 10.1002/hep.27951. [DOI] [PubMed] [Google Scholar]
- 50.Hartner JC, et al. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- 51.Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 52.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma S, et al. Widely divergent transcriptional patterns between SLE patients of different ancestral backgrounds in sorted immune cell populations. J Autoimmun. 2015;60:51–58. doi: 10.1016/j.jaut.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Börnsen L, et al. Endogenous interferon-β-inducible gene expression and interferon-β-treatment are associated with reduced T cell responses to myelin basic protein in multiple sclerosis. PLoS ONE. 2015;10:e0118830. doi: 10.1371/journal.pone.0118830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheler M, et al. Indoleamine 2,3-dioxygenase (IDO): the antagonist of type I interferon-driven skin inflammation? Am J Pathol. 2007;171:1936–1943. doi: 10.2353/ajpath.2007.070281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Indraccolo S, et al. Identification of genes selectively regulated by IFNs in endothelial cells. J Immunol. 2007;178:1122–1135. doi: 10.4049/jimmunol.178.2.1122. [DOI] [PubMed] [Google Scholar]
- 57.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jackson SW, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol. 2014;192:4525–4532. doi: 10.4049/jimmunol.1400098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Data that support the findings of this study are available from the corresponding author upon request.